

Abstract
Although Epstein-Barr virus (EBV) is present in the malignant Hodgkin/Reed-Sternberg (HRS) cells of a proportion of cases of classical Hodgkin lymphoma (cHL), how the virus contributes to the pathogenesis of this disease remains poorly defined. It is clear from the studies of other EBV-associated cancers that the virus is usually not sufficient for tumor development and that other oncogenic co-factors are required. This article reviews what is known about the contribution of EBV to the pathogenesis of cHL and focuses on emerging evidence implicating chronic inflammation as a potential oncogenic co-factor in this malignancy.
Keywords: Epstein-Barr virus, Hodgkin lymphoma, chronic inflammation
Hodgkin lymphoma (HL) is an unusual malignancy that is characterized by the presence of a minority of malignant Hodgkin/Reed Sternberg (HRS) cells surrounded by a non-neoplastic inflammatory infiltrate. There are two distinct forms of HL, known as classical Hodgkin lymphoma (cHL) and nodular lymphocyte- predominant Hodgkin lymphoma (NLPHL), that are separated on the basis of morphologic, immunophenotypic, and clinical differences. Despite earlier reports to the contrary, recent studies suggest that a small proportion of NLPHL cases harbor Epstein-Barr virus (EBV)[1],[2]. However, this review focuses on cHL where the link with EBV is most clearly established.
Origin of HRS Cells in cHL
The identification of clonally rearranged and somatically mutated immunoglobulin genes in single isolated HRS cells provides evidence that these cells are malignant and derived from B cells that have undergone a germinal center (GC) reaction[3]. However, HRS cells lack a functional B-cell receptor (BCR). In some cases, this lack of BCR expression is the result of somatic mutations that destroy the coding capacity of originally functional immunoglobulin genes (so called “crippling” mutations), whereas in others, it can be caused by the loss of the immunoglobulin-specific transcription factors POU class 2 associating factor 1 (POU2AF1/BOB1), POU class 2 homeobox 2 (POU2F2/OCT2), and Spi-1 proto-oncogene (SPI1/PU.1) or mutations in the immunoglobulin gene promoter[4]–[11]. Because apoptosis is the normal fate of GC B cells lacking a functional BCR, the survival of the BCR-negative HRS cell precursor must depend upon the acquisition of novel anti-apoptotic functions.
In addition to the loss of a functional BCR, HRS cells also display a characteristic loss of B-cell lineage gene expression, including the down-regulation of components of the BCR signaling machinery[12]. This phenotype has been attributed in part to the overexpression in HRS cells of transcription factors such as inhibitor of DNA-binding 2 (ID2), which has been shown to negatively regulate B-cell-specific transcription factors, including transcription factor 3 (TCF3/E2A) and paired box 5 (PAX5)[13],[14].
EBV and cHL
EBV was first implicated in the pathogenesis of cHL when it was shown that patients had raised antibody titers to EBV antigens and that these preceded the development of cHL by several years[15],[16]. Subsequently, EBV DNA and RNA were detected in HRS cells[17],[18]. The viral episomes present in HRS cells were also shown to be monoclonal, suggesting that infection of the tumor progenitor occurred prior to its clonal expansion[19].
Epidemiologic studies suggest there are three forms of cHL: pediatric HL (EBV-positive, mixed cellularity type), HL of young adults (EBV-negative, nodular sclerosis type), and HL of older adults (EBV-positive, mixed cellularity type)[20],[21]. The development of EBV-positive cHL in children is thought to be a consequence of a rare, abnormal response to early primary infection, whereas EBV-positive cHL in older adults has been attributed to a decline in EBV-specific immunity associated with advancing age[22],[23]. Although senescence of EBV immunity is also suspected in a related tumor, known as “EBV-positive diffuse large B-cell lymphoma (DLBCL) of the elderly,” the defects in EBV-specific immunity in these patients have yet to be defined[24],[25].
Contribution of EBV to the Survival of HRS Cell Precursors
As described by Rowe et al.[26], EBV contributes to the pathogenesis of Burkitt's lymphoma (BL) by providing the anti-apoptotic signals necessary to override c-myc-induced cell death. In the case of cHL, anti-apoptotic stimuli are also required, but this time to overcome the cell death that would otherwise occur in the absence of a functional BCR. Two pieces of evidence support a role for EBV in providing this anti-apoptotic function. First, the so-called “crippling” mutations in immunoglobulin genes described above are almost exclusively found in EBV-positive cases, and second, EBV has been shown to immortalize BCR-negative GC B cells in vitro[27]–[30]. To begin to understand how EBV contributes to this anti-apoptotic phenotype, we need to revisit the functions of some of the latent EBV genes expressed in HRS cells.
