

ABSTRACT
Objectives:
Severe congenital diarrhea occurs in approximately half of patients with Aristaless-Related Homeobox (ARX) null mutations. The cause of this diarrhea is unknown. In a mouse model of intestinal Arx deficiency, the prevalence of a subset of enteroendocrine cells is altered, leading to diarrhea. Because polyalanine expansions within the ARX protein are the most common mutations found in ARX-related disorders, we sought to characterize the enteroendocrine population in human tissue of an ARX(GGC)7 mutation and in a mouse model of the corresponding polyalanine expansion (Arx(GCG)7).
Methods:
Immunohistochemistry and quantitative real-time polymerase chain reaction were the primary modalities used to characterize the enteroendocrine populations. Daily weights were determined for the growth curves, and Oil-Red-O staining on stool and tissue identified neutral fats.
Results:
An expansion of 7 alanines in the first polyalanine tract of both human ARX and mouse Arx altered enteroendocrine differentiation. In human tissue, cholecystokinin, glucagon-like peptide 1, and somatostatin populations were reduced, whereas the chromogranin A population was unchanged. In the mouse model, cholecystokinin and glucagon-like peptide 1 populations were also lost, although the somatostatin-expressing population was increased. The ARX(GGC)7 protein was present in human tissue, whereas the Arx(GCG)7 protein was degraded in the mouse intestine.
Conclusions:
ARX/Arx is required for the specification of a subset of enteroendocrine cells in both humans and mice. Owing to protein degradation, the Arx(GCG)7 mouse recapitulates findings of the intestinal Arx null model, but is not able to further the study of the differential effects of the ARX(GCG)7 protein on its transcriptional targets in the intestine.
Keywords: Arx, enteroendocrine dysgenesis, polyalanine
Loss of enteroendocrine cells (enteric anendocrinosis) related to NEUROGENIN3 (NEUROG3) mutations is a recognized cause of congenital malabsorptive diarrhea (1). The intestinal endocrine system secretes more than a dozen different hormones that are involved in digestion, absorption, and motility of the bowel (reviewed in (2)). Mouse models of Neurog3 mutations first demonstrated the loss of enteroendocrine cells, although the mechanism of the malabsorptive diarrhea is not completely understood (3–5). At present, no treatments are available for this rare disorder.
Autoimmune-polyendocrine-candidiasis-ectodermal-dystrophy (APECED) syndrome includes malabsorptive diarrhea related to autoimmune destruction of enteroendocrine cells (6,7). Both APECED and NEUROG3 mutations lead to the loss of the majority of enteroendocrine cells, whereas proprotein convertase 1/3 (PC1/3) deficiency causes early congenital diarrhea with normal chromogranin A staining (8). Although PC1/3 is expressed in the majority of enteroendocrine cells, the full extent of hormonal populations that are affected by PC1/3 processing, beyond glucagon-like peptide (GLP)-1 and GLP-2, is unclear (9–11). Furthermore, changes in enteroendocrine cell function are involved in other chronic diarrheal cases (12), although they may be overlooked because histologic features are frequently normal and enteroendocrine staining is not necessarily part of the routine pathologic assessment.
Several transcription factors have been identified in mice that specify distinct lineages of the intestinal endocrine population (2). ARX (Aristaless-Related Homeobox) is a paired domain transcription factor on the X chromosome associated with neurologic disease (13), loss of pancreatic α cells (14), and early-onset, severe diarrhea (15). Approximately half of patients with missense or nonsense mutations present with congenital diarrhea that leads to early mortality. The mouse model of endodermal Arx deficiency recapitulates this intestinal phenotype, with diarrhea and failure to thrive as a result of a loss of enteroendocrine subpopulations (16,17). Although the chromogranin A cell number is unchanged, GLP-1, glucose-dependent insulinotropic peptide, cholecystokinin (CCK), secretin, and gastrin-producing cells are reduced, and somatostatin (SST)-expressing cells are increased in this model. Interestingly, both Arx null and Neurog3 null mice die within a few days of birth, compared with PC1/3 null mice that have reduced survival and growth impairment similar to mice with endodermal Arx deficiency (14,18,19). The effects of these genes on multiple tissues, however, make the contribution of intestinal disease to early mortality difficult to determine. Thus far, human intestinal tissue from patients with ARX loss-of-function mutations has not been examined.
