

Abstract
Poly(ADP-ribose) polymerase-1 (PARP1) is a key facilitator of DNA repair. PARP inhibitors have gained recent attention as promising therapeutic agents for the treatment of solid tumours including breast cancer (BC). However, the biological and clinical significance of PARP1 expression in BC and its role in DNA-damage response (DDR) remain to be defined. We investigated the expression of PARP1 expression, cleaved (PARP1c) and non-cleaved (PAR1nc) forms, in a large and well-characterised cohort of clinically annotated stage I–III operable BCs (n = 1,269) and 43 BRCA1-mutated BCs using immunohistochemistry. PARP1 expression was correlated to clinicopathological variables, outcome and expression of other key DNA repair proteins (BRCA1, RAD51, Ku70/80, PIASγ and CHK1). Expression of PARP1 was exclusively nuclear. 49 and 85 % of sporadic BC showed expression PARP1nc and PARP1c, respectively. In BRCA1-mutated tumours, PARP1nc/PARP1c was highly expressed (95 and 79 %, respectively). PARP1nc expression was positively associated with premenopausal younger age patients, larger size and higher tumour grade. PARP1 was positively associated with DDR-proteins; RAD51, BRCA1, CHK1 and PIASγ (p < 0.001). Negative association was found between PARP1nc and Ki67. PARP1c was associated with ER (p < 0.001). Different associations between PARP1 and DDR-proteins were observed when stratified based on ER/BRCA1 status. PARP1 was not an independent predictor of outcome in sporadic or BRCA1-mutated BC. Our results demonstrate a potential biological role for PARP1c and PARP1nc in DNA repair in BC based on the significant association with other key DNA damage repair proteins. These associations were not restricted to ER-negative or triple-negative subgroup.
Keywords: PARP1, Breast cancer, DNA repair gene defects, BRCA1, Biology, Clinical significance
Introduction
DNA repair pathways play key roles in maintaining genomic stability and influence carcinogenesis and tumour biology. Impaired DNA repair also impacts upon response to DNA damaging radiotherapy and chemotherapeutics [1]. Poly[ADP-ribose] polymerase (PARP) is a key DNA repair factor. PARP is an abundant, highly conserved, cell signalling protein that exclusively catalyses poly ADP-ribosylation of DNA-binding proteins, thereby modulating their activity. PARP is essential for DNA single strand break (SSB) repair [2], a sub-pathway related to base excision repair. Loss of PARP are associated persistent SSBs that get converted to DNA double-strand breaks (DSBs) following collapse of replication forks. DSBs generated during the S-phase of the cell cycle is repaired via homologous recombination (HR) pathway whereas DSBs generated outside the S-phase are processed through the error-prone non-homologous end joining (NHEJ) repair pathway. PARP1 is also involved in HR and NHEJ pathways [3–5].
The BRCA genes encode BRCT repeat containing proteins that facilitate the efficient resolution of DSBs generated during the S-phase through HR. Cells lacking functional BRCA proteins are deficient in HR, and thus dependent on the more error-prone NHEJ pathway. This transition results in chromosomal instability and drive a malignant phenotype. Women carrying deleterious germline mutations in the BRCA1 and BRCA2 genes have a high risk of developing breast and ovarian cancers. It was recently demonstrated that HR impaired BRCA deficient cells are hypersensitive to PARP inhibitors. Although the precise mechanism for synthetic lethality is not fully known, SSB repair inhibition may result in the formation and accumulation of toxic DSBs at replication forks in BRCA deficient cells and induces synthetic lethality. Emerging data from clinical trials using PARP inhibitors in BRCA deficient breast and ovarian tumours has provided promising evidence that synthetic lethality by targeting PARP has clinical potential [6–9].
PARP1 is the main member of the PARP family. PARP1 induce cell survival through DNA repair; however during apoptosis, PARP1 is cleaved into two fragments by caspases resulting in its inactivation [10]. This caspase-mediated PARP1 inactivation suggests that blocking PARP1 activity is vital for the proper function of the apoptotic machinery by the ensuing DNA fragmentation [11]. Although overexpression of PARP1 is found in different primary human tumours compared to normal tissue counterparts [12–14], the biological and clinical significance of PARP1 protein expression in breast cancer remains to be fully elucidated. In this study we have assessed the expression of PARP1 (both cleaved and non-cleaved forms) in a large and well-characterised clinically annotated series of breast cancer with a long term follow-up data. Its association with clinicopathological variable, molecular variables and patients outcome was evaluated.
Materials and methods
Study patients
This retrospective study was conducted using two independent cohorts of patients; an initial biomarker discovery cohort consisting of 1902 ER-negative and ER-positive patients and a control cohort of 43 breast cancer from patients with confirmed BRCA1-germline mutations. The discovery cohort comprises a well-characterised consecutive series of early stage (TNM Stage I–III) sporadic primary operable invasive breast carcinoma from patients (age ≤70 years) enrolled into the Nottingham Tenovus Primary Breast Carcinoma Series that presented at Nottingham City Hospital between 1989 and 2004 (n = 1,502) and managed in accordance to a uniform protocol. Patients’ clinical history, tumour characteristics, information on therapy and outcomes are prospectively maintained. Outcome data include survival status, survival time, cause of death and development and time to local and regional recurrence and distant metastasis (DM). Breast cancer specific survival (BCSS) is defined as the time (in months) from the date of primary surgery to the date of breast cancer-related death. Disease free survival (DFI) is defined as the time (in months) from the date of primary surgery to the appearance of a recurrence. In this study, overall relapse free survival was not considered in the analysis as the development of locoregional recurrence is influenced by local management factors and tumour stage, which may confound the effect of the primary tumour biology.
