

Abstract
Base excision repair (BER) is a primary mechanism for repair of base lesions in DNA such as those formed by exposure to the DNA methylating agent methyl methanesulfonate (MMS). Both DNA polymerase β (pol β)- and XRCC1-deficient mouse fibroblasts are hypersensitive to MMS. This is linked to a repair deficiency as measured by accumulation of strand breaks and poly(ADP-ribose) (PAR). The interaction between pol β and XRCC1 is important for recruitment of pol β to sites of DNA damage. Endogenous DNA damage can substitute for MMS-induced damage such that BER deficiency as a result of either pol β- or XRCC1-deletion is associated with sensitivity to PARP inhibitors. Pol β shRNA was used to knock down pol β in Xrcc1+/+ and Xrcc1−/− mouse fibroblasts. We determined whether pol β-mediated cellular resistance to MMS and PARP inhibitors resulted entirely from coordination with XRCC1 within the same BER sub-pathway. We find evidence for pol β- dependent cell survival independent of XRCC1 expression for both types of agents. The results suggest a role for pol β-dependent, XRCC1-independent repair. PAR immunofluorescence data are consistent with the hypothesis of a decrease in repair in both pol β knock down cell variants.
Keywords: DNA polymerase β, XRCC1, methyl methanesulfonate, PARP inhibitor;, camptothecin
1. Introduction
Base excision repair (BER) is a primary mechanism for repair of endogenous and exogenous base lesions in DNA. Cellular lesions formed by exposure to the DNA methylating agent methyl methanesulfonate (MMS), are removed by a damage specific monofunctional glycosylase producing abasic (AP) sites that are cleaved by AP endonuclease 1. DNA polymerase β (pol β) lyase activity removes the resulting 5´- deoxyribose (dRP) flap, and the polymerase domain performs gap-filling DNA synthesis leaving a DNA nicked substrate for DNA ligation.
XRCC1 is a multi-domain protein with no known catalytic activity. XRCC1 interacts with a number of repair proteins, e.g., PARP-1, pol β and lig-IIIα, and is thought to modulate and coordinate steps of BER [1]. XRCC1-deficient cell extracts are reported to have ligation defects [2, 3], but not a general BER deficiency [4]. The interaction between pol β and XRCC1 is important for recruitment of pol β to sites of DNA damage [5]. Both pol β- and XRCC1-deficient mouse fibroblasts are hypersensitive to MMS (Fig. S1), and this is linked to repair deficiency as measured by accumulation of strand breaks [6] and of poly (ADP-ribose) (PAR) [5, 7]. The greater MMS hypersensitivity phenotype of Xrcc1−/− cells than pol β−/− cells (Fig. S1) provides evidence for XRCC1-dependent, pol β-independent protection in addition to the pol β/XRCC1 coordinated BER pathway.
PARP-1 protein is critical for DNA damage recognition and binds to strand-breaks in DNA [8], including intermediates of BER [9]. Binding to damaged DNA stimulates synthesis of PAR polymers onto itself, as well as other repair proteins, and mediates recruitment of XRCC1 [10]. In the presence of an inhibitor of PARP catalytic activity, binding of PARP-1 to DNA is stabilized [11]. DNA-bound and inhibited PARP-1 protein is cytotoxic as a function of formation of replication-dependent double-strand breaks [12]. PARP inhibitor-induced enhancement of alkylating agent cytotoxicity in mouse fibroblasts is observed where damage is repaired by a BER sub-pathway involving pol β and XRCC1 [13]. Sensitization to MMS is consistent with increased PARP binding sites in DNA in the absence of efficient BER. Endogenous DNA damage can substitute for MMS-induced damage such that BER deficiency as a result of either pol β- or XRCC1-deletion is associated with sensitivity to PARP inhibitors [14] (Fig. S1).
Utilizing reversible covalent binding, topoisomerase 1 (Top1) nicks DNA and relaxes DNA supercoiling ahead of transcription and replication. Top1 can become trapped during formation of the cleavage complex by an inhibitor such as camptothecin [15]. Repair of the trapped complex involves removal of Top1 by tyrosyl DNA phosphodiesterase (Tdp1) and end-cleaning by polynucleotide kinase/phosphatase (PNKP) prior to religation. XRCC1 is known to bind and enhance the activities of both Tdp1 and PNKP, and to facilitate repair [16, 17]. Also, there may be a link between repair of the Top1 complex and XRCC1-mediated single-strand break repair. Hypersensitivity to camptothecin is observed in XRCC1-deficient cells (Fig. S1) [17]. Previously it was proposed that pol β was involved in gap-filling during repair [17]. Currently it is believed that camptothecin generates a nick rather than a gap in DNA consistent with the absence of significant hypersensitivity in pol β-deficient mouse fibroblasts (Fig. S1).