As described by Young et al.[31] in this issue, switching between different forms of latency in the B-cell system may allow the virus to regulate its own life cycle and redirect the fate of the infected B cells towards long-term persistence in the memory pool. Some evidence suggests that the EBV-infected GC B cells of asymptomatic virus carriers express several virus genes designed to drive their transit through a GC reaction and subsequent differentiation into memory B cells[32]–[34]. Critical to this process are the two viral latent membrane proteins, latent membrane protein-1 (LMP1) and LMP2A, which are commonly expressed in EBV-positive HRS cells[35],[36]. LMP1 has been shown to replace survival and differentiation signals that are similar to those provided by an activated CD40 receptor[37],[38]. When expressed in the proposed precursor cells of cHL, LMP1 contributes up to 25% of the transcriptional changes found in cHL[39]. LMP1 may contribute to the survival of apoptosis prone GC B cells by activating several survival pathways, including the nuclear factor-kappa B (NF-κB), Janus activated kinase/signal transducers and activators of transcription (JAK/STAT), and phosphatidylinositol 3-kinase (PI3K)/AKT pathways, signaling pathways that have been found to be constitutively active in cHL[40]–[46]. LMP2A, on the other hand, mimics an activated BCR signal and is able to drive B cell survival in the absence of a functional BCR[47]–[52]. In transgenic mice, LMP2A expression interferes with normal B-cell development and contributes to cell survival involving activation of the RAS/PI3K/AKT pathway[53],[54].
Loss of BCR Functions During the Evolution of EBV-positive cHL
Although EBV can provide anti-apoptotic functions that contribute to the survival of BCR-negative HRS cell progenitors, it is not clear how the loss of a functional BCR is involved in the pathogenesis of EBV-associated cHL. To answer this question, we need to return to another aspect of EBV biology, namely the regulation of the viral replicative cycle.
In addition to existing in various latent states, EBV can also induce its replicative cycle in B cells, a process that eventually leads to the production of new viral particles or virions. The switch from latency to the replicative cycle is triggered by two distinct mechanisms—activation of BCR signaling and plasma cell differentiation, and the switch is regulated in part by the two latent membrane proteins. By providing a BCR-like signal, LMP2A induces entry into the viral replicative cycle[55],[56] (Figure 1). On the other hand, LMP1 prevents entry into the replicative cycle by suppressing plasma cell differentiation[57],[58].
Figure 1. Loss of viral replication functions may promote the deve-lopment of Epstein-Barr virus (EBV)-positive classical Hodgkin lymphoma (cHL).

Upper panel: in normal B cells, either B-cell receptor (BCR)- or latent membrane protein 2A (LMP2A)-mediated signaling can induce the EBV lytic cycle, ultimately leading to the release of infectious virions and eventual cell death. Lower panel: Hodgkin/Reed-Sternberg (HRS) cells lack not only a functional BCR but also essential components of the BCR signaling machinery such as Lyn and Syk and the transcription factor Egr1. Thus, BCR-negative germinal center (GC) B cells that also lack essential BCR signaling components might be positively selected during the development of cHL because they are protected from entry into the EBV replicative cycle and the ensuing cell death. LMP2A expression is retained in EBV-positive HRS cells, but it is not known how LMP2A signals in the absence of BCR signaling components.
Because virus release results in cell death, eventual completion of the EBV replicative cycle is most likely incompatible with tumor development[59]. In the context of the development of cHL, the loss of a functional BCR could be important because it would be expected to prevent BCR-mediated entry into the replicative cycle and thus cell death. Although LMP2A can activate EBV replication even in the absence of a functional BCR, we have shown that LMP2A cannot induce the replicative cycle when essential components of the BCR signaling machinery are missing[60]. Thus, the loss of BCR as well as of BCR signaling components combine to prevent both BCR- and LMP2A-induced virus replication and might explain why cells lacking BCR signaling functions are positively selected during the development of EBV-associated cHL (Figure 1). However, HRS cells retain LMP2A expression, suggesting that this virus protein has BCR-independent functions that are important for tumor development and/or maintenance.
Human Immunodeficiency Virus (HIV), Chronic Immune Stimulation, and cHL
Compared to the general population, the incidence of cHL is 5-15 times higher among people with HIV and acquired immune deficiency syndrome (AIDS), and most cases of cHL in HIV-positive patients are EBV-positive and of mixed cellularity type[61]–[64]. However, in contrast to other forms of HIV-associated lymphomas, the incidence of cHL is the highest when CD4+ T-cell counts are only modestly reduced[63]. Furthermore, the incidence of cHL in HIV-positive patients has not fallen in the post-highly active anti-retroviral therapy (HAART) era[65]–[67]; indeed, some studies suggest HL risk may be increased in the first few months following immune reconstitution on HAART[64],[68]. Two interpretations might explain these data. First, at very low CD4+ T-cell counts, the morphologic presentation of cHL may shift to an appearance more similar to non-Hodgkin lymphoma (in which there are fewer CD4+ T cells in the tumor microenvironment), resulting in a diagnostic misclassification. A second, more likely explanation is that CD4+ T cells may be required for the development of cHL. Indeed, the inflammatory infiltrate of cHL is known to be rich in CD4+ T cells that can promote HRS survival through direct and cytokine-mediated interactions[69]. EBV genes, particularly LMP1, have been shown to contribute to this microenvironment by producing cytokines and chemokines that recruit and modify the chronic inflammatory cells, including CD4+ T cells[69]–[71].
