

Abstract
Aim
The current study was designed to determine whether ET-1 derived from endothelial cells contributes to oxidative stress in the glomerulus of mice subjected to a high salt diet and/or hypoxia.
Methods
C57BL6/J control mice or vascular endothelial cell ET-1 knockout (VEET KO) mice were subjected to three-hour exposure to hypoxia (8% O2) and or 2 weeks of high salt diet (4% NaCl) prior to metabolic cage assessment of renal function and isolation of glomeruli for determination of reactive oxygen species (ROS).
Results
In control mice, hypoxia significantly increased urinary protein excretion during the initial 24 hrs, but only in animals on a high salt diet. Hypoxia increased glomerular ET-1 mRNA expression in control, but not in vascular endothelial cell ET-1 knockout (VEET KO) mice. Under normoxic conditions, mice on a high salt diet had approximately 150% higher glomerular ET-1 mRNA expression compared to a normal salt diet (p<0.05). High salt diet administration significantly increased glomerular ROS production in flox control, but not in glomeruli isolated from VEET KO mice. In C57BL6/J mice, the ETA receptor selective antagonist, ABT-627, significantly attenuated the increase in glomerular ROS production produced by high salt diet. In addition, chronic infusion of C57BL6/J mice with a sub-pressor dose of ET-1 (osmotic pumps) significantly increased levels of glomerular ROS that were prevented by ETA antagonist treatment.
Conclusion
These data suggest that both hypoxia and a high salt diet increases glomerular ROS production via endothelial derived ET-1-ETA receptor activation and provide a potential mechanism for ET-1 induced nephropathy.
Keywords: endothelin, hypoxia, reactive oxygen species, high salt diet, glomerulus, sodium, kidney
Introduction
Two established mechanisms that can increase ET-1 production and activity are hypoxia and high dietary salt. Hypoxia is understood to be an integral part of the pathogenesis of pulmonary hypertension, sickle cell disease, and acute kidney injury among others. ET-1 is produced in cultured endothelial cells in response to hypoxia (Kourembanas et al., 1991), while plasma levels of ET-1 are known to be elevated after a hypoxic episode (Li et al., 1994). Experimental ischemia induced by renal artery clamping is reported to stimulate ET-1 predominantly in peritubular capillaries as well as renal tubular cells (Wilhelm et al., 1999). Accumulating evidence over the past 20 years has also indicated that ET-1 contributes to post-ischemic renal injury (Zager et al., 2013), although the etiology of ET-1 during hypoxia in the kidney is not well defined. Furthermore, whether hypoxia and/or high salt diet have an influence on glomerular ET-1 activity is not known.
Because of the profound potential for vasoconstriction, many investigators have been surprised to learn that the physiological role of ET-1, especially within the kidney, is to function as a pro-natriuretic factor (Kohan et al., 2011). High salt intake increases ET-1 production in both renal tubular cells as well as within vascular elements (Kohan and Padilla, 1993, Pollock and Pollock, 2001). While increases in ET-1 may be important for facilitating the excretion of excess salt in the diet, it could also increase the potential sensitivity to additional stimuli such as hypoxia. The renal response to hypoxia in a setting of high dietary salt intake has not been investigated.
Increasing evidence has accumulated in recent years to suggest that a high salt diet increases oxidative stress (Williams et al., 2004). It is not clear whether this is due to the associated increases in ET-1. ET-1 activates NADPH oxidase in isolated arterial tissue and increases the production of reactive oxygen species (ROS) in arterial smooth muscle cells (Loomis et al., 2005, Wedgwood et al., 2001). ET-1 also stimulates superoxide production by the same pathway in mineralocorticoid-induced hypertensive rats independent of blood pressure (Callera et al., 2003).
Given the prominent role of ET-1 in response to high dietary salt intake and the potential for high dietary salt to exacerbate many cardiovascular disorders, we sought to test the hypothesis that hypoxia effects on the kidney would be more evident under conditions of high salt intake when the ET-1 system is most active and that ET-1 stimulates glomerular ROS formation in the mouse.
Methods
Animals
Studies used 12-wk old male C57BL6/J mice purchased from Jackson Laboratories. Where indicated, experiments also used 12-wk old male vascular endothelial cell specific ET-1 knockout (VEETKO) mice [ET-1f/f;Tie2-cre(+)] and floxed controls [ET-1f/f;Tie2-Cre(-)] obtained from a colony established with breeders provided by Masashi Yanagisawa (Kisanuki et al., 2010). The Georgia Regents University Institutional Animal Care and Use Committee in accordance with the National Research Council Guide for the Care and Use of Laboratory Animals approved all experiments and procedures.
Treatments
Mice were maintained on either a 4% NaCl diet (high salt) or 0.4% NaCl (normal salt) commercially available diets (Harlan Teklad) given ad libitium for 2 weeks prior to any experimental procedure. Where indicated, the ETA antagonist, ABT-627 (AbbVie, Inc., North Chicago, IL), was administered via drinking water and concentration adjusted daily according to intake to maintain consistent dosing (5mg/kg/day) for at least 7 days. Chronic ET-1 administration was performed using osmotic mini-pumps (model 1002, Alzet, Cupertino, CA) loaded with either ET-1 (American Peptide Inc.) or saline (0.9% NaCl) and implanted subcutaneously. The concentration was adjusted to deliver ET-1 at a rate of 2pmol/kg/day a dose that previous investigations in our laboratory showed do not produce blood pressure elevations (Saleh et al., 2010).
