

Abstract
Objectives
To describe the natural history of pulmonary hypertension (PH) and the risk of death and pulmonary morbidity associated with the persistence of PH through the neonatal hospitalization for these infants.
Study design
We performed a retrospective cohort study of infants with CDH cared for at UCSF (2002-12). Infants with other major anomalies or syndromes were excluded (n=43). Clinical echocardiograms were performed weekly for up to 6 weeks or until PH resolved off respiratory support or until hospital discharge. Echocardiograms were re-read by a blinded reviewer and categorized by severity of elevation in estimated pulmonary arterial pressure. PH was defined as ≥2/3 systemic blood pressure. Severity was determined by a hierarchy of ductus arteriosus level shunt, interventricular septal position, and tricuspid regurgitant jet velocity.
Results
Of 140 infants with ≥1 echo, 98 resolved their PH prior to death/discharge. Mean time to resolution was 18d (median 14d, IQR 8, 21d). Those with persistence of PH had a higher rate of ECMO (p<0.001) and death (p<0.001), and fewer ventilator-free days (p<0.001). Persistence of PH at 14d predicted mortality (AUC 0.87) and adverse respiratory outcomes (AUC 0.80-0.83).
Conclusions
The majority of infants with congenital diaphragmatic hernia (CDH) resolve PH between 1 and 3 weeks of life. At 2 weeks of age, severity of PH by echocardiogram strongly predicts short-term pulmonary morbidity and death. Further evaluation of physiological alterations during that time may lead to novel therapies for severe CDH.
Keywords: Pulmonary vascular resistance, Bronchopulmonary dysplasia, Chronic lung disease
Congenital diaphragmatic hernia (CDH) is one of the most common major congenital anomalies, occurring in 1 in 4,000-5,000 live births[1, 2]. CDH is associated with significant morbidity and mortality, primarily due to lung hypoplasia and pulmonary hypertension (PH) [3-7]. However, little is known about the natural history of PH in this population. Previous studies that have attempted to characterize PH in infants with CDH have limited utility due to the inclusion of data that were collected during an earlier period of management strategies or used echocardiograms associated with clinical events, potentially biasing the results[5, 8, 9]. We previously demonstrated, with a limited sample size, that PH at two weeks was predictive of short-term clinical outcomes, with infants discharged on room air resolving their PH to a greater extent than those with persistence of PH[10]. In contrast, healthy term neonates have a rapid fall in pulmonary vascular resistance (PVR) by 72 hours of life [11].
Although echocardiography for the assessment of pulmonary hypertension and prediction of outcomes in infants with CDH has previously been studied[4, 5, 9, 10], a longitudinal evaluation of standard clinical echocardiography in a large, contemporary cohort has not been reported. Our aim was to evaluate the natural history of PH in infants with CDH, based on routine echocardiography over the first 6 weeks of life. We also to evaluated PH on routine echocardiography at various time points as a biomarker for predicting the risk of death and short-term pulmonary morbidity among infants with CDH. Finally, we determined the time point that was the best predictor of a poor outcome.
Methods
This retrospective cohort study of infants cared for at the University of California San Francisco (UCSF) Benioff Children's Hospital (2002-12) included patients with ≥1 echocardiogram and a posterolateral Bochdalek-type CDH. Patients with multiple congenital anomalies or a confirmed or suspected syndromes were excluded. During this period, all infants with CDH had routine weekly echocardiograms per clinical protocol. Demographic and clinical data were collected by chart review. For this study, echocardiograms were re-reviewed by a reader blinded to clinical outcomes of the infants. Outcomes of interest were death prior to discharge, prolonged intubation (≥28 days), and chronic lung disease (any respiratory support at ≥56 days of life or discharge on oxygen prior to 56 days, based on the NICHD proposed definition of moderate-to-severe bronchopulmonary dysplasia for infants ≥32 weeks gestational age)[12]. The UCSF Institutional Review Board approved this study.
Beginning in 2002, infants with CDH had routine echocardiograms (Acuson Sequoia C256 and C512 and SC2000, Mountain View CA) within the first 48 hours of life, then weekly for the first 6 weeks or until death, discharge, or resolution of PH off respiratory support. Infants did not routinely receive additional sedation for echocardiography. Blood pressure, heart rate, and weight at the time of the scan were recorded. Standard clinical echocardiographic views were used to estimate PAp compared with systemic systolic blood pressure at the time of the scan, to categorize degree of PH. Infants were classified as follows: <2/3 systemic systolic pressures (no/mild PH), ≥2/3 systemic-to-systemic pressure (moderate PH), or systemic-to-suprasystemic pressure (severe PH), as previously described [10]. Classification was by a hierarchy of assessments: (1) direction and velocity of ductus arteriosus flow using the Bernoulli equation, (2) interventricular septum position (parasternal short axis), graded as normal, flattened, or D-shaped and (3) right ventricular pressures using peak tricuspid regurgitant (TR) jet velocity estimated by the modified Bernoulli equation (assuming a right atrial pressure of 0mmHg).
