

Abstract
In 1964, a new herpesvirus, Epstein-Barr virus (EBV), was discovered in cultured tumor cells derived from a Burkitt lymphoma (BL) biopsy taken from an African patient. This was a momentous event that reinvigorated research into viruses as a possible cause of human cancers. Subsequent studies demonstrated that EBV was a potent growth-transforming agent for primary B cells, and that all cases of BL carried characteristic chromosomal translocations resulting in constitutive activation of the c-MYC oncogene. These results hinted at simple oncogenic mechanisms that would make Burkitt lymphoma paradigmatic for cancers with viral etiology. In reality, the pathogenesis of this tumor is rather complicated with regard to both the contribution of the virus and the involvement of cellular oncogenes. Here, we review the current understanding of the roles of EBV and c-MYC in the pathogenesis of BL and the implications for new therapeutic strategies to treat this lymphoma.
Keywords: Epstein-Barr virus, Burkitt lymphoma, tumor virus, Malaria, c-MYC
Historical Perspective
Fifty years ago, the idea that viruses might be causative agents of human cancers was controversial. Moreover, all of the then-known viruses in animal models were RNA viruses, raising questions about the discovery by Epstein, Barr, and Achong of a herpesvirus, a DNA virus, in Burkitt lymphoma (BL). Proving that Epstein-Barr virus (EBV) was a driver and not simply a passenger in BL was a difficult task, and it initially relied heavily on epidemiological observations[1],[2].
Henle et al.[3] provided the first evidence that all African patients with BL were infected with EBV. To do so, they used serological assays they developed to detect the presence of antibodies to EBV capsid antigens. While these assays indicated EBV infection in all African children with BL, the virus was also prevalent in approximately 90% of healthy adults and 30% of children in the United States, where BL was not endemic[3]. Clearly, EBV infection was not, by itself, sufficient to cause BL. It was notable, however, that the incidence of EBV infection in African patients with BL was markedly higher than that in healthy children in the West, suggesting that most African children acquire EBV in the first 2 years of life whereas infection was more likely to be delayed by several years among affluent American populations[4]. The serological responses to EBV were qualitatively and quantitatively different between BL and control groups and during clinical progression, response, and relapse, each in a way that strongly implicated a role for EBV in BL[4]–[7]. Importantly, African patients with BL not only carried EBV, but the virus was also present in all of the malignant cells in their tumors, as demonstrated by DNA hybridization to detect viral genomes[8] and immunofluorescence to detect the latent infection EBV-encoded nuclear antigen (EBNA)[9],[10].
One key piece of evidence complementing the epidemiologic features of BL in Africans was that EBV is able to growth-transform normal resting B lymphocytes into lymphoblastoid cell lines (LCLs) that grow indefinitely in the laboratory[11],[12]. EBV clearly has oncogenic potential[13]. The growth-transforming function of EBV is now known to require coexpression of at least 6 latent infection viral genes that encode the nuclear antigens (EBNA1, EBNA2, EBNA3A, EBNA3C, and EBNA-LP) and the latent membrane protein 1 (LMP1)[14],[15]. Paradoxically, BL tumors usually express a far more restricted pattern of latent genes (Figure 1), with EBNA1 being the only viral protein regularly expressed, along with various non-coding RNA species that are dispensable for transforming function[16]–[19]. This more restricted pattern of EBV gene expression is insufficient to growth-transform primary B cells, which begs the question, what role does EBV play in the pathogenesis and maintenance of BL?
Figure 1. Epstein-Barr virus (EBV) gene expression in Burkitt lymphoma.