Tissue arrays and immunohistochemistry
Tumour samples were arrayed as previously described [15]. In brief, tissue cores with a diameter of 0.6 mm were punched from the representative tumour regions of each donor block. Cores were precisely arrayed into a new recipient paraffin blocks (TMA) using a tissue microarrayer (Beecher Instruments). Immunohistochemical staining was performed on 4 μm thick sections using Novolink polymer detection system (Leica Biosystems, RE7150-K), composed of Peroxidase Block, Post Primary Block, Novolink Polymer, DAB chromogen and substrate buffer and Novolink haematoxylin. Table 1 shows the dilution, source and clone of antibodies used in this study. Briefly, tissue slides were deparaffinised with xylene and rehydrated through 3 changes of alcohol. Antigen retrieval (except for EGFR and HER2) was performed in citrate buffer (pH 6.0) for 20 min using a microwave oven. Endogenous peroxidase activity was blocked by Peroxidase Block for 5 min. Slides were washed with Tris-Buffered Saline (TBS, pH 7.6), followed by application of Protein Block for 5 min. Following another TBS wash, primary antibody, optimally diluted in Leica antibody diluent (RE7133), was applied and incubated for 60 min. Slides were washed with TBS followed by incubation with Post Primary Block for 30 min followed by a TBS wash. Novolink polymer was applied for 30 min. DAB working solution made up of 1:20 DAB chromogen in DAB substrate buffer was prepared and applied for 5 min. Slides were counterstained with Novolink haematoxylin for 6 min, dehydrated and coverslipped. Negative (omission of the primary antibody) and Positive controls were included according to manufacturer datasheet of each antibody. Table 1 shows the different biomarkers included in this study. Antibody specificity was tested using Western blotting and correct sized band was obtained for the different proteins. HER2 status was also confirmed using in situ hybridisation in borderline cases. Ki67 was assessed using full face section as previously described [17].
Table 1.
Source, dilution and pre-treatment of antibodies used
| Antibody | Dilution | Source/clone | Pretreatment/solution | Cut-off |
|---|---|---|---|---|
| Hormone receptor | ||||
| ERa | 1:200 | DAKO, ID5 | Microwave, citrate pH 6.0 | ≥1 % |
| DNA damage repair molecules | ||||
| PARP1 (Cleaved) | 1:1,000 | Abnova, A6.4.12 | Microwave, citrate pH 6.0 | >200 H-score |
| PARP1 (Non-cleaved) | 1:1,000 | BD Pharmingen, 7D3-6 | Microwave, citrate pH 6.0 | >10 H-score |
| RAD51 | 1:70 | Abcam, mouse polyclonal | Microwave, citrate pH 6.0 | |
| BRCA1 | 1:150 | Oncogene research, MS110 | Microwave, citrate pH 6.0 | >100 H-score |
| Ku70/Ku80 | 1:2,500 | Abcam, mouse monoclonal | Microwave, citrate pH 6.0 | >90 H-score |
| PIASγ | 1:50 | Abcam, anti E3 (ab61405) | Microwave, citrate pH 6.0 | >160 H-score |
| BRCA1 transcriptional suppressor molecule | ||||
| ID4 | 1:300 | Abcam, ab 20988- 100 | Microwave, citrate pH 6.0 | >12 H-score |
| EGFR family members | ||||
| EGFR | 1:30 | Zymed, 31G7 | Proteinase K | >10 % |
| c-erbB-2 (HER2) | 1:250 | DAKO, c-erbB-2 | None | 3+ (2+ with CISH) |
| Cell cycle, proliferation and apoptosis related proteins | ||||
| p53 | 1:50 | Novocstra, DO7 | Microwave, citrate pH 6.0 | >10 % |
| MDM2 | 1:50 | Novocastra, IB10 | Microwave, citrate pH 6.0 | ≥10 % |
| Ki-67/clone MIB1a | 1:100 | DAKO, M7240 | Microwave, citrate pH 6.0 | >10 % |
| Bcl2 | 1:100 | DAKO, 124 | Microwave, citrate pH 6.0 | >10 % |
| CHK1 | 1:150 | Abcam, rabbit polyclonal | Microwave, citrate pH 6.0 | >30 H-score |
| CK5/6 | 1:100 | Boehringer biochemica, D5/16134 | Microwave, citrate pH 6.0 | >10 % |
| CK14 | 1:100 | Novocastra, LL002 | Microwave, citrate pH 6.0 | >10 % |
| CK17 | 1:20 | Abcam, E3 | Microwave, citrate pH 6.0 | >10 % |
aPerformed on full face formalin fixed, paraffin embedded tissue sections
Evaluation of immunohistochemical staining
Two cores were evaluated from each tumour. Each core was scored individually and the mean of the two readings was calculated. If one core was uninformative, the overall score applied was that of the remaining core. Assessment of staining was based on a semi-quantitative approach using a modified histochemical score (H-score) was used which includes an assessment of both the intensity of staining and the percentage of stained cells [16]. For the intensity, a score index of 0, 1, 2 and 3 corresponding to negative, weak, moderate and strong staining intensity was used and the percentage of positive cells at each intensity was estimated subjectively. A final score of 0–300 is the product of both the intensity and the percentage. Cut-off of expression of PARP1 and ID4 was chosen based on the median: H-score of 200 for PARP1 cleaved (PARP1c) and 10 for PARP1 non-cleaved (PARP1nc). Cut-offs of the other biomarkers included in this study (Table 1) were chosen as per previous publications [17–19].
Statistical analysis
Statistical analysis was performed using SPSS 21.0 statistical software. Univariate and multivariate analyses were performed by Chi-squared test, Log rank and Cox regression analysis, respectively. Survival curves were analysed by the method of Kaplan–Meier (Kaplan and Meier, 1958). A p value < 0.01 was considered significant. This study complied with reporting recommendations for tumour marker prognostic studies (REMARK) criteria.
Results
In this study, the expression of PARP1 protein including the non-cleaved/active (PARP1nc) and the cleaved (PARP1c) forms were assessed immunohistochemically and correlated with clinicopathological variables, expression of key biomarkers with relevance to breast cancer, DNA repair markers and patient outcome. Both PARP1 protein forms (PARP1c and PARP1nc) were observed only in the nuclei with no cytoplasmic or membranous expression. Expression of PARP1c and PARP1nc were observed in the normal parenchymal cells of the entrapped normal tissue but their expression was upregulated in malignant cells (Fig. 1). Only nuclear expression in the malignant cells was considered in this study Fig. 2.