ARX-related neurologic disorders comprise a spectrum of phenotypes of X-linked lissencephaly with abnormal genitalia (XLAG; OMIM #300215; (20,21)), X-linked infantile spasms (ISSX; OMIM 308350; (22)), and X-linked intellectual disability (XLID; (23,24)). The loss of function, missense, and protein truncation mutations have been identified. Interestingly, approximately half of the identified disease-causing mutations are expansions of the polyalanine tract within the ARX protein, of which ARX/Arx has 4 (25,26). Polyalanine expansions have become increasingly recognized as disease-causing mutations in a variety of diseases (reviewed in (27)). For example, a small expansion of a polyalanine tract in PHOX2B can cause central hypoventilation syndrome with Hirschsprung disease (28).
Here, we report a case of enteroendocrine dysgenesis in a patient with an ARX polyalanine expansion. The chromogranin A population was unchanged. Duodenal biopsies, however, revealed a reduction in CCK, SST, and GLP-1 cell number. In the mouse model with the corresponding polyalanine insertion, the enteroendocrine changes mimicked those of the intestinal loss-of-function model, that is, loss of CCK and GLP-1 cells, but an increase in the SST-expressing population. Thus, ARX/Arx is required for the enteroendocrine development in mice and humans.
METHODS
Mice and Tissue Preparation
The mice used for these experiments were a kind gift from Kunio Kitamura (29). Seven (GCG) triplets were placed into the first polyalanine tract at residue 330, resulting in Arx(GCG)7 mice. Hemizygous mice (Arx(GCG)7/Y) were obtained by crossing heterozygous females (Arx(GCG)7/+) with C57BL/6J wild-type males. All mice were cared and handled according to The Children's Hospital of Philadelphia's institutional animal care and use committee–approved.
All dissections were performed in cold 1× phosphate-buffered saline, and tail snips were used for determining genotypes. Genotyping primers were as follows: 5′-AAAGGCGAAAAGGACGAGGAAAGG-3′ and 5′-TGTTCAATGGCCGATCCCAT-3′ and 5′-CTTTAGCTCCCCTTCCTGGCACAC-3′, resulting in a wild-type band of 500 base pairs (bp) and a mutant product of 236 bp. Following dissection, tissues were fixed in fresh 4% paraformaldehyde overnight at 4°C, embedded in paraffin or optimal cutting temperature freezing medium, and sectioned at 8 μm.
Human Slides
The genetic analysis for the patient was performed at Genetic Services Laboratories at University of Chicago. In the ARX gene, all 5 coding exons were polymerase chain reaction (PCR) amplified and sequenced. An insertion of 21 bp, 335–336ins(GGC)7, was detected in exon 2 of the ARX gene. The insertion is in-frame, resulting in the insertion of 7 alanine residues at amino acid position 112. Of note, the triplet repeat GCG codes for alanine; although the insertion in human ARX is termed (GGC)7, it is the same sequence shifted by 1 bp. Duodenal tissue was obtained during upper endoscopy for the evaluation of his pseudo-obstruction. For this article, additional slides were obtained from paraffin blocks in storage in our pathology department. Control slides were obtained from age-matched controls viewed to be histologically normal and without a diagnosis of celiac, eosinophilic, or inflammatory bowel disease. The P-values were obtained by comparing the 2 temporally distinct biopsies of the patient with the ARX(GGC)7 mutation and 3 to 4 age-matched controls.
Real-Time PCR Analysis
Total RNA was extracted with TRIZOL (Invitrogen, Grand Island, NY) using the RNeasy kit (Qiagen, Valencia, CA). Oligo-dT, SuperScript, and other reagents were used to synthesize complementary DNA. Brilliant SYBR Green PCR Master Mix (Sigma, St Louis, MO) was used to set up PCR reactions in the Stratagene MX3005P (La Jolla, CA) real-time PCR machine. Primer sequences are available upon request. All reactions were performed in triplicate with reference dye normalization. Each primer set was normalized to a housekeeping gene, either glyceraldehyde 3-phosphate dehydrogenase or hypoxanthine-guanine phosphoribosyltransferase. Fold change relative to control values and standard deviation were calculated and then plotted on a bar graph. The P-values were obtained using Student t test.
Immunohistochemistry and Histology
Slides were subjected to microwave antigen retrieval in 10 mmol/L sodium citrate buffer (pH 6.0). Endogenous peroxidase activity was quenched with 3% H2O2 in phosphate-buffered saline for 15 minutes. Sections were then blocked with avidin block, biotin block, and CAS Block reagent (Invitrogen). The sections were incubated with primary antibodies overnight at 4°C and biotinylated secondary antibodies for 40 minutes at 37°C. Immunohistochemical detection was performed with the VECTASTAIN ABC kit (Vector Laboratories, Burlingame, CA) and diaminobenzidine tetrahydrochloride as the substrate. For immunofluorescence, secondary antibodies were directly conjugated to Cy3 or Cy2 and incubated for 4 hours at room temperature.