A, organization of the viral genes express during latent, non-productive infection. The schematic is not to scale, and for simplicity the double-stranded DNA episomal viral genome is represented here as a linear genome. The 6 nuclear protein-encoding genes (EBNAs; in red) and the latent membrane protein-2A and -2B genes (LMP2A/B; in blue) are transcribed in a rightward direction. The latent membrane protein-1 gene (LMP1; in blue) is transcribed from the reverse DNA strand. The relative locations of the non-coding RNAs, which include the EBV-encoded non-polyadenylated RNAs (EBERs) and numerous microRNAs, are depicted in purple. The origin of plasmid replication (OriP), which contains dyad repeat and direct repeat elements, is indicated downstream of the EBER gene. For reference, the terminal repeat (TR) region is indicated. The TRs are the site of genome linearization during lytic virus replication and re-ligation following latent infection. Because the first coding exon of LMP2A (and the first non-coding exon of LMP2B) is located to the left of the TR and the common LMP2A/B-coding exons are located to the right of the TR, transcription of LMP2A from the TP1 promoter and LMP2B from the TP2 promoter can only occur from the episomal genome. B, different latent gene transcription patterns. All of the coding and non-coding latent genes are expressed in normal B cells that are directly growth-transformed by EBV. This form of latency (Latency III) is observed in lymphoblastoid cell lines (LCLs) in vitro and in post-transplantation lymphoproliferative disease in vivo. All 6 EBNAs and BHRF1 vBCL2 are transcribed from the Wp and/or Cp promoters during Latency III. The majority of EBV-positive Burkitt lymphoma tumor biopsies display a Latency I pattern of gene expression in which the non-coding RNAs are expressed, but EBNA1 is the only protein-encoding gene transcribed. EBNA1 gene transcription in Latency I is driven from the Qp promoter (in contrast to Latency III in which Cp/Wp-driven transcription occurs). A minority of Burkitt lymphomas carry genomes with a deletion spanning their EBNA2 gene, from which they display a Wp-restricted form of latency. Wp-driven transcription allows expression of all of the remaining 5 EBNAs and also of BHRF1, which is expressed at much higher levels than in Latency III LCLs. The absence of EBNA2 in Wp-restricted latency results in repression of EBNA2-dependent LMP1 and LMP2 gene expression.
The high incidence of BL in Africa, ranging from around 3–6 new cases per year per 100,000 children aged 0–14 years[2], was shown by Denis Burkitt to be confined to geographic and climatic areas consistent with involvement of an insect-borne agent[20],[21]. This agent was subsequently identified as the Plasmodium falciparum malarial parasite, which is transmitted by mosquitoes and is holoendemic in the equatorial “tumour belt” mapped by Burkitt[22],[23]. Suppression of malaria infection associated with reduced incidence of BL[24], and there is a link between the incidence of BL in Africa and high antibody titers to both EBV and malaria[25],[26]. The evidence was, and remains, strongly suggestive that coinfection with EBV and Plasmodium falciparum malaria is somehow important in the development of BL[27].
Both EBV and Plasmodium falciparum malaria infections are widespread among children in Africa, yet only a relatively small number of coinfected children develop BL. Clearly other factors are involved. The search for features unique to the tumor revealed characteristic reciprocal chromosomal translocations, always involving chromosome 8 at what is now known to be the c-MYC oncogene locus, and most frequently chromosome 14 at the immunoglobulin heavy chain locus or, less commonly, chromosomes 2 or 22 at the immunoglobulin light chain loci[28]–[30].
While this lymphoma in Africa attracted much attention due to its unusually high incidence and its association with EBV, Gregory O'Conor, who was a colleague of Denis Burkitt in Africa, recognized early on that clinically and histologically indistinguishable lymphomas occurred outside Africa, albeit at much lower and variable incidence, and were not confined to children[31]–[34]. These so-called sporadic tumors are not associated with malaria and typically show only 10%–20% association with EBV, although this may increase to 30%–60% in BLs arising in human immunodeficiency virus (HIV)-positive individuals[35],[36]. Regardless of EBV association and geographic origin, all BLs contain c-MYC translocations and a consistent cellular gene expression profile[34],[37],[38].