Hypoxia
Conscious animals in groups of 3-6 were exposed to whole-body hypoxia in a constant-flow chamber. The chamber was perfused with 8% oxygen, 5% carbon dioxide, and 87% nitrogen electronically pre-mixed and delivered at a flow rate of 50 ml/min. Expired chamber air was monitored to ensure the veracity of delivered air. Animals were either exposed to hypoxia for 3 hours beginning in the morning between 8 and 10 AM and then sacrificed immediately following, or exposed to 3 hours of hypoxia then 6 hours of room air (re-oxygenation) and sacrificed.
For metabolic studies, mice were housed in metabolic cages 24 hours before quantitative measurement, allowing mice to adapt to the cage environment. Baseline urine was collected before exposure to hypoxia and for 2 subsequent days after hypoxia. A final 24-hour urine collection was obtained 1-week after hypoxia.
Isolation of glomeruli
Animals were anesthetized with isoflurane (2.5%) followed by sodium methohexital (5mg/kg). Kidneys were removed, de-capsulated, and placed in ice-cold Hanks balanced salt solution (HBSS 5.33mM potassium chloride, 0.44mM potassium phosphate monobasic, 138mM sodium chloride, 4mM sodium bicarbonate, 0.3mM sodium phosphate dibasic, 5.6mM glucose). The cortex was carefully dissected and cut into small slices. Using a spatula, the pieces were first passed through a 200-μm wire sieve to separate glomeruli, and then filtered through a 200-μm cellulose filter. The filtrate was then passed through a smaller 70 μm cellulose filter. Glomeruli were retained on the top of the 70 μM sieve and washed with cold HBSS into a 50 mL vial. The glomerular suspension was then centrifuged (10min, 1000g) and the pellet re-suspended in HBSS. The glomeruli were spun again and re-suspended. Tubular contamination was less than 5% as visually assessed by light microscopy of the final suspension. The pellet was re-suspended in HBSS, snap frozen in liquid nitrogen and stored at -80°C for later analysis.
Glomerular ROS Assay
Fresh glomerular pellets were re-suspended in 1 ml of 1mM L-012, a luminol derivative (Wako Chemicals) in HBSS and aliquoted into three wells on a clear, flat-bottom 96-well plate. Luminescence of L-012 was used as an index of ROS production. Luminescence was measured at 37°C on a FLUOstar Omega plate reader (BMG Labtech, Germany) for baseline recording at 450nm. Phorbol 12-myristate 13-acetate (PMA, LC Labs), a PKC activator, was added to each well at recording cycle 6 to yield a final concentration of 1mM. Luminescence was measured every minute for 25 minutes. Data was quantitated using Graph Pad Prism Software (San Diego, CA) and area under the curve analysis using the first 5 data points as the baseline.
Superoxide Assay
Freshly isolated tissue was rinsed with chilled Krebs-HEPES buffer (NaCl 99mM KCl 4.69 mM, CaCl 3.5mM, MgSO4 1.2mM, NaHCO3 25mM, monopotassium phosphate 1.03mM, D-glucose 5.6mM, HEPES 20mM) and then exposed to 25 μM dihydroethidium (DHE) for 20 min at 37°C in Krebs-HEPES buffer containing 0.1% DMSO. Glomeruli were placed in 300 μL of cold methanol, homogenized, and filtered (0.22 μm). Separation of ethidium, oxyethidium, and DHE was performed using a Hitachi LaChrom Elite 2300 HPLC, (Tokyo, Japan) and C-18 reverse phase column (Nucleosil 250, 4.5 mm; Sigma-Aldrich, St. Louis, MO). Oxyethidium production was measured via fluorescence detection at 580 nm (emission) and 480 nm (excitation). UV absorption at 355 nm was used for the detection of DHE as previously described (Lau et al., 2006). Superoxide quantitation was determined by calculating the area under the oxyethidium curve.
Urinary Albumin and Nephrin Assay
Nephrin and albumin were measured using an indirect competitive ELISA (Exocell, Philadelphia, PA). 10× diluted urine from samples was added in duplicate to a microplate with immobilized nephrin or albumin antigen. Soluble nephrin or albumin antibodies were added in the fluid phase, washed, and a horseradish peroxidase conjugate was used to detect bound nephrin or albumin antibody in a chromogenic reaction. Concentrations in experimental samples are determined from a standard curve provided by the manufacturer.
Urinary Protein Assay
Bradford assay utilizing the absorbance of Coomassie blue dye in the presence of urinary protein was used to determine protein excretion over a 24-hour period. Absorbance was measured at 595nm and compared to a known quantity of protein.
Urinary ET-1 Assay
ET-1 was determined by radioimmunoassay (Peninsula Laboratories Inc., San Carlos, CA). According to the manufacturers' instructions, there was 100% cross reactivity with ET-1, 19% cross reactivity with Big ET-1 and 7% cross reactivity with ET-3.
mRNA quantification
mRNA was isolated from up to 100mg of tissue and homogenized in 1mL of lysis buffer reagent provided in the Pure Link Mini RNA extraction kit (Ambion, Austin, TX) as previously described (Boesen et al., 2008). Following isolation, mRNA concentration was determined using a Nanodrop spectrophotometer (Model 2000, Thermo Scientific, Wilmington, Delaware). cDNA was made using the Biorad cDNA synthesis kit using 1ug of RNA. Finally, real time PCR was carried out using iTaq Universal Probes Mastermix (BioRad) and Taqman primer gene expression assays (Applied Biosystems). Gene expression was quantified using 2ΔΔCT method.