Fig. 1.
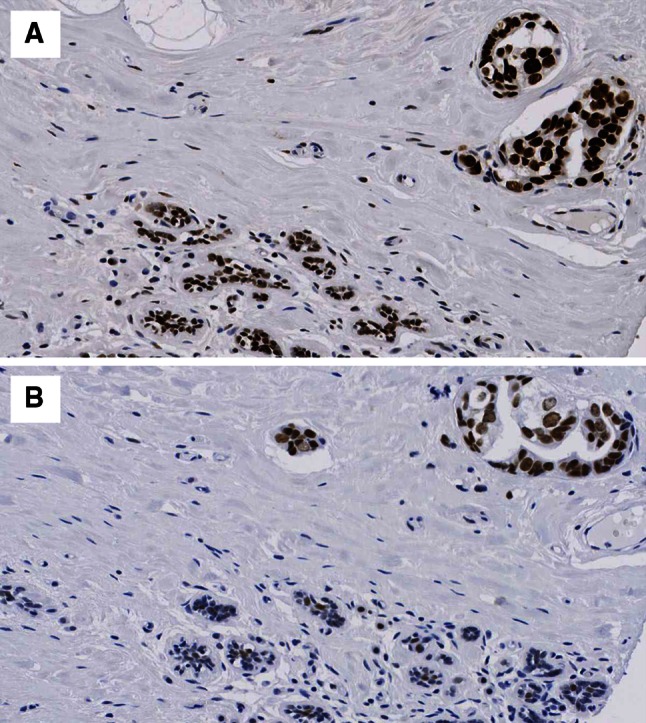
Expression of a PARP1 non-cleaved b PARP1 cleaved protein expression. Normal breast tissue showing weak expression with tumour showing strong expression ×20
Fig. 2.

Cell cycle regulators (p21 and p27), BRCA1 transcriptional suppressor marker (ID4) and DNA repair proteins [PARP1 (cleaved), RAD51, Ku70/80, PIASγ and CHK1] immunohistochemical expressions in invasive breast carcinomas: a P21 positive IHC expression, b P27 positive IHC expression, c ID4 positive IHC expression, d PARP1 (cleaved) positive IHC expression, e RAD51 positive IHC expression, f Ku70/80 positive IHC expression, g PIASγ positive IHC expression and h CHK1 positive IHC expression (×400)
In unselected sporadic breast cancer, reduced or absent expression of PARP1c was observed in 15.4 % while negative expression of PARP1nc (active form) was observed in 51.3 % (Table 2). There was a highly significant correlation between both proteins (p < 0.001). PARP1nc was negative in 2/43 (4.7 %) BRCA1 germline-mutated tumours whereas PARP1c was negative in 5/43 (10.4 %, Table 2) tumours.
Table 2.
Immunohistochemical expression of PARP1 cleaved and non-cleaved in breast cancer
| PARP1 non-cleaved n (%) | PARP1 cleaved n (%) | ||||||
|---|---|---|---|---|---|---|---|
| Negative | Positive | p value | Negative | Positive | p value | ||
| Unselected sporadic series | 552 (51.3) | 524 (48.7) | N/A | 194 (15.4) | 1075 (84.6) | N/A | |
| BRCA1-mutated tumours | 2 (4.7) | 41 (95.3) | 5 (10.4) | 38 (79.2) | |||
| Classes | |||||||
| ER | Negative | 140 (49.6) | 142 (50.4) | NS | 59 (18.9) | 253 (81.1) | 0.041 |
| Positive | 405 (51.7) | 379 (48.3) | 134 (14.1) | 815 (85.9) | |||
| HER2 | Negative | 469 (52.1) | 432 (47.9) | NS | 165 (15.3) | 913 (84.7) | NS |
| Positive | 63 (46.3) | 73 (53.7) | 22 (14.0) | 135 (86.0) | |||
| Triple negative | No | 446 (52.1) | 410 (47.9) | NS | 145 (14.1) | 883 (85.9) | 0.030 |
| Yes | 90 (47.6) | 99 (52.4) | 42 (20.0) | 168 (80.0) | |||
| Basal phenotype | No | 414 (52.0) | 382 (48.0) | NS | 141 (14.8) | 810 (85.2) | NS |
| Yes | 138 (48.9) | 132 (51.1) | 53 (16.8) | 263 (83.2) | |||
Bold values indicate significance
ER oestrogen receptor, NS not significant
Association with clinicopathological and molecular variables sporadic breast cancer
PARP1nc
PARP1nc expression was positively associated with premenopausal younger age patients and higher tumour grade with nuclear pleomorphism, mitosis and a poorer prognosis (Table 3). There was no association with histological tumour type, vascular invasion, size or stage.
Table 3.