All patients were intubated immediately after birth and managed with a gentle ventilation strategy of pressure limitation and permissive hypercapnea. High frequency oscillatory ventilation was used for patients with refractory hypercapnea or high peak inspiratory pressures. Inhaled nitric oxide (iNO) was initiated at 20 ppm for poor oxygenation secondary to PH. Patients received extracorporeal membrane oxygenation (ECMO) support if they continued to have refractory hypoxemia or evidence of inadequate oxygen delivery despite optimal support. Surgical repair followed clinical stabilization. Clinical practice evolved over the study years, with increasing use of assisted ventilation (earlier extubation to nasal CPAP), and a decrease in duration of ECMO support, following an internal review demonstrating poor survival to discharge among infants with CDH on ECMO support for ≥10d.
Statistical Analyses
Data were analyzed by chi squared and rank sum or t-tests as appropriate. Time to resolution of PH (<2/3 systemic pressure) was assessed by Kaplan-Meier survival curves. Sensitivity, specificity, positive predictive value (PPV), and negative predictive value (NPV) were calculated to assess the utility of PH severity classification by echocardiogram at 1, 2, 3, 4, and 6 weeks. Area under the receiver-operator characteristic curves (AUC) was used to determine the overall predictive accuracy at these time points (Stata 12.0, College Station, TX).
Results
One hundred forty infants met inclusion criteria (including 39 infants previously reported in our prospective study [10]); 43 were excluded due to multiple anomalies, a genetic syndrome, or Morgagni hernia and 5 infants were excluded for having no available echocardiogram (Figure 2; available at www.jpeds.com). Of those included, most infants were term (mean gestational age 38±2.4 weeks) with a predominance of male infants and left-sided defects. There was a range of severity; lung-to-head ratio was <1 in 36% of fetuses with measurements and liver was herniated into the thorax in 58% of 133 newborns. Prostaglandin E1 was used to maintain ductal patency in the setting of severe suprasystemic pulmonary hypertension in only 10% of infants, though this practice was not routinely considered for use until 2008 (Table I).
Figure 2. Study subject flow diagram.
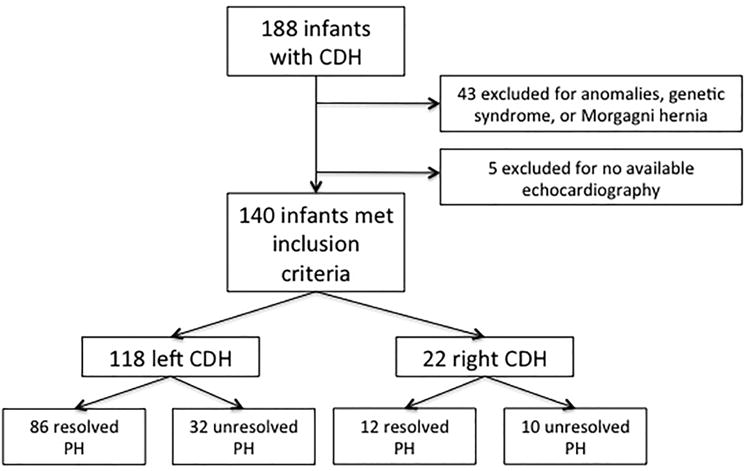
Table 1. Cohort characteristics and outcomes in 140 infants with CDH and ≥1 echocardiogram.