Statistics
Student's t test was used when comparing the means of 2 groups. A two-way ANOVA was used with Bonferroni post hoc tests to compare individual means as appropriate. Results are expressed as means ± SEM, with p<0.05 being considered statistically significant.
Results
High salt exacerbates hypoxia-induced renal dysfunction
Male C57BL6/J mice were fed a high or normal salt diet for two weeks then exposed to 3 hours of hypoxia. Animals treated with a high salt diet had significantly increased urinary protein excretion 24 hours after hypoxia, a response that was abolished by one week of in vivo administration of the ETA receptor antagonist, ABT-627 (Figure 1a). Neither control nor ABT-627 treated mice on a normal salt diet had any changes in proteinuria as a result of hypoxia (Figure 1b). Neither salt status nor administration of ABT-627 had any effect on albumin excretion after exposure to hypoxia (Figure 1c,d). Animals on a high salt diet had significantly elevated levels of urinary nephrin excretion following hypoxia, an effect that was prevented by ETA receptor antagonism while animals on normal salt exhibited no change in nephrin excretion as a result of hypoxic exposure (Figure 2a and 2b). ET-1 excretion in both the control and ABT-627 treated animals on a high salt diet was unchanged by hypoxia exposure (Figure 3).
Figure 1.
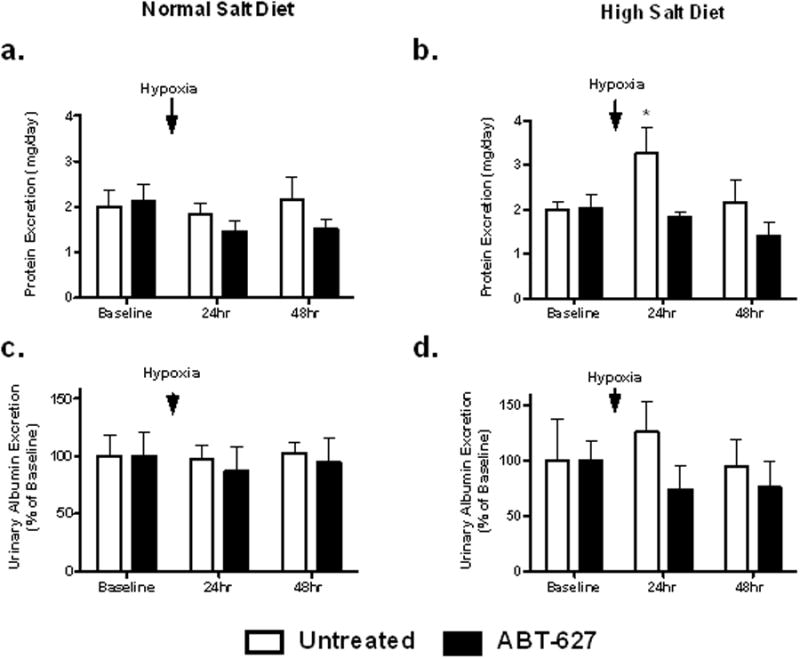
Protein and albumin excretion in C57BL6/J mice maintained on a normal (a,c; 0.4% NaCl) or high (b,d;4% NaCl) salt diet; 24-hr urine collections were obtained before (baseline) and after a 3-hr exposure to hypoxia. Separate groups of mice on each diet were also treated with the ETA selective receptor antagonist, ABT-627, for the duration of the study. *P<0.05 vs. ABT-627 mice on the same diet at the same time point.
Figure 2.
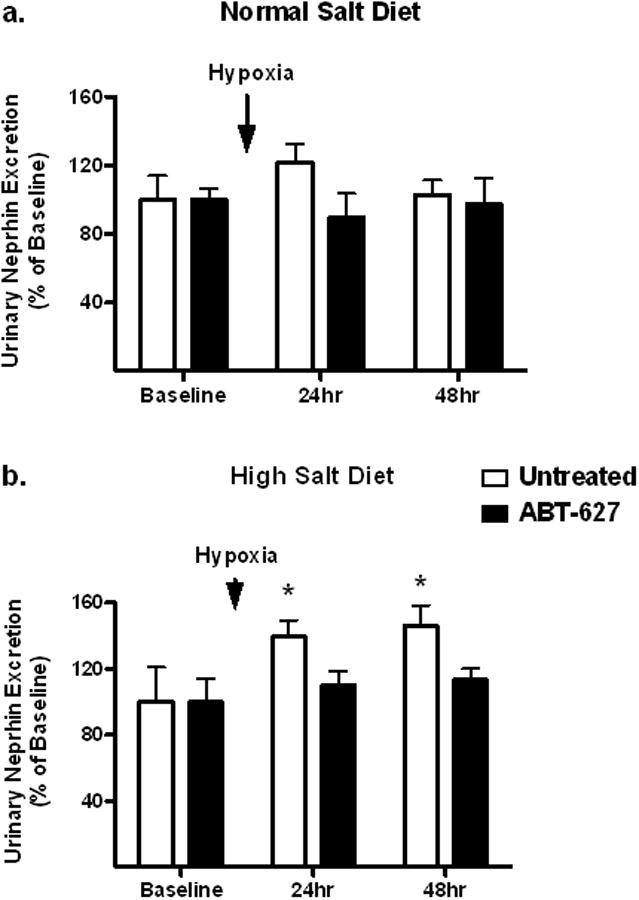
Nephrin excretion in C57BL6/J mice maintained on a normal (a) or high salt (b) diet as measured in 24 hr urine collections before (baseline) and after 3-hr of hypoxia (8% O2). A separate group of mice were also treated with the ETA receptor antagonist, ABT-627. *P<0.05 vs. ABT-627 mice on the same diet at the same time point.