Relationship between PARP1 cleaved and non-cleaved with clinicopathological parameters
| Variables | PARP1 non-cleaved | PARP1 cleaved | ||||
|---|---|---|---|---|---|---|
| Negative n (%) | Positive n (%) | p value | Negative n (%) | Positive n (%) | p value | |
| Menopausal status | ||||||
| Pre | 182 (33.2) | 216 (41.7) | 0.004 | 65 (33.5) | 409 (38.5) | NS |
| Post | 366 (66.8) | 302 (58.3) | 129 (66.5) | 653 (61.5) | ||
| Age | ||||||
| <50 | 163 (29.7) | 193 (36.9) | 0.012 | 61 (31.4) | 365 (34) | NS |
| ≥50 | 386 (70.3) | 330 (63.1) | 133 (68.6) | 710 (66) | ||
| Tumour size | ||||||
| ≤2 cm | 279 (51.2) | 240 (46.1) | NS | 96 (50) | 523 (48.8) | NS |
| >2 cm | 266 (48.8) | 281 (53.9) | 96 (50) | 548 (51.2) | ||
| Tumour stage | ||||||
| 1 | 346 (62.9) | 310 (59.5) | NS | 129 (66.5) | 634 (59.2) | NS |
| 2 | 159 (28.9) | 164 (31.5) | 51 (26.3) | 342 (31.9) | ||
| 3 | 45 (8.2) | 47 (9.0) | 14 (7.2) | 95 (8.9) | ||
| Tumour grade | ||||||
| 1 | 113 (20.7) | 69 (13.3) | 0.001 | 32 (16.7) | 180 (17.9) | NS |
| 2 | 180 (33) | 163 (31.3) | 61 (31.8) | 368 (34.5) | ||
| 3 | 253 (46.3) | 288 (55.4) | 99 (51.6) | 519 (48.6) | ||
| Tubule formation | ||||||
| 1 | 35 (6.7) | 24 (4.7) | NS | 11 (5.9) | 62 (6) | NS |
| 2 | 181 (34.6) | 160 (31.6) | 57 (30.3) | 336 (32.6) | ||
| 3 | 307 (58.7) | 323 (63.7) | 120 (63.8) | 634 (61.4) | ||
| Pleomorphism | ||||||
| 1 | 14 (2.7) | 10 (2) | <0.001 | 1 (0.5) | 29 (2.8) | NS |
| 2 | 233 (44.6) | 168 (33.1) | 76 (40.4) | 404 (39.2) | ||
| 3 | 275 (52.7) | 329 (64.9) | 111 (59) | 598 (58) | ||
| Mitosis | ||||||
| 1 | 201 (38.4) | 145 (28.6) | 0.001 | 66 (35.1) | 373 (36.1) | NS |
| 2 | 103 (19.7) | 95 (18.7) | 30 (16) | 200 (19.4) | ||
| 3 | 219 (41.9) | 267 (52.7) | 92 (48.9) | 459 (44.5) | ||
| Vascular invasion | ||||||
| Negative | 185 (34) | 174 (33.5) | NS | 65 (34) | 355 (33.2) | NS |
| Positive | 359 (66) | 346 (66.5) | 126 (66) | 714 (66.8) | ||
| NPI groups | ||||||
| Good | 190 (34.9) | 134 (25.7) | 0.005 | 63 (32.8) | 326 (30.4) | NS |
| Moderate | 269 (49.4) | 295 (56.6) | 94 (49) | 566 (52.8) | ||
| Poor | 86 (15.8) | 92 (17.7) | 35 (18.2) | 179 (16.7) | ||
Bold values indicate significance
NS not significant
Expression of PARP1nc showed positive association with the expression of other DNA repair proteins involved in DDR (BRCA1, RAD51, check-point proteins, (CHK1 and CHK2) PIASγ and DNA PK), together with the proliferation marker Ki67 and p53 (Table 4). No association was observed with ER, HER2, the BRCA1 transcriptional suppressor ID4, BARD1, BRCA2, Ku70/Ku80 or APE1.
Table 4.
Relationship between PARP1 cleaved and non-cleaved with other tumour markers
| Variables | PARP1 non-cleaved | PARP1 cleaved | |||||
|---|---|---|---|---|---|---|---|
| Negative no. (%) | Positive no. (%) | p value | Negative no. (%) | Positive no. (%) | p value | ||
| APE1 | Negative | 31 (16.2) | 36 (15.9) | NS | 16 (19.8) | 59 (14.4) | NS |
| Positive | 160 (83.8) | 190 (84.1) | 65 (80.2) | 352 (85.6) | |||
| BARD1 | Negative | 299 (87.4) | 314 (88.2) | NS | 115 (89.8) | 620 (86.8) | NS |
| Positive | 43 (12.6) | 42 (11.8) | 13 (10.2) | 86 (12.2) | |||
| BRCA1 | Negative | 218 (51.8) | 178 (41.9) | 0.004 | 74 (50.3) | 370 (43.3) | NS |
| Positive | 203 (48.2) | 247 (58.1) | 73 (49.7) | 485 (56.7) | |||
| BRCA2 | Negative | 348 (92.6) | 333 (92.5) | NS | 111 (92.5) | 666 (92.4) | NS |
| Positive | 28 (7.4) | 27 (7.5) | 9 (7.5) | 55 (7.6) | |||
| RAD51 | Negative | 275 (68.9) | 191 (48.1) | <0.001 | 107 (84.3) | 391 (53.8) | <0.001 |
| Positive | 124 (31.1) | 206 (51.9) | 20 (15.7) | 336 (46.2) | |||
| CHK1 | Negative | 360 (79.3) | 323 (69.3) | <0.001 | 127 (84.7) | 607 (71.9) | <0.001 |
| Positive | 94 (20.7) | 143 (30.7) | 23 (15.3) | 237 (28.1) | |||
| Ku70/Ku80 | Negative | 78 (17.3) | 65 (14.6) | NS | 61 (38.4) | 101 (12.5) | <0.001 |
| Positive | 373 (82.7) | 379 (85.4) | 98 (61.6) | 708 (87.5) | |||
| PIASγ | Negative | 156 (33.3) | 74 (15.7) | <0.001 | 80 (50) | 164 (18.3) | <0.001 |
| Positive | 113 (66.7) | 396 (84.3) | 80 (50) | 730 (81.7) | |||
| DNA-PKcs | Negative | 81 (25.9) | 38 (11.3) | <0.001 | 44 (36.7) | 88 (13.8) | <0.001 |
| Positive | 232 (74.1) | 299 (88.7) | 76 (63.3) | 552 (86.2) | |||
| Ki67 | Negative | 145 (35.5) | 122 (28.2) | 0.022 | 49 (33.6) | 284 (33.1) | NS |
| Positive | 263 (64.5) | 311 (71.8) | 97 (66.4) | 573 (66.9) | |||
| ID4 | Negative | 424 (87.4) | 410 (89.5) | NS | 148 (85.1) | 839 (89.4) | NS |
| Positive | 61 (12.6) | 48 (10.5) | 26 (14.9) | 99 (10.6) | |||
| P53 | Negative | 413 (77.1) | 328 (65.3) | <0.001 | 137 (75.3) | 748 (71.2) | NS |
| Positive | 123 (22.9) | 174 (34.7) | 45 (24.7) | 302 (28.8) | |||
Bold values indicate significance
NS not significant
Although there was a positive association between PARP1nc and BRCA1, both proteins showed opposite association with proliferation and with other DNA repair proteins; the majority of PARP1nc+/BRCA1− was associated with high expression of DNA PK (p = 0.003), p53 (p < 0.001), and ki67 (p < 0.001) compared with PARP1nc−/BRCA1+ tumours (Table 5). Positive CHK1 expression (p = 0.004) and negative expression of PIASγ (p = 0.013) were associated with PARP1nc−/BRCA1+ tumours.