| All infants (N=140) | PH Status Prior to Death/Dishcarge | P value | ||
|---|---|---|---|---|
|
| ||||
| Resolved (N=98) | Unresolved (N=42) | |||
| Characteristics | ||||
|
| ||||
| Gestational age at birth (weeks) | 38 ± 2.4 | 38 ± 2.4 | 38 ± 2.4 | 0.98 |
| Birth weight (kilograms) | 3.1 ± 0.6 | 3.1 ± 0.6 | 3.1 ± 0.5 | 0.88 |
| Male | 78 (56%) | 50 (51%) | 28 (67%) | 0.09 |
| Left-sided hernia | 118 (84%) | 86 (88%) | 32 (76%) | 0.085 |
| Intra-thoracic liver | 77/133 (58%) | 44 (47%) | 33 (83%) | <0.001 |
| Fetal lung-to-head ratio (n=56)* | 1.2 ±0.5 | 1.3 ± 0.5 | 1.03 ± 0.4 | 0.05 |
| Lung-to-head ratio ≤1 | 31/85 (36%) | 13 (13%) | 18 (43%) | <0.001 |
| Fetal surgery | 12 (9%) | 5 (5%) | 7 (17%) | 0.04 |
| Prostoglandin E1 for ductal patency | 14 (10%) | 5 (5%) | 9 (21%) | 0.006 |
|
| ||||
| Outcomes | ||||
|
| ||||
| Death | 27 (19%) | 2 (2%) | 25 (60%) | <0.001 |
| Extracorporeal membrane oxygenation (ECMO) | 15 (11%) | 4 (4%) | 11 (26%) | <0.001 |
| Non-primary repair§ | 79 (59%) | 51 (52%) | 28 (80%) | 0.004 |
| Inhaled nitric oxide (iNO) | 78 (56%) | 42 (43%) | 36 (86%) | <0.001 |
| Ventilator-free days¶ | 13 ± 9 | 16 ± 8 | 5 ± 8 | <0.001 |
| Home on supplemental oxygen** | 27/113 (24%) | 23/91 (25%) | 4/12 (33%) | 0.51 |
| Time to discharge (days) | 39 ±27 | 39 ±26 | 40 ±32 | 0.86 |
All results reported as mean(±SD) or N(%)
Measured on fetal ultrasound between 20 and 29 weeks gestational age
Surgical repair requiring patch or muscle flap
Ventilator-free days by 28 days of age; Wilcoxon rank sum test
Does not include infants who died prior to DC
PH was classified for 597 echocardiograms. Of these, only 38% of scans had a quantifiable TR jet. Studies demonstrating severe PH were significantly more likely to have a measurable TR envelope than those with mild/no PH (81% versus 19%, p<0.001). The ductus arteriosus was open with measurable direction/velocity in 58% of scans, specifically, 85% in the first 48 hours of life and 43%, 26%, 23%, and 19% at 1, 2, 3 and 4 weeks of life, respectively. Septal position was interpretable in 97% of scans. All three variables were evaluable in only 20% of echocardiograms. Week one echos were performed at a median of 8 days of life (IQR 7-9d), week two at a median of 15d (IQR 14-17d), week three at a median of 22d (IQR 21-28d), week four at a median of 29.5d (IQR 28-37d), and week six at a median of 49d (IQR 42-64d).
Of the 140 patients, 42 (30%) never resolved their PH prior to death or discharge. Among those who did resolve, the mean time to resolution was 17 days (median 14 days, IQR 7, 21; Figure 1). Although overall mortality was not high (19%), the risk of death was significantly lower among infants who resolved their PH as compared with those who did not (1% versus 60%, p<0.001). Only two infants died after their PH resolved. One was a preterm infant with necrotizing enterocolitis. The other was a term infant who died of respiratory failure after resolution of PH. Patients who resolved their PH had decreased rates of iNO and prostaglandin E1 use, need for ECMO support and non-primary repair, and an increase in ventilator-free days by 28 days of age (Table I). Right-sided defects were more common among those with no resolution of PH, although this did not reach statistical significance.
Figure 1.
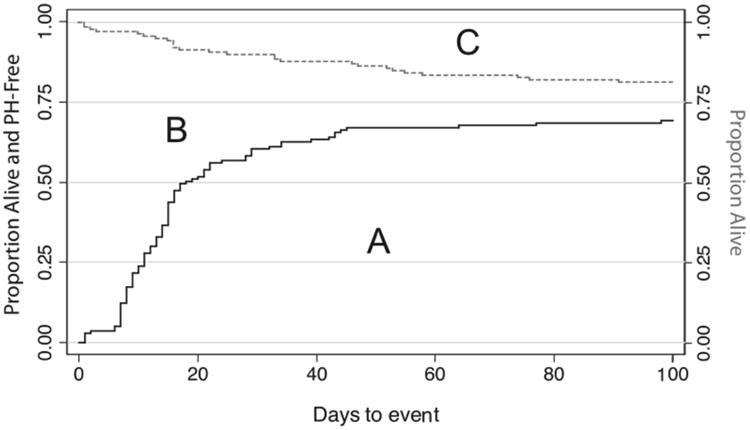
Kaplan-Meier curves showing the time to resolution of pulmonary hypertension (black solid line), and time to death (grey dashed line) among study infants. Area A Represents the proportion of infants alive and PH-free; (B) represents the proportion of infants alive with PH; (C) represents the proportion of infants died.