There is emerging evidence that other aspects of HIV infection, in addition to the reduced EBV-specific immunity, might contribute to lymphomagenesis. Increased EBV loads are usually observed only during the early stages of HIV infection when there is little or no T-cell impairment and are attributed to a generalized activation by HIV of the B-cell system[72]. The increased risk of BL that occurs in the early stages of HIV infection is thought to be a consequence of the expansion of the pool of EBV-infected B-cell precursors that results from this chronic B-cell stimulation (reviewed in Ref. [73]). This could eventually lead to the development of BL if one or more of these infected GC precursors acquires the necessary genetic alterations characteristic of BL (reviewed in Ref. [73]). It remains to be established if hyper-stimulation of the B-cell system also contributes in a similar fashion to the pathogenesis of cHL.
Modulation of EBV Gene Expression and Function by the Microenvironment
There is increasing evidence that the microenvironment of the EBV-infected B cell can regulate virus gene expression. The regulation of virus latency is of course not only important in dictating the fate of the EBV-infected B cell transiting through normal lymphoid tissues such as the tonsil but could also be a critical determinant of gene expression in tumors in which there is disruption of the normal microenvironment. For example, the cytokines interleukin-21 (IL-21) and IL-2, along with intercellular interactions such as CD40 ligation, all present in the GC of the tonsil, have been shown to down-regulate the expression of Epstein-Barr virus nuclear antigen-2 (EBNA2) and up-regulate the expression of LMP1, thus imposing a type II expression profile similar to that observed in cHL[74],[75]. Furthermore, our recent data have identified a significant role for Notch ligation in the regulation of LMP1[76]. Activated Notch inhibits the initiation of LMP1 expression from the conventional LMP1 promoter by EBNA2 during primary infection of resting B cells. Activated Notch also inhibits LMP2A expression during primary B-cell infection but only transiently down-regulates LMP2A in established lymphoblastoid cell lines (LCLs). This is of particular interest given that LMP2A expression is often observed in cHL. LMP2A can apparently induce its own promoter, and several reports have demonstrated that LMP2A constitutively activates the Notch pathway[77],[78].
In addition to regulating EBV expression, there is emerging evidence that the microenvironment can also modulate the function of individual virus proteins. For example, we have shown that LMP1 can induce the expression of the collagen receptor, discoidin domain receptor 1 (DDR1). This is important because collagen is a major constituent of the chronic inflammatory microenvironment of cHL[79]–[82]. Ligation of DDR1 by collagen promotes the survival of lymphoma cells in vitro, suggesting that the excess collagen present in lymphomas could drive some oncogenic functions of LMP1[82] (Figure 2). Changes to the microenvironment of the infected B cell might help to explain why on the one hand LMP1 expression in the asymptomatic host provides only those signals required to drive the differentiation of EBV-infected GC B cells, yet on the other hand, LMP1 has potentially oncogenic functions. An important area of future research will be to unravel the complex interactions between the EBV-infected B cell and its microenvironment and to determine how disruption or modification of these interactions might be tumor-promoting.
Figure 2. Potential role for collagen in activating the oncogenic functions of LMP1.
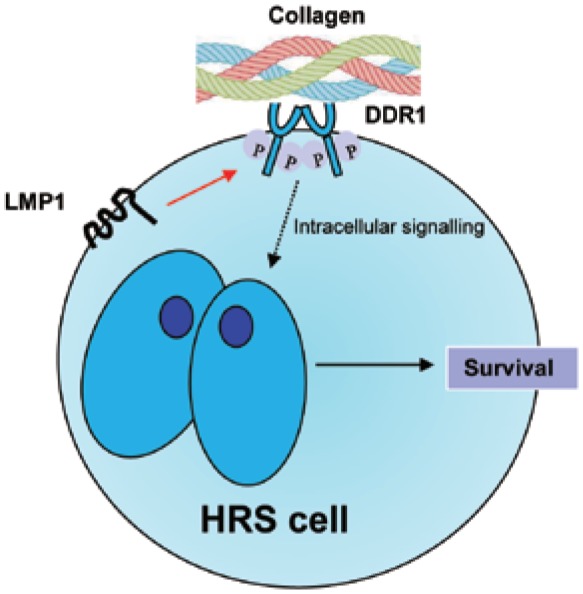
LMP1 can induce expression of the collagen receptor, discoidin domain receptor 1 (DDR1), in the normal EBV-infected GC B cell (indicated by red arrow). During a chronic inflammatory response, collagen is deposited in the microenvironment of the EBV-infected cell and can activate DDR1, leading to growth-promoting signaling.