Table 5.
Relationship between PARP1 non-cleaved and BRCA1 with DNA repair markers
| Marker | PARP1nc+/BRCA1− n (%) | PARP1nc−/BRCA1+ n (%) | p value | |
|---|---|---|---|---|
| CHK1 | Negative | 137 (52.3) | 125 (47.7) | 0.004 |
| Positive | 21 (32.3) | 44 (67.7) | ||
| DNA-PKcs | Negative | 12 (32.4) | 25 (67.6) | 0.003 |
| Positive | 116 (59.2) | 80 (40.8) | ||
| P53 | Negative | 99 (37.1) | 168 (62.9) | <0.001 |
| Positive | 78 (69.0) | 25 (31.0) | ||
| PIASγ | Negative | 25 (36.2) | 44 (63.8) | 0.013 |
| Positive | 143 (53.0) | 127 (47.0) | ||
| Ki67 | Negative | 25 (29.8) | 59 (70.2) | <0.001 |
| Positive | 130 (61.3) | 82 (47.6) |
Bold values indicate significance
When the series was classified based on ER expression, positive correlations between PARP1nc and PIASγ, DNA-PKcs and p53 were seen in both ER + and ER− tumours while positive associations with BRCA1, RAD51, CHK1 and CHK2 were seen only in ER+ but not in the ER− tumours (Table 6). In ER− tumours, associations between PARP1nc and ki67 (positive) and ID4 (negative) were observed but not in the ER+ tumours.
Table 6.
Relationship between DNA repair biomarkers PARP1ceaved and non-cleaved with biomarkers in ER-positive and negative populations
| Variables | ER-positive | ER negative | |||||
|---|---|---|---|---|---|---|---|
| Negative n (%) | Positive n (%) | p value | Negative n (%) | Positive n (%) | p value | ||
| PARP1 non-cleaved | |||||||
| BRCA1 | Negative | 138 (44.5) | 105 (33.9) | 0.007 | 80 (72.1) | 73 (64) | NS |
| Positive | 172 (55.5) | 205 (66.1) | 31 (27.9) | 41 (36) | |||
| RAD51 | Negative | 197 (67) | 113 (39.9) | <0.001 | 77 (75.5) | 78 (69.6) | NS |
| Positive | 97 (33) | 170 (60.1) | 25 (24.5) | 34 (30.4) | |||
| CHK1 | Negative | 256 (77.3) | 216 (64.1) | <0.001 | 102 (85) | 106 (83.5) | NS |
| Positive | 75 (22.7) | 121 (35.9) | 18 (15) | 21 (16.5) | |||
| CHK2 | Negative | 135 (57.4) | 94 (39.8) | <0.001 | 66 (64.1) | 54 (53.5) | NS |
| Positive | 100 (42.6) | 142 (60.2) | 37 (35.9) | 47 (46.5) | |||
| PIASγ | Negative | 111 (32.5) | 48 (14.3) | <0.001 | 45 (36) | 26 (19.7) | 0.003 |
| Positive | 231 (67.5) | 288 (85.7) | 80 (64) | 106 (80.3) | |||
| DNA-PKcs | Negative | 58 (26) | 25 (10.5) | <0.001 | 23 (25.8) | 13 (13.3) | 0.029 |
| Positive | 165 (74) | 214 (89.5) | 66 (74.2) | 85 (86.7) | |||
| Ki67 | Negative | 126 (42.3) | 114 (36.4) | NS | 18 (16.7) | 8 (6.7) | 0.019 |
| Positive | 172 (57.7) | 199 (63.6) | 90 (83.3) | 111 (93.3) | |||
| ID4 | Negative | 323 (91.2) | 302 (90.1) | NS | 98 (77.2) | 106 (87.6) | 0.031 |
| Positive | 31 (8.8) | 33 (9.9) | 29 (22.8) | 15 (12.4) | |||
| P53 | Negative | 339 (85.2) | 277 (75.7) | 0.001 | 73 (53.3) | 51 (37.5) | 0.009 |
| Positive | 59 (14.8) | 89 (24.3) | 64 (46.7) | 85 (62.5) | |||
| PARP1 cleaved | |||||||
| RAD51 | Negative | 70 (81.4) | 264 (49.1) | <0.001 | 37 (90.2) | 126 (68.9) | 0.010 |
| Positive | 16 (18.6) | 274 (50.9) | 4 (9.8) | 57 (31.1) | |||
| CHK1 | Negative | 85 (82.5) | 428 (67.9) | 0.003 | 42 (89.4) | 175 (84.1) | NS |
| Positive | 18 (17.5) | 202 (32.1) | 5 (10.6) | 33 (15.9) | |||
| CHK2 | Negative | 52 (66.7) | 214 (45.2) | <0.001 | 27 (61.4) | 106 (58.9) | NS |
| Positive | 26 (33.3) | 259 (54.8) | 17 (38.6) | 75 (41.1) | |||
| PIASγ | Negative | 55 (50.9) | 113 (17) | <0.001 | 25 (48.1) | 51 (22.9) | <0.001 |
| Positive | 53 (49.1) | 552 (83) | 27 (51.9) | 172 (77.1) | |||
| DNA-PKcs | Negative | 26 (34.2) | 65 (13.6) | <0.001 | 18 (40.9) | 23 (14.3) | <0.001 |
| Positive | 50 (65.8) | 413 (86.4) | 26 (59.1) | 138 (85.7) | |||
| P53 | Negative | 112 (87.5) | 637 (79.8) | 0.040 | 25 (46.3) | 108 (43.4) | NS |
| Positive | 16 (12.5) | 161 (20.2) | 29 (53.7) | 141 (56.6) | |||
Bold values indicate significance
In triple-negative tumour, PARP1nc was positively associated with expression of PIASγ (p = 0.002) and p53 (p = 0.022, Table 7). No association with the expression of the other markers was found.