The primary antibodies used were as follows: anti-SST (1:3000; Santa Cruz sc-7819 [Santa Cruz Laboratories, Santa Cruz, CA]), anti-ghrelin (1:200; Santa Cruz sc-10368), anti–5-hydroxytryptophan (5-HT/Serotonin; 1:50,000; ImmunoStar 20080 [Hudson, WI]); anti-chromogranin A (1:3,000; DiaSorin 20085 [Stillwater, MN]), anti-GLP-1 (1:500; Abcam ab26278 [Cambridge, UK]), and anti-CCK (1:100; Santa Cruz sc-21617). Rabbit anti-ARX polyclonal antibody (1:500) was a gift from Dr Kanako Miyabayashi (Kyushu University, (21)). Sections were stained with hematoxylin and eosin (H&E) or Oil-Red-O according to standard protocols. Oil-Red-O staining was performed using frozen sections.
Hormone-positive cells from different regions of the intestine were counted and normalized to the respective epithelial area of the same or adjacent sections yielding cell numbers per square millimeter tissue area. Epithelial area was measured with an Aperio Image Analysis System (Leica, Germany). At least 3 control and 3 mutant animals were used for each hormone analysis in the intestine. P-values were obtained using a Student t test.
RESULTS
ARX Polyalanine Expansion Related to Pseudo-Obstruction
To determine the intestinal consequence of an ARX polyalanine expansion, we identified a patient with a 335-336ins(GGC)7 mutation in ARX who presented with infantile spasms, hypotonia, and severe intellectual disability, and was also diagnosed with chronic intestinal pseudo-obstruction. This expansion in the first polyalanine tract is one of the more common in the ARX gene (25). For most of his life, this patient had feeding intolerance manifesting as abdominal pain and vomiting. He had multiple abdominal surgeries to place feeding tubes and had a Nissen fundoplication that was repeated 3 times. At the age of 8, his inability to tolerate enteral feeds and weight loss became so severe that he required total parenteral nutrition, which has been his maintenance nutrition for the past 5 years. No mechanical obstruction was ever identified. Antroduodenal manometry revealed a diagnosis of neuropathic intestinal dysmotility based on antral hypomotility, abnormal phase 3 migrating motility complexes during fasting, and cluster contractions in the duodenum. In the process of his evaluation, 2 upper endoscopies with biopsies were performed before initiation of total parenteral nutrition. No pathologic diagnosis was identified in the esophagus, antrum, or duodenum by H&E staining.
Because Arx regulates enteroendocrine development in mice (17,30), we analyzed the enteroendocrine populations in the duodenum from the patient biopsies (Fig. 1). Immunohistochemistry from 2 temporally distinct biopsies for this patient were compared with 3 or 4 age-matched control patients (no diagnosis of celiac, eosinophilic, or inflammatory bowel disease). Of note, the CCK and GLP-1 populations were dramatically reduced in the ARX(GGC)7 patient biopsies; only 4 CCK cells and 2 GLP-1 cells were detected (Fig. 1B, C). The SST population was also significantly reduced (Fig. 1D). The chromogranin A population was unchanged (Fig. 1A).
FIGURE 1.

Enteroendocrine dysgenesis in a patient with an ARX(GGC)7 mutation. Control human tissue is represented in A–D and patient tissue (ARXGGC7) in E–H. Hormones stained were CgA in A and F; CCK in B and G; GLP-1 in C and H; and SST in D and I. The cell counts are listed below each panel, with the P value for each hormone. ARX = aristaless-related homeobox; CCK = cholecystokinin; CgA = chromogranin A; GLP = glucagon-like peptide; SST = somatostatin.
In the intestinal null mouse model, the chromogranin A population is also unchanged, with a significant decrease in CCK and GLP-1 cells. In the mouse model, SST cells are, however, significantly upregulated (16,17). To explore whether these phenotypic differences were caused by null versus polyalanine expansion mutations or interspecies differences, we next analyzed the corresponding polyalanine expansion mouse model (Arx(GCG)7, (29)).
Arx Polyalanine Expansion Mice Have Failure to Thrive and Fat Malabsorption
First, we determined the growth characteristics of the male Arx(GCG)7 mice compared with male littermate controls. Starting at P5, the mutant Arx(GCG)7 mice are significantly smaller than their littermate controls (Fig. 2A). This difference persists into adulthood (Fig. 2B). The adult animals have a seizure disorder as previously described, and die between 2 and 3 months of age ((29), Eric Marsh, personal communication). The tissue histology is normal by H&E staining (supplemental Fig. 1).
FIGURE 2.