EBV and the Pathogenesis of BL
To recap, EBV is known to be widespread in all human communities, and therefore, only a very small minority of infected individuals develop BL or indeed any of the other cancers now linked to the virus. Additionally, not all cases of BL are EBV-associated. By themselves, these facts do not rule out a causative role for EBV in oncogenesis, as cancer arises from multistep genetic events and different genetic events and molecular mechanisms may lead to the same cancer. Consistent with this, the role of EBV in BL pathogenesis is not a simple result of the virus' ability to growth-transform B lymphocytes, as most of the growth-transforming genes necessary for establishing and maintaining LCLs are repressed in BL tumors.
These observations can be reconciled with a role for EBV in BL pathogenesis by supposing that EBV somehow increases the likelihood of generating cellular genetic changes, most importantly MYC-activating translocations, which drive lymphoma development. The opportunities for EBV to promote such genetic events are apparent when one considers the mechanisms by which the virus colonizes and persists within the B-lymphocyte compartment of healthy infected people. These events have been extensively reviewed elsewhere[39]–[42]. While the details are still under debate[42]–[45], it is broadly agreed that the virus normally enters the B-cell compart-ment after crossing the epithelial barrier of oropharyngeal lymphoid tissues. Here, infected B cells may express transformation-associated viral genes and proliferate[44],[46],[47], but the only infected cells that enter the circulating peripheral blood are those with a non-proliferating memory B-cell phenotype and those with repressed expression of transformation-associated viral genes[48],[49].
The ability of EBV to induce cell division in lymphoid tissues represents a mechanism for expanding the pool of virus-infected cells, albeit at the cost of rendering the cells susceptible to elimination by virus-specific immune T cells[50]. Conversely, the ability to establish a silent infection in resting memory B cells allows persistence in immune-competent individuals. Thorley-Lawson[41] developed the idea that EBV-transformed, naïve B cells follow the normal B-cell development pathway to generate memory B cells via germinal center reactions, during which EBV gene expression is sequentially repressed. There is some dispute as to whether circulating memory B cells carrying EBV as a silent infection are derived only from infected naïve B cells that subsequently participate in a germinal center reaction, and whether EBV modulates the germinal center reaction or responds to it[42],[43],[51]. However, it is not disputed that small numbers of EBV-infected cells with more restricted patterns of latent gene expression are found within germinal centers, and in numbers that fit mathematical models of germinal center involvement in EBV persistence[43],[45],[52].
The significance of EBV-infected cells passing through germinal centers is that these anatomical sites of genetic instability are necessary for B-cell maturation. In germinal center B cells, there is up-regulation of activation-induced cytidine deaminase (AID), which is essential for somatic hypermutation (SHM) and immunoglobulin class-switching, the two genetic events that generate the vast repertoire of B-cell receptors from which clones with highest antigen affinity are selected. EBV-infected naïve B cells participating in germinal center reactions are subject to these genetic events. In addition, as revealed by in vitro infection experiments, EBV infection of naïve B cells can induce AID activation and SHM and, with additional T-cell help, immunoglobulin class-switching, raising the possibility that EBV can potentially drive memory B-cell maturation independently of germinal centers[51]. Regardless of whether or not EBV-infected naïve B cells enter the memory B-cell pool via germinal center-dependent or -independent routes, these infected B cells will be subject to genetic trauma en route to maturation. The pathogenesis of BL, in common with many other B-cell malignancies, is likely to involve genetic accidents arising from normal processes of B-cell maturation[53], with the most critical, or driver, mutation in the genesis of BL being the c-MYC oncogene translocation.
How, then, might EBV contribute to the generation of aberrantly mutated cells? One likely possibility is that its transforming potential allows the survival of aberrantly mutated cells that, in the absence of B-cell receptor (BCR) ligation through high-affinity cognate antigen, would otherwise be scheduled to die by apoptosis. This is evident in vitro, where EBV can transform germinal center B cells lacking immunoglobulin expression[54]–[57], and in vivo, where some EBV-positive lymphoproliferations have crippling immunoglobulin gene mutations[58]–[60]. In this context, expression of the latent membrane protein-2A (LMP2A) viral gene might override the need for high-affinity antigen/BCR survival signals, as LMP2A contains an immunoreceptor tyrosine-based activation motif that can replace BCR signals[61]–[64]. In addition, the EBV-encoded LMP1 protein, which functions similarly to a constitutively active CD40 receptor, can provide prosurvival signals to infected B cells within the germinal center[65]–[69].
