

Abstract
Spontaneous retinal activity mediated by glutamatergic neurotransmission—so-called “Stage 3” retinal waves—drives anti-correlated spiking in ON and OFF RGCs during the second week of postnatal development of the mouse. In the mature retina, the activity of a retinal interneuron called the AII amacrine cell is responsible for anti-correlated spiking in ON and OFF α-RGCs. In mature AIIs, membrane hyperpolarization elicits bursting behavior. Here, we postulated that bursting in AIIs underlies the initiation of glutamatergic retinal waves. We tested this hypothesis by using two-photon calcium imaging of spontaneous activity in populations of retinal neurons and by making whole-cell recordings from individual AIIs and α-RGCs in in vitro preparations of mouse retina. We found that AIIs participated in retinal waves, and that their activity was correlated with that of ON α-RGCs and anti-correlated with that of OFF α-RGCs. Though immature AIIs lacked the complement of membrane conductances necessary to generate bursting, pharmacological activation of the M-current, a conductance that modulates bursting in mature AIIs, blocked retinal wave generation. Interestingly, blockade of the pacemaker conductance Ih, a conductance absent in AIIs but present in both ON and OFF cone bipolar cells, caused a dramatic loss of spatial coherence of spontaneous activity. We conclude that during glutamatergic waves, AIIs act to coordinate and propagate activity generated by BCs rather than to initiate spontaneous activity.
Keywords: development, spontaneous activity, two-photon calcium imaging
Introduction
Spontaneous retinal activity, termed retinal waves, shapes visual system development (Huberman et al., 2008; Kirkby et al., 2013; Ackman and Crair, 2014). The circuitry underlying these waves changes as the retina matures. Initially, waves are generated and propagated by cholinergic interneurons called starburst amacrine cells; later, they depend upon glutamatergic transmission (Bansal et al., 2000; Zhou and Zhao, 2000; Wong and Wong, 2001; Maccione et al., 2014).
Starburst cells, which make reciprocal excitatory connections, depolarize spontaneously to initiate cholinergic waves (Zheng et al., 2006). Their activity then is propagated to other neurons, at least in part, by volume release of acetylcholine (Ford et al., 2012). Analogously, spontaneous depolarizations of glutamatergic interneurons, primarily cone bipolar (CB) cells, should initiate glutamatergic waves that are propagated by volume release of glutamate (Firl et al., 2013).
The generation of glutamatergic waves, however, is only partially understood. Although CBs are depolarized by waves (Akrouh and Kerschensteiner, 2013; Firl et al., 2013), it is unknown if they initiate waves. Moreover, the mechanism(s) responsible for propagating these waves is yet to be fully elucidated. The two mechanisms that have been proposed are glutamate spillover (Blankenship et al., 2009; Firl et al., 2013) and gap-junction coupling (Akrouh and Kerschensteiner, 2013). Interestingly, a recent study demonstrated that pharmacological blockade of either ionotropic glutamate receptors or gap junctions was sufficient to prevent glutamatergic waves (Akrouh and Kerschensteiner, 2013), implicating both mechanisms in wave generation.
Synaptic inhibition also has been shown to shape glutamatergic waves (Sernagor et al., 2003; Maccione et al., 2014) by limiting the number of neurons activated (Firl et al., 2013) and by affecting wave timing. ON RGCs depolarize ∼1 s before OFF RGCs, and this offset is eliminated when synaptic inhibition is blocked (Kerschensteiner and Wong, 2008). This anti-correlation of activity in ON and OFF RGCs caused by synaptic—particularly glycinergic—inhibition suggests that AII amacrine cells play a critical role in the underlying circuitry (Kerschensteiner and Wong, 2008; Akrouh and Kerschensteiner, 2013).
AIIs are glycinergic interneurons in the INL. They receive excitatory input from rod bipolar cells, make inhibitory synapses onto some OFF ganglion cells (GCs) and OFF CBs, and are coupled by gap junctions to ON CBs and to each other (Famiglietti and Kolb, 1975; Demb and Singer, 2012). Initially thought to participate only in rod-mediated vision (Kolb and Nelson, 1983; Strettoi et al., 1992), they are now known to mediate “crossover” inhibition between ON and OFF pathways during cone-mediated vision: depolarization of ON CBs depolarizes AIIs and elicits inhibition of OFF RGCs (Manookin et al., 2008; Münch et al., 2009; van Wyk et al., 2009; Ke et al., 2014). This crossover inhibition generates anti-correlated spiking in ON and OFF α-GCs that receive significant input from AIIs during both rod- and cone-mediated vision (Margolis and Detwiler, 2007; Murphy and Rieke, 2008; van Wyk et al., 2009).
Since membrane hyperpolarization elicits bursting behavior in mature AIIs (Cembrowski et al., 2012) and since bursting in AIIs underlies rhythmic spiking anti-correlated in ON and OFF α-GCs of the degenerating retina (Choi et al., 2014; Margolis et al., 2014), we hypothesized that glutamatergic waves are initiated by AIIs. To test this hypothesis, we used two-photon calcium imaging and targeted whole-cell recordings to determine how circuits in the inner retina—in particular, the network of coupled AIIs and ON CBs—generate and propagate glutamatergic retinal waves.
Materials and Methods
Mice.
Retinas were isolated from C57BL/6 wild-type (Harlan Laboratories), Fbxo32-eGFP, and Cdh1-eGFP mice of either sex and aged P9–P12. Fbxo32-eGFP and Cdh1-eGFP mice express eGFP in AIIs (Gong et al., 2003; Siegert et al., 2009; Cembrowski et al., 2012; Kay et al., 2012). They were obtained from the Mutant Mouse Regional Resource Center [Stock (Tg:Fbxo32-EGFP)IM138Gsat/Mmucd, Stock #030719-UCD and STOCK Tg(Cdh1-EGFP)AR201Gsat/Mmucd, Stock #011775-UCD] and bred into the C57BL/6 background. All procedures involving animals were approved by the Institutional Animal Care and Use Committees of the University of California (Berkeley, CA) and of the University of Maryland (College Park, MD) and conformed to the guidelines of the National Institutes of Health Guide for the Care and Use of Laboratory Animals, the Public Health Service Policy, and the Society for Neuroscience Policy on the Use of Animals in Neuroscience Research.
Retinal preparation.
P9–P12 C57BL/6, Fbxo32-eGFP, and Cdh1-eGFP mice of either sex were deeply anesthetized with isoflurane and decapitated. Eyes were removed, and retinas were isolated in ACSF bubbled with carbogen (95% O2/5% CO2) containing the following (in mm):119.0 NaCl, 26.2 NaHCO3, 11 glucose, 2.5 KCl, 1.0 K2HPO4, 2.5 CaCl2, and 1.3 MgCl2. Retinal whole-mount preparations were prepared for electrophysiological and fluorescence-imaging recordings of retinal waves by mounting retinas' GC layer up on Anodisc filter paper (Whatman). Retinal slices (300–400 μm thick) were prepared by embedding retinas in 3% agarose (Sigma type VIIA) in ACSF with HEPES substituted for NaHCO3 before cutting on a vibrating microtome (Leica). Retinal whole mounts and slices were stored in carbogen-bubbled ACSF at room temperature. During experiments, retinal preparations were superfused continuously with carbogen-bubbled ACSF heated to near-physiological temperature (30–34°C) at a rate of 1–2 ml/min.
Immunohistochemistry.