Conclusions
EBV is present in a proportion of cHL cases and probably provides important anti-apoptotic signals that prevent cell death in HRS progenitors lacking a functional BCR. Loss of BCR and of other key components of the BCR signaling machinery could be important for the pathogenesis of cHL because they might protect HRS progenitors from entry into the EBV replicative cycle and subsequent cell death. Chronic inflammation in the microenvironment of cHL might not only dictate the pattern of EBV gene expression but also modulate the oncogenic functions of individual EBV genes such as LMP1.
References
- 1.Wang S, Medeiros LJ, Xu-Monette ZY, et al. Epstein-Barr virus-positive nodular lymphocyte predominant Hodgkin lymphoma. Ann Diagn Pathol. 2014;18:203–209. doi: 10.1016/j.anndiagpath.2014.03.007. [DOI] [PubMed] [Google Scholar]
- 2.Huppmann AR, Nicolae A, Slack GW, et al. EBV may be expressed in the LP cells of nodular lymphocyte-predominant Hodgkin lymphoma (NLPHL) in both children and adults. Am J Surg Pathol. 2014;38:316–324. doi: 10.1097/PAS.0000000000000107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Küppers R, Rajewsky K, Zhao M, et al. Hodgkin disease: Hodgkin and Reed-Sternberg cells picked from histological sections show clonal immunoglobulin gene rearrangements and appear to be derived from B cells at various stages of development. Proc Natl Acad Sci U S A. 1994;91:10962–10966. doi: 10.1073/pnas.91.23.10962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kanzler H, Küppers R, Hansmann ML, et al. Hodgkin and Reed-Sternberg cells in Hodgkin's disease represent the outgrowth of a dominant tumor clone derived from (crippled) germinal center B cells. J Exp Med. 1996;184:1495–1505. doi: 10.1084/jem.184.4.1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Re D, Muschen M, Ahmadi T, et al. Oct-2 and Bob-1 deficiency in Hodgkin and Reed Sternberg cells. Cancer Res. 2001;61:2080–2084. [PubMed] [Google Scholar]
- 6.Jundt F, Kley K, Anagnostopoulos I, et al. Loss of PU.1 expression is associated with defective immunoglobulin transcription in Hodgkin and Reed-Sternberg cells of classical Hodgkin disease. Blood. 2002;99:3060–3062. doi: 10.1182/blood.v99.8.3060. [DOI] [PubMed] [Google Scholar]
- 7.Torlakovic E, Tierens A, Dang HD, et al. The transcription factor PU.1, necessary for B-cell development, is expressed in lym-phocyte predominance, but not classical Hodgkin's disease. Am J Pathol. 2001;159:1807–1814. doi: 10.1016/S0002-9440(10)63027-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Marafioti T, Hummel M, Foss HD, et al. Hodgkin and Reed-Sternberg cells represent an expansion of a single clone originating from a germinal center B-cell with functional immunoglobulin gene rearrangements but defective immunoglobulin transcription. Blood. 2000;95:1443–1450. [PubMed] [Google Scholar]
- 9.Jox A, Zander T, Kuppers R, et al. Somatic mutations within the untranslated regions of rearranged Ig genes in a case of classical Hodgkin's disease as a potential cause for the absence of Ig in the lymphoma cells. Blood. 1999;93:3964–3972. [PubMed] [Google Scholar]
- 10.Theil J, Laumen H, Marafioti T, et al. Defective octamer-dependent transcription is responsible for silenced immunoglobulin tran-scription in Reed-Sternberg cells. Blood. 2001;97:3191–3196. doi: 10.1182/blood.v97.10.3191. [DOI] [PubMed] [Google Scholar]
- 11.Ushmorov A, Ritz O, Hummel M, et al. Epigenetic silencing of the immunoglobulin heavy-chain gene in classical Hodgkin lymphoma-derived cell lines contributes to the loss of immunoglobulin expression. Blood. 2004;104:3326–3334. doi: 10.1182/blood-2003-04-1197. [DOI] [PubMed] [Google Scholar]
- 12.Schwering I, Brauninger A, Klein U, et al. Loss of the B-lineage-specific gene expression program in Hodgkin and Reed-Sternberg cells of Hodgkin lymphoma. Blood. 2003;101:1505–1512. doi: 10.1182/blood-2002-03-0839. [DOI] [PubMed] [Google Scholar]
- 13.Renne C, Martin-Subero JI, Eickernjager M, et al. Aberrant expression of ID2, a suppressor of B-cell-specific gene expression, in Hodgkin's lymphoma. Am J Pathol. 2006;169:655–664. doi: 10.2353/ajpath.2006.060020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mathas S, Janz M, Hummel F, et al. Intrinsic inhibition of tran-scription factor E2A by HLH proteins ABF-1 and Id2 mediates reprogramming of neoplastic B cells in Hodgkin lymphoma. Nat Immunol. 2006;7:207–215. doi: 10.1038/ni1285. [DOI] [PubMed] [Google Scholar]