In addition to enhancing the survival of mutated cells, EBV might also be directly involved in promoting genetic instability[70]–[72]. Various potential mechanisms have been identified, including induction of oxidative stress[71], induction of DNA damage and telomere dysfunction[70],[72],[73], activation of recombinases[74], and activation of AID and SHM[51],[75].
Malaria, HIV, and the Pathogenesis of BL
The high incidence of BL in Africa is restricted to those geographic areas where Plasmodium falciparum malaria is holoendemic. In these areas, EBV is present in tumor cells in virtually all BL cases. By contrast, the association with EBV is less frequent in sporadic BL that occurs worldwide. These observations are consistent with EBV and malaria cooperating to increase the incidence of this lymphoma[36],[76]. To a lesser extent, coinfection with HIV also increases both the overall incidence of BL and the frequency of EBV-positive BL[35],[36],[77]–[79]. Indeed, Rochford et al.[27] have argued that endemic BL is a polymicrobial disease.
Both malaria and HIV induce hypergammaglobulinemia through polyclonal B-cell activation and cause a sustained increase in EBV loads. It was postulated early that the contribution of malaria to BL pathogenesis involves its immunostimulatory effects on B cells[80] and possibly selective activation of the memory B-cell compartment[81],[82]. Malaria also impairs T-cell immunity to EBV infection[83]–[86]. Although this impairment is transient, the substantially increased EBV loads in circulating memory B cells associated with malarial infection are long-lived[87],[88]. Malaria's immunostimulatory effects on B cells, which include AID activation, and immunosuppressive effects on T-cell responses, combine to increase both EBV loads and the likelihood of generating and selecting for c-MYC/IG gene translocations[27],[89].
The Ongoing Contribution of EBV to the Evolved Tumor
Once established, most EBV-associated BL tumors display a Latency I pattern of viral gene expression. Coupled with inherent impairments in antigen processing functions in BL cells, the repressed viral gene expression probably benefits tumors insomuch as they can evade recognition by virus-specific immune T cells[17],[90],[91]. However, the virus is not simply a subdued passenger at this stage; rather, it continues to contribute to the malignant phenotype. Thus, rare subclones of some BL cell lines that have lost the viral genome display increased sensitivity to apoptosis-inducing agents, reduced growth in low serum or soft agar cultures, and loss of in vivo tumorigenicity as xenografts in mouse models[92]–[94]. The virus appears therefore to provide some survival advantage to malignant cells, even in the absence of most transformation-associated genes.
How EBV effects this enhanced survival advantage is unresolved. Reports on the role of EBNA1[93]–[96], EBERs[97]–[99], and the BART microRNAs[100] are sometimes contradictory but generally support the idea that one or more of these viral transcripts contribute to survival in BL cell lines. Most of these studies have been performed on a few select subclones of the Akata-BL cell line, and the generality of the observed protective effects is not clear. Our own recent data on a larger panel of cell lines with EBV loss derived from 5 different parental BL cell lines suggests that the survival advantage of Latency I parental BL lines over derivatives lacking EBV cannot be fully restored by physiological levels of any one of EBNA1, EBERs, or BART microRNAs, but rather requires the cooperative activity of these genes (unpublished data).
Although BL tumors generally display a Latency I form of EBV gene expression, an alternative pattern of viral gene expression was recently identified in a minority of BL cell lines and in their corresponding tumor biopsies[101],[102]. Such lines contain EBV genomes with deletions of around 6–8 kb that span the EBNA2 gene. Here, instead of Q-promoter (Qp)-driven EBNA1 expression, the W-promoter (Wp) is highly active and drives expression of all EBNAs except EBNA2 (Figure 1). Importantly, the broader pattern of latent viral gene expression in these Wp-restricted BLs includes high levels of the BHRF1 gene encoding a vBCL2 homolog that was previously thought to be exclusively a lytic cycle protein[103]. Interestingly, Wp-restricted BL cell lines usually contain both wild-type genomes and EBNA2-deleted genomes, and gene expression seems to be derived entirely from the deleted genomes while the wild-type genomes remain silenced[101]. The mechanism of this selective genome expression is unknown. Because the deleted genomes are derived from wild-type viruses that reside in the same tumor cells, it is possible that such deletions may also arise in other cases of BL that display classical Latency I gene expression. However, the deleted genomes would not be apparent unless the wild-type genomes were silenced and the Wp on the deleted genomes was activated.