In this study, we used shRNA-mediated knock down of pol β in Xrcc1+/+ and Xrcc1−/− mouse fibroblasts. We aimed to determine whether pol β-mediated cellular resistance to MMS and PARP inhibitors resulted entirely from coordination with XRCC1 within the same BER sub-pathway. We find evidence for pol β-dependent cell survival independent of XRCC1 expression for both types of agents. The results suggest a role for pol β-dependent, XRCC1-independent repair. PAR immunofluorescence data are consistent with the hypothesis of decreased repair in both pol β knock down cell variants. Camptothecin sensitivity, not dependent on pol β expression, was studied as a negative control.
2. Materials and methods
2.1. Cell culture - pol β and XRCC1 cell variants
Wild type and pol β null (termed pol β −/−) SV40-transformed mouse embryonic fibroblasts (16.3 and 19.4, respectively) have been described previously [18]. Xrcc1+/+ and Xrcc1−/− p53-deficient mouse embryonic fibroblasts were obtained from Dr. Robert Tebbs [19]. Mycoplasma testing was performed routinely on all cell lines using a MycoAlert® Mycoplasma detection kit (Lonza, Rockland, ME).
2.2. Lentiviral knock down of pol β
Lentiviral plasmid constructs for stable depletion of mouse pol β mRNA were obtained from Sigma-Aldrich (MISSION shRNA). Two different shRNAs against pol β were used: pol β shRNA-468 (TRCN0000348053): CCGGTCAACGAATTGGGCTGAAATACTCGAGTATTTCAGCCCAATTCGTTGATTTTTG pol β shRNA-662 (TRCN0000331721): CCGGCCAAAGTTGTTACATCGTGTTCTCGAGAACACGATGTAACAACTTTGGTTTTTG Lentivirus particles were produced by the NIEHS Viral Vector Core and packaged in HEK293T/17 cells (ATCC # CRL-11268) according to published Current Protocols in Neuroscience by P. Salmon and D. Trono. Briefly, 293T cells were transiently transfected with pMD2G, psPAX2 and transfer vector containing shRNA using Lipofectamine 2000. Supernatant was collected 48 h post transfection and concentrated by centrifugation at 50,000 g for 2 h. Pellets were resuspended in phosphate-buffered saline (PBS) and used for infection. All titers were determined by quantitative PCR to measure the number of lentiviral particles that integrated into the host genome. In addition, biological titration of viruses co-expressing fluorescent moieties was determined by flow cytometry. Xrcc1+/+ and Xrcc1−/− cells were transduced with lentiviral particles at multiplicity of infection (MOI) of 5 and 25 per 50,000 cells, and stable cell lines were recovered after puromycin selection (6 µg/ml, Life Technologies). After characterization by western blot analysis, single cell clones were isolated from those demonstrating significant pol β knock down. Clones were again characterized by western blotting and also by RT-PCR. Lentiviral plasmid pLKO.1 transcribes nonspecific shRNA.
2.3. Western blot analysis
Whole cell extracts were prepared as described previously [14]. Extract samples (60 µg) were loaded onto 4–12% Bis-Tris NuPAGE gels (Invitrogen) and electrophoresed in NuPAGE MES running buffer at 4°C. Proteins were transferred to nitrocellulose filters for 4 h at 25 V, in the cold. Filters were blocked overnight at 4°C in 5% nonfat dry milk in Tris-buffered saline (TBS) with 0.1% Tween 20 (TBST), then incubated for 2 h at room temperature or overnight at 4°C with either pol β 18S monoclonal antibody [20] or mouse monoclonal anti-XRCC1 primary antibody (Thermo Fisher Scientific, 33-2-5). After washing, filters were incubated with anti-mouse IgG-horseradish peroxidase (HRP) conjugated secondary antibody (1:2,000–1:20,000 dilution, Bio-Rad) and visualized using Super Signal (Thermo Scientific). Blots were stripped in Restore Western Blot Stripping Buffer (Thermo Scientific), washed three times in TBST, and blocked in 5% nonfat dry milk/TBST overnight. Anti α-tubulin (Sigma T-9026) was used as a loading control.
2.4. Measurement of cellular pol β gene expression by RT-PCR
Total RNA was isolated from clonal cell lines for Xrcc1+/+ and Xrcc1−/− transfected with pol β shRNA using the QIAGEN RNeasy mini kit. RT-PCR was performed using the 7900HT sequence detection system and predesigned primer/probe sets from Applied Biosystems. The signal obtained from each gene was normalized to the housekeeping gene cyclophilin B.
2.5. Cytotoxicity studies by growth inhibition assay
Cells were seeded (20,000 per well for XRCC1 and 40,000 per well for pol β) in six-well dishes in medium without selection antibiotic. The next day, cells were treated for 1 h with a range of concentrations of MMS (Sigma-Aldrich). Alternatively, cells were treated for 24 h with the Top1 inhibitor camptothecin (Sigma-Aldrich), or continuously with the PARP inhibitors, 4-amino-1,8-naphthalimide (4-AN) (Acros) and olaparib (Selleckchem). After washing as needed, growth medium was added and cells further incubated.