Figure 3.
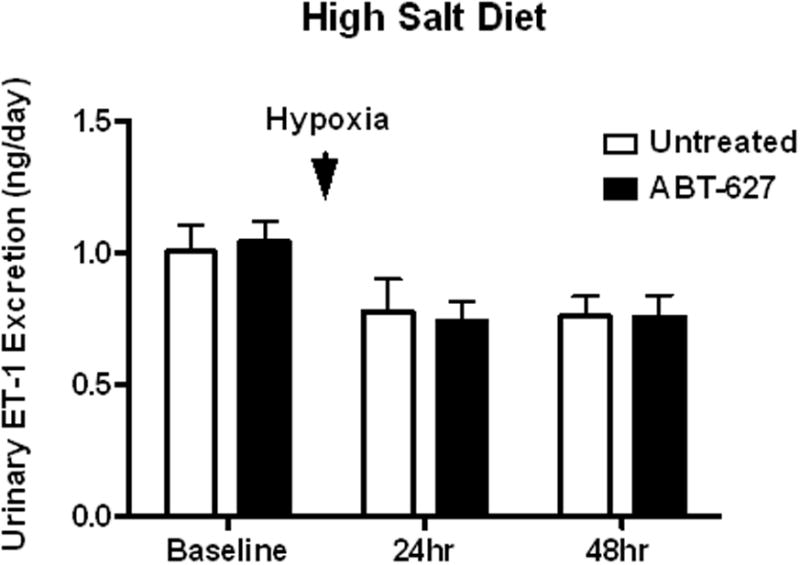
Urinary ET-1 excretion in mice maintained on a high salt diet with or without treatment with BT-627.
Hypoxia stimulates ET-1 and superoxide
We sought to determine whether hypoxia specifically increases glomerular endothelial cell ET-1 production. Vascular endothelial ET-1 knockout (VEET KO) mice and flox controls on a normal salt diet were exposed to whole body hypoxia for 3 hours. Floxed mice had significant (208%) increases in glomerular ET-1 mRNA after hypoxia compared to non-hypoxic mice, whereas VEET KO mice did not exhibit any significant change in glomerular ET-1 as a result of hypoxia (Figure 4a). Hypoxia had no effect on ET-1 mRNA expression in the inner or outer medulla of floxed or VEET KO mice (Figure 4b and 4c) consistent with the effect of hypoxia manifested specifically in the glomeruli. As expected, renal medullary ET-1 mRNA expression was significantly lower in VEET KO mice compared to flox controls. Mice exposed to hypoxia exhibited significantly higher levels of superoxide in their cortex compared to the normoxic controls (p<0.01; n=5, Figure 4d).
Figure 4.
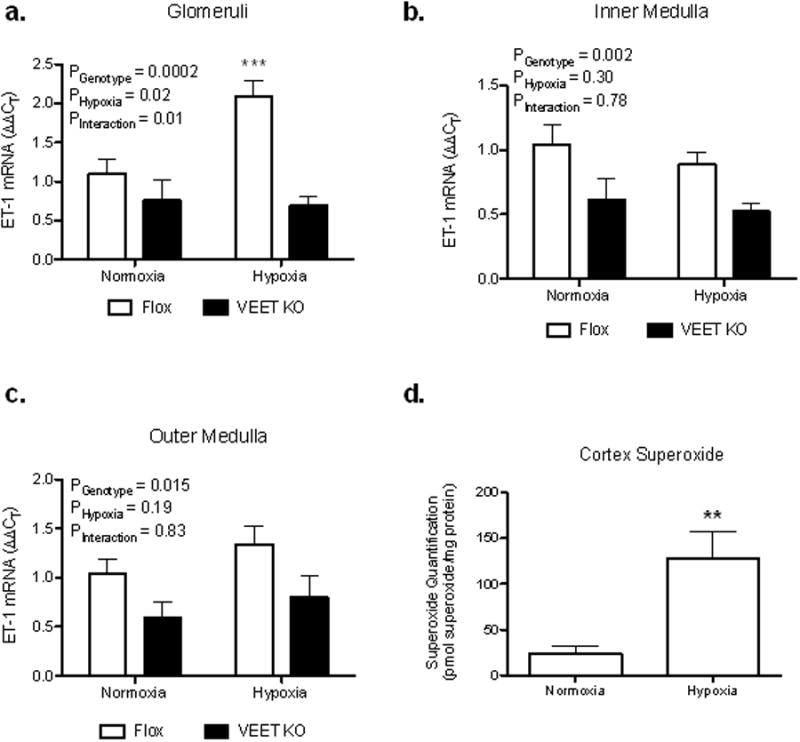
ET-1 mRNA measured in the glomerulus (a), outer (b), and inner (c) medulla of VEET KO or flox control mice. Separate groups of mice were subjected to 3-hr of hypoxia (8% O2) immediately prior to harvesting tissue. *** P < 0.001 vs. VEET KO hypoxia. Renal cortical superoxide levels measured by HPLC from C57BL6/J mice exposed to hypoxia (8% O2) and normoxic controls (d). **P<0.01 vs. normoxia.