Retinas from Fbxo32-eGFP mice were fixed in 4% paraformaldehyde for 1 h, rinsed in PBS, sunk in 30% sucrose, and then finally equilibrated in a 50:50 mix of 30% sucrose and OCT freezing media. Retinas were then frozen in OCT in a liquid nitrogen bath. Sections (10 μm) were cut at on a cryostat. Sections were incubated in blocking solution (7.5% normal donkey serum, 1× PBS, pH7.4, 0.1% Triton X-100) for 30 min. Primary and secondary antibodies were diluted in this blocking solution. Primary antibodies were used at a concentration of 1:500 (DAB1; generous gift from Brian Howell) or 1:1000 (mouse anti-GFP; NeuroMab). Sections were incubated in primary antibody overnight at 4°C. Sections were washed three times in PBS for 10 min each wash and then incubated in secondary antibodies (goat anti-mouse IgG2A, Invitrogen; donkey anti-rabbit cy3, Jackson ImmunoResearch) at dilutions of 1:1000. The nuclear dye Draq5 (Cell Signaling Technology) was incorporated with the secondary antibodies at a dilution of 1:2000. Sections were incubated in secondary antibodies for 2 h at room temperature. Sections were then washed three times in PBS for 10 min each wash and mounted in 80% glycerol. Whole retinas were stained in a similar fashion to sections except that the blocking solution contained 0.4% Triton X-100, the primary incubation was performed for 5 d, the secondary incubation was performed for 2 d at 4°C, and the washes were extended to 2 h each. Three retinas were imaged at either P8 or P11–P12 and consistent staining was observed between and within retinas. Tissue was imaged using an Olympus DSU spinning disk microscope. Any changes to the images, for example to brightness, were done across the image in accordance with journal policies.
To quantify the colocalization of GFP and DAB1, four fields intermediate between the optic disk and retina periphery were imaged in P8 and P12 retinas (N = 6, three retinas for each age). The total number of DAB1+ GFP+ and DAB+ GFP− AIIs in each field was counted to generate measurements of cell density (cells/mm2 ± deviation). From these counts, we generated an estimate of the percentage of AIIs that did not express GFP (DAB+ GFP−/DAB+ total).
Two-photon calcium imaging.
Retinas were loaded with Oregon green 488 BAPTA-1 AM (OGB) using the multicell bolus loading technique (Stosiek et al., 2003; Blankenship et al., 2009). For identifying AIIs in Fbxo32-eGFP mice, retinas were imaged with the laser tuned to 920 nm to preferentially excite GFP before bolus loading. Two-photon calcium imaging of neurons in the INL and GCL was performed using a custom-modified two-photon microscope (FluoView 300; Olympus America). XYZ scans were used to localize neurons in the GCL and INL. Time series images were acquired at 1 Hz using a 60× objective (Olympus LUMPlanFl/IR ×60/0.90W) with the excitation laser tuned to 790 nm. Images were corrected for motion artifacts using the TurboReg ImageJ plugin. The 10 × 10 pixel (12 × 12 μm) regions of interest were selected manually within all cells in the field of view. Fluorescent signals were averaged within these regions over time. Cell events were identified when change in fluorescence exceeded 15% of the cell's baseline fluorescence within 1 s. Cells were categorized as participating in a retinal wave if a cell's events were correlated with those of neighboring cells.
Electrophysiology.
Both retinal slices and whole mounts were placed in a recording chamber mounted below an upright video microscope so that cells of interest could be visualized and targeted for whole-cell recordings with pipettes containing the following (in mm): 110 K-gluconate, 5 NaCl, 10 HEPES, 1 BAPTA, 8 Tris-phosphocreatine, 4 MgATP, 0.4 NaGTP, and 0.05 Alexa 488, 594, or 647 hydrazide to permit visualization of the cells by epifluorescence or laser-scanning (confocal or two-photon; Thorlabs) imaging after recording (pH adjusted to 7.4 by KOH and osmolarity to ∼280 mOsm with sucrose). Drugs (from Tocris Bioscience) were added to the bath solution as follows: during recordings from retinal slices, synaptic transmission was blocked with DNQX (25 μm), CPP (5 μm), strychnine (1 μm), picrotoxin (50 μm), and TPMPA (50 μm), which block AMPA/KARs, NMDARs, GlyRs, GABAARs, and GABACRs, respectively; M-type K conductances were blocked with linopirdine (LP; 50 μm) and activated with flupirtine (30 μm); and Ih was blocked with ZD7288 (30 μm).
For recordings from RGCs, α-cells were targeted based on soma size (diameter 18–25 μm). AIIs in retinal whole-mount preparations prepared from Cdh1-eGFP mice were targeted for recording by visualizing eGFP fluorescence. In retinal slice preparations, AIIs were identified by soma size and shape and position at the border of the INL and inner plexiform layer (IPL). Access resistances were <25 MΩ and were not compensated. Recordings were made using a single Multiclamp 700B amplifier. Recorded voltages were low-pass filtered at 1–2 kHz and digitized at 1–10 kHz by an ITC-18 A/D board (InstruTech) controlled by software written in Igor Pro (WaveMetrics). Analyses were performed in Igor Pro.
Waves were detected as follows: first, data were low-pass filtered digitally (Bessel filter; 10 Hz cutoff) and smoothed (50 point sliding average) to remove spikes and other fast membrane voltage fluctuations; second, waves were detected as voltage excursions above a threshold (usually +2 mV from baseline) lasting >100 ms. All data processed by this algorithm were inspected by eye to ensure their reliability. Differences between samples were assessed for significance using a two-tailed Wilcoxon signed-rank test (called Wilcoxon test, below) for paired samples. Significance was taken as p < 0.05.
Results
AIIs participate in glutamatergic waves
In mature Fbxo32-eGFP mice, AIIs, which are uniformly distributed in the inner part of the inner nuclear layer, as well as other neuronal types deeper in the inner nuclear layer express GFP (Siegert et al., 2009; Cembrowski et al., 2012). To confirm that developing AIIs of Fbxo32-eGFP mice expressed GFP, we assessed colocalization of anti-GFP and anti-Dab1 immunofluorescence (Fig. 1A); Dab1 (disabled 1) is a protein that, in the retina, is expressed specifically by AIIs (Rice and Curran, 2000; Lee et al., 2003, 2004; Fuerst et al., 2009). We found that nearly all AIIs were GFP+ (P8: 88 ± 4%, n = 3 retinas; Fig. 1B; P12: 90 ± 1%, n = 3 retinas; data not shown).
Figure 1.

AIIs are depolarized during waves. A, Fluorescence confocal images from sections (left) and whole-mount retina (right) from P8 (top) and P12 (bottom) Fbxo32-GFP mice stained with antibodies to GFP and disabled (Dab1), a marker for AII amacrine cells. Dotted line depicts z-plane. Scale bar, 15 μm. B, Schematic of inner nuclear circuit for retinal waves. Wave starts when ON-CBs depolarize, AIIs are depolarized either via gap junction coupling or glutamate spillover (gray gradient). AIIs inhibit OFF-CBs. AIIs then release inhibition onto OFF-CBs, causing them to release glutamate and starting the wave OFF phase. C, INL loaded with OGB, GFP-expressing AIIs overlaid in orange (right), and optical reconstruction of retinal cross section (left), GCL on bottom. Dotted line depicts z-plane. Scale bar, 15 μm. D, Example of wave propagation in the INL observed with two-photon calcium imaging at a frame rate of 1 Hz. Orange circles are identified AII amacrine cells, blue circles are unidentified cells in INL, and black indicates INL neurons with ΔF/F of >15% for the first time in that frame. E, Left, Sample ΔF/F traces from representative cells, in AIIs (top) and other unidentified INL neurons (bottom). Right, Raster plots of neuronal calcium transients of >15% ΔF/F for all AII amacrine cells in the field of view (top), and other unidentified INL neurons (bottom). The total imaging duration was 8 min. F, Boxplot comparison of proportion of cells depolarized by waves in AIIs, GCL cells, and unidentified INL neurons. Medians plotted with whiskers extending to the most extreme values that are still considered not to be outliers. G, Histogram of interwave intervals for calcium imaging from the both the GCL and the INL. In a subset of experiments, recordings were performed in Fbxo32-eGFP mice in which AIIs express eGFP and therefore interwave intervals for AIIs were determined.
Given the AIIs' abilities to synchronize activity in electrically coupled ON CBs and to generate crossover inhibition that anti-correlates activity in ON and OFF RGCs (Demb and Singer, 2012), it would seem that these cells are well positioned to play a significant role in the generation and/or propagation of glutamatergic waves (Akrouh and Kerschensteiner, 2013). As a starting point for our study, we schematized a hypothetical circuit that generates and propagates glutamatergic retinal waves (Fig. 1B). Since previous calcium-imaging experiments indicate that rod bipolar cell somas do not depolarize during waves (Firl et al., 2013), we assume that depolarization of AIIs is either intrinsic or mediated by glutamate spillover and/or electrical synapses with ON CBs.