When untreated cells were 80% confluent, triplicate wells for each drug concentration were counted by a cell lysis procedure as utilized previously [21, 22]. Briefly, the cell culture medium was aspirated, and cells washed with 0.9% saline. Hypotonic buffer (10 mM Hepes and 1.5 mM MgCl2.6H2O, pH 7.4) was added to each well (1 ml) and the cells incubated at room temperature with gentle shaking for 5 min. Lysing solution (200 µl) containing 5 g ethylhexadecyldimethylammonium bromide (Sigma-Aldrich) and 3 ml glacial acetic acid per 100 ml [21] was then added to each well and the cells incubated with agitation for 7 min. Saline + formaldehyde (4 ml) was added to the wells and the counting solutions were mixed vigorously. An aliquot (4 ml) of the mix was removed and added to a counting vial containing 10 ml 0.9% saline. Cell nuclei were counted using a Multisizer 3 Coulter Counter (Beckman Coulter). Results were expressed as % growth of drug-treated cells relative to control untreated cells. Fold hypersensitivity was determined at IC90 concentrations, the dose required for 90% decrease in cell growth. For experiments with mouse fibroblasts, we find this growth inhibition assay to be more reliable and consistent than clongenic or short-term cytotoxicity assays. Results obtained are in agreement with alternate assay methods [14].
2.6. Micro-irradiation and immunofluorescence
Cells were seeded (2×105 cells per dish) in 35 mm glass bottomed petri dishes containing an etched grid (MatTek, Ashland, MA) and incubated in medium supplemented with 10 µM BrdU. After 24 h, DNA single strand breaks and base lesions were introduced by low power UV laser micro-irradiation at 364 nm [7]. Cells were imaged using a 40× C-Apochromat (numerical aperture 1.2) water immersion objective coupled to a Zeiss LSM510 META confocal microscope (Carl Zeiss MicroImaging).
Cells were fixed in 4% paraformaldehyde 1 min after UV damage, permeabilized with 0.25% Triton X-100 in PBS for 10 min, washed three times in PBS, then further permeabilized and blocked with PBS + 1% BSA for 30 min. Following incubation with anti- XRCC1 antibody (1:50; Abcam ab1838) and anti-PADPR antibody (1:100; Abcam ab14460) for 1 h, cells were washed three times with PBS, then incubated with Alexa 488 conjugated anti-mouse and Alexa 647 conjugated anti-chicken secondary antibodies (1:2,000; Life Technologies) for 1 h. Cells were then washed three times with PBS, and the nucleus was stained with NucBlue® Fixed Cell Stain ReadyProbes™ (Life Technologies) for 5 min. Fluorescence images were acquired with the same 40× water immersion objective on the LSM510. Recruitment of XRCC1 and accumulation of PAR at the site of DNA damage were measured using IMAGEJ as described previously [7].
3. Results
3.1. Characterization of pol β knock down in XRCC1 cell populations
Xrcc1+/+ and Xrcc1−/− cells were transduced with control (pLKO.1) and two different pol β shRNA lentiviral particles, and stable cell lines were recovered after puromycin selection. Whole cell extracts were prepared and analyzed by western blotting. Control PLKO Xrcc1+/+ (Fig. 1A) and Xrcc1−/− cells (Fig. 1B) retained expression of pol β protein, while all of the pol β shRNA-transduced (pol β shRNA-468 and pol β shRNA-662) cells had undetectable levels of pol β. Cells with plasmid 662 at a MOI of 5 (X+662/5 and X-662/5) were chosen for further development (boxed in Fig. 1A and B). Single cell clones were isolated and again analyzed by western blotting. X+662/5 clones demonstrated variable levels of pol β knock down (Fig. 1C). In clone 7 (cl.7), pol β was undetectable and was selected for survival studies (boxed in Fig. 1C). All of the X-662/5 clones had undetectable pol β levels (Fig. 1D). Clone 1 (cl.1, boxed in Fig. 1D) was chosen for further study. Western blotting was repeated frequently to check for any reversal of pol β knock down.
Figure 1.

Western blotting of control and pol β shRNA transduced cells lines. (A) Xrcc1+/+ (X+) and (B) Xrcc1−/− (X-) cells were transduced with contol plasmid (PLKO) or pol β shRNA vectors (468 or 622) at a MOI of 5 or 25. After selection in puromycin, cell lines were compared with the original Xrcc1+/+ and Xrcc1−/− and analyzed by western blotting for pol β knock down. All pol β constructs produced significant knock down. Single cell clones were isolated from X+662/5 and X-662/5 cell populations and further subjected to western blotting. (C) X+662/5 cl.7 and (D) X-662/5 cl.1 were chosen for further study. Isolation of cell lines and western blotting were performed as described in Section 2.