- 15.Levine PH, Ablashi DV, Berard CW, et al. Elevated antibody titers to Epstein-Barr virus in Hodgkin's disease. Cancer. 1971;27:416–421. doi: 10.1002/1097-0142(197102)27:2<416::aid-cncr2820270227>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- 16.Mueller N, Evans A, Harris NL, et al. Hodgkin's disease and Epstein-Barr virus. Altered antibody pattern before diagnosis. N Engl J Med. 1989;320:689–695. doi: 10.1056/NEJM198903163201103. [DOI] [PubMed] [Google Scholar]
- 17.Weiss LM, Movahed LA, Warnke RA, et al. Detection of Epstein-Barr viral genomes in Reed-Sternberg cells of Hodgkin's disease. N Engl J Med. 1989;320:502–506. doi: 10.1056/NEJM198902233200806. [DOI] [PubMed] [Google Scholar]
- 18.Wu TC, Mann RB, Charache P, et al. Detection of EBV gene expression in Reed-Sternberg cells of Hodgkin's disease. Int J Cancer. 1990;46:801–804. doi: 10.1002/ijc.2910460509. [DOI] [PubMed] [Google Scholar]
- 19.Anagnostopoulos I, Herbst H, Niedobitek G, et al. Demonstration of monoclonal EBV genomes in Hodgkin's disease and Ki-1-positive anaplastic large cell lymphoma by combined Southern blot and in situ hybridization. Blood. 1989;74:810–816. [PubMed] [Google Scholar]
- 20.Glaser SL, Lin RJ, Stewart SL, et al. Epstein-Barr virus-associated Hodgkin's disease: epidemiologic characteristics in international data. Int J Cancer. 1997;70:375–382. doi: 10.1002/(sici)1097-0215(19970207)70:4<375::aid-ijc1>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- 21.Cader FZ, Kearns P, Young L, et al. The contribution of the Epstein-Barr virus to the pathogenesis of childhood lymphomas. Cancer Treat Rev. 2010;36:348–353. doi: 10.1016/j.ctrv.2010.02.011. [DOI] [PubMed] [Google Scholar]
- 22.Jarrett RF, Gallagher A, Jones DB, et al. Detection of Epstein-Barr virus genomes in Hodgkin's disease: relation to age. J Clin Pathol. 1991;44:844–848. doi: 10.1136/jcp.44.10.844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Armstrong AA, Alexander FE, Cartwright R, et al. Epstein-Barr virus and Hodgkin's disease: further evidence for the three disease hypothesis. Leukemia. 1998;12:1272–1276. doi: 10.1038/sj.leu.2401097. [DOI] [PubMed] [Google Scholar]
- 24.Oyama T, Ichimura K, Suzuki R, et al. Senile EBV+ B-cell lympho-proliferative disorders: a clinicopathologic study of 22 patients. Am J Surg Pathol. 2003;27:16–26. doi: 10.1097/00000478-200301000-00003. [DOI] [PubMed] [Google Scholar]
- 25.Oyama T, Yamamoto K, Asano N, et al. Age-related EBV-associated B-cell lymphoproliferative disorders constitute a distinct clinicopathologic group: a study of 96 patients. Clin Cancer Res. 2007;13:5124–5132. doi: 10.1158/1078-0432.CCR-06-2823. [DOI] [PubMed] [Google Scholar]
- 26.Rowe M, Fitzsimmons L, Bell AI. Epstein-Barr virus and Burkitt's lymphoma. Chin J Cancer. 2014;33:609–619. doi: 10.5732/cjc.014.10190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Brauninger A, Spieker T, Willenbrock K, et al. Survival and clonal expansion of mutating “forbidden” (immunoglobulin receptor-deficient) Epstein-Barr virus-infected B cells in angioimmunoblastic T cell lymphoma. J Exp Med. 2001;194:927–940. doi: 10.1084/jem.194.7.927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bechtel D, Kurth J, Unkel C, et al. Transformation of BCR-deficient germinal center B cells by EBV supports a major role of the virus in the pathogenesis of Hodgkin and post transplant lymphoma. Blood. 2005;106:4345–4350. doi: 10.1182/blood-2005-06-2342. [DOI] [PubMed] [Google Scholar]
- 29.Mancao C, Altmann M, Jungnickel B, et al. Rescue of “crippled” germinal center B cells from apoptosis by Epstein-Barr virus. Blood. 2005;106:4339–4344. doi: 10.1182/blood-2005-06-2341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chaganti S, Bell AI, Begue-Pastor N, et al. Epstein-Barr virus infection in vitro can rescue germinal centre B cells with inactivated immunoglobulin genes. Blood. 2005;106:4249–4252. doi: 10.1182/blood-2005-06-2327. [DOI] [PubMed] [Google Scholar]