We verified that AII cells were depolarized by retinal waves by two-photon calcium imaging of retinas from Fbxo32-eGFP mice in which AIIs express eGFP. Our two-photon calcium imaging offered a limited field of view relative to that observed in previous epifluorescence imaging-based studies (Blankenship et al., 2009; Ford et al., 2012). The technique, however, allowed us to image the INL and GCL independently (Briggman and Euler, 2011; Firl et al., 2013) and to record calcium transients in different neuronal populations by varying the depth of the imaging plane without contamination from out-of-focus fluorescence.
We found the same pattern of expression of GFP+ cells in the live retina as determined with two-photon imaging (Fig. 1C). The GFP+ cells closest to the IPL displayed wave-related calcium transients in conjunction with other INL neurons (Fig. 1C–E). We found that ∼50% of these AIIs participated in retinal waves (Fig. 1F) and exhibited calcium transients at the same frequency as other neurons in the INL and GCL (Fig. 1G). Thus AIIs appeared to be depolarized by retinal waves. It is important to note that the ability of two-photon calcium imaging to detect depolarization of a cell is limited by the signal-to-noise ratio of the transients, which in turn vary with the magnitude of the depolarization. Immature AIIs depolarize by only a few millivolts during waves (see below) and do not fire action potentials, limiting the activation of voltage-gated Ca channels subsequent to Ca2+ influx. Hence, calcium imaging is likely to underestimate the participation of AIIs in waves.
To better ascertain the participation of AIIs in retinal waves, we made simultaneous, whole-cell current-clamp recordings from AIIs and RGCs (Fig. 2). In all dual recordings (n = 8 AII-ON RGC pairs, n = 11 AII-OFF RGCs) depolarizations in AIIs were largely correlated with depolarizations in RGCs (Fig. 2A), and waves in both cell types were blocked by iGluR antagonists (n = 3; Fig. 2A). On average, RGCs depolarized every 27.6 ± 4.2 s and AIIs every 26.6 ± 2.9 s (mean ± SEM; difference is not significant; p = 0.74; Fig. 2B). Waves recorded in AIIs varied widely in amplitude (average = 6.3 ± 3.4 mV, mean ± SD; range = 2.1–15.0 mV; n = 22). The smaller depolarizations (average amplitude in 12/22 AIIs <6 mV) observed likely account for the 50% participation rate established with calcium imaging; i.e., a subset of AIIs does not depolarize sufficiently to generate [Ca2+] sufficient to visualize.
Figure 2.

AIIs are correlated with ON-RGCs and anti-correlated with OFF-RGCs during waves. A, Top, Concurrent current-clamp recordings of spontaneous depolarizations in an AII (gold) and an ON RGC (black). Note that spiking in the RGC is accompanied by depolarization of the AII; the AII appears to hyperpolarize during the interwave interval. Middle, CPP and DNQX, antagonists of ionotropic GluRs, block activity in both cells. Bottom, Concurrent current-clamp recordings from an AII (gold) and an OFF RGC (black) illustrate spontaneous depolarizations in the two cell types. Note that the RGC stops spiking and hyperpolarizes when the AII depolarizes; the AII is hyperpolarized while the RGC spikes. B, Summary data of interwave intervals for AIIs and in RGCs. C, Normalized histograms showing timing differences between waves in AIIs and waves in RGCs. Differences were measured for each recorded AII-RGC pair, and data from AII-ON RGC pairs and AII-OFF RGC pairs were pooled. Depolarizations in AIIs preceded closely depolarizations in ON RGCs; depolarizations in AIIs preceded depolarizations in OFF RGCs >800 ms.
To examine the temporal relationship between waves in AIIs in RGCs, we measured the times between each depolarization in the AII and the closest (temporally) depolarization in the RGC for every paired AII-RGC recording and found that depolarizations of the two cells in a pair were well coordinated: 61% of depolarizations in an AII was accompanied by depolarizations in an ON RGC, and 48% of depolarizations in an AII was accompanied by depolarizations in an OFF RGC (148/238 waves for n = 8 AII-ON RGC pairs and 132/275 waves for n = 12 AII-OFF RGC pairs). The absence of completely coordinated activity in the two cells likely reflects the fact that waves have a finite propagation distance and propagate in multiple directions across the retina; the cells we recorded from were displaced laterally up to ∼100 μm from each other.
Measurements of AII-RGC timing differences pooled from AII-ON RGC pairs (n = 8) and from AII-OFF RGC pairs (n = 12) revealed that depolarizations in AIIs were followed closely by depolarizations in ON RGCs (median delay = 295 ms), while depolarizations in OFF RGCs occurred with a more substantial delay (median delay = 855 ms; Fig. 2C). These data support the conclusion that activity in AIIs is correlated with activity in the ON pathway (Akrouh and Kerschensteiner, 2013).
AIIs in the developing retina do not burst but rather alter the timing of crossover inhibition
Our observation that spontaneous depolarizations of AIIs are absent in the presence of iGluR antagonists suggests that the cells are not intrinsically oscillatory, as they are in the degenerated retina (Choi et al., 2014; Margolis et al., 2014). Rather, AIIs appeared to be driven by chemical and/or electrical synaptic inputs.
Given this observation, we wished to determine whether developing AIIs are capable of generating intrinsic bursts. Therefore, we made whole-cell current-clamp recordings from AIIs in a retinal slice preparation and examined their responses to injected current when chemical synaptic transmission was blocked (see Materials and Methods). Current steps (generally ±30 pA; Fig. 3A) or ramps (±50 pA over 2 s; Fig. 3B) were injected into AIIs and voltage changes recorded.
Figure 3.

Blocking M-current alters the frequency of spontaneous depolarizations. A, Current-clamp recording of AII responses to hyperpolarizing and depolarizing current injection. Note the lack of hyperpolarizing sag or of any spiking upon depolarization. B, Current-clamp recordings from AIIs show responses to injected current ramps in the absence and presence of the M-current antagonist linopirdine (LP). This protocol was used to measure the input resistance (RN) as a function of membrane potential. C, Summary data for effects of LP on resting potential of AIIs. D, Example recording of effects of LP on spontaneous depolarization in both AIIs and RGCs. Control trace same as that in Figure 2A. E, Summary data illustrate a reduction in average interwave interval. Dotted line represents distribution from Figure 2B.
We found that immature AIIs differed substantially from the mature ones described previously. Unlike AIIs in the mature retina (Boos et al., 1993; Tian et al., 2010; Cembrowski et al., 2012), immature AIIs did not exhibit Na channel-dependent spikes when depolarized above resting VM, which averaged −55.9 ± 3.0 mV (n = 12 cells); resting VM in these AIIs was ∼10 mV hyperpolarized relative to that of mature AIIs (Fig. 3B,C; Cembrowski et al., 2012). Additionally, immature AIIs had high input resistances [RN = 1.15 ± 0.1 GΩ as measured between −80 and −70 mV (n = 12 cells)]; this is approximately twice the RN measured from mature AIIs (Cembrowski et al., 2012).
Immature AIIs, however, appeared to exhibit M-type K-currents, just as mature AIIs do (Cembrowski et al., 2012). Depolarizing AIIs above −40 mV resulted in a significant reduction in RN, to 0.80 ± 0.1 GΩ (p = 0.006; n = 12 cells), indicative of the opening of a voltage-gated conductance at these potentials. Addition of the M-current antagonist LP (50 μm) blocked this reduction in RN (in LP, RN measured at −80 to −70 mV = 1.23 ± 0.1 GΩ, and RN measured at −40 to −30 mV = 1.08 ± 0.1 GΩ; this difference is not significant; Fig. 3B; p = 0.08; n = 12 cells) and depolarized the cells by ∼5 mV, to VM = 50.0 ± 3.1 mV (Fig. 3B,C; p = 0.0005; n = 12).