ET-1 infusion activates glomerular ROS
To directly examine the influence of chronic increases in ET-1 on glomerular ROS production, C57BL6/J mice were implanted with osmotic pumps containing saline or ET-1 for 2 weeks with a subgroup being treated with ABT-627. Animals chronically infused with ET-1 had significantly higher rates of glomerular ROS production compared to glomeruli from untreated saline-infused mice (Figure 5). Interestingly, administration of the ETA antagonist, ABT-627, abolished increases in glomerular ROS in response to chronic ET-1 infusion indicating that ET-1 stimulates glomerular ROS production via activation of the ETA receptor.
Figure 5.
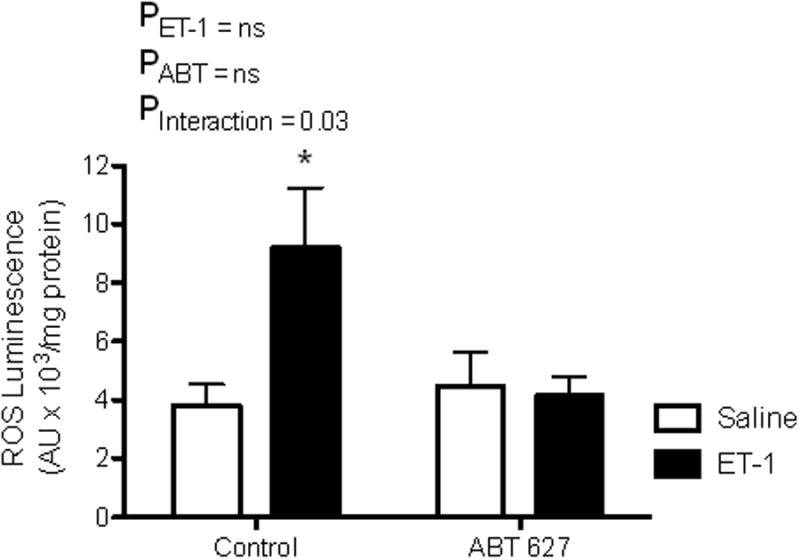
PMA-stimulated ROS production in glomeruli from C57BL6/J mice infused subcutaneously with ET-1 (2pmol/kg/min) or saline for one week (osmotic mini-pump). Half of the animals were also treated with the ETA receptor antagonist, ABT-627 (5mg/kg/day) in drinking water. *P<0.05 vs. saline treated animals not treated with ABT-627.
High salt stimulates ET-1 and superoxide
To test the effect of high dietary salt on cortical ET-1 mRNA expression, control C57BL6/J mice were given a high or normal salt diet for two weeks. High salt fed mice had significantly elevated cortical ET-1 mRNA compared to normal salt controls (149.8±0.17% of control, n=5-6, p<0.05).
VEET KO and flox controls were fed either a normal or high salt diet for one week. Animals with an intact ET-1 system had significantly elevated levels of glomerular ROS production compared to KO mice (Figure 6a). VEET KO and flox mice had similar levels of ROS production on a normal salt diet (Figure 6a).
Figure 6.

PMA-stimulated ROS production in glomeruli of mice maintained for 2 wks. on a normal (0.4% NaCl) or high (4% NaCl) diet. Results in panel a are from VEET KO and Flox control mice while results in panel b are from C57BL6/J mice treated with our without the ETA receptor antagonist, ABT-627. *P<0.05 vs. control mice on high salt.
A separate series of C57BL6/J mice were placed on either a normal or high salt diet with half of the animals being treated with the ETA antagonist for one week prior to assessment of glomerular ROS production. Untreated animals on a high salt diet had significantly increased levels of glomerular ROS production compared to normal salt controls (Figure 6b). High salt animals treated with ABT-627 had a blunted increase in glomerular ROS production (Figure 6b).
To test the etiology of ROS production, we incubated isolated glomeruli with 100 μM apocynin, a NADPH oxidase inhibitor. Apocynin completely prevented ROS production to sub-control levels in normal and high salt treated animals (1.0±0.3 and 1.3±0.4 × 104 ALU/mg protein, n=5). These results indicate that specifically ETA receptor activation increases glomerular NADPH oxidase activity in response to a high salt diet.
Hypoxia and high salt produce increased glomerular ROS through an endothelial ET-1 dependent mechanism
VEET KO and flox mice were fed a high salt diet and evaluated for glomerular ROS formation after hypoxic exposure to determine the effects of endothelial derived ET-1 on glomerular ROS production. Flox mice with intact endothelial ET-1 had significantly elevated glomerular ROS across both the normoxia and hypoxia groups in addition to having enhanced glomerular ROS production in response to hypoxia (Figure 7).
Figure 7.