Among the 8 infants who resolved their PH within the first week of life, there were no subsequent adverse outcomes including death, prolonged intubation, or prolonged respiratory support at 56d. However, the overall predictive accuracy of PH for death, death or prolonged intubation, and death or prolonged respiratory support was moderate at one week of life (AUC 0.73, 0.72 and 0.7, respectively; Table II). Thirty-nine additional infants resolved their PH between weeks 1 and 2. The best predictive accuracy of severity of PH was at 2 weeks, with AUC for each of the three outcomes of 0.87, 0.83 and 0.8, respectively. There was ongoing improvement at 3 weeks of life with another 26 infants resolving their PH. At this time point, the AUC for the three outcomes ranged from 0.74 to 0.83 (Table II). Infants with both right and left-sided CDH and persistence of PH at 3 weeks were equally likely to avoid adverse outcomes (31% vs. 36%, respectively, p=1.0). However, those infants with liver herniation were less likely to avoid adverse outcomes than those without liver herniation (23% vs. 70%, p=0.008). Following the third week of life, there was a drop in the rate of resolution of PH; only 7 and 9 additional infants resolve their PH by 4 and 6 weeks of life. Test diagnostics for severity of PH and outcome, among those infants still hospitalized, are only moderate at 4 weeks of age but patients with persistence of severe PH at 4 and 6 weeks of life had a high risk of death (71% and 60% respectively).
Table 2. Outcomes in infants alive with and without PH at 1,2,3,4, and 6 weeks.
| Death | Death or Prolonged Intubation (≥28 d) | Death or Prolonged Respiratory Support (≥56 d) or Home Oxygen* | |||||||
|---|---|---|---|---|---|---|---|---|---|
|
| |||||||||
| % Outcome (N) | P value | AUROC (95% CI) | % Outcome (N) | P value | AUROC (95% CI) | % Outcome (N) | P value | AUROC (95% CI) | |
|
| |||||||||
| Week 1 (n=136) § | |||||||||
|
| |||||||||
| With PH ¶(n=128) | 18% (23/128) | p=0.35 | 0.73 | 25% (32/128) | p=0.20 | 0.72 | 41% (52/128) | p=0.023 | 0.7 |
| Without PH (n=8) | 0% (0/8) | (0.64, 0.82) | 0% (0/8) | (0.65, 0.81) | 0% (0/8) | (0.62, 0.79) | |||
|
| |||||||||
| Week 2 (n=133) | |||||||||
|
| |||||||||
| With PH (n=86) | 23% (20/86) | p<0.001 | 0.87 | 34% (29/86) | p<0.001 | 0.83 | 55% (47/86) | p<0.001 | 0.8 |
| No PH (n=47) | 0% (0/47) | (0.81, 0.94) | 0% (0/47) | (0.75, 0.91) | 6% (3/47) | (0.72, 0.88) | |||
|
| |||||||||
| Week 3 (n=128) | |||||||||
|
| |||||||||
| With PH (n=55) | 27% (15/55) | p<0.001 | 0.83 | 40% (22/55) | p<0.001 | 0.77 | 65% (36/55) | p<0.001 | 0.74 |
| No PH (n=73) | 0% (0/73) | (0.70, 0.95) | 3% (2/73) | (0.64, 0.89) | 12% (9/73) | (0.64, 0.83) | |||
|
| |||||||||
| Week 4 (n=126) | |||||||||
|
| |||||||||
| With PH (n=46) | 28% (13/46) | p<0.001 | 0.76 | 43% (20/46) | p<0.001 | 0.72 | 65% (30/46) | p<0.001 | 0.73 |
| No PH (n=80) | 0% (0/80) | (0.59, 0.93) | 3% (2/80) | (0.58, 0.86) | 16% (13/80) | (0.64, 0.83) | |||
|
| |||||||||
| Week 6 (n=123) | |||||||||
|
| |||||||||
| With PH (n=34) | 26% (9/34) | p<0.001 | 0.91 | - - | - | - | 62% (21/34) | p<0.001 | 0.63 |
| No PH (n=89) | 1% (1/89) | (0.80, 1.00) | 21% (19/89) | (0.42, 0.82) | |||||
Per definition of bronchopulmonary dysplasia in infants >32 weeks gestational age [16]. Nine infants were discharged on O2 prior to 56 days median 37d, range 20-55)
Four infants died prior to one week echocardiogram
PH is defined as as estimated pulmonary arterial pressures ≥2/3 systemic pressures
Abbreviations: AUROC - Area under the receiver operating curve; CI - confidence interval; PH - pulmonary hypertension
The majority of infants who resolved their PH did so between 1 and 3 weeks of age (65/98), and there were no deaths among those infants who resolved their PH by 3 weeks of life. Severity of PH by echo had the strongest relationship to outcome at 2 weeks. Detailed test diagnostics at this time point for various thresholds of severity of PH are presented in Table III.