Blockade of the M-current also affected the structure and frequency of spontaneous depolarizations recorded in AIIs and RGCs (Fig. 3D). In the presence of LP, the distribution of interevent intervals was shifted toward smaller intervals for AIIs and for both ON and OFF RGCs (Fig. 3E; AII wave interval: control 15.86 ± 26.40 s, n = 271 waves; LP 8.14 ± 20.75 s, n = 144 waves; p = 0.60 Wilcoxon signed-rank test; ZD7288 5.17 ± 5.73, n = 436 waves; p < 0.001 Wilcoxon signed-rank test; RGC wave intervals: control 12.80 ± 20.75 s, n = 218 waves; LP 3.62 ± 3.33, n = 143 waves; p < 0.001 Wilcoxon signed-rank test; ZD7288 4.40 ± 6.15 s, n = 389 waves; p < 0.001 Wilcoxon signed-rank test).
To monitor the effects of LP and an M-current activator, flupirtine (FL; 10 μm) across a larger population of neurons in both the GCL and INL, we used two-photon calcium imaging. Application of LP produced more frequent transients in INL and GCL neurons (Fig. 4B,D; Wilcoxon signed-rank test p = 0.01; wave interval: control 202.4 ± 90.6 s, N = 21 waves, 5 retinas; LP 92.7 ± 32.8 s, N = 43 waves). LP also induced longer calcium transients in the AIIs (Fig. 4B), consistent with previous reports of prolonged depolarization induced by LP in AIIs of normal and rd1 retinas (Cembrowski et al., 2012; Choi et al., 2014). LP, however, did not affect the proportion of AIIs or GCL cells that participated in waves (Fig. 4C; control 0.84 ± 0.010, LP 0.92 ± 0.071; n = 4 retinas; Student's t test p = 0.19).
Figure 4.

Altering the M-current alters the properties of retinal waves. A, Sample ΔF/F traces from representative cells, in control ACSF (left) and after flupirtine (right), and in AIIs (top), other unidentified INL neurons (middle), and GCL neurons (bottom). B, Sample ΔF/F traces from representative cells, in control ACSF (left) and after LP (right) in AIIs (top), other unidentified INL neurons (middle), and GCL neurons (bottom). C, Summary of effects of LP on the average proportion of cells that participated per wave. Lines connect values of average cell participation per wave for one retina in control versus LP among AIIs (left), INL neurons (middle), and GCL (right). Open circles are group means and SD. D, Summary of effects of LP on the distribution of interwave interval. Dotted lines represent distributions from Figure 1F.
In contrast, FL blocked waves, abolishing calcium transients in AIIs and in other neurons in the INL and GCL (Fig. 4A). We also saw a blockade of spontaneous depolarization in whole-cell current-clamp recordings from AIIs and RGCs (n = 3 paired recordings; data not shown). These data indicate that strong hyperpolarization of AIIs was sufficient to stop the generation of retinal waves.
We conclude that intrinsic membrane conductances of AIIs do not support intrinsic bursting; therefore it is unlikely that glutamatergic waves originate with spontaneous depolarizations of AIIs. Therefore, we wished to determine whether immature CBs exhibited intrinsic properties consistent with spontaneous depolarization and/or bursting.
CB cells possess Ih, the blockade of which significantly alters wave properties
We recorded from ON and OFF CBs (identified by visualization of fluorescent tracer following recording; see Materials and Methods) in retinal slice preparations in the current-clamp configuration and examined cells' responses to injected currents (generally ±30 pA; Fig. 5A,B). In 4/5 ON cells and in 7/8 OFF cells, we observed a significant depolarizing “sag” in response to hyperpolarizing current injection as well as a depolarizing potential following the termination of hyperpolarizing current injection; this sag and afterdepolarization are indicative of the activation of Ih, a hyperpolarization-activated depolarizing conductance (Lüthi and McCormick, 1998). In all cases, this sag and the following afterdepolarization were blocked by the Ih antagonist ZD7288 (Fig. 5A,B). Ih also has been observed in recordings from mature CBs (Ma and Pan, 2003); therefore it would seem that the intrinsic membrane properties of CBs, unlike those of AIIs, are established early in development.
Figure 5.

Ih blockade of HCN channels in CBs modulates frequency of spontaneous depolarizations. A, Current-clamp recording from ON CB in response to injected current in the absence and presence of Ih antagonist ZD7288. Arrows, hyperpolarizing sag and rebound depolarization indicative of Ih. B, Current-clamp recordings from an OFF CB in response to injected current in the absence and presence of ZD7288. C, Concurrent current-clamp recordings of spontaneous depolarizations in an AII and OFF RGC in whole-mount retina in the absence and presence of ZD7288. Note that the AII and the OFF GC remain anti-correlated but that the hyperpolarization of the AII in the interwave interval is abolished by ZD7288. Control recording same as Figure 2. D, Summary data of effects on interwave interval distributions. Dotted lines are control distributions from Figure 2.
Next, we examined the effects of ZD7288 on retinal waves. ZD7288 increased the frequencies of spontaneous depolarization in AIIs and RGCs, as assessed by whole-cell recordings (Fig. 5C,D). Two-photon calcium imaging revealed a dramatic effect of ZD7288 on the spatial correlations of glutamatergic waves (Fig. 6A,B): GCL and INL neurons exhibited more frequent bursts (reported as median, lower quartile/upper quartile; control: 36, 20/82 s, N = 150 wave intervals; ZD7288: 6, 3/12 s, N = 62 wave intervals; Fig. 6E). The proportion of cells active during waves increased for cell types in both the INL and GCL, including AIIs (GCL: control 0.16 ± 0.12, ZD7288 0.68 ± 0.12; INL: control 0.38 ± 0.11, ZD7288 0.57 ± 0.065; AIIs: control 0.50 ± 0.27, ZD7288 0.87 ± 0.12; Fig. 6C). The proportion of cells that never displayed significant calcium transients decreased, most notably for AIIs, for which it dropped to zero (GCL: control 0.69 ± 0.24, ZD7288 0.089 ± 0.072; INL: control 0.37 ± 0.21, ZD7288 0.059 ± 0.049; AIIs: control 0.36 ± 0.27, ZD7288 0 ± 0; Fig. 6D). Hence, blockade of Ih significantly increased the participation of INL neurons in retinal waves.
Figure 6.