Wp-restricted tumors all show characteristic BL morphology, c-MYC/IG gene translocations and high c-MYC expression. Genome-wide expression arrays of early passage lines and isogenic subclones with different latencies confirmed that Latency I and Wp-restricted BL tumors both fall within the “molecular BL” cellular gene signature, although Wp-restricted tumors were identifiable through up-regulation of certain plasmacytoid differentiation genes[104]. This plasmacytoid shift does not reflect the involvement of a different progenitor cell but, rather, is the phenotype imposed on a common BL progenitor by Wp-restricted latency. Despite expressing many immunogenic latent proteins that are not found in classical Latency I BLs, Wp-restricted BLs are insensitive to CD8+ immune T cells specific for these latent proteins[102]. All BL tumors, whether EBV-negative or EBV-positive, are therefore defective for HLA class I antigen processing, a phenotype that appears to be driven predominantly by overexpression of c-MYC and the absence of EBV LMP1[90],[105].
Another remarkable consequence of Wp-restricted latency in BL is that it confers a substantial anti-apoptotic phenotype on tumor lines relative to Latency I BL cell lines or EBV-negative BL cell lines[101]. The enhanced resistance to a broad range of apoptotic stimuli is predominantly mediated by Wp-driven expression of the vBCL2 encoded by BHRF1[103], aided by contributions from the EBNA3 proteins[106] and miR-BHRF1 miRNAs[107]. The greatly enhanced survival advantage of Wp-restricted BLs raises the question of whether this subset of BL tumors might show different clinical presentation and responses to therapy. This is the subject of ongoing collaborative studies initiated by our group. At present, there is no reliable estimate of the frequency of Wp-restricted tumors. Analysis of our large panel of Ugandan and Kenyan BL cell lines indicates that approximately 15% of the lines are Wp-restricted, but this is likely an overestimate because not all cultured biopsies generate cell lines and there is likely to be a bias toward successful establishment of vBCL2-expressing lines over Latency I lines. In addition, it is possible that there will be geographical variations in the incidence of Wp-restricted BL due to genetic traits in cellular and/or viral genomes.
The Impact of c-MYC Translocation and Other Mutations
The evidence is overwhelming that EBV both increases the incidence of BL and continues to provide a growth and/or survival advantage to the established tumor. What, then, can we say about the majority of sporadic tumors that are EBV-negative? One hypothesis is that EBV is involved in the pathogenesis of most BLs but malignant cells are obliged to either repress most latent viral gene expression or lose the viral genome completely, because of incompatibility between c-MYC and EBNA2/LMP1 expression[108] and of immune-selection against EBV transformation–associated proteins[17],[90]. Such a “hit-and-run” hypothesis for EBV is hard to prove or disprove, but the available evidence suggests that EBV-negative BLs arise independently of EBV involvement. Most notably, EBV-positive and EBV-negative cases of BL differ in the number of somatic mutations in their immunoglobulin heavy chain (VH) genes and the involvement of antigen selection, suggesting that virus-associated and virus-independent forms of BL differ in their cells of origin and probably in their pathogenesis[109]. Consistent with this observation, there are also striking differences in the translocation breakpoints in the c-MYC locus between EBV-positive and EBV-negative BLs[110]–[112].