Table 3. Test characteristics of echocardiography at 2 weeks of life.
| Outcome | Sensitivity | Specificity | Positive Predictive Value | Negative Predictive Value | AUROC (95%CI) |
|---|---|---|---|---|---|
| Death | |||||
|
| |||||
| Any PH | 100% | 48% | 31% | 100% | 0.81 (0.76, 0.86) |
| Severe PH | 74% | 68% | 36% | 92% | 0.73 (0.60, 0.86) |
| Echocardiography Overall§ | - | - | - | - | 0.87 (0.80, 0.94) |
|
| |||||
| Death or Prolonged Intubation (≥28 d) | |||||
|
| |||||
| Any PH | 97% | 51% | 43% | 98% | 0.81 (0.74, 0.88) |
| Severe PH | 58% | 67% | 40% | 81% | 0.64 (0.53, 0.75) |
| Echocardiography Overall | - | - | - | - | 0.83 (0.75, 0.91) |
|
| |||||
| Prolonged Respiratory Support (≥56 d) or Home Oxygen | |||||
|
| |||||
| Any PH | 84% | 54% | 58% | 82% | 0.79 (0.72, 0.88) |
| Severe PH | 47% | 63% | 49% | 61% | 0.61 (0.54, 0.69) |
| Echocardiography Overall | - | - | - | - | 0.80 (0.72, 0.88) |
“Any PH” defined as estimated pulmonary arterial pressures ≥2/3 systemic blood pressures
“Severe PH” defined as estimated pulmonary pressures systemic-to-suprasystemic blood pressures
“Echocardiography overall” defined as three-level classification of estimated pulmonary arterial pressure: < 2/3 systemic blood pressure, ≥ 2/3 systemic blood pressure, systemic-to-suprasystemic blood pressure
Abbreviations: AUROC - area under the reveiver operating curve; CI - confidence interval; PH - pulmonary hypertension
To investigate the effects of potential variations in treatment practice over time, we assessed the rates of outcomes over three separate 3-4 year time periods: 2002-2005 (n=41), 2006-2008 (n=36), and 2009-2012 (n=63). There was no difference by era in mortality (22%, 17%, and 19%, p=0.84), chronic lung disease at 56d (28%, 25%, and 27%, p=0.98), or home O2 use (25%, 23%, and 24%, p=0.99). However, there was a decrease in the proportion of infants with prolonged intubation over time (19%, 12%, and 0%, p=0.007) likely secondary to increasing use of non-invasive modes of ventilatory support, and a decrease in the mean age of death (48±20d, 34±19d, and 18±25d, p=0.02).
Discussion
Pulmonary hypertension is a major cause of morbidity and mortality in patients with CDH[3-5, 13]. Persistence of PH in infants with CDH is associated with mortality [5, 8, 10], but it is also true that elevated PVR is part of the normal newborn transition, and the time course of resolution of PH in infants with CDH who do well has not been well established. We report the largest group of infants with isolated CDH who have undergone routine weekly echocardiograms, regardless of disease severity. This approach provides an unbiased natural history of the persistence of PH by week of life. In this study, persistence of PH in infants with CDH after 2-3 weeks of age is abnormal and associated with a worse prognosis with respect to survival and adverse short-term pulmonary outcomes.
In healthy infants, estimated PAp falls rapidly after birth, achieving a level <2/3 systemic pressure by 24 hours of life[11]. This normal fall in PVR is delayed in infants with CDH, even in those who go on to do well. In our cohort, estimated PAp fell to < 2/3 systemic pressure by 1 week of life in only 6% (8/140 infants). In fact, half of our cohort (73/140) achieved this milestone by 3 weeks of age, and all of these infants survived with low rates of respiratory morbidity (9/73, or 12% with prolonged respiratory support). This timeline suggests that the usual decrease in pulmonary vascular tone seen with the transition to extrauterine life is impaired in infants with CDH. Whether this is due to persistence of abnormalities in fetal biochemical pathways, or to subsequent postnatal insults related to supportive treatment, is unknown[14-18].