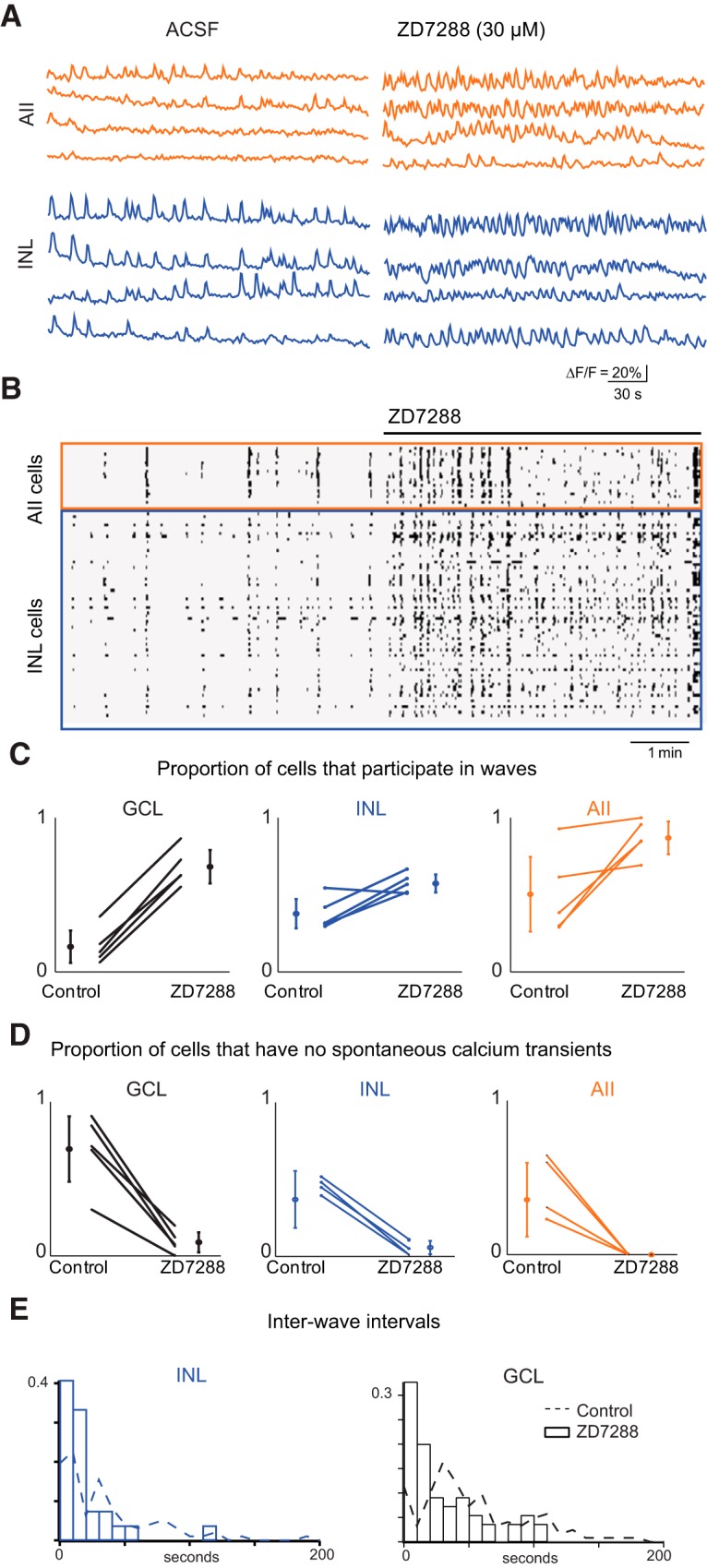
Blockade of HCN channels with ZD7288 abolishes wave structure. A, Sample ΔF/F traces from representative cells, in control ACSF (left) and after ZD7288 (right), and in AIIs (top) and other unidentified INL neurons (bottom). B, Raster plots of neuronal calcium transients of >15% ΔF/F for all INL cells in the field of view, in control ACSF (left) and 50 μm ZD7288 (right). The total imaging duration was 10 min. C, Summary of effects of ZD7288 on the average proportion of cells that participated per wave. Lines connect values of average cell participation per wave for one retina in control versus ZD7288 among GCL (left), INL neurons that are not AIIs amacrine cells (center), and INL neurons that are AII amacrine cells (right). Open circles are group means and SD. D, Summary of effects of ZD7288 on the number of cells that never displayed calcium transients of >15% ΔF/F. Lines connect values of number of inactive cells per retina in control versus ZD7288 among GCL (left), INL neurons that are not AIIs amacrine cells (center), and INL neurons that are AII amacrine cells (right). Open circles are group means and SD. E, Histogram of interwave intervals as assayed by calcium imaging for control ACSF (black) and ZD7288 (red). Inset is normalized cumulative distribution for both conditions (control: N = 150 wave intervals and ZD7288: N = 62 wave intervals).
Absence of gap junctions does not limit AIIs' participation in waves
AIIs are coupled electrically to other AIIs and to ON CBs by gap junctions comprising connexin (Cx) proteins, of which there are many subtypes (Demb and Singer, 2012; Völgyi et al., 2013). AII→AII gap junctions are Cx36 homotypic (Deans et al., 2002), and AII→ON CB gap junctions are Cx36/45 heterotypic (Güldenagel et al., 2000; Feigenspan et al., 2001, 2004; Lin et al., 2005; Maxeiner et al., 2005). To determine how electrical coupling within the AII network shapes glutamatergic waves, we used two-photon calcium imaging to examine the spatiotemporal pattern of activity in retinas from Cx36 ko and Cx36/45 dko mice.
Previously we used a multi-electrode array (MEA) to characterize the spontaneous firing patterns of retinas from Cx36 ko and Cx36/45 dko mice (Hansen et al., 2005; Torborg et al., 2005; Blankenship et al., 2011). In these studies, we recorded retinal waves from both Cx36 ko and Cx36/45 dko retinas; RGCs from these retinas also exhibited many uncorrelated action potentials in between waves. Here we assessed the spontaneous calcium transients of the neurons in the INL in these retinas. Similar to observations of firing patterns of RGCs, waves were detected in the INL, and the interwave intervals did not vary significantly across genotypes (data not shown; Kruskal–Wallis test; INL: WT, n = 18 waves; Cx36 ko, n = 97 waves; Cx36/45 dko, n = 19 waves; GCL: WT, n = 16 waves; Cx36 ko, n = 49 waves; Cx36/45 dko, n = 17 waves). Cx36 ko and Cx36/45 dko retinas, however, exhibited higher frequencies of uncorrelated interwave calcium transients in the INL and GCL layers (Fig. 7A), consistent with previous MEA recordings (Blankenship et al., 2011). In the case of Cx36 ko, AIIs still participated in waves (Fig. 7B). Compared with WT, the Cx36 ko and Cx36/Cx45 dko mice showed a higher proportion of cells in the GCL displaying calcium transients during waves(Figure 7C,D; Kruskal–Wallis test, p = 0.035; INL: WT, n = 9 retinas; Cx36 ko, n = 21 retinas; Cx36/45 dko, n = 5 retinas; GCL: WT, n = 4 retinas; Cx36 ko, n = 6 retinas; Cx36/45 dko, n = 4 retinas; differences found between Cx36 ko and WT in GCL; Tukey–Kramer post hoc tests). Thus, gap junctions play a role in controlling cell participation during waves.
Figure 7.

Connexin ko does not reduce AIIs' participation in waves. A, Raster plots of neuronal calcium transients of >15% ΔF/F for all cells in the field of view for Cx36 ko (left) and Cx36/45 dko (right). The total imaging duration was 5 min. B, Raster plots of neuronal calcium transients of >15% ΔF/F for all AIIs in the field of view (top), and other unidentified INL neurons (bottom). Recordings were performed in Cx36 ko/Fbxo32-eGFP mice in which AIIs express eGFP but lack Cx36. The total imaging duration was 10 min. C, Boxplot comparisons of proportion of cells that displayed transients of >15% ΔF/F during at least one wave. Cx36 GCL was found to be significantly different (Kruskal–Wallis test, p = 0.03). D, Proportion of cells that displayed calcium transients >15% ΔF/F during waves (left), versus proportion of cells that never displayed calcium transients of >15% ΔF/F (right). Lines connect values of proportion of AIIs and other INL neurons per retina.
Discussion
We have clarified the role that AIIs play in the generation and propagation of glutamateric retinal waves. Using both two-photon calcium imaging (Fig. 1) and targeted whole-cell recordings (Fig. 2), we demonstrated that AIIs participate in retinal waves and that they are depolarized concurrent with neighboring RGCs. AIIs' intrinsic membrane properties, however, do not support a role in wave initiation for these cells, and we conclude that AIIs do not serve as the pacemakers for glutamatergic retinal waves. Rather AIIs appear to provide inhibition necessary to anti-correlate activity in ON and OFF ganglion cells (Akrouh and Kerschensteiner, 2013), and M-type K+ conductances in AIIs modulate signal propagation through the electrically coupled network of AIIs and ON CBs (Figs. 3, 4). We hypothesize that wave initiation depends upon oscillations in the membrane potentials of CBs, and that the hyperpolarization-activated, depolarizing Ih permits these oscillations to be synchronized by synaptic inhibition (Figs. 5, 6). This conclusion is supported by previous observations in the adult retina that CBs possess both Ih and calcium channel-dependent oscillatory activity (Protti et al., 2000; Ma and Pan, 2003; Ma et al., 2005; Toychiev et al., 2013). Of course, the channels underlying Ih and M-type IK are likely to be found in interneurons other than CBs and AIIs, respectively (Cangiano et al., 2007); our data do not preclude contributions from such neurons to wave generation and/or propagation. Indeed, we observe many other neurons within the inner nuclear layer that are depolarized during retinal waves (Figs. 1, 4, 6, 7) indicating that there are potentially several other elements in the network generating Stage 3 waves.