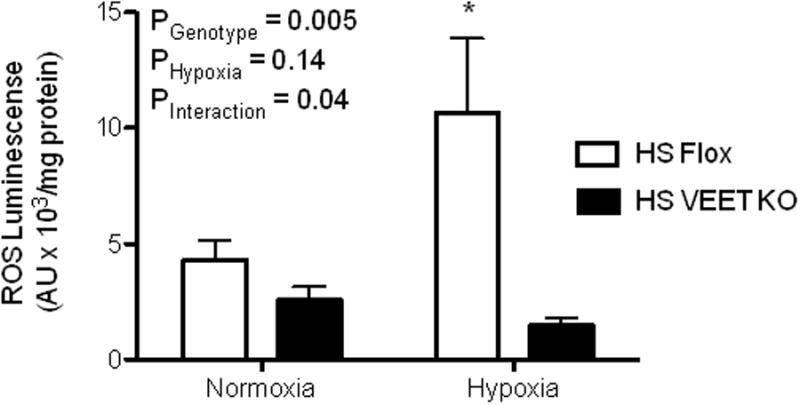
Glomerular ROS production in Flox and VEET KO mice on a high salt diet. A subset of animals were exposed to 3 hours of hypoxia (8% O2) *P<0.05 vs. HS VEET KO
Discussion
The current study provides new evidence that endothelial cell-derived ET-1, via the ETA receptor, contributes to renal dysfunction associated with hypoxia and a high salt diet in the mouse. Acute hypoxia caused proteinuria and nephrinuria in mice on a high salt diet, an effect that was blocked through ETA receptor antagonism. Hypoxia also increased ET-1 mRNA expression, but this effect was limited to the glomerulus and not renal medullary tissue where ET-1 expression is normally high; hypoxia-induced ET-1 expression was absent in animals lacking ET-1 in endothelial cells (VEET KO) suggesting that hypoxia stimulates ET-1 production by the endothelium. Since hypoxia produces changes in many factors aside from just ET-1, we observed that direct chronic administration of exogenous ET-1 increased glomerular ROS through the ETA receptor. It is well established that hypoxia/re-perfusion injury occurs through an ETA dependent mechanism. We extend this information by demonstrating that ROS production was specifically elevated in glomeruli of mice on a high salt diet or exposed to hypoxia, both known stimulators of ET-1 production. Importantly, deletion of ET-1 specifically from endothelial cells or ETA receptor blockade prevented high salt and hypoxia-induced effects on glomerular ROS.
High salt diet stimulates glomerular ET-1 and ROS
Elevated dietary salt increases renal medullary ET-1 to promote natriuresis; however, the role of concomitant increases in ET-1 elsewhere in the kidney as a result of high salt intake are unclear (Kohan et al., 2011). Previous studies showed that increased salt intake leads to enhanced ROS production via NADPH oxidase in the renal cortex of rats (Kitiyakara et al., 2003). In our experiments, one week of high salt diet increased ROS production specifically within glomeruli of control mice. These increases were most likely due in part to stimulation of NADPH oxidase. Apocynin, at relatively low concentrations, completely prevented ROS production in glomeruli from C57BL6/J mice. This effect was also preventable through ETA receptor antagonism in vivo as well as being absent in mice lacking ET-1 gene expression specifically in vascular endothelial cells. These experiments allow us to conclude that ET-1 via the ETA receptor plays a critical role in mediating the stimulation of ROS in glomeruli from mice fed a high salt diet.
Hypoxia stimulates glomerular ET-1 and ROS
We observed that brief exposure to hypoxia increased glomerular ET-1 expression, but not in renal medullary tissue. Studies where ET-1 expression is knocked out of the collecting duct demonstrate substantial reductions in urinary ET-1 excretion (Ahn et al., 2004). Also, chronic ETA and/or ETB receptor blockade has no effect on urinary ET-1 (Pollock and Pollock, 2001). In light of these observations and the fact that we observed increases in glomerular ET-1 expression without a change in urinary ET-1 excretion, we hypothesize that glomerular endothelial cells are more susceptible to hypoxia-induced ET-1 production compared to epithelial sources of ET-1. Our findings are somewhat at odds with a previous study reporting that hypoxia increases in both plasma and urinary ET-1 (Nir et al., 1994). Urinary ET-1 is derived from within the kidney, as filtered ET-1 is irreversibly bound to its receptors and any filtered ET-1 is degraded by neutral endopeptidase activity in the proximal tubule (Janas et al., 1994). Again, our data are consistent with urinary ET-1 being derived from renal tubular sources since hypoxic animals only showed increases in ET-1 within the glomerulus and not within the renal medulla. Consequently, our experiments further indicate that hypoxia-dependent increases ET-1 in endothelial cells are not reflected into the final urine.
Synergism of high salt and hypoxia
Endothelial derived ET-1 was responsible for glomerular ROS production in both the setting of high salt and to a greater extent in high salt with concomitant hypoxic exposure. Furthermore, the podocyte derived protein nephrin (Kestilä et al., 1998) was elevated in urine of mice on a high salt diet at 24 hours post hypoxia, but not in animals on a normal salt diet demonstrating a potential synergistic effect of hypoxia and salt diet on glomerular injury. ET-1 was previously reported to cause nephrin shedding independent of increased glomerular capillary pressure (Collino et al., 2008, Saleh et al., 2010) and thus may contribute to glomerulosclerotic and nephrotic pathological processes (Dhaun et al., 2006).