- 31.Young LS, Dawson CW. Epstein-Barr virus and nasopharyngeal carcinoma. Chin J Cancer. 2014;33:581–590. doi: 10.5732/cjc.014.10197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Thorley-Lawson DA. Epstein-Barr virus: exploiting the immune system. Nat Rev Immunol. 2001;1:75–82. doi: 10.1038/35095584. [DOI] [PubMed] [Google Scholar]
- 33.Thorley-Lawson DA, Gross A. Persistence of the Epstein-Barr virus and the origins of associated lymphomas. N Engl J Med. 2004;350:1328–1337. doi: 10.1056/NEJMra032015. [DOI] [PubMed] [Google Scholar]
- 34.Thorley-Lawson DA, Duca KA, Shapiro M. Epstein-Barr virus: a paradigm for persistent infection—for real and in virtual reality. Trends Immunol. 2008;29:195–201. doi: 10.1016/j.it.2008.01.006. [DOI] [PubMed] [Google Scholar]
- 35.Niedobitek G, Kremmer E, Herbst H, et al. Immunohistochemical detection of the Epstein-Barr virus-encoded latent membrane protein 2A in Hodgkin's disease and infectious mononucleosis. Blood. 1997;90:1664–1672. [PubMed] [Google Scholar]
- 36.Murray PG, Young LS, Rowe M, et al. Immunohistochemical demonstration of the Epstein-Barr virus-encoded latent membrane protein in paraffin sections of Hodgkin's disease. J Pathol. 1992;166:1–5. doi: 10.1002/path.1711660102. [DOI] [PubMed] [Google Scholar]
- 37.Uchida J, Yasui T, Takaoka-Shichijo Y, et al. Mimicry of CD40 signals by Epstein-Barr virus LMP1 in B lymphocyte responses. Science. 1999;286:300–303. doi: 10.1126/science.286.5438.300. [DOI] [PubMed] [Google Scholar]
- 38.Rastelli J, Homig-Holzel C, Seagal J, et al. LMP1 signaling can replace CD40 signaling in B cells in vivo and has unique features of inducing class-switch recombination to IgG1. Blood. 2008;111:1448–1455. doi: 10.1182/blood-2007-10-117655. [DOI] [PubMed] [Google Scholar]
- 39.Vockerodt M, Morgan SL, Kuo M, et al. The Epstein-Barr virus oncoprotein, latent membrane protein-1, reprograms germinal centre B cells towards a Hodgkin's Reed-Sternberg-like phenotype. J Pathol. 2008;216:83–92. doi: 10.1002/path.2384. [DOI] [PubMed] [Google Scholar]
- 40.Huen DS, Henderson SA, Croom-Carter D, et al. The Epstein-Barr virus latent membrane protein-1 (LMP1) mediates activation of NF-kappa B and cell surface phenotype via two effector regions in its carboxy-terminal cytoplasmic domain. Oncogene. 1995;10:549–560. [PubMed] [Google Scholar]
- 41.Kieser A, Kilger E, Gires O, et al. Epstein-Barr virus latent membrane protein-1 triggers AP-1 activity via the c-Jun N-terminal kinase cascade. EMBO J. 1997;16:6478–6485. doi: 10.1093/emboj/16.21.6478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gires O, Zimber-Strobl U, Gonnella R, et al. Latent membrane protein 1 of Epstein-Barr virus mimics a constitutively active receptor molecule. EMBO J. 1997;16:6131–6140. doi: 10.1093/emboj/16.20.6131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bargou RC, Emmerich F, Krappmann D, et al. Constitutive nuclear factor-kappaB-RelA activation is required for proliferation and survival of Hodgkin's disease tumor cells. J Clin Invest. 1997;100:2961–2969. doi: 10.1172/JCI119849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kube D, Holtick U, Vockerodt M, et al. STAT3 is constitutively activated in Hodgkin cell lines. Blood. 2001;98:762–770. doi: 10.1182/blood.v98.3.762. [DOI] [PubMed] [Google Scholar]
- 45.Skinnider BF, Elia AJ, Gascoyne RD, et al. Signal transducer and activator of transcription 6 is frequently activated in Hodgkin and Reed-Sternberg cells of Hodgkin lymphoma. Blood. 2002;99:618–626. doi: 10.1182/blood.v99.2.618. [DOI] [PubMed] [Google Scholar]
- 46.Dutton A, Reynolds GM, Dawson CW, et al. Constitutive activation of phosphatidyl-inositide 3 kinase contributes to the survival of Hodgkin's lymphoma cells through a mechanism involving Akt kinase and mTOR. J Pathol. 2005;205:498–506. doi: 10.1002/path.1725. [DOI] [PubMed] [Google Scholar]
- 47.Brielmeier M, Mautner J, Laux G, et al. The latent membrane protein 2 gene of Epstein-Barr virus is important for efficient B cell immortalization. J Gen Virol. 1996;77:2807–2818. doi: 10.1099/0022-1317-77-11-2807. [DOI] [PubMed] [Google Scholar]
- 48.Caldwell RG, Wilson JB, Anderson SJ, et al. Epstein-Barr virus LMP2A drives B cell development and survival in the absence of normal B cell receptor signals. Immunity. 1998;9:405–411. doi: 10.1016/s1074-7613(00)80623-8. [DOI] [PubMed] [Google Scholar]