Table 7.
Relationship between PARP1 cleaved and non-cleaved with other tumour markers in triple-negative tumour
| Variables | PARP1 non-cleaved | PARP1 cleaved | |||||
|---|---|---|---|---|---|---|---|
| Negative n (%) | Positive n (%) | p value | Negative n (%) | Positive n (%) | p value | ||
| PIASγ | Negative | 35 (42.7) | 19 (20.7) | 0.002 | 19 (51.4) | 38 (25) | 0.002 |
| Positive | 47 (57.3) | 73 (79.3) | 18 (48.6) | 114 (75) | |||
| DNA-PKcs | Negative | 15 (25.9) | 10 (15.2) | NS | 12 (41.4) | 16 (14.4) | 0.001 |
| Positive | 43 (74.1) | 56 (84.8) | 17 (58.6) | 95 (85.6) | |||
| P53 | Negative | 48 (54.5) | 37 (37.8) | 0.022 | 18 (45) | 71 (42.5) | NS |
| Positive | 40 (45.5) | 61 (62.2) | 22 (55) | 96 (57.5) | |||
Bold values indicate significance
NS not significant
PARP1c
PARP1c expression was positively associated with the expression of ER, non Triple-Negative tumour (Table 2) and DDR proteins including RAD51, Ku70/80, CHK1, CHK2, DNA-PKcs, and PIASγ (Table 4). No association was observed with the clinicopathological variables (Table 3), expression of other DDR proteins (BRCA1, BRCA2, ID4, BARD1 and APE1), HER2, p53, Ki67 or basal phenotype (Tables 3, 4).
When the series was classified based on ER expression, the correlation between PARP1c, and RAD51, PIASγ and DNA-PKcs was seen in both ER+ and ER− cohorts while its associations with CHK1 and p53 were seen only in ER+ but not in the ER− subgroups (Table 6). No association with PARP1c was seen in the ER− subgroup only. In the triple-negative tumour, PARP1c showed positive association with the expression of PIASγ and DNA-PKcs (Table 7).
Combined expression of PARP1nc and PARP1c showed a positive association between PARP1nc +/PARP1c + tumours and BRCA1 nuclear (p = 0.027), CHK1 (p < 0.001), DNA-PKcs (p < 0.001), PIASγ (p < 0.001) and p53 (p < 0.001) expression and between PARP1nc−/PARP1c+ tumours and APE1 (p = 0.016) and KU70/KU80 (p < 0.001) expression.
Association with patient outcome
Although no association between PARP1 and patient outcome (BCSS or DFI) was observed in the sporadic breast cancer series or in subgroups based on ER, an association was identified when PARP1nc was combined with BRCA1 (Χ 2 = 13.8, p = 0.003). The association between PARP1nc and DFI in the context of BRCA1 expression was observed in BRCA1-positive tumours (Χ 2 = 5.4, p = 0.020) (Fig. 3) but not in the BRCA1-negative tumours. There were no significant associations between PARP1 expression and patient outcome according to systemic treatment.
Fig. 3.

Kaplan–Meier curve for DMFS with respect to PARP1 (non-cleaved) expression in the BRCA1 positive-tumours
Discussion
This study investigated the expression levels of PARP1 in breast cancer using immunohistochemistry. Our main aim was to better understand the biological role of PARP1 with particular emphasis on DNA repair mechanisms and in the different subgroups of breast cancer. We found that PARP1nc expression was increased in BRCA1-mutated tumours compared with invasive sporadic breast cancer. PARP1nc was also correlated with BRCA1 protein expression in sporadic tumours. High nuclear PARP1, incorporating both the cleaved and non-cleaved expression, is found in the majority of BRCA1-associated BCs which is similar to that observed in our study [20]. Conversely, we have previously shown PARP1nc is relatively low in BRCA1-protein deficient ovarian high grade serous carcinoma [21].
Inherent defects in DNA repair pathways represent a common feature characterising susceptibility to PARP inhibitors [22, 23]. This observation was consistent with the hypothesis that loss of one critical DNA repair pathway, such as defect in the HR-pathway in the BRCA-mutated hereditary breast cancer, can be compensated by upregulation of the other double-strand DNA repair mechanisms, namely NHEJ or alternative pathway involved in single-stand DNA repair mechanisms including nucleotide (NER) and base (BER) excision repair. Some authors [24, 25] have therefore postulated that when the BRCA-dependent HR, is lost or dysfunctional, repair shifts toward alternate DNA repair mechanisms which are dependent on PARPs. This hypothesis was also supported by the study of Wang et al. [26], who demonstrated that PARP1 downregulates BRCA2 gene expression and the study of Ossovskaya et al. [12] who found an inverse relationship between PARP1 and BRCA1 expression in a subset of ovarian cancer. However, current evidence indicates that the specific driver of PARP1 expression in breast cancer still unknown. The application of PARP inhibitors to a subset of sporadic breast cancer namely triple-negative class, was mainly based on (1) the morphologic and molecular similarities between these tumours and BRCA1-mutated breast cancers, and (2) the results from preclinical studies demonstrating sensitivity to PARP inhibition in tumours characterised by deficient HR but not by active PARP DNA related pathways or functional status of PARP proteins [22, 25]. Interestingly, our observation in the BRCA1-mutated tumours support the hypothesis that in tumours with defective HR pathways, the expression of PARP1 proteins are upregulated.