Somewhat surprising was the observation that endothelial-specific knockout of ET-1 reduced ET-1 mRNA expression at basal levels by only 50% or less in the glomerulus, outer medulla, and inner medulla. VEET KO mice lack ET-1 gene expression in their endothelial cells but retain the ability to produce ET-1 in all other cell types. The maintenance of ET-1 expression in our tissue preparations, especially in the glomeruli, would most likely be ascribed to mesangial cell or podocyte origin although there is the possibility of a small amount of contaminating tubular epithelium in our preparations. In contrast, ET-1 is primarily derived from collecting duct cells in the renal medulla (Kohan et al., 2011) so it was not at all surprising to detect robust expression in these preparations. Nonetheless, our data still indicate that hypoxia-induced changes in glomerular ET-1 expression are of endothelial origin since there were no effects of hypoxia on ET-1 mRNA in the VEET KO animals.
Hypoxia and high salt intake stimulate a wide range of factors in addition to ET-1 and reactive oxygen species. Therefore, we sought to test the effects of ET-1 in the absence of other factors that might be occurring during both hypoxia and high salt intake. Chronic administration of low-dose exogenous ET-1 via osmotic minipumps produced significant increases in glomerular ROS production and this effect was again prevented with in vivo treatment of ABT-627. These results further support the hypothesis that chronically elevated levels of ET-1 contributes to increased ROS production in vivo. Given the strong vasoconstrictor capabilities of ET-1 through the ETA receptor, we sought to give a sub-pressor dose of ET-1 to prevent any alterations in the hemodynamic profile of our experimental mice (Elmarakby et al., 2005, Saleh et al., 2010). Minor hemodynamic differences secondary to treatment with ABT-627 cannot be ruled out; however, the low dose of ET-1 given subcutaneously has been demonstrated to elevate plasma levels of ET-1 without causing concomitant increases in blood pressure. We observed that chronic infusion of ET-1 significantly increased glomerular ROS, thus providing further confirmation that ET-1 is capable of increasing oxidative stress within the glomerulus through an ETA dependent pathway.
Overall, our findings suggest a critical role for endothelial derived ET-1 in circumstances where endogenous production is high (e.g., high salt diet) and hypoxia is prevalent. This could explain reports of ET-1 involvement in glomerular injury in models such as sickle cell nephropathy (Sabaa et al., 2008) or acute ischemic renal injury (Zager et al., 2013). We report that both exogenous and endogenous sources of increased ET-1 lead to increased glomerular ROS formation that were prevented with blockade of the ETA receptor. Furthermore, elevated ROS formation was due to enhanced activity of NADPH oxidase, illuminating a mechanistic role for endothelin via ETA receptors. Combined, these results provide further evidence that endothelin receptor antagonists are an attractive therapeutic modality for future studies in progressive renal disease.
Acknowledgments
This work was supported by a institutional training grant from the National Heart Lung and Blood Institute, T32 HL076146 (J.B.H.) and research grants from the National Heart Lung and Blood Institute, P01 HL09455 and U01 HL117684 (D.M.P. and J.S.P.)
Footnotes
Conflict of Interest: None.
References
- Ahn D, Ge Y, Stricklett PK, Gill P, Taylor D, Hughes AK, Yanagisawa M, Miller L, Nelson RD, Kohan DE. Collecting duct-specific knockout of endothelin-1 causes hypertension and sodium retention. J Clin Invest. 2004;114:504–511. doi: 10.1172/JCI21064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boesen EI, Sasser JM, Saleh MA, Potter WA, Woods M, Warner TD, Pollock JS, Pollock DM. Interleukin-1beta, but not interleukin-6, enhances renal and systemic endothelin production in vivo. Am J Physiol Renal Physiol. 2008;295:F446–F453. doi: 10.1152/ajprenal.00095.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callera GE, Touyz RM, Teixeira SA, Muscara MN, Carvalho MH, Fortes ZB, Nigro D, Schiffrin EL, Tostes RC. ETA receptor blockade decreases vascular superoxide generation in DOCA-salt hypertension. Hypertension. 2003;42:811–817. doi: 10.1161/01.HYP.0000088363.65943.6C. [DOI] [PubMed] [Google Scholar]
- Collino F, Bussolati B, Gerbaudo E, Marozio L, Pelissetto S, Benedetto C, Camussi G. Preeclamptic sera induce nephrin shedding from podocytes through endothelin-1 release by endothelial glomerular cells. Am J Physiol Renal Physiol. 2008;294:F1185–F1194. doi: 10.1152/ajprenal.00442.2007. [DOI] [PubMed] [Google Scholar]
- Dhaun N, Goddard J, Webb DJ. The endothelin system and its antagonism in chronic kidney disease. J Am Soc Nephrol. 2006;17:943–955. doi: 10.1681/ASN.2005121256. [DOI] [PubMed] [Google Scholar]