Arx(GCG)7 mice have poor postnatal growth and lipid malabsorption. A, Growth curves for P0-21. B, Growth curves for postnatal weeks 3–6. Oil-Red-O stains of stool (C, G, K, L) and intestinal tissue (D–F and H–J). Samples from P5 control are in C–F and P5 ArxGCG7 in G–J, whereas 4-week-old control is K and ArxGCG7 is L. ARX = aristaless-related homeobox.
Because fat malabsorption has been described in mice lacking enteroendocrine cells as a result of Neurog3 mutations (5), we analyzed stool and tissue by Oil-Red-O. Before weaning, when the neonatal mice are on a high-fat diet while nursing, there was excess fat in the stool smear by qualitative analysis (Fig. 2C,G) correlating with poor weight gain. Furthermore, when investigating tissue morphology, we found a large amount of Oil-Red-O staining in the ileum and colon of mutant Arx(GCG)7 mice, whereas the control littermates had minimal lipid present in those areas (Fig. 2D–F,H–J). Once mice were weaned onto a standard low-fat diet, the stool smears were comparable between control and mutant Arx(GCG)7 littermates (Fig. 2K,L).
Arx Polyalanine Tract Expansion Impairs Enteroendocrine Development
Arx is expressed specifically in subpopulations of enteroendocrine cells (30,31). To determine the changes in enteroendocrine populations as a consequence of the Arx polyalanine expansion, we determined the messenger RNA (mRNA) and protein expression of the intestinal endocrine subpopulations at several time points: postnatal days 0–1 (P0), postnatal day 14 (P14), and adult (5–7 weeks of age).
At birth, the Arx(GCG)7 mutants had significantly reduced numbers of CCK and GLP-1 containing cells in the duodenum (Fig. 3I–P). This change corresponded to reduced mRNA expression of CCK and preproglucagon, the precursor to GLP-1. SST expression was significantly increased by mRNA and the number of hormone-positive cells (Fig. 3Q–T). Both chromogranin A and serotonin (5-HT) cell number and mRNA levels were unchanged (Fig. 3A–H).
FIGURE 3.

Enteroendocrine population changes in the P0 duodenum of Arx(GCG)7 mice. Hormone staining is pictured for ChrA (A, B), 5-HT (E, F), CCK (I, J), GLP-1 (M, N), and SST (Q, R). Control tissue is in the left panel (A, E, I, M, Q) and ArxGCG7 tissue in the left-middle panel (B, F, J, N, R). Expression for mRNA was quantified by RT-PCR for the right-middle panels (C, G, K, O, S) and cell counts for protein expression on the far right panel (D, H, L, P, T) for each respective hormone: ChrA (C, D), 5-HT/Tph1 (G, H), CCK (K, L), GLP-1/preproglucagon (O, P), and SST (S, T). The dark bars designate controls, whereas the open bars designate ArxGCG7. ∗∗∗Designated P value is <0.05. ARX = aristaless-related homeobox; CCK = cholecystokinin; ChrA = chromogranin A; GLP = glucagon-like peptide; mRNA = messenger RNA; RT-PCR = real-time polymerase chain reaction; SST = somatostatin.
In the P14 duodenum (supplemental Fig. 2), the polyalanine expansion mice demonstrated continued depletion of CCK and GLP-1–producing cells (Fig. S2I–P). SST was significantly upregulated (Fig. S2Q–T). Although chromogranin A expression was unchanged (Fig. S2A–D), there was a significant, though mild, increase in 5-HT-expressing cells (Fig. S2E–H).
These hormone changes were also present in the ileum, with increased SST and decreased GLP-1 at P0 and P14 by cell counts and qRT-PCR (supplemental Fig. 3). We also assayed mRNA expression in the duodenum of older animals (5–7 weeks) to find the same downregulation of preproglucagon and CCK and upregulation of SST mRNAs without a change in chromogranin A (Fig. 4).
FIGURE 4.

Enteroendocrine hormone expression changes in adult mouse duodenum. Expression of mRNA was quantified by RT-PCR for chromogranin A (A), SST (B), preproglucagon (C), and CCK (D). The dark bars designate controls, whereas the open bars designate ArxGCG7. ∗∗∗Designated P value is <0.05. ARX = aristaless-related homeobox; CCK = cholecystokinin; mRNA = messenger RNA; RT-PCR = real-time polymerase chain reaction; SST = somatostatin.