- 49.Caldwell RG, Brown RC, Longnecker R. Epstein-Barr virus LMP2A-induced B-cell survival in two unique classes of EmuLMP2A transgenic mice. J Virol. 2000;74:1101–1113. doi: 10.1128/jvi.74.3.1101-1113.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Engels N, Merchant M, Pappu R, et al. Epstein-Barr virus latent membrane protein 2A (LMP2A) employs the SLP-65 signaling module. J Exp Med. 2001;194:255–264. doi: 10.1084/jem.194.3.255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Casola S, Otipoby KL, Alimzhanov M, et al. B cell receptor signal strength determines B cell fate. Nat Immunol. 2004;5:317–327. doi: 10.1038/ni1036. [DOI] [PubMed] [Google Scholar]
- 52.Engels N, Yigit G, Emmerich CH, et al. Epstein-Barr virus LMP2A signalling in statu nascendi mimics a B cell antigen receptor-like activation signal. Cell Commun Signal. 2012;10:9. doi: 10.1186/1478-811X-10-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Portis T, Dyck P, Longnecker R. Epstein-Barr virus (EBV) LMP2A induces alterations in gene transcription similar to those observed in Reed-Sternberg cells of Hodgkin lymphoma. Blood. 2003;102:4166–4178. doi: 10.1182/blood-2003-04-1018. [DOI] [PubMed] [Google Scholar]
- 54.Portis T, Longnecker R. Epstein-Barr virus (EBV) LMP2A mediates B-lymphocyte survival through constitutive activation of the Ras/PI3K/Akt pathway. Oncogene. 2004;23:8619–8628. doi: 10.1038/sj.onc.1207905. [DOI] [PubMed] [Google Scholar]
- 55.Laichalk LL, Thorley-Lawson DA. Terminal differentiation into plasma cells initiates the replicative cycle of Epstein-Barr virus in vivo. J Virol. 2005;79:1296–1307. doi: 10.1128/JVI.79.2.1296-1307.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Schaadt E, Baier B, Mautner J, et al. Epstein-Barr virus latent membrane protein 2A mimics B-cell receptor-dependent virus reactivation. J Gen Virol. 2005;86:551–559. doi: 10.1099/vir.0.80440-0. [DOI] [PubMed] [Google Scholar]
- 57.Prince S, Keating S, Fielding C, et al. Latent membrane protein 1 inhibits Epstein-Barr virus lytic cycle induction and progress via different mechanisms. J Virol. 2003;77:5000–5007. doi: 10.1128/JVI.77.8.5000-5007.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Vrzalikova K, Vockerodt M, Leonard S, et al. Down-regulation of BLIMP1alpha by the EBV oncogene, LMP-1, disrupts the plasma cell differentiation program and prevents viral replication in B cells: implications for the pathogenesis of EBV-associated B-cell lymphomas. Blood. 2011;117:5907–5917. doi: 10.1182/blood-2010-09-307710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yates JL. EBV DNA replication: how do viral genomes become amplified during latent infection? In: Tursz T, Pagano JS, Ablashi DV, et al., editors. The Epstein-Barr virus and associated diseases. John Libbey Eurotext; 1993. [Google Scholar]
- 60.Vockerodt M, Wei W, Nagy E, et al. Suppression of the LMP2A target gene, EGR-1, protects Hodgkin's lymphoma cells from entry to the EBV lytic cycle. J Pathol. 2013;230:399–409. doi: 10.1002/path.4198. [DOI] [PubMed] [Google Scholar]
- 61.Uccini S, Monardo F, Stoppacciaro A, et al. High frequency of Epstein-Barr virus genome detection in Hodgkin's disease of HIV-positive patients. Int J Cancer. 1990;46:581–585. doi: 10.1002/ijc.2910460405. [DOI] [PubMed] [Google Scholar]
- 62.Glaser SL, Clarke CA, Gulley ML, et al. Population-based patterns of human immunodeficiency virus-related Hodgkin lymphoma in the Greater San Francisco Bay Area, 1988-1998. Cancer. 2003;98:300–309. doi: 10.1002/cncr.11459. [DOI] [PubMed] [Google Scholar]
- 63.Biggar RJ, Jaffe ES, Goedert JJ, et al. Hodgkin lymphoma and immunodeficiency in persons with HIV/AIDS. Blood. 2006;108:3786–3791. doi: 10.1182/blood-2006-05-024109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lanoy E, Rosenberg PS, Fily F, et al. HIV-associated Hodgkin lymphoma during the first months on combination antiretroviral therapy. Blood. 2011;118:44–49. doi: 10.1182/blood-2011-02-339275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Powles T, Robinson D, Stebbing J, et al. Highly active antiretroviral therapy and the incidence of non-AIDS-defining cancers in people with HIV infection. J Clin Oncol. 2009;27:884–890. doi: 10.1200/JCO.2008.19.6626. [DOI] [PubMed] [Google Scholar]