AII-CB interactions govern glutamatergic wave generation and propagation
Our observations support the following model: bipolar cells oscillate intrinsically (Protti et al., 2000; Ma et al., 2005; Toychiev et al., 2013), periodically depolarizing AIIs. Since AII depolarization persists in the Cx36 ko (Fig. 7), this depolarization is likely to be mediated by glutamate spillover originating from ON CB terminals. By virtue of their glycinergic output (which, in the mature retina, is to some types of OFF CBs and to some types of OFF RGC, primarily α-type cells; Demb and Singer, 2012), AIIs inhibit OFF cells and anti-correlate activity in the developing ON and OFF pathways (Akrouh and Kerschensteiner, 2013). Thus, oscillations in ON and OFF CBs are normally anti-correlated by synaptic inhibition: on depolarization it produces OFF inhibition, which acts to hyperpolarize OFF CBs and activate Ih; when ON CBs hyperpolarize, OFF inhibition is relieved and Ih serves to depolarize OFF CBs. This model is consistent with the findings that OFF CBs hyperpolarize as a result of glycinergic inhibition when ON RGCs receive excitatory input during a glutamatergic wave and that synaptic inhibition generates the temporal structure of RGC firing patterns during waves (Kerschensteiner and Wong, 2008; Akrouh and Kerschensteiner, 2013).
What remains to be determined is the source of the large-scale coupling that inhibits activity in between waves. Our first hypothesis was that by virtue of their electrical coupling to each other and to ON CBs, AIIs would function to coordinate activity among neighboring ON CBs and propagate activity within the inner retina. The observation that AIIs continued to depolarize in a correlated fashion during waves in the Cx36 ko mouse (Fig. 7), however, indicated that gap junction coupling was not the primary source of depolarization during glutamatergic waves; rather, depolarization appeared to be propagated primarily by glutamate spillover (Blankenship et al., 2009; Firl et al., 2013). Gap junctions, then, may serve to correlate activity of AIIs and ON CBs on a shorter timescale that can be assessed by calcium imaging.
It is important to note that previous studies have shown that glutamatergic waves are blocked completely by meclofenamic acid (MFA), a gap junction antagonist (Akrouh and Kerschensteiner, 2013). In contrast, RGCs from retinas of Cx36 ko, Cx45 ko, and Cx36/45 dko mice exhibit increased uncorrelated firing between waves as well as increase temporally correlated firing between spatially distant neurons during waves (Hansen et al., 2005; Torborg et al., 2005; Blankenship et al., 2011). The discrepancy between these two findings—waves with altered spontaneous spiking in Cx36/45 dko mice versus a complete blockade of activity by a gap junction antagonist—could have multiple causes: nonspecific effects of MFA, developmental compensation in the Cx36/45 dko, or involvement of connexins other than Cx36 and Cx45 in glutamatergic waves. The relative strength of these arguments has been discussed previously (Blankenship et al., 2011).
We propose that decreased neural activity during the interwave interval arises from synaptic inhibition. Based on the dramatic effect that blocking Ih has on the spatial correlations of neural activity during waves, we hypothesize that blocking Ih decouples the intrinsic activity of CBs from synaptic inhibition. In normal retinas, glutamate released during a wave excites inhibitory interneurons, which in turn synchronously hyperpolarizes CB terminals. As this inhibition is relieved, Ih causes CBs to depolarize, initiating the next wave. We propose that gap junctions containing Cx36 and Cx45 coordinate this inhibition and, therefore, coordinated inhibition underlies the silences between waves. This idea is consistent with our observation that uncorrelated activity in the interwave interval is increased in the absence of gap junction coupling.
How do glutamatergic waves compare with “wave-like” phenomena in the developed retina?
It is instructive to compare glutamatergic waves with examples of wave-like activities observed in the mature retina. In the normal, developed retina, blockade of synaptic inputs to bipolar cells induces calcium channel-dependent oscillations in bipolar cell-membrane potential; a potential role for Ih in this process has not been investigated (Toychiev et al., 2013). These changes to membrane potential appear to induce wave-like activity that propagates across the inner retina via a combination of electrical transmission and glutamate spillover that drives spiking in ganglion cells via glutamatergic CB→RGC synapses (Toychiev et al., 2013). These mechanisms of propagation are similar to those that appear to underlie glutamatergic waves (Blankenship et al., 2009, 2011; Firl et al., 2013). Thus, it would seem that the neural substrate of the retina is designed to propagate activity induced by a variety of cellular processes; indeed, it was found that in the absence of synaptic transmission, pharmacological activation of calcium channels could induce propagating waves of neural activity in the developing retina (Singer et al., 2001; Torborg et al., 2004).
In the degenerating retina of the rd1 mouse, a model for human retinitis pigmentosa, oscillatory spontaneous activity is observed in RGCs (Margolis et al., 2008, 2014; Stasheff, 2008; Menzler and Zeck, 2011; Yee et al., 2012;Menzler et al., 2014). Though the spatiotemporal patterns of activity in the rd mouse differs from that of retinal waves (Menzler and Zeck, 2011; Maccione et al., 2014), the activity does share many circuit features. This activity originates in the inner retina and is propagated to RGCs via CB→GC synapses (Borowska et al., 2011; Trenholm et al., 2012; Choi et al., 2014; Margolis et al., 2014). Activity in neurons in the INL does not appear to result solely from calcium channel-mediated bursting (Borowska et al., 2011). As it is blocked by antagonists of gap junctions, it has been suggested that this activity is an emergent property of a degenerated network (Trenholm et al., 2012; Yee et al., 2012) and can be induced in healthy retinas in response to photoreceptor bleaching (Menzler et al., 2014). More recently, however, an alternate hypothesis has emerged: oscillations in the rd1 retina emerge from intrinsic bursting in AIIs; gap junctions are relevant only insofar as they permit ON CBs, hyperpolarized because of the lack of depolarizing photoreceptor input, to hyperpolarize the AII membrane potential to a level that permits bursting (Cembrowski et al., 2012; Choi et al., 2014; Margolis et al., 2014). The idea that electrical coupling allows ON CBs and AIIs to influence each other's behaviors is reinforced by a recent study of light-evoked signaling through the AII network (Grimes et al., 2014).
Together, observations from developed normal and rd1 retinas indicate that activity arising in the inner retina as a result of a variety of cellular processes can become oscillatory. This oscillatory activity can be propagated through the retina by a number of mechanisms, and the AII, by virtue of its position in both gap junction-coupled and inhibitory circuits, can play a significant role in this propagation.
Footnotes
This work was supported by National Institutes of Health grants R01EY019498, R01EY013528, and P30EY003176 (M.B.F.); R01EY017836 (J.H.S.); and NIBIB T32 EB005586-05 (A.J.F.). We thank P. Han, C. Gainer, and M. Tong for technical assistance; members of the Feller lab for commenting on this manuscript; and James McFarland for a helpful discussion.
The authors declare no competing financial interests.