Arx Protein is Lost in Polyalanine Expansion Mouse Mutants
The hormone changes in the polyalanine expansion mouse mutants phenocopy the Arx loss of function in the intestine (16,17). To determine whether the similarity is because of changes in expression of Arx, we first tested whether Arx was transcribed in the polyalanine expansion mutants (Fig. 5A). At P0 and P14, the mRNA levels were the same as control littermates. In adult mutant Arx(GCG)7 animals, Arx mRNA was, however, significantly downregulated. Next, we tested protein expression in control and mutant littermates. The Arx antibody used recognizes both wild-type and Arx(GCG)7 protein, as previously reported (29,32). We did not detect any Arx-positive cells in the P0 or adult duodenum of Arx(GCG)7 mutants (Fig. 5B–G), suggesting that the Arx(GCG)7 mouse model approaches an intestinal null situation. To determine whether this loss of ARX protein was also found in human tissue, we stained the patient slides. In the human ARX(GGC)7 tissue, ARX protein was present at the same levels as in control tissue, despite the polyalanine expansion (Fig. 5H, I).
FIGURE 5.
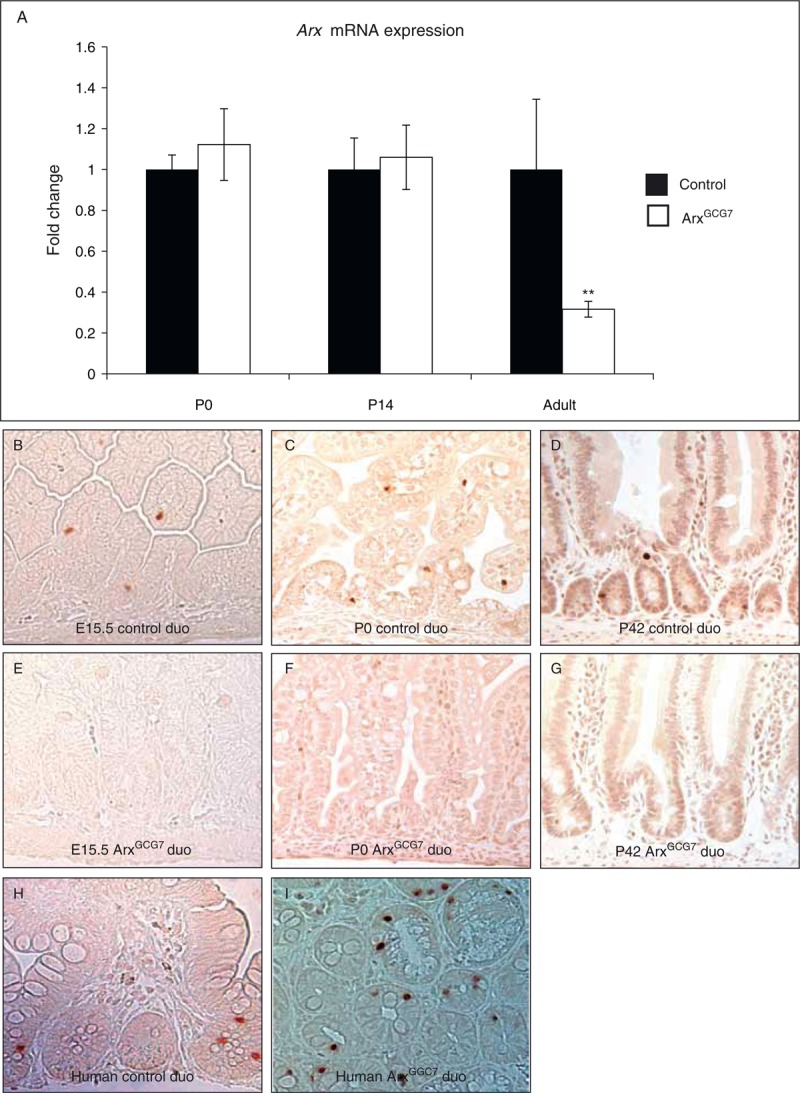
Expression of ARX/Arx mRNA and protein. mRNA expression is depicted in (A), with the dark bars for control samples and the open bars for ArxGCG7 mouse model. Staining for Arx protein in the control mouse duodenal tissue (B–D) and ArxGCG7 mouse model (E–G) at E15.5 (B, E), P0 (C, F), and P42 (D,G). Staining for Arx protein in control human duodenal tissue (H) and patient ArxGGC7 tissue (I). ∗∗Designated P value is <0.05. ARX = aristaless-related homeobox; mRNA = messenger RNA.