Similarly, among those infants who fail to transition to a lower estimated PAp due to decrease in PVR by 2 weeks of age, we previously demonstrated fetal derangements of a variety of inflammatory markers that are implicated in pulmonary vascular remodeling[18], and substantial neonatal elevations in endothelin-1 (ET1)[5]. These differences in ET1 levels emerge at 1 week of age, and become prominent at 2 weeks, suggesting that, in addition to more severe lung hypoplasia (as suggested by evaluating the characteristics of those with resolved versus unresolved PH; Table I), secondary effects on the vasculature may be responsible for persistence of PH in this population, with further delay in the transition toward normal pulmonary vascular tone, or a failure to transition altogether. A substantial proportion of those with PH at 3 weeks either die or remain ventilated beyond 28 days (22/55; Table II). Thus, the effects of ventilation and oxygen exposure due to the underlying lung hypoplasia may account for this observation. Further, those infants with persistence of PH at 3 weeks who avoid adverse outcomes are also less likely to have liver herniation, suggesting less severe lung hypoplasia in that group of infants.
By assessing the risk of important outcomes including death and pulmonary morbidity as predicted by presence or absence of PH at weekly time points, we have determined the optimal timing, at 2 weeks of age, for severity of PH by echocardiography as a prognostic biomarker. A similar time point is suggested by data from Dillon et al, who found 100% survival among infants with CDH who had estimated PAp <50% systemic pressure by 3 weeks of life, but only 50% among those with persistence of PH[5]. For purposes of further research, PH at 2 weeks of age could 1) provide a robust classification of severity of CDH[18], 2) identify infants for early novel therapeutic interventions, and 3) serve as a surrogate, or interim, outcome, for fetal intervention trial design. This time point is consistent with what we observed in our earlier, prospective study, and it may represent a “critical period” for biological changes in the lung and pulmonary vascular bed, similar to what has been described in experimental models of hypoalveolarization and impaired vascular development[10, 19, 20].
In contrast to Dillon et al, we found that the persistence of severe PH (systemic–to-suprasystemic) at 4-6 weeks of age was not universally fatal[5]. This may be due to evolving strategies of respiratory management that have preserved the potential for lung and vascular growth, or emerging therapies for PH, which we consider in infants with persistence of PH beyond 6-8 weeks in our practice.
A strength of this study is the large number of infants, as well as the standardized approach to echocardiography. The weekly routine echocardiogram protocol removes the selection bias inherent from echocardiograms used for clinical indications. However, our study was limited by the inability to accurately measure PAp as right heart catheterization remains the gold standard for evaluation of pulmonary arterial pressure and resistance. Previous studies in infants with congenital heart disease have shown Doppler echocardiography to produce accurate estimates of PAp[21-24], but TR was frequently not quantifiable in our studies. Regardless, in infants with chronic lung disease, Mourani et al found TR jet alone did not accurately predict PH by cardiac catheterization [25]. However, multiple measurements together predicted PH in that study. By design, our study also used multiple variables to estimate PAp. Despite these limitations, we have shown that these non-invasive measures are predictive of future morbidity and mortality, particularly at 2 weeks of life. This study sought to assess the utility of routine clinical echocardiography as a predictive biomarker and thus more complex quantitative assessments of pulmonary hemodynamics such as right ventricular strain, pulmonary acceleration time and Tei index were not evaluated, but are an area of future longitudinal studies in this patient population.
Another potential limitation of this study is the extended period over which data were collected. Although general practice guidelines have remained similar over the study period at our institution, there has been an evolution toward earlier extubation and increasing use of non-invasive assisted ventilation, as well as a decrease in the duration of ECMO support. Other evolving practices have included increased use of prostaglandin infusions to maintain ductal patency in infants with concern for persistence of suprasystemic PVR and milrinone infusions to potentiate iNO effects on the pulmonary vasculature[26]. These changes did not, however, translate into changes in rates of death, chronic lung disease, ECMO use, severity of PH, or other outcomes across 3-4 year eras of the study. The age of death did decrease, however. The use of emerging oral therapeutic treatments for PH also likely did not influence outcomes substantially as these are generally reserved for infants with persistence of PH after 6-8 weeks of age.
In conclusion, we have demonstrated that the majority of PH that resolves in the CDH population does so within the first 2 to 3 weeks of life. Persistence of PH as early as 2 weeks is a biomarker for severe disease, predicting mortality and short-term pulmonary morbidity. In contrast, early resolution of PH by this time point is an indicator of mild disease, with almost universally good outcomes. This information could guide clinicians in early prognostication and target infants for early, specific interventions to optimize pulmonary vascular outcomes.