However, the expression in the sporadic tumours appeared to be different with positive correlation between PARP1 and most of the other DNA repair proteins. These findings were also observed in the different molecular subclasses of breast cancer including triple-negative tumour. Our findings in addition to the cytoplasmic expression of some DNA repair genes may reflect the complex mechanism of DNA damage repair in sporadic tumours. Consistent with our findings, it has been reported that transfection of the BRCA1-mutated cell line with wild-type BRCA1 resulted in a significant increase in BER, whereas knock-down of BRCA1 resulted in decreased BER [27, 28].
Consistent with our study, associations between PARP1 and PIASγ [29] and Ku70/80 [30, 31] have been reported. Another important observation in our study was the lack of association between PARP1 and HER2 and basal-associated marker expression in breast cancer.
It is reported that PARP1c is not active as the protein undergoes apoptotic/necrotic cleavage (caspase cleavage) and that PARP1 cleavage is a hallmark of apoptosis [32]. However, our results indicate that PARP1c plays an active role in DNA repair with significant association with other DNA repair proteins. This is consistent with a recent study which demonstrated other physiological functions for the cleaved form [33].
Although PARP1 has been thoroughly investigated and its role in the repair of DNA break is well documented, a model representing its exact role and placing this enzyme in a network with other known DNA damage repair mechanisms remains unavailable. Our results demonstrate that expression of PARP1 proteins are associated with the expression of other DNA repair proteins involved in both HR and NHEJ mechanisms in addition to other markers involved in check-point control and cell proliferation. Our findings also indicate that the association between different DNA damage repair proteins is observed in ER-negative and ER-positive breast cancer suggesting variable activity of the different mechanisms of DNA damage repair in these tumours. Unlike BRCA1, RAD51 and ID4, no association was observed between PARP1 and basal- phenotype in breast cancer [34]). This lack of association was seen in the whole series as well as in ER-negative and ER-positive tumours. Importantly, if PARP inhibitors are possibly targeting PARP1 proteins, other subsets of sporadic breast cancer rather than basal-like triple-negative class may be potential candidate for this promising therapy.
In conclusion, our results demonstrate the biological role of both forms of PARP1 (cleaved and non-cleaved) in DNA repair in breast cancer based on the significant association with other key DNA damage repair proteins. These associations were not restricted to ER-negative or triple-negative subgroup. This study also emphasises the fact that the mechanisms controlling DNA repair in sporadic breast cancer are complex particularly among the different molecular classes of sporadic breast cancer; emphasising the need for further investigation for the target gene that can be used as predictor for PARP inhibitors.
Acknowledgments
Funding was received from the Nottingham University Hospitals Trust and University of Nottingham. DC was sponsored by The Vito Distante Project in Breast Cancer Clinical Research, Florence, Italy.
Conflict of interest
None declared
References
- 1.Lord CJ, Ashworth A. The DNA damage response and cancer therapy. Nature. 2012;481(7381):287–294. doi: 10.1038/nature10760. [DOI] [PubMed] [Google Scholar]
- 2.Burkle A. Poly(APD-ribosyl)ation, a DNA damage-driven protein modification and regulator of genomic instability. Cancer Lett. 2001;163(1):1–5. doi: 10.1016/S0304-3835(00)00694-7. [DOI] [PubMed] [Google Scholar]
- 3.Ame JC, Spenlehauer C, de Murcia G. The PARP superfamily. BioEssays. 2004;26(8):882–893. doi: 10.1002/bies.20085. [DOI] [PubMed] [Google Scholar]
- 4.Krishnakumar R, Kraus WL. The PARP side of the nucleus: molecular actions, physiological outcomes, and clinical targets. Mol Cell. 2010;39(1):8–24. doi: 10.1016/j.molcel.2010.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schreiber V, et al. Poly(ADP-ribose): novel functions for an old molecule. Nat Rev Mol Cell Biol. 2006;7(7):517–528. doi: 10.1038/nrm1963. [DOI] [PubMed] [Google Scholar]
- 6.Sultana R, et al. Synthetic lethal targeting of DNA double-strand break repair deficient cells by human apurinic/apyrimidinic endonuclease inhibitors. Int J Cancer. 2012;131(10):2433–2444. doi: 10.1002/ijc.27512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cerbinskaite A, et al. Defective homologous recombination in human cancers. Cancer Treat Rev. 2012;38(2):89–100. doi: 10.1016/j.ctrv.2011.04.015. [DOI] [PubMed] [Google Scholar]
- 8.Orlando L, et al. Poly(ADP-ribose) polymerase (PARP): rationale, preclinical and clinical evidences of its inhibition as breast cancer treatment. Expert Opin Ther Targets. 2012;16(Suppl 2):S83–S89. doi: 10.1517/14728222.2011.648925. [DOI] [PubMed] [Google Scholar]
- 9.Tutt A, et al. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and advanced breast cancer: a proof-of-concept trial. Lancet. 2010;376(9737):235–244. doi: 10.1016/S0140-6736(10)60892-6. [DOI] [PubMed] [Google Scholar]
- 10.Germain M, et al. Cleavage of automodified poly(ADP-ribose) polymerase during apoptosis. Evidence for involvement of caspase-7. J Biol Chem. 1999;274(40):28379–28384. doi: 10.1074/jbc.274.40.28379. [DOI] [PubMed] [Google Scholar]