3.2. Cellular pol β gene expression in shRNA-transduced clones
For comparison with the knock down clones, the level of pol β gene expression was first measured in pol β wild type and null cells developed in the lab [18]. As expected, null cells had undetectable levels of pol β mRNA (Fig. 2). Expression of mRNA was slightly (1.4- fold) higher in Xrcc1−/− compared with Xrcc1+/+ cells (Fig. 2) in contrast to the lower pol β protein level observed in Xrcc1−/− cells (see all panels of Fig. 1). Decreased pol β protein has been documented in other XRCC1-deficient cells and it has been reported that expression of XRCC1 is required to maintain the stability of cellular pol β, as well as ligase III protein [23]. An elevation of pol β mRNA in cells in the absence of XRCC1 may be a feedback mechanism designed to increase cellular levels of pol β protein. In agreement with the results of western blot analysis, pol β mRNA expression was reduced by approximately 90% in the knock down clones (Fig. 2).
Figure 2.

Measurement of cellular pol β gene expression by RT-PCR. Xrcc1+/+ and Xrcc1−/− control and clonal knock down (pol βkd) cell lines transfected with pol β shRNA were cultured until confluent, and total RNA was isolated using the QIAGEN RNeasy mini kit. RT-PCR was performed and the signal obtained was normalized to the housekeeping gene cyclophilin B as outlined in Section 2. As a reference, pol β−/− cells were compared with pol β+/+ mouse fibroblast cell lines. Pol βkd clones were compared with the original Xrcc1+/+ and Xrcc1−/− cell lines.
3.3. Effect of pol β knock down on cellular sensitivity to MMS
Cellular sensitivity to MMS was compared in Xrcc1+/+ cells expressing control vector (X+ PLKO) and those (Fig. 1C and Fig. 2) selected for significant pol β knockdown (X+/βkd). As expected, since MMS hypersensitivity in pol β null variants is well known [18], pol β knockdown cells were 1.5-fold hypersensitive to MMS (Fig. 3A). This hypersensitivity of the knock down clone was less than that of pol β null cells (generally 2.5–3-fold; [24]) but it should be noted that gene expression remained significantly higher than in cells with the deletion mutation (Fig. 2). Surprisingly, the X−/βkd clone (Fig. 1D) was similarly hypersensitive to MMS (1.8-fold) compared with the control X- PLKO clone (Fig. 3B). Thus, the effect of pol β knock down on hypersensitivity to MMS did not depend on XRCC1 expression. Control vector-transduced Xrcc1−/− cells (X- PLKO) were 8.1-fold hypersensitive to MMS compared with control vector-transduced Xrcc1+/+ cells (X+ PLKO) (Table 1). Knock down of pol β in Xrcc1−/− cells (X−/βkd), resulted in an increased (14.7-fold) hypersensitivity phenotype (Table 1). Alternative X+/βkd (X+468/5 cl. 2) and X−/βkd (X-662/5 cl. 3) clones with knock down of pol β (Fig. S2A and B) were likewise hypersensitive to MMS (Fig. S2C and D). The results from pol β knock down in XRCC1 wild type and Xrcc1−/− cells are consistent with a mechanism for pol β-dependent cell survival after MMS that is independent of XRCC1 expression.
Figure 3.

MMS and camptothecin hypersensitivity of pol βkd clones. Clones selected as shown in Figure 1 were treated for 1 h with a range of concentrations of MMS or for 24 h with a range of concentrations of camptothecin as described in Section 2. (A) Hypersensitivity (1.5-fold) of X+/βkd compared with control X+ PLKO. (B) Hypersensitivity (1.8-fold) of X/-βkd compared with control X- PLKO. Plotted are mean ± SEM values obtained from at least 5–7 independent experiments. (C) Minimal resistance of X+/βkd compared with control X+ PLKO. (B) Minimal hypersensitivity of X-/βkd compared with control X- PLKO. Plotted are mean ± SEM values obtained from 3 independent experiments.
Table 1.
Sensitization by pol β knock down in Xrcc1−/− cells
| Hypersensitivity (fold) relative to X+ PLKO* |
||
|---|---|---|
| DNA damaging agent | X- PLKO | X-/βkd |
| MMS | 8.1 ± 0.28 (7) | 14.7 ± 0.70 (5) |
| Camptothecin | 3.6 ± 0.43 (3) | 4.0 ± 0.44 (3) |
Measured at IC90
3.4. Effect of pol β knock down on cellular sensitivity to camptothecin
Sensitivity to camptothecin was examined as a negative control since pol β null cells have only a minor camptothecin hypersensitivity phenotype [24] and only at the highest doses (Fig. S1C). As predicted, pol β knock down only slightly enhanced camptothecin cytotoxicity in Xrcc1−/− cells (X−/βkd; Fig 3D) while X+/βkd became slightly more resistant than the X+ PLKO clone (Fig. 3C). Comparing clone X−/βkd with control X+ PLKO, there was a small (1.1-fold) enhancement of sensitivity relative to X- PLKO control cells (Table 1).