Acknowledgments
L.L. was support by the National Institute of Child Health and Human Development (T32 HD-07162).
Abbreviations and Acronyms
- CDH
congenital diaphragmatic hernia
- PH
pulmonary hypertension
- PVR
pulmonary vascular resistance
- iNO
inhaled nitric oxide
- ECMO
extracorporeal membrane oxygenation
- PPV
positive predictive value
- NPV
negative predictive value
- AUC
area under the receiver operating characteristic curve
- CI
confidence interval
Footnotes
The authors declare no conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Yang W, Carmichael SL, Harris JA, Shaw GM. Epidemiologic characteristics of congenital diaphragmatic hernia among 2.5 million California births, 1989-1997. Birth Defects Res A Clin Mol Teratol. 2006;76:170–4. doi: 10.1002/bdra.20230. [DOI] [PubMed] [Google Scholar]
- 2.Balayla J, Abenhaim HA. Incidence, predictors and outcomes of congenital diaphragmatic hernia: a population-based study of 32 million births in the United States. J Matern Fetal Neonatal Med. 2013 doi: 10.3109/14767058.2013.858691. [DOI] [PubMed] [Google Scholar]
- 3.Thibeault DW, Haney B. Lung volume, pulmonary vasculature, and factors affecting survival in congenital diaphragmatic hernia. Pediatrics. 1998;101:289–95. doi: 10.1542/peds.101.2.289. [DOI] [PubMed] [Google Scholar]
- 4.Iocono JA, Cilley RE, Mauger DT, Krummel TM, Dillon PW. Postnatal pulmonary hypertension after repair of congenital diaphragmatic hernia: predicting risk and outcome. J Pediatr Surg. 1999;34:349–53. doi: 10.1016/s0022-3468(99)90207-5. [DOI] [PubMed] [Google Scholar]
- 5.Dillon PW, Cilley RE, Mauger D, Zachary C, Meier A. The relationship of pulmonary artery pressure and survival in congenital diaphragmatic hernia. J Pediatr Surg. 2004;39:307–12. doi: 10.1016/j.jpedsurg.2003.11.010. [DOI] [PubMed] [Google Scholar]
- 6.Bos A, Tibboel D, Koot V, Hazebroek F, Molenaar J. Persistent Pulmonary Hypertension in High-Risk Congenital Diaphragmatic Hernia: Incidence and Vasodilator Therapy. J Pediatr Surg. 1993;28:1463–5. doi: 10.1016/0022-3468(93)90431-j. [DOI] [PubMed] [Google Scholar]
- 7.Kinsella JP, Truog WE, Walsh WF, Goldberg RN, Bancalari E, Mayock DE, et al. Randomized, multicenter trial of inhaled nitric oxide and high-frequency oscillatory ventilation in severe persistent pulmonary hypertension of the newborn. J Pediatr. 1997;131:55–62. doi: 10.1016/s0022-3476(97)70124-0. [DOI] [PubMed] [Google Scholar]
- 8.Al-Hathlol K, Elmahdy H, Nawaz S, Ali I, Al-Saif S, Tawakol H, et al. Perioperative course of pulmonary hypertension in infants with congenital diaphragmatic hernia: impact on outcome following successful repair. J Pediatr Surg. 2011;46:625–9. doi: 10.1016/j.jpedsurg.2010.11.046. [DOI] [PubMed] [Google Scholar]
- 9.Wynn J, Krishnan U, Aspelund G, Zhang Y, Duong J, Stolar CJ, et al. Outcomes of congenital diaphragmatic hernia in the modern era of management. J Pediatr. 2013;163:114–9 e1. doi: 10.1016/j.jpeds.2012.12.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Keller RL, Tacy TA, Hendricks-Munoz K, Xu J, Moon-Grady AJ, Neuhaus J, et al. Congenital diaphragmatic hernia: endothelin-1, pulmonary hypertension, and disease severity. Am J Respir Crit Care Med. 2010;182:555–61. doi: 10.1164/rccm.200907-1126OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Skinner JR, Boys RJ, Hunter S, Hey E. Non-invasive assessment of pulmonary arterial pressure in healthy neonates. Arch Dis Child. 1991;66:386–90. doi: 10.1136/adc.66.4_spec_no.386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jobe AH, Bancalari E. Bronchopulmonary dysplasia. Am J Respir Crit Care Med. 2001;163:1723–9. doi: 10.1164/ajrccm.163.7.2011060. [DOI] [PubMed] [Google Scholar]