- Elmarakby AA, Loomis ED, Pollock JS, Pollock DM. NADPH oxidase inhibition attenuates oxidative stress but not hypertension produced by chronic ET-1. Hypertension. 2005;45:283–287. doi: 10.1161/01.HYP.0000153051.56460.6a. [DOI] [PubMed] [Google Scholar]
- Janas J, Sitkiewicz D, Warnawin K, Janas RM. Characterization of a novel, high-molecular weight, acidic, endothelin-1 inactivating metalloendopeptidase from the rat kidney. J Hypertension. 1994;12:1155–1162. [PubMed] [Google Scholar]
- Kestilä M, Lenkkeri U, Männikkö M, Lamerdin J, McCready P, Putaala H, Ruotsalainen V, Morita T, Nissinen M, Herva R, Kashtan C, Peltonen L, Holmberg C, Olsen A, Tryggvason K. Positionally cloned gene for a novel glomerular protein-nephrin-is mutated in congenital nephrotic syndrome. Mol Cell. 1998;1:575–582. doi: 10.1016/s1097-2765(00)80057-x. [DOI] [PubMed] [Google Scholar]
- Kisanuki YY, Emoto N, Ohuchi T, Kedzierski RM, Hammer RE, Yanagisawa H, Williams SC, Richardson JA, Suzuki T, Yanagisawa M. Low blood pressure in endothelial cell-specific endothelin-1 knockout mice. Hypertension. 2010;56:121–128. doi: 10.1161/HYPERTENSIONAHA.109.138701. [DOI] [PubMed] [Google Scholar]
- Kitiyakara C, Chabrashvili T, Chen Y, Blau J, Karber A, Aslam S, Welch WJ, Wilcox CS. Salt intake, oxidative stress, and renal expression of NADPH oxidase and superoxide dismutase. J Am Soc Nephrol. 2003;14:2775–2782. doi: 10.1097/01.asn.0000092145.90389.65. [DOI] [PubMed] [Google Scholar]
- Kohan DE, Inscho EW, Wesson D, Pollock DM. Physiology of endothelin and the kidney. Comprehensive Physiol. 2011;1:883–919. doi: 10.1002/cphy.c100039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohan DE, Padilla E. Osmolar regulation of endothelin-1 production by rat inner medullary collecting duct. J Clin Invest. 1993;91:1235–1240. doi: 10.1172/JCI116286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kourembanas S, Marsden PA, McQuillan LP, Faller DV. Hypoxia induces endothelin gene expression and secretion in cultured human endothelium. J Clin Invest. 1991;88:1054–1057. doi: 10.1172/JCI115367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau YE, Galligan JJ, Kreulen DL, Fink GD. Activation of ETB receptors increases superoxide levels in sympathetic ganglia in vivo. Am J Physiol Regul Integr Comp Physiol. 2006;290:R90–R95. doi: 10.1152/ajpregu.00505.2005. [DOI] [PubMed] [Google Scholar]
- Li H, Chen SJ, Chen YF, Meng QC, Durand J, Oparil S, Elton TS. Enhanced endothelin-1 and endothelin receptor gene expression in chronic hypoxia. J Appl Physiol. 1994;77:1451–1459. doi: 10.1152/jappl.1994.77.3.1451. [DOI] [PubMed] [Google Scholar]
- Loomis ED, Sullivan JC, Osmond DA, Pollock DM, Pollock JS. Endothelin mediates superoxide production and vasoconstriction through activation of NADPH oxidase and uncoupled nitric-oxide synthase in the rat aorta. J Pharm Exptl Therap. 2005;315:1058–1064. doi: 10.1124/jpet.105.091728. [DOI] [PubMed] [Google Scholar]
- Nir A, Clavell AL, Heublein D, Aarhus LL, Burnett JCJ. Acute hypoxia and endogenous renal endothelin. J Am Soc Nephrol. 1994;4:1920–1924. doi: 10.1681/ASN.V4111920. [DOI] [PubMed] [Google Scholar]
- Pollock DM, Pollock JS. Evidence for endothelin involvement in the response to high salt. Am J Physiol Renal Physiol. 2001;281:F144–F150. doi: 10.1152/ajprenal.2001.281.1.F144. [DOI] [PubMed] [Google Scholar]
- Sabaa N, de Franceschi L, Bonnin P, Castier Y, Malpeli G, Debbabi H, Galaup A, Maier-Redelsperger M, Vandermeersch S, Scarpa A, Janin A, Levy B, Girot R, Beuzard Y, Leboeuf C, Henri A, et al. Endothelin receptor antagonism prevents hypoxia-induced mortality and morbidity in a mouse model of sickle-cell disease. J Clin Invest. 2008;118:1924–1933. doi: 10.1172/JCI33308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saleh MA, Boesen EI, Pollock JS, Savin VJ, Pollock DM. Endothelin-1 increases glomerular permeability and inflammation independent of blood pressure in the rat. Hypertension. 2010;56:942–949. doi: 10.1161/HYPERTENSIONAHA.110.156570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wedgwood S, Dettman RW, Black SM. ET-1 stimulates pulmonary arterial smooth muscle cell proliferation via induction of reactive oxygen species. Am J Physiol Lung Cell Mol Physiol. 2001;281:L1058–L1067. doi: 10.1152/ajplung.2001.281.5.L1058. [DOI] [PubMed] [Google Scholar]
- Wilhelm SM, Simonson MS, Robinson AV, Stowe NT, Schulak JA. Endothelin up-regulation and localization following renal ischemia and reperfusion. Kidney Int. 1999;55:1011–1018. doi: 10.1046/j.1523-1755.1999.0550031011.x. [DOI] [PubMed] [Google Scholar]
- Williams JM, Pollock JS, Pollock DM. Arterial pressure response to the antioxidant tempol and ETB receptor blockade in rats on a high-salt diet. Hypertension. 2004;44:770–775. doi: 10.1161/01.HYP.0000144073.42537.06. [DOI] [PubMed] [Google Scholar]
- Zager RA, Johnson AC, Andress D, Becker K. Progressive endothelin-1 gene activation initiates chronic/end-stage renal disease following experimental ischemic/reperfusion injury. Kidney Int. 2013;84:703–12. doi: 10.1038/ki.2013.157. [DOI] [PMC free article] [PubMed] [Google Scholar]