DISCUSSION
With recognition of the neurologic phenotype of ARX-related disorders, it was also noted that approximately 50% of patients with XLAG with ARX loss-of-function mutations have a severe congenital enteropathy that is fatal in some cases (15). The case highlighted here demonstrates changes in the enteroendocrine population with a polyalanine expansion of the ARX protein, the more common type of mutation (25,26). In the presence of the ARX(GGC)7 protein, the CCK, SST, and GLP-1 lineages are not specified, although the chromogranin A population is present at normal density. The role of ARX was previously tested in human intestinal organoids derived from embryonic stem cells, using small hairpin RNA-mediated intestinal loss of function (16). With 60% to 80% knockdown of ARX, the preproglucagon and CCK populations were lost and the SST population was unchanged. Thus, the influence of ARX on the SST population appears to differ in human tissue compared with the Arx loss-of-function mouse model, wherein the SST population is increased (16,17).
Arx protein acts as both a transcriptional activator and transcriptional repressor (33). In the mouse brain, complete loss of Arx results in impaired tangential and radial migration of GABAergic interneurons. Only tangential migration is, however, impaired in the Arx(GCG)7 mouse model, which could explain the less severe phenotype (29). Several downstream targets have been identified that are differentially affected by the Arx(GCG)7 protein as opposed to an Arx null in the mouse brain (34). In the pancreas, Arx activates the α cell program while repressing the β cell program (35,36). In the Arx(GCG)7 mouse model, all α cells are still lost, but without any increase in β cells, suggesting that the Arx(GCG)7 protein in early development is still capable of repression of β cells, but not activation of the α cell program (35).
Unfortunately, the mouse model of the corresponding Arx first tract polyalanine expansion does not fully recapitulate the human disease because the Arx(GCG)7 protein is degraded in the mouse intestine. In contrast, the ARX(GGC)7 protein is still present in human tissue, although it is not fully functional. The hormone changes in the Arx(GCG)7 mouse model are similar to those found in the Arx intestinal null model, consistent with the fact that all Arx(GCG)7 protein is lost (16,17). The reduced levels of the Arx(GCG)7 protein have also been described in the brain of the mouse model (29,32), although some Arx(GCG)7 protein is still present.
The patient described here demonstrates a unique phenotype of pseudo-obstruction without congenital diarrhea, compared with patients with ARX loss-of-function mutations. At this time, we are not able to determine whether the enteroendocrine population changes are directly responsible for the motility disorder. The role of various enteroendocrine subpopulations in gut motility is, however, well-recognized through exogenous agonist and antagonist studies (37). Many of the intestinal hormones inhibit gastric or small bowel motility. The relation is, however, often complex and dynamic. For example, in pediatric patients, exogenous octreotide (an SST analogue) inhibits gastric motility and promotes small intestine migrating motility complexes (38).
Motility studies on mouse models with alterations in the enteroendocrine cells are necessary to further understand the contribution of these cells in regulation of how the bowel moves in fasting and fed states. Although expression of Arx by cross-sectional analysis in the bowel is limited to the enteroendocrine cells (16,17), it is possible that a small subset of enteric nervous system cells expresses ARX/Arx and contributes to the phenotype, or, alternatively, exerts direct or indirect effects in the muscular layers of the bowel. Another confounding variable for this case is the history of abdominal surgeries; it is difficult to determine whether his bowel disorder led to the multiple surgeries or what dysfunction was attributable to multiple surgeries. Finally, his long-standing seizure disorder and medications could also contribute to the phenotype.
Enteroendocrine dysgenesis is becoming increasingly recognized for its role in congenital diarrhea, irritable bowel syndrome, and inflammatory bowel disease (39). With NEUROG3 mutations (1) or AIRE mutations associated with APECED (6,7) almost all enteroendocrine cells are lost, leading to congenital diarrhea. Unique to Arx loss of function in the mouse intestine (16,17) and PC1/3 mutations in humans, loss of only a subset of hormone producing cells can lead to congenital diarrhea (9) despite normal chromogranin A and serotonin/5-HT staining. The determination of which enteroendocrine subsets are responsible for the malabsorptive or motility phenotype in enteroendocrine dysgenesis will provide an excellent step forward in identifying therapeutic targets.
Supplementary Material
Acknowledgments
The authors thank members of the Molecular Pathology and Imaging Core in the Center for Molecular Studies in Digestive and Liver Disease (P30-DK050306) for their assistance and providing reagents. The authors also thank members of The Children's Hospital of Philadelphia Pathology Core Laboratories for their assistance in slide processing, especially Dr Tricia R. Bhatti. They also thank Dr Eric D. Marsh for interesting discussions, sharing reagents, referring the patient, and review of this manuscript, and Almedia McCoy for assistance with mouse breeding and handling.
Footnotes
N.A.T was supported by NIH K12-HD043245, Children's Hospital of Philadelphia Foerderer Grant; K.H.K. by NIH R37-DK053839; C.L.M. by NIH-DK078606, NIH-DK019525, and JDRF2-2007-730.