- 13.Bos AP, Tibboel D, Koot VC, Hazebroek FW, Molenaar JC. Persistent pulmonary hypertension in high-risk congenital diaphragmatic hernia patients: incidence and vasodilator therapy. J Pediatr Surg. 1993;28:1463–5. doi: 10.1016/0022-3468(93)90431-j. [DOI] [PubMed] [Google Scholar]
- 14.Bos AP, Tibboel D, Hazebroek FW, Stijnen T, Molenaar JC. Congenital diaphragmatic hernia: impact of prostanoids in the perioperative period. Arch Dis Child. 1990;65:994–5. doi: 10.1136/adc.65.9.994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nakayama DK, Motoyama EK, Evans R, Hannakan C. Relation between arterial hypoxemia and plasma eicosanoids in neonates with congenital diaphragmatic hernia. J Surg Res. 1992;53:615–20. doi: 10.1016/0022-4804(92)90263-y. [DOI] [PubMed] [Google Scholar]
- 16.Kobayashi H, Yamataka A, Okazaki T, Lane GJ, Puri P, Miyano T. Increased levels of circulating adhesion molecules in neonates with congenital diaphragmatic hernia complicated by persistent pulmonary hypertension. Pediatr Surg Int. 2004;20:19–23. doi: 10.1007/s00383-003-1072-8. [DOI] [PubMed] [Google Scholar]
- 17.Okawada M, Kobayashi H, Tei E, Okazaki T, Lane GJ, Yamataka A. Serum monocyte chemotactic protein-1 levels in congenital diaphragmatic hernia. Pediatr Surg Int. 2007;23:487–91. doi: 10.1007/s00383-006-1858-6. [DOI] [PubMed] [Google Scholar]
- 18.Fleck S, Bautista G, Keating SM, Lee TH, Keller RL, Moon-Grady AJ, et al. Fetal production of growth factors and inflammatory mediators predicts pulmonary hypertension in congenital diaphragmatic hernia. Pediatr Res. 2013;74:290–8. doi: 10.1038/pr.2013.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McGrath-Morrow SA, Cho C, Cho C, Zhen L, Hicklin DJ, Tuder RM. Vascular endothelial growth factor receptor 2 blockade disrupts postnatal lung development. Am J Respir Cell Mol Biol. 2005;32:420–7. doi: 10.1165/rcmb.2004-0287OC. [DOI] [PubMed] [Google Scholar]
- 20.Todd L, Mullen M, Olley PM, Rabinovitch M. Pulmonary toxicity of monocrotaline differs at critical periods of lung development. Pediatr Res. 1985;19:731–7. doi: 10.1203/00006450-198507000-00019. [DOI] [PubMed] [Google Scholar]
- 21.Kosturakis D, Goldberg SJ, Allen HD, Loeber C. Doppler echocardiographic prediction of pulmonary arterial hypertension in congenital heart disease. Am J Cardiol. 1984;53:1110–5. doi: 10.1016/0002-9149(84)90646-5. [DOI] [PubMed] [Google Scholar]
- 22.Skinner JR, Stuart AG, O'Sullivan J, Heads A, Boys RJ, Hunter S. Right heart pressure determination by Doppler in infants with tricuspid regurgiation. Arch Dis Child. 1993;69:216–20. doi: 10.1136/adc.69.2.216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chotivittayatarakorn P, Pathmanand C, Thisyakorn C, Sueblinvong V. Doppler echocardiographic predictions of pulmonary artery pressure in children with congenital heart disease. J Med Assoc Thai. 1992;75:79–84. [PubMed] [Google Scholar]
- 24.Kirkpatrick EC. Echocardiography in pediatric pulmonary hypertension. Paediatr Respir Rev. 2013;14:157–64. doi: 10.1016/j.prrv.2012.12.008. [DOI] [PubMed] [Google Scholar]
- 25.Mourani PM, Sontag MK, Younoszai A, Ivy DD, Abman SH. Clinical utility of echocardiography for the diagnosis and management of pulmonary vascular disease in young children with chronic lung disease. Pediatrics. 2008;121:317–25. doi: 10.1542/peds.2007-1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Keller RL. Management of the Infant with Congenital Diaphragmatic Hernia. In: Bancalari E, editor. The Newborn Lung. Philadelphia, PA: Elsevier Saunders; 2012. pp. 381–406. [Google Scholar]