- 11.Virag L, Szabo C. The therapeutic potential of poly(ADP-ribose) polymerase inhibitors. Pharmacol Rev. 2002;54(3):375–429. doi: 10.1124/pr.54.3.375. [DOI] [PubMed] [Google Scholar]
- 12.Ossovskaya V, et al. Upregulation of poly(ADP-Ribose) polymerase-1 (PARP1) in triple-negative breast cancer and other primary human tumor types. Genes Cancer. 2010;1(8):812–821. doi: 10.1177/1947601910383418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Quiles-Perez R, et al. Inhibition of poly adenosine diphosphate-ribose polymerase decreases hepatocellular carcinoma growth by modulation of tumor-related gene expression. Hepatology. 2010;51(1):255–266. doi: 10.1002/hep.23249. [DOI] [PubMed] [Google Scholar]
- 14.Nosho K, et al. Overexpression of poly(ADP-ribose) polymerase-1 (PARP-1) in the early stage of colorectal carcinogenesis. Eur J Cancer. 2006;42(14):2374–2381. doi: 10.1016/j.ejca.2006.01.061. [DOI] [PubMed] [Google Scholar]
- 15.Kononen J, et al. Tissue microarrays for high-throughput molecular profiling of tumor specimens. Nat Med. 1998;4(7):844–847. doi: 10.1038/nm0798-844. [DOI] [PubMed] [Google Scholar]
- 16.McCarty KS, Jr, McCarty KS., Sr Histochemical approaches to steroid receptor analyses. Semin Diagn Pathol. 1984;1(4):297–308. [PubMed] [Google Scholar]
- 17.Aleskandarany MA, et al. Prognostic value of proliferation assay in the luminal, HER2 positive and triple negative biological classes of breast cancer. Breast Cancer Res. 2011;14(1):R3. doi: 10.1186/bcr3084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rakha EA, et al. Triple-negative breast cancer: distinguishing between basal and nonbasal subtypes. Clin Cancer Res. 2009;15(7):2302–2310. doi: 10.1158/1078-0432.CCR-08-2132. [DOI] [PubMed] [Google Scholar]
- 19.Rakha EA, et al. Prognostic markers in triple-negative breast cancer. Cancer. 2007;109(1):25–32. doi: 10.1002/cncr.22381. [DOI] [PubMed] [Google Scholar]
- 20.Domagala P, et al. PARP-1 expression in breast cancer including BRCA1-associated, triple negative and basal-like tumors: possible implications for PARP-1 inhibitor therapy. Breast Cancer Res Treat. 2011;127(3):861–869. doi: 10.1007/s10549-011-1441-2. [DOI] [PubMed] [Google Scholar]
- 21.Gan A, et al. Poly(adenosine diphosphate-ribose) polymerase expression in BRCA-proficient ovarian high-grade serous carcinoma; association with patient survival. Hum Pathol. 2013;44(8):1638–1647. doi: 10.1016/j.humpath.2013.01.015. [DOI] [PubMed] [Google Scholar]
- 22.Farmer H, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434(7035):917–921. doi: 10.1038/nature03445. [DOI] [PubMed] [Google Scholar]
- 23.McCabe N, et al. Deficiency in the repair of DNA damage by homologous recombination and sensitivity to poly(ADP-ribose) polymerase inhibition. Cancer Res. 2006;66(16):8109–8115. doi: 10.1158/0008-5472.CAN-06-0140. [DOI] [PubMed] [Google Scholar]
- 24.Plummer R. Poly(ADP-ribose) polymerase inhibition: a new direction for BRCA and triple-negative breast cancer? Breast Cancer Res. 2010;13(4):218. doi: 10.1186/bcr2877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bryant HE, et al. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature. 2005;434(7035):913–917. doi: 10.1038/nature03443. [DOI] [PubMed] [Google Scholar]
- 26.Wang J, et al. Poly(ADP-ribose) polymerase-1 down-regulates BRCA2 expression through the BRCA2 promoter. J Biol Chem. 2008;283(52):36249–36256. doi: 10.1074/jbc.M803693200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Alli E, et al. Defective repair of oxidative dna damage in triple-negative breast cancer confers sensitivity to inhibition of poly(ADP-ribose) polymerase. Cancer Res. 2009;69(8):3589–3596. doi: 10.1158/0008-5472.CAN-08-4016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Anders CK, et al. Poly(ADP-Ribose) polymerase inhibition: “targeted” therapy for triple-negative breast cancer. Clin Cancer Res. 2010;16(19):4702–4710. doi: 10.1158/1078-0432.CCR-10-0939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McCool K, Miyamoto S. A PAR-SUMOnious mechanism of NEMO activation. Mol Cell. 2009;36(3):349–350. doi: 10.1016/j.molcel.2009.10.022. [DOI] [PubMed] [Google Scholar]
- 30.Idogawa M, et al. Ku70 and poly(ADP-ribose) polymerase-1 competitively regulate beta-catenin and T-cell factor-4-mediated gene transactivation: possible linkage of DNA damage recognition and Wnt signaling. Cancer Res. 2007;67(3):911–918. doi: 10.1158/0008-5472.CAN-06-2360. [DOI] [PubMed] [Google Scholar]
- 31.Cheng Q, et al. Ku counteracts mobilization of PARP1 and MRN in chromatin damaged with DNA double-strand breaks. Nucleic Acids Res. 2011;39(22):9605–9619. doi: 10.1093/nar/gkr656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yu SW, et al. Apoptosis-inducing factor mediates poly(ADP-ribose) (PAR) polymer-induced cell death. Proc Natl Acad Sci USA. 2006;103(48):18314–18319. doi: 10.1073/pnas.0606528103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Erener S, et al. Inflammasome-activated caspase 7 cleaves PARP1 to enhance the expression of a subset of NF-kappaB target genes. Mol Cell. 2012;46(2):200–211. doi: 10.1016/j.molcel.2012.02.016. [DOI] [PubMed] [Google Scholar]
- 34.Rakha EA, et al. Breast carcinoma with basal differentiation: a proposal for pathology definition based on basal cytokeratin expression. Histopathology. 2007;50(4):434–438. doi: 10.1111/j.1365-2559.2007.02638.x. [DOI] [PubMed] [Google Scholar]