3.5. Effect of pol β knock down on cellular sensitivity to PARP inhibitors
Recently, it was shown that the absence of either pol β or XRCC1 resulted in cellular hypersensitivity to PARP inhibitors as a function of BER deficiency [14]. We further determined the PARP inhibitor phenotype for pol β knock down in XRCC1 cells. As anticipated, pol β knock down in Xrcc1+/+ cells (X+/βkd) enhanced sensitivity to 4-AN (Fig. 4A). Similar to MMS, pol β knock down in Xrcc1−/− cells (X−/βkd) also resulted in hypersensitivity to 4-AN (Fig. 4B). This resulted in a greater 4-AN hypersensitivity in X−/βkd than in control X- PLKO (Fig. 4C), consistent with a pol β-dependent deficiency in BER. Interestingly, the 4-AN sensitivity of X+/βkd and X- PLKO was very similar (Fig. 4C). Studies with the clinically used PARP inhibitor olaparib produced comparable results to those with 4-AN (Fig. S3).
Figure 4.
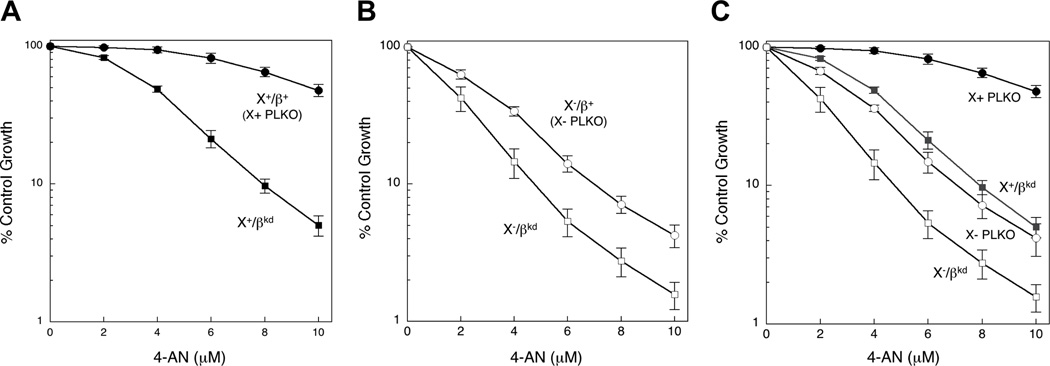
4-AN hypersensitivity of pol βkd clones. Clones selected as shown in Figure 1 were treated continuously with a range of concentrations of 4-AN as described in Section 2. (A) Hypersensitivity of X+/βkd compared with control X+ PLKO. (B) Hypersensitivity of X-/βkd compared with control X- PLKO. (C) 4-AN hypersensitivity of X+/βkd and X-/βkd compared with X+ PLKO and X- PLKO. Plotted are mean ± SEM values obtained from 4 independent experiments.
3.6. PAR synthesis after low energy laser-induced damage
Micro-irradiation has been used to demonstrate recruitment of repair proteins to nuclear DNA strand breaks [7, 25]. XRCC1 protein was recruited to sites of DNA damage in Xrcc1+/+ cells at 1 min after irradiation (Fig. 5A). As expected, recruitment of XRCC1 was seen in Xrcc1+/+-derived (Fig. 5B) but not in Xrcc1−/−-derived cells (data not shown). Levels of XRCC1 were not significantly different (p=0.292) in X+ PLKO and X+/βkd clones (Fig. 5B). Elevated PAR after DNA damage has been associated with a repair deficiency in cells [7]. The observed PAR increase reported previously was higher in pol β−/− than in Xrcc1−/− compared with the respective wild type cell lines [5, 7]. PAR was observed in Xrcc1+/+ and Xrcc1−/− variants 1 min after irradiation (Fig. 5A). Consistent with earlier data in the parental cell lines [5], the level of PAR was significantly greater (p=0.001) in X- PLKO compared with X+ PLKO vector control cell lines (Fig. 5C). Similar to the phenotype of pol β null cells deficient in pol β-dependent BER, knock down of pol β in Xrcc1+/+ cells (X+/βkd) and in Xrcc1−/− cells (X-/βkd) resulted in significant increases in levels of PAR (Fig. 5C). These results suggest a decrease in repair efficiency as a result of pol β knock down and parallel the increase in MMS (Fig. 3) and PARP inhibitor sensitivity (Fig. 4).