- 66.Shiels MS, Pfeiffer RM, Gail MH, et al. Cancer burden in the HIV-infected population in the United States. J Natl Cancer Inst. 2011;103:753–762. doi: 10.1093/jnci/djr076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Goedert JJ, Bower M. Impact of highly effective antiretroviral therapy on the risk for Hodgkin lymphoma among people with human immunodeficiency virus infection. Curr Opin Oncol. 2012;24:531–536. doi: 10.1097/CCO.0b013e3283560697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kowalkowski MA, Mims MP, Amiran ES, et al. Effect of immune reconstitution on the incidence of HIV-related Hodgkin lymphoma. PLoS One. 2013;8:e77409. doi: 10.1371/journal.pone.0077409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Teruya-Feldstein J, Tosato G, Jaffe ES. The role of chemokines in Hodgkin's disease. Leuk Lymphoma. 2000;38:363–371. doi: 10.3109/10428190009087027. [DOI] [PubMed] [Google Scholar]
- 70.Baumforth KR, Birgersdotter A, Reynolds GM, et al. Expression of the Epstein-Barr virus-encoded Epstein-Barr virus nuclear antigen 1 in Hodgkin's lymphoma cells mediates up-regulation of CCL20 and the migration of regulatory T cells. Am J Pathol. 2008;173:195–204. doi: 10.2353/ajpath.2008.070845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Küppers R. New insights in the biology of Hodgkin lymphoma. Hematology. 2012;2012:328–334. doi: 10.1182/asheducation-2012.1.328. [DOI] [PubMed] [Google Scholar]
- 72.Piriou ER, van Dort K, Nanlohy NM, et al. Altered EBV viral load setpoint after HIV seroconversion is in accordance with lack of predictive value of EBV load for the occurrence of AIDS-related non-Hodgkin lymphoma. J Immunol. 2004;172:6931–6937. doi: 10.4049/jimmunol.172.11.6931. [DOI] [PubMed] [Google Scholar]
- 73.Rickinson AB. Co-infections, inflammation and oncogenesis: future directions for EBV research. Semin Cancer Biol. 2014;26:99–115. doi: 10.1016/j.semcancer.2014.04.004. [DOI] [PubMed] [Google Scholar]
- 74.Kis LL, Salamon D, Persson EK, et al. IL-21 imposes a type II EBV gene expression on type III and type I B cells by the repression of C- and activation of LMP-1-promoter. Proc Natl Acad Sci U S A. 2010;107:872–877. doi: 10.1073/pnas.0912920107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Nagy N, Adori M, Rasul A, et al. Soluble factors produced by activated CD4+ T cells modulate EBV latency. Proc Natl Acad Sci U S A. 2012;109:1512–1517. doi: 10.1073/pnas.1120587109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Rowe M, Raithatha S, Shannon-Lowe C. Counteracting effects of cellular Notch and Epstein-Barr virus EBNA2: implications for stromal effects on virus-host interactions. J Virol. 2014 doi: 10.1128/JVI.01431-14. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Anderson LJ, Longnecker R. EBV LMP2A provides a surrogate pre-B cell receptor signal through constitutive activation of the ERK/MAPK pathway. J Gen Virol. 2008;89:1563–1568. doi: 10.1099/vir.0.2008/001461-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Anderson LJ, Longnecker R. Epstein-Barr virus latent membrane protein 2A exploits Notch1 to alter B-cell identity in vivo. Blood. 2009;113:108–116. doi: 10.1182/blood-2008-06-160937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Crocker J, Overton SP, Smith PJ. Type IV collagen in Hodgkin's disease. An immunohistochemical study. Am J Clin Pathol. 1988;89:57–62. doi: 10.1093/ajcp/89.1.57. [DOI] [PubMed] [Google Scholar]
- 80.Poppema S, Potters M, Emmens R, et al. Immune reactions in classical Hodgkin's lymphoma. Sem Hematol. 1999;36:253–259. [PubMed] [Google Scholar]
- 81.Enblad G, Molin D, Glimelius I, et al. The potential role of innate immunity in the pathogenesis of Hodgkin's lymphoma. Hematol Oncol Clin North Am. 2007;21:805–823. doi: 10.1016/j.hoc.2007.07.007. [DOI] [PubMed] [Google Scholar]
- 82.Cader FZ, Vockerodt M, Bose S, et al. The EBV oncogene LMP1 protects lymphoma cells from cell death through the collagen-mediated activation of DDR1. Blood. 2013;122:4237–4245. doi: 10.1182/blood-2013-04-499004. [DOI] [PubMed] [Google Scholar]