Regardless of the origins of sporadic and endemic BLs, a consistent hallmark of all BLs is translocation and dysregulation of the c-MYC oncogene. Indeed, the molecular signature of all types of BL[34],[37],[38] appears to be largely but not entirely due to expression of c-MYC[113]. Furthermore, overexpression of c-MYC in human EBV-infected B cells can reproduce some key phenotypic characteristics of BL-derived tumor lines[105],[108],[114]. However, studies in both humans and murine models of c-MYC-induced tumors show that c-MYC/IG translocations can be detected in normal preneoplastic cells[115]–[117], inferring that dysregulation of the c-MYC oncogene is not by itself sufficient to cause Burkitt lymphoma. Indeed, the evidence now suggests that rather than being an initiator of oncogenic events, c-MYC has pleiotropic effects because it acts as a universal amplifier of transcriptionally active genes[118]–[120]. Therefore, while c-MYC can drive cell proliferation through up-regulation of cyclins D and E and down-regulation of the negative regulators, it also enhances normal negative regulation of proliferation, including apoptosis[121] (Figure 2).
Figure 2. Cellular gene mutations in Burkitt lymphoma.
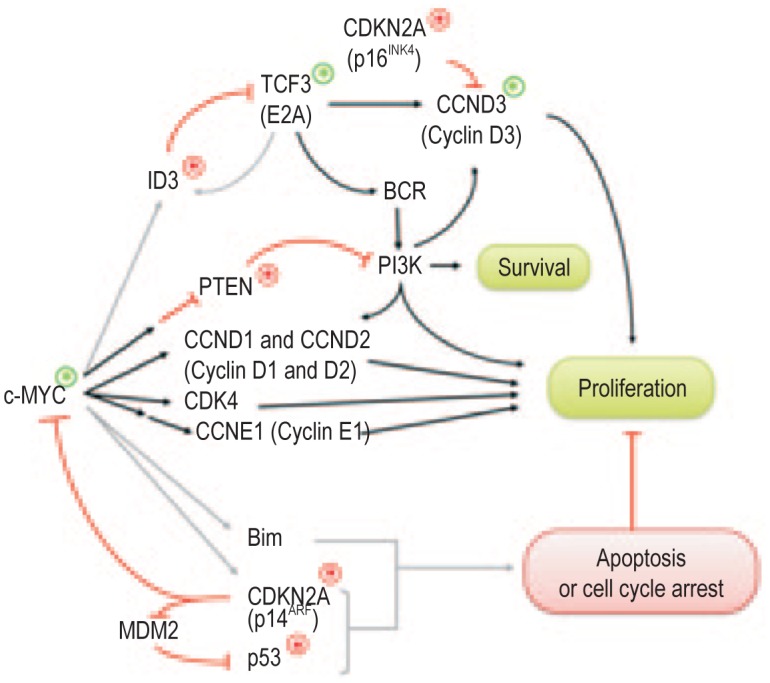
Key proliferative and apoptotic pathways induced by c-MYC are indicated in this schematic adapted from Schmitz et al.[136] and Kelly & Rickinson[159]. Genes that are frequently mutated in Burkitt lymphoma are indicated. Green asterisks denote gene- activating mutations, while red asterisks denote inhibitory mutations.
Animal models of c-MYC–induced tumorigenesis have demon-strated synergy between c-MYC and suppression of apoptosis through overexpression of anti-apoptotic BCL2 family genes[122], inactivation of the pro-apoptotic BH3-only Bcl2 family members, Puma, Noxa, and Bim[123],[124], modulation of PI3K signaling[125], or disruption of the ARF/MDM2/p53 pathway[126]–[128]. There is also evidence that EBNA1 can synergize with c-MYC in lymphomagenesis[129].
Consistent with experimental models showing that inhibition of apoptotic pathways is necessary for c-MYC-induced tumorigenesis, TP53 mutations are frequently observed in BL[130]–[134]. The overall incidence of TP53 mutations is approximately 30%–40% in tumor biopsies and approximately 60%–70% in established BL cell lines, regardless of the EBV status of the tumors. Other genetic and epigenetic alterations involved in complementing c-MYC activation in BL have been identified in the p14ARF/MDM2/p53 pathway [135].