Figure 5.

Immunofluorescence imaging of XRCC1 and PAR in XRCC1 cell variants. (A) Representative images of XRCC1 recruitment and PAR accumulation in PLKO and pol βkd cell variants are shown after irradiation and 1 min repair as described in Section 2. DAPI was used as a nuclear stain. Bar represents 10 µm. Quantification of (B) XRCC1 recruitment and (C) PAR synthesis at 1 min. Results represent mean ± SEM from analysis of 20–30 individual cells. Statistical significance was determined using the Student t test. The significance level of PAR synthesis comparing X+ PLKO and X-PLKO is 0.001.
4. Discussion
Cellular deficiency of either pol β or XRCC1 results in hypersensitivity to DNA methylating agents such as MMS and to PARP inhibitors such as 4-AN [6, 13]. Hypersensitivity is associated with BER deficiency. Here we determined whether pol β coordinates with XRCC1 within the same BER sub-pathway to provide cellular resistance or whether pol β can function independently. The effects of lentiviral shRNA knock down of pol β were examined in XRCC1 wild type and XRCC1-deficient cells.
Knock down of pol β in Xrcc1+/+ cells (Fig. 3A) and in Xrcc1−/− cells (Fig. 3B), resulted in sensitization to MMS. The latter results suggest that pol β can act independently of XRCC1 in the protection of cells against MMS cytotoxicity. An enhanced level of cellular PAR after laser irradiation is an indicator of cellular BER deficiency [7]. Both pol β knock down variants had significantly elevated PAR after micro-irradiation (Fig. 5). Taken together, the results provide evidence that a deficiency in pol β-dependent repair is responsible for the MMS hypersensitivity of the knock down clones. For 4-AN, the survival data in Figure 4C correlate well with the PAR synthesis data shown in Figure 5C. X+ PLKO repair-proficient cells are most resistant to 4-AN, and have the least PAR synthesis following damage by micro-irradiation. At the opposite end of the spectrum, X-/βkd cells are most hypersensitive to 4-AN and have the highest PAR accumulation. X+/βkd and XPLKO cells have similar intermediate levels of PAR synthesis (Fig. 5C) and similar sensitivities to 4-AN (Fig. 4C).
In previous experiments, it was determined that XRCC1 was required for pol β accumulation at sites of micro-irradiation-induced single-strand breaks and DNA base lesions [5, 25]. In contrast, the current results indicate that pol β can function in repair of MMS-induced damage in the absence of the XRCC1. A proposed direct interaction with PARP-1 [26] could enhance pol β accumulation in the absence of XRCC1. Alternatively, pol β might interact directly with the 5´-dRP intermediate of BER. It is the 8-kDa-associated dRP lyase activity of pol β and the removal of the dRP group that is essential for repair of DNA methylation damage [27]. Interestingly, recruitment of the 8-kDa domain can occur in an XRCC1-independent manner [25]. A point mutation in mouse XRCC1 (V88R) prevents binding to pol β [5, 28]. This mutant protein stably expressed in Xrcc1−/− cells only partially complemented the MMS hypersensitivity phenotype [5]. This result indicates the importance of the pol β/XRCC1 interaction, but also the ability of non-interacting XRCC1 to partially rescue cells from MMS-induced cytotoxicity. The greater MMS hypersensitivity phenotype of Xrcc1−/− cells than pol β−/− cells (Fig. S1) also suggests a role for XRCC1- dependent, pol β-independent cellular protection.
Experiments with camptothecin failed to show sensitization following knock down of pol β in either of the XRCC1 cell lines (Fig. 3C and D). These results are consistent with cytotoxicity studies implicating XRCC1, but not pol β, in repair of camptothecin-Top1 cleavage complexes (Fig. S1). Data obtained for the PARP inhibitors 4-AN and olaparib in the pol β knock down clones were similar to those obtained with MMS. These results are consistent with the previously published hypothesis that PARP inhibitor hypersensitivity is a function of inefficient BER of endogenous DNA damage involving the AP-site intermediate [14]. Repair of camptothecin-blocked Top1 cleavage complexes does not involve BER or production of this intermediate.
Previous results have suggested XRCC1 is necessary for recruitment of pol β and that the interaction of pol β and XRCC1 is required for BER [5]. MMS sensitivity experiments in pol β knock down Xrcc1−/− cells demonstrated a role for pol β in the absence of XRCC1 expression. Elevated PAR in knock down cells after micro-irradiation implicates BER deficiency as the cause of the sensitization to MMS. Here, we demonstrate repair by pol β in the absence of XRCC1, just as XRCC1 can protect cells from MMS damage in the absence of interaction with pol β [5].
Supplementary Material
Highlights.