The authors report no conflicts of interest.
REFERENCES
- 1.Wang J, Cortina G, Wu SV, et al. Mutant neurogenin-3 in congenital malabsorptive diarrhea. N Engl J Med 2006; 355:270–280. [DOI] [PubMed] [Google Scholar]
- 2.May CL, Kaestner KH. Gut endocrine cell development. Mol Cell Endocrinol 2010; 323:70–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lee CS, Perreault N, Brestelli JE, et al. Neurogenin 3 is essential for the proper specification of gastric enteroendocrine cells and the maintenance of gastric epithelial cell identity. Genes Dev 2002; 16:1488–1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jenny M, Uhl C, Roche C, et al. Neurogenin3 is differentially required for endocrine cell fate specification in the intestinal and gastric epithelium. EMBO J 2002; 21:6338–6347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mellitzer G, Beucher A, Lobstein V, et al. Loss of enteroendocrine cells in mice alters lipid absorption and glucose homeostasis and impairs postnatal survival. J Clin Invest 2010; 120:1708–1721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hogenauer C, Meyer RL, Netto GJ, et al. Malabsorption due to cholecystokinin deficiency in a patient with autoimmune polyglandular syndrome type I. N Engl J Med 2001; 344:270–274. [DOI] [PubMed] [Google Scholar]
- 7.Posovszky C, Lahr G, von Schnurbein J, et al. Loss of enteroendocrine cells in autoimmune-polyendocrine-candidiasis-ectodermal-dystrophy (APECED) syndrome with gastrointestinal dysfunction. J Clin Endocrinol Metab 2012; 97:E292–300. [DOI] [PubMed] [Google Scholar]
- 8.Scopsi L, Gullo M, Rilke F, et al. Proprotein convertases (PC1/PC3 and PC2) in normal and neoplastic human tissues: their use as markers of neuroendocrine differentiation. J Clin Endocrinol Metab 1995; 80:294–301. [DOI] [PubMed] [Google Scholar]
- 9.Bandsma RH, Sokollik C, Chami R, et al. From diarrhea to obesity in prohormone convertase 1/3 deficiency: age-dependent clinical, pathologic, and enteroendocrine characteristics. J Clin Gastroenterol 2013; 47:834–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jackson RS, Creemers JW, Farooqi IS, et al. Small-intestinal dysfunction accompanies the complex endocrinopathy of human proprotein convertase 1 deficiency. J Clin Invest 2003; 112:1550–1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Martin MG, Lindberg I, Solorzano-Vargas RS, et al. Congenital proprotein convertase 1/3 deficiency causes malabsorptive diarrhea and other endocrinopathies in a pediatric cohort. Gastroenterology 2013; 145:138–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ohsie S, Gerney G, Gui D, et al. A paucity of colonic enteroendocrine and/or enterochromaffin cells characterizes a subset of patients with chronic unexplained diarrhea/malabsorption. Hum Pathol 2009; 40:1006–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Marsh ED, Golden JA. Noebels JL, Avoli MA, Rogawski RW, et al. Developing models of aristaless-related homeobox mutations. Jasper's Basic Mechanisms of the Epilepsies. Bethesda, MD: National Center for Biotechnology Information; 2012. [PubMed] [Google Scholar]
- 14.Collombat P, Mansouri A, Hecksher-Sorensen J, et al. Opposing actions of Arx and Pax4 in endocrine pancreas development. Genes Dev 2003; 17:2591–2603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kato M, Dobyns WB. X-linked lissencephaly with abnormal genitalia as a tangential migration disorder causing intractable epilepsy: proposal for a new term, “interneuronopathy”. J Child Neurol 2005; 20:392–397. [DOI] [PubMed] [Google Scholar]
- 16.Du A, McCracken KW, Walp ER, et al. Arx is required for normal enteroendocrine cell development in mice and humans. Dev Biol 2012; 365:175–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Beucher A, Gjernes E, Collin C, et al. The homeodomain-containing transcription factors Arx and Pax4 control enteroendocrine subtype specification in mice. PLoS One 2012; 7:e36449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhu X, Zhou A, Dey A, et al. Disruption of PC1/3 expression in mice causes dwarfism and multiple neuroendocrine peptide processing defects. Proc Natl Acad Sci U S A 2002; 99:10293–10298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gradwohl G, Dierich A, LeMeur M, et al. neurogenin3 is required for the development of the four endocrine cell lineages of the pancreas. Proc Natl Acad Sci U S A 2000; 97:1607–1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dobyns WB, Berry-Kravis E, Havernick NJ, et al. X-linked lissencephaly with absent corpus callosum and ambiguous genitalia. Am J Med Genet 1999; 86:331–337. [PubMed] [Google Scholar]