References
- Ackman JB, Crair MC. Role of emergent neural activity in visual map development. Curr Opin Neurobiol. 2014;24:166–175. doi: 10.1016/j.conb.2013.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akrouh A, Kerschensteiner D. Intersecting circuits generate precisely patterned retinal waves. Neuron. 2013;79:322–334. doi: 10.1016/j.neuron.2013.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bansal A, Singer JH, Hwang BJ, Xu W, Beaudet A, Feller MB. Mice lacking specific nicotinic acetylcholine receptor subunits exhibit dramatically altered spontaneous activity patterns and reveal a limited role for retinal waves in forming ON and OFF circuits in the inner retina. J Neurosci. 2000;20:7672–7681. doi: 10.1523/JNEUROSCI.20-20-07672.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blankenship AG, Ford KJ, Johnson J, Seal RP, Edwards RH, Copenhagen DR, Feller MB. Synaptic and extrasynaptic factors governing glutamatergic retinal waves. Neuron. 2009;62:230–241. doi: 10.1016/j.neuron.2009.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blankenship AG, Hamby AM, Firl A, Vyas S, Maxeiner S, Willecke K, Feller MB. The role of neuronal connexins 36 and 45 in shaping spontaneous firing patterns in the developing retina. J Neurosci. 2011;31:9998–10008. doi: 10.1523/JNEUROSCI.5640-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boos R, Schneider H, Wässle H. Voltage- and transmitter-gated currents of AII-amacrine cells in a slice preparation of the rat retina. J Neurosci. 1993;13:2874–2888. doi: 10.1523/JNEUROSCI.13-07-02874.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borowska J, Trenholm S, Awatramani GB. An intrinsic neural oscillator in the degenerating mouse retina. J Neurosci. 2011;31:5000–5012. doi: 10.1523/JNEUROSCI.5800-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briggman KL, Euler T. Bulk electroporation and population calcium imaging in the adult mammalian retina. J Neurophysiol. 2011;105:2601–2609. doi: 10.1152/jn.00722.2010. [DOI] [PubMed] [Google Scholar]
- Cangiano L, Gargini C, Della Santina L, Demontis GC, Cervetto L. High-pass filtering of input signals by the Ih current in a non-spiking neuron, the retinal rod bipolar cell. PLoS One. 2007;2:e1327. doi: 10.1371/journal.pone.0001327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cembrowski MS, Logan SM, Tian M, Jia L, Li W, Kath WL, Riecke H, Singer JH. The mechanisms of repetitive spike generation in an axonless retinal interneuron. Cell Rep. 2012;1:155–166. doi: 10.1016/j.celrep.2011.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi H, Zhang L, Cembrowski MS, Sabottke CF, Markowitz AL, Butts DA, Kath WL, Singer JH, Riecke H. Intrinsic bursting of AII amacrine cells underlies oscillations in the rd1 mouse retina. J Neurophysiol. 2014;112:1491–1504. doi: 10.1152/jn.00437.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deans MR, Volgyi B, Goodenough DA, Bloomfield SA, Paul DL. Connexin36 is essential for transmission of rod-mediated visual signals in the mammalian retina. Neuron. 2002;36:703–712. doi: 10.1016/S0896-6273(02)01046-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demb JB, Singer JH. Intrinsic properties and functional circuitry of the AII amacrine cell. Vis Neurosci. 2012;29:51–60. doi: 10.1017/S0952523811000368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Famiglietti EV, Jr, Kolb H. A bistratified amacrine cell and synaptic circuitry in the inner plexiform layer of the retina. Brain Res. 1975;84:293–300. doi: 10.1016/0006-8993(75)90983-X. [DOI] [PubMed] [Google Scholar]
- Feigenspan A, Teubner B, Willecke K, Weiler R. Expression of neuronal connexin36 in AII amacrine cells of the mammalian retina. J Neurosci. 2001;21:230–239. doi: 10.1523/JNEUROSCI.21-01-00230.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feigenspan A, Janssen-Bienhold U, Hormuzdi S, Monyer H, Degen J, Söhl G, Willecke K, Ammermüller J, Weiler R. Expression of connexin36 in cone pedicles and OFF-cone bipolar cells of the mouse retina. J Neurosci. 2004;24:3325–3334. doi: 10.1523/JNEUROSCI.5598-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Firl A, Sack GS, Newman ZL, Tani H, Feller MB. Extrasynaptic glutamate and inhibitory neurotransmission modulate ganglion cell participation during glutamatergic retinal waves. J Neurophysiol. 2013;109:1969–1978. doi: 10.1152/jn.00039.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ford KJ, Félix AL, Feller MB. Cellular mechanisms underlying spatiotemporal features of cholinergic retinal waves. J Neurosci. 2012;32:850–863. doi: 10.1523/JNEUROSCI.5309-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuerst PG, Bruce F, Tian M, Wei W, Elstrott J, Feller MB, Erskine L, Singer JH, Burgess RW. DSCAM and DSCAML1 function in self-avoidance in multiple cell types in the developing mouse retina. Neuron. 2009;64:484–497. doi: 10.1016/j.neuron.2009.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong S, Zheng C, Doughty ML, Losos K, Didkovsky N, Schambra UB, Nowak NJ, Joyner A, Leblanc G, Hatten ME, Heintz N. A gene expression atlas of the central nervous system based on bacterial artificial chromosomes. Nature. 2003;425:917–925. doi: 10.1038/nature02033. [DOI] [PubMed] [Google Scholar]
- Grimes WN, Schwartz GW, Rieke F. The synaptic and circuit mechanisms underlying a change in spatial encoding in the retina. Neuron. 2014;82:460–473. doi: 10.1016/j.neuron.2014.02.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Güldenagel M, Söhl G, Plum A, Traub O, Teubner B, Weiler R, Willecke K. Expression patterns of connexin genes in mouse retina. J Comp Neurol. 2000;425:193–201. doi: 10.1002/1096-9861(20000918)425:2<193::AID-CNE3>3.3.CO;2-E. [DOI] [PubMed] [Google Scholar]
- Hansen KA, Torborg CL, Elstrott J, Feller MB. Expression and function of the neuronal gap junction protein connexin 36 in developing mammalian retina. J Comp Neurol. 2005;493:309–320. doi: 10.1002/cne.20759. [DOI] [PubMed] [Google Scholar]
- Huberman AD, Feller MB, Chapman B. Mechanisms Underlying Development of Visual Maps and Receptive Fields. Annu Rev Neurosci. 2008;31:479–509. doi: 10.1146/annurev.neuro.31.060407.125533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kay JN, Chu MW, Sanes JR. MEGF10 and MEGF11 mediate homotypic interactions required for mosaic spacing of retinal neurons. Nature. 2012;483:465–469. doi: 10.1038/nature10877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ke JB, Wang YV, Borghuis BG, Cembrowski MS, Riecke H, Kath WL, Demb JB, Singer JH. Adaptation to background light enables contrast coding at rod bipolar cell synapses. Neuron. 2014;81:388–401. doi: 10.1016/j.neuron.2013.10.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerschensteiner D, Wong RO. A precisely timed asynchronous pattern of on and off retinal ganglion cell activity during propagation of retinal waves. Neuron. 2008;58:851–858. doi: 10.1016/j.neuron.2008.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkby LA, Sack GS, Firl A, Feller MB. A role for correlated spontaneous activity in the assembly of neural circuits. Neuron. 2013;80:1129–1144. doi: 10.1016/j.neuron.2013.10.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolb H, Nelson R. Rod pathways in the retina of the cat. Vision Res. 1983;23:301–312. doi: 10.1016/0042-6989(83)90078-0. [DOI] [PubMed] [Google Scholar]
- Lee EJ, Kim HJ, Kim IB, Park JH, Oh SJ, Rickman DW, Chun MH. Morphological analysis of disabled-1-immunoreactive amacrine cells in the guinea pig retina. J Comp Neurol. 2003;466:240–250. doi: 10.1002/cne.10870. [DOI] [PubMed] [Google Scholar]
- Lee EJ, Kim HJ, Lim EJ, Kim IB, Kang WS, Oh SJ, Rickman DW, Chung JW, Chun MH. AII amacrine cells in the mammalian retina show disabled-1 immunoreactivity. J Comp Neurol. 2004;470:372–381. doi: 10.1002/cne.20010. [DOI] [PubMed] [Google Scholar]