Pol β- and XRCC1-deficient mouse fibroblasts are hypersensitive to MMS
Pol β and XRCC1 interaction is important for recruitment of pol β to DNA damage
Pol β shRNA was used to knock down pol β in Xrcc1+/+ and Xrcc1−/− mouse fibroblasts
Studies provide evidence for pol β-dependent survival independent of XRCC1
PAR imaging data are consistent with decreased repair in pol β knock down variants
Acknowledgements
The authors thank Lois Wyrick for help with figure preparation and members of the NIEHS Viral Vector Core Facility for assistance with the lentivirus expression vectors. We thank C. Jeff Tucker of the NIEHS Fluorescence Microscopy Imaging Center for assistance with the micro-irradiation studies.
Grant Support
This work was supported by the Intramural Research Program of the NIH, National Institute of Environmental Health Sciences (project number Z01 ES050158 and ES050159).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest Statement: The authors declare that there is no conflict of interest.
References
- 1.Caldecott KW. XRCC1 and DNA strand break repair. DNA Repair. 2003:955–969. doi: 10.1016/s1568-7864(03)00118-6. [DOI] [PubMed] [Google Scholar]
- 2.Cappelli E, Taylor R, Cevasco M, Abbondandolo A, Caldecott K, Frosina G. Involvement of XRCC1 and DNA ligase III gene products in DNA base excision repair. J Biol Chem. 1997;272:23970–23975. doi: 10.1074/jbc.272.38.23970. [DOI] [PubMed] [Google Scholar]
- 3.Thompson LH, West MG. XRCC1 keeps DNA from getting stranded. Mutat Res. 2000;459:1–18. doi: 10.1016/s0921-8777(99)00058-0. [DOI] [PubMed] [Google Scholar]
- 4.Wong H-K, Kim D, Hogue BA, McNeill DR, Wilson DM., 3rd DNA damage levels and biochemical repair capacities associated with XRCC1 deficiency. Biochemistry. 2005;44:14335–14343. doi: 10.1021/bi051161o. [DOI] [PubMed] [Google Scholar]
- 5.Horton JK, Stefanick DF, Gassman NR, Williams JG, Kedar PS, Wilson SH. Preventing oxidation of cellular XRCC1 affects PARP-mediated DNA damage responses. DNA Repair. 2013;12:774–785. doi: 10.1016/j.dnarep.2013.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Horton JK, Watson M, Stefanick DF, Shaughnessy DT, Taylor JA, Wilson SH. XRCC1 and DNA polymerase β in cellular protection against cytotoxic DNA singlestrand breaks. Cell Res. 2008;18:48–63. doi: 10.1038/cr.2008.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gassman NR, Stefanick DF, Kedar PS, Horton JK, Wilson SH. Hyperactivation of PARP triggers nonhomologous end-joining in repair-deficient mouse fibroblasts. PLoS ONE. 2012;7:e49301. doi: 10.1371/journal.pone.0049301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Amé JC, Spenlehauer C, de Murcia G. The PARP superfamily. Bioessays. 2004;26:882–893. doi: 10.1002/bies.20085. [DOI] [PubMed] [Google Scholar]
- 9.Lavrik OI, Prasad R, Sobol RW, Horton JK, Ackerman EJ, Wilson SH. Photoaffinity labeling of mouse fibroblast enzymes by a base excision repair intermediate. Evidence for the role of poly(ADP-ribose) polymerase-1 in DNA repair. J Biol Chem. 2001;276:25541–25548. doi: 10.1074/jbc.M102125200. [DOI] [PubMed] [Google Scholar]
- 10.El-Khamisy SF, Masutani M, Suzuki H, Caldecott KW. A requirement for PARP-1 for the assembly or stability of XRCC1 nuclear foci at sites of oxidative DNA damage. Nucleic Acids Res. 2003;31:5526–5533. doi: 10.1093/nar/gkg761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Murai J, Huang S-yN, Das BB, Renaud A, Zhang Y, Doroshow JH, Ji J, Takeda S, Pommier Y. Trapping of PARP1 and PARP2 by clinical PARP inhibitors. Cancer Res. 2012;72:5588–5599. doi: 10.1158/0008-5472.CAN-12-2753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Heacock ML, Stefanick DF, Horton JK, Wilson SH. Alkylation DNA damage in combination with PARP inhibition results in formation of S-phase-dependent double-strand breaks. DNA Repair. 2010;9:929–936. doi: 10.1016/j.dnarep.2010.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Horton JK, Wilson SH. Predicting enhanced cell killing through PARP inhibition. Mol Cancer Res. 2013;11:13–18. doi: 10.1158/1541-7786.MCR-12-0512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Horton JK, Stefanick DF, Prasad R, Gassman NR, Kedar PS, Wilson SH. Base excision repair defects invoke hypersensitivity to PARP inhibition. Mol Cancer Res. 2014;12:1128–1139. doi: 10.1158/1541-7786.MCR-13-0502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pommier Y. Topoisomerase I inhibitors: camptothecins and beyond. Nat Rev Cancer. 2006;6:789–802. doi: 10.1038/nrc1977. [DOI] [PubMed] [Google Scholar]