- 21.Kitamura K, Yanazawa M, Sugiyama N, et al. Mutation of ARX causes abnormal development of forebrain and testes in mice and X-linked lissencephaly with abnormal genitalia in humans. Nat Genet 2002; 32:359–369. [DOI] [PubMed] [Google Scholar]
- 22.Stromme P, Mangelsdorf ME, Scheffer IE, et al. Infantile spasms, dystonia, and other X-linked phenotypes caused by mutations in Aristaless related homeobox gene, ARX. Brain Dev 2002; 24:266–268. [DOI] [PubMed] [Google Scholar]
- 23.Bonneau D, Toutain A, Laquerriere A, et al. X-linked lissencephaly with absent corpus callosum and ambiguous genitalia (XLAG): clinical, magnetic resonance imaging, and neuropathological findings. Ann Neurol 2002; 51:340–349. [DOI] [PubMed] [Google Scholar]
- 24.Bienvenu T, Poirier K, Friocourt G, et al. ARX, a novel Prd-class-homeobox gene highly expressed in the telencephalon, is mutated in X-linked mental retardation. Hum Mol Genet 2002; 11:981–991. [DOI] [PubMed] [Google Scholar]
- 25.Shoubridge C, Fullston T, Gecz J. ARX spectrum disorders: making inroads into the molecular pathology. Hum Mutat 2010; 31:889–900. [DOI] [PubMed] [Google Scholar]
- 26.Gecz J, Cloosterman D, Partington M. ARX: a gene for all seasons. Curr Opin Genet Dev 2006; 16:308–316. [DOI] [PubMed] [Google Scholar]
- 27.Hughes JN, Thomas PQ. Molecular pathology of polyalanine expansion disorders: new perspectives from mouse models. Methods Mol Biol 2013; 1017:135–151. [DOI] [PubMed] [Google Scholar]
- 28.Amiel J, Laudier B, Attie-Bitach T, et al. Polyalanine expansion and frameshift mutations of the paired-like homeobox gene PHOX2B in congenital central hypoventilation syndrome. Nat Genet 2003; 33:459–461. [DOI] [PubMed] [Google Scholar]
- 29.Kitamura K, Itou Y, Yanazawa M, et al. Three human ARX mutations cause the lissencephaly-like and mental retardation with epilepsy-like pleiotropic phenotypes in mice. Hum Mol Genet 2009; 18:3708–3724. [DOI] [PubMed] [Google Scholar]
- 30.Du A, McCracken KW, Walp ER, et al. Arx is required for normal enteroendocrine cell development in mice and humans. Dev Biol 2012; 365:175–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Beucher A, Gjernes E, Collin C, et al. The homeodomain-containing transcription factors Arx and Pax4 control enteroendocrine subtype specification in mice. PLoS One 2012; 7:e36449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lee K, Mattiske T, Kitamura K, et al. Reduced polyalanine-expanded Arx mutant protein in developing mouse subpallium alters Lmo1 transcriptional regulation. Hum Mol Genet 2014; 23:1084–1094. [DOI] [PubMed] [Google Scholar]
- 33.Fulp CT, Cho G, Marsh ED, et al. Identification of Arx transcriptional targets in the developing basal forebrain. Hum Mol Genet 2008; 17:3740–3760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nasrallah MP, Cho G, Simonet JC, et al. Differential effects of a polyalanine tract expansion in Arx on neural development and gene expression. Hum Mol Genet 2012; 21:1090–1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wilcox CL, Terry NA, Walp ER, et al. Pancreatic alpha-cell specific deletion of mouse Arx leads to alpha-cell identity loss. PLoS One 2013; 8:e66214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Courtney M, Gjernes E, Druelle N, et al. The inactivation of Arx in pancreatic alpha-cells triggers their neogenesis and conversion into functional beta-like cells. PLoS Genet 2013; 9:e1003934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wu T, Rayner CK, Young RL, et al. Gut motility and enteroendocrine secretion. Curr Opin Pharmacol 2013; 13:928–934. [DOI] [PubMed] [Google Scholar]
- 38.Di Lorenzo C, Lucanto C, Flores AF, et al. Effect of sequential erythromycin and octreotide on antroduodenal manometry. J Pediatr Gastroenterol Nutr 1999; 29:293–296. [DOI] [PubMed] [Google Scholar]
- 39.Moran GW, Leslie FC, Levison SE, et al. Enteroendocrine cells: neglected players in gastrointestinal disorders? Therap Adv Gastroenterol 2008; 1:51–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.