- Lin B, Jakobs TC, Masland RH. Different functional types of bipolar cells use different gap-junctional proteins. J Neurosci. 2005;25:6696–6701. doi: 10.1523/JNEUROSCI.1894-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lüthi A, McCormick DA. H-current: properties of a neuronal and network pacemaker. Neuron. 1998;21:9–12. doi: 10.1016/S0896-6273(00)80509-7. [DOI] [PubMed] [Google Scholar]
- Ma YP, Pan ZH. Spontaneous regenerative activity in mammalian retinal bipolar cells: roles of multiple subtypes of voltage-dependent Ca2+ channels. Vis Neurosci. 2003;20:131–139. doi: 10.1017/S0952523803202042. [DOI] [PubMed] [Google Scholar]
- Ma YP, Cui J, Pan ZH. Heterogeneous expression of voltage-dependent Na+ and K+ channels in mammalian retinal bipolar cells. Vis Neurosci. 2005;22:119–133. doi: 10.1017/S0952523805222010. [DOI] [PubMed] [Google Scholar]
- Maccione A, Hennig MH, Gandolfo M, Muthmann O, van Coppenhagen J, Eglen SJ, Berdondini L, Sernagor E. Following the ontogeny of retinal waves: pan-retinal recordings of population dynamics in the neonatal mouse. J Physiol. 2014;592:1545–1563. doi: 10.1113/jphysiol.2013.262840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manookin MB, Beaudoin DL, Ernst ZR, Flagel LJ, Demb JB. Disinhibition combines with excitation to extend the operating range of the OFF visual pathway in daylight. J Neurosci. 2008;28:4136–4150. doi: 10.1523/JNEUROSCI.4274-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margolis DJ, Detwiler PB. Different mechanisms generate maintained activity in ON and OFF retinal ganglion cells. J Neurosci. 2007;27:5994–6005. doi: 10.1523/JNEUROSCI.0130-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margolis DJ, Newkirk G, Euler T, Detwiler PB. Functional stability of retinal ganglion cells after degeneration-induced changes in synaptic input. J Neurosci. 2008;28:6526–6536. doi: 10.1523/JNEUROSCI.1533-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margolis DJ, Gartland AJ, Singer JH, Detwiler PB. Network oscillations drive correlated spiking of on and off ganglion cells in the rd1 mouse model of retinal degeneration. PLoS One. 2014;9:e86253. doi: 10.1371/journal.pone.0086253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maxeiner S, Dedek K, Janssen-Bienhold U, Ammermüller J, Brune H, Kirsch T, Pieper M, Degen J, Krüger O, Willecke K, Weiler R. Deletion of connexin45 in mouse retinal neurons disrupts the rod/cone signaling pathway between AII amacrine and on cone bipolar cells and leads to impaired visual transmission. J Neurosci. 2005;25:566–576. doi: 10.1523/JNEUROSCI.3232-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menzler J, Zeck G. Network oscillations in rod-degenerated mouse retinas. J Neurosci. 2011;31:2280–2291. doi: 10.1523/JNEUROSCI.4238-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menzler J, Channappa L, Zeck G. Rhythmic ganglion cell activity in bleached and blind adult mouse retinas. PLoS One. 2014;9:e106047. doi: 10.1371/journal.pone.0106047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Münch TA, da Silveira RA, Siegert S, Viney TJ, Awatramani GB, Roska B. Approach sensitivity in the retina processed by a multifunctional neural circuit. Nat Neurosci. 2009;12:1308–1316. doi: 10.1038/nn.2389. [DOI] [PubMed] [Google Scholar]
- Murphy GJ, Rieke F. Signals and noise in an inhibitory interneuron diverge to control activity in nearby retinal ganglion cells. Nat Neurosci. 2008;11:318–326. doi: 10.1038/nn2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Protti DA, Flores-Herr N, von Gersdorff H. Light evokes Ca2+ spikes in the axon terminal of a retinal bipolar cell. Neuron. 2000;25:215–227. doi: 10.1016/S0896-6273(00)80884-3. [DOI] [PubMed] [Google Scholar]
- Rice DS, Curran T. Disabled-1 is expressed in type AII amacrine cells in the mouse retina. J Comp Neurol. 2000;424:327–338. doi: 10.1002/1096-9861(20000821)424:2<327::AID-CNE10>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- Sernagor E, Young C, Eglen SJ. Developmental modulation of retinal wave dynamics: shedding light on the GABA saga. J Neurosci. 2003;23:7621–7629. doi: 10.1523/JNEUROSCI.23-20-07621.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegert S, Scherf BG, Del Punta K, Didkovsky N, Heintz N, Roska B. Genetic address book for retinal cell types. Nat Neurosci. 2009;12:1197–1204. doi: 10.1038/nn.2370. [DOI] [PubMed] [Google Scholar]
- Singer JH, Mirotznik RR, Feller MB. Potentiation of L-type calcium channels reveals nonsynaptic mechanisms that correlate spontaneous activity in the developing mammalian retina. J Neurosci. 2001;21:8514–8522. doi: 10.1523/JNEUROSCI.21-21-08514.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stasheff SF. Emergence of sustained spontaneous hyperactivity and temporary preservation of OFF responses in ganglion cells of the retinal degeneration (rd1) mouse. J Neurophysiol. 2008;99:1408–1421. doi: 10.1152/jn.00144.2007. [DOI] [PubMed] [Google Scholar]
- Stosiek C, Garaschuk O, Holthoff K, Konnerth A. In vivo two-photon calcium imaging of neuronal networks. Proc Natl Acad Sci U S A. 2003;100:7319–7724. doi: 10.1073/pnas.1232232100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strettoi E, Raviola E, Dacheux RF. Synaptic connections of the narrow-field, bistratified rod amacrine cell (AII) in the rabbit retina. J Comp Neurol. 1992;325:152–168. doi: 10.1002/cne.903250203. [DOI] [PubMed] [Google Scholar]
- Tian M, Jarsky T, Murphy GJ, Rieke F, Singer JH. Voltage-gated Na channels in AII amacrine cells accelerate scotopic light responses mediated by the rod bipolar cell pathway. J Neurosci. 2010;30:4650–4659. doi: 10.1523/JNEUROSCI.4212-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torborg C, Wang CT, Muir-Robinson G, Feller MB. L-type calcium channel agonist induces correlated depolarizations in mice lacking the beta2 subunit nAChRs. Vision Res. 2004;44:3347–3355. doi: 10.1016/j.visres.2004.08.015. [DOI] [PubMed] [Google Scholar]
- Torborg CL, Hansen KA, Feller MB. High frequency, synchronized bursting drives eye-specific segregation of retinogeniculate projections. Nat Neurosci. 2005;8:72–78. doi: 10.1038/nn1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toychiev AH, Yee CW, Sagdullaev BT. Correlated spontaneous activity persists in adult retina and is suppressed by inhibitory inputs. PLoS One. 2013;8:e77658. doi: 10.1371/journal.pone.0077658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trenholm S, Borowska J, Zhang J, Hoggarth A, Johnson K, Barnes S, Lewis TJ, Awatramani GB. Intrinsic oscillatory activity arising within the electrically coupled AII amacrine-ON cone bipolar cell network is driven by voltage-gated Na+ channels. J Physiol. 2012;590:2501–2517. doi: 10.1113/jphysiol.2011.225060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Wyk M, Wässle H, Taylor WR. Receptive field properties of ON- and OFF-ganglion cells in the mouse retina. Vis Neurosci. 2009;26:297–308. doi: 10.1017/S0952523809990137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Völgyi B, Pan F, Paul DL, Wang JT, Huberman AD, Bloomfield SA. Gap junctions are essential for generating the correlated spike activity of neighboring retinal ganglion cells. PLoS One. 2013;8:e69426. doi: 10.1371/journal.pone.0069426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong WT, Wong RO. Changing specificity of neurotransmitter regulation of rapid dendritic remodeling during synaptogenesis. Nat Neurosci. 2001;4:351–352. doi: 10.1038/85987. [DOI] [PubMed] [Google Scholar]
- Yee CW, Toychiev AH, Sagdullaev BT. Network deficiency exacerbates impairment in a mouse model of retinal degeneration. Front Syst Neurosci. 2012;6:8. doi: 10.3389/fnsys.2012.00008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng J, Lee S, Zhou ZJ. A transient network of intrinsically bursting starburst cells underlies the generation of retinal waves. Nat Neurosci. 2006;9:363–371. doi: 10.1038/nn1644. [DOI] [PubMed] [Google Scholar]
- Zhou ZJ, Zhao D. Coordinated transitions in neurotransmitter systems for the initiation and propagation of spontaneous retinal waves. J Neurosci. 2000;20:6570–6577. doi: 10.1523/JNEUROSCI.20-17-06570.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]