- 16.Whitehouse CJ, Taylor RM, Thistlethwaite A, Zhang H, Karimi-Busheri F, Lasko DD, Weinfeld M, Caldecott KW. XRCC1 stimulates human polynucleotide kinase activity at damaged DNA termini and accelerates DNA single-strand break repair. Cell. 2001;104:107–117. doi: 10.1016/s0092-8674(01)00195-7. [DOI] [PubMed] [Google Scholar]
- 17.Plo I, Liao ZY, Barcelo JM, Kohlhagen G, Caldecott KW, Weinfeld M, Pommier Y. Association of XRCC1 and tyrosyl DNA phosphodiesterase (Tdp1) for the repair of topoisomerase I-mediated DNA lesions. DNA Repair. 2003;2:1087–1100. doi: 10.1016/s1568-7864(03)00116-2. [DOI] [PubMed] [Google Scholar]
- 18.Sobol RW, Horton JK, Kuhn R, Gu H, Singhal RK, Prasad R, Rajewsky K, Wilson SH. Requirement of mammalian DNA polymerase-β in base-excision repair. Nature. 1996;379:183–186. doi: 10.1038/379183a0. [DOI] [PubMed] [Google Scholar]
- 19.Tebbs RS, Flannery ML, Meneses JJ, Hartmann A, Tucker JD, Thompson LH, Cleaver JE, Pedersen RA. Requirement for the Xrcc1 DNA base excision repair gene during early mouse development. Dev Biol. 1999;208:513–529. doi: 10.1006/dbio.1999.9232. [DOI] [PubMed] [Google Scholar]
- 20.Singhal RK, Prasad R, Wilson SH. DNA polymerase β conducts the gap-filling step in uracil-initiated base excision repair in a bovine testes nuclear extract. J Biol Chem. 1995;270:949–957. doi: 10.1074/jbc.270.2.949. [DOI] [PubMed] [Google Scholar]
- 21.Butler WB. Preparing nuclei from cells in monolayer cultures suitable for counting and for following synchronized cells through the cell cycle. Anal Biochem. 1984;141:70–73. doi: 10.1016/0003-2697(84)90426-3. [DOI] [PubMed] [Google Scholar]
- 22.Horton JK, Joyce-Gray DF, Pachkowski BF, Swenberg JA, Wilson SH. Hypersensitivity of DNA polymerase β null mouse fibroblasts reflects accumulation of cytotoxic repair intermediates from site-specific alkyl DNA lesions. DNA Repair. 2003;2:27–48. doi: 10.1016/s1568-7864(02)00184-2. [DOI] [PubMed] [Google Scholar]
- 23.Parsons JL, Tait PS, Finch D, Dianova, Allinson SL, Dianov GL. CHIP-mediated degradation and DNA damage-dependent stabilization regulate base excision repair proteins. Mol Cell. 2008;29:477–487. doi: 10.1016/j.molcel.2007.12.027. [DOI] [PubMed] [Google Scholar]
- 24.Horton JK, Wilson SH. Hypersensitivity phenotypes associated with genetic and synthetic inhibitor-induced base excision repair deficiency. DNA Repair. 2007;6:530–543. doi: 10.1016/j.dnarep.2006.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lan L, Nakajima S, Oohata Y, Takao M, Okano S, Masutani M, Wilson SH, Yasui A. In situ analysis of repair processes for oxidative DNA damage in mammalian cells. Proc Natl Acad Sci USA. 2004;101:13738–13743. doi: 10.1073/pnas.0406048101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dantzer F, Schreiber V, Niedergang C, Trucco C, Flatter E, De La Rubia G, Oliver J, Rolli V, Ménissier-de Murcia J, de Murcia G. Involvement of poly(ADP-ribose) polymerase in base excision repair. Biochimie. 1999;81:69–75. doi: 10.1016/s0300-9084(99)80040-6. [DOI] [PubMed] [Google Scholar]
- 27.Sobol RW, Prasad R, Evenski A, Baker A, Yang X-P, Horton JK, Wilson SH. The lyase activity of the DNA repair protein β-polymerase protects from DNA-damageinduced cytotoxicity. Nature. 2000;405:807–810. doi: 10.1038/35015598. [DOI] [PubMed] [Google Scholar]
- 28.Marintchev A, Gryk MR, Mullen GP. Site-directed mutagenesis analysis of the structural interaction of the single-strand-break repair protein, X-ray crosscomplementing group 1, with DNA polymerase β. Nucleic Acids Res. 2003;31:580–588. doi: 10.1093/nar/gkg159. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.