A shift from targeted gene analysis to global genetic analysis, made possible by a new generation of sequencing technologies, has transformed our understanding of the genetic landscape of BL. Independent studies by Schmitz et al.[136], Richter et al.[137], and Love et al.[138] have integrated structural and functional genomics to catalog the broad range of somatic mutations in BL. The most commonly mutated gene was c-MYC itself, which was mutated in approximately 70% of BL[136]–[138]. This observation was consistent with a previous study that showed c-MYC to be hypermutated in BL, possibly due to translocation-induced juxtaposition with the immunoglobulin gene[139]. Activating mutations in c-MYC can dramatically alter target gene responses to increase c-MYC's oncogenic potential in BL[140], again highlighting the fact that the c-MYC/IG translocation alone is insufficient to drive lymphomagenesis.
A surprising observation from the game-changing structural and functional genomic studies was the frequent involvement of ID3, TCF3 (encoding E2A), and CCND3 (encoding cyclin D3) mutations, implicating a new pathway of oncogenic cooperation in the pathogenesis of BL. A summary of how some of the more common mutations in BL tumors affect the proliferative and apoptotic functions of c-MYC is shown schematically in Figure 2. The ID3 gene, a direct transcriptional target of c-MYC[141], is a negative regulator of TCF3. In BL, inactivating mutations of ID3 and activating mutations of TCF3 result in enhanced E2A function, which activates proliferation through induced cyclin D3 expression and survival through activation of PI3K signaling[136]. Interestingly, the mutations in ID3 and/or TCF3 were more common in sporadic BL (70%) than in endemic BL (40%), providing the first convincing evidence that the pathogenesis of EBV-negative BL requires more oncogenic mutations than EBV-positive BL.
New Therapeutic Targets
While Burkitt[142] initially found BL in African children to be exquisitely sensitive to chemotherapy, relapses of this aggressive lymphoma do occur and the initial promise of curative therapy faded. Certainly, the prospects for sporadic adult BLs were poor. The development of multi-agent chemotherapy and immunotherapy has led to marked improvements, although toxicities remain significant[143]–[149].
The scope for new targeted therapies has been widened by recent advances in identifying new pathways essential for the pathogenesis of BL, most notably the ID3/E2A/cyclinD3 pathway[136]–[138]. In addition, the recent detailed genetic mapping of BL also confirmed the importance of previously recognized targets that are ripe for exploitation. These include inhibitors of c-MYC[150]–[152], PI3 kinase[153]–[155], and BCL2 family members[156],[157]. Finally, while the effectiveness of EBV-targeted immunotherapy for virus-associated cases of EBV may be diminished by the inherent impaired antigen presentation functions of malignant cells[90],[102], small-molecule inhibitors of EBNA1 function represent a promising strategy in combination with other treatments[158].
Acknowledgments
The study was supported by a grant from the Cancer Research UK, London (Programme Award C5575/A15032).
References
- 1.De-The G. The epidemiology of Burkitt's lymphoma: evidence for a causal association with Epstein-Barr virus. Epidemiol Rev. 1979;1:32–54. doi: 10.1093/oxfordjournals.epirev.a036213. [DOI] [PubMed] [Google Scholar]
- 2.Magrath I. Epidemiology: clues to the pathogenesis of Burkitt lymphoma. Br J Haematol. 2012;156:744–756. doi: 10.1111/j.1365-2141.2011.09013.x. [DOI] [PubMed] [Google Scholar]
- 3.Henle G, Henle W. Immunofluorescence in cells derived from Burkitt's lymphoma. J Bacteriol. 1966;91:1248–1256. doi: 10.1128/jb.91.3.1248-1256.1966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Henle G, Henle W, Clifford P, et al. Antibodies to Epstein-Barr virus in Burkitt's lymphoma and control groups. J Natl Cancer Inst. 1969;43:1147–1157. [PubMed] [Google Scholar]
- 5.Klein G, Clifford P, Henle G, et al. EBV-associated serological patterns in a Burkitt lymphoma patient during regression and recurrence. Int J Cancer. 1969;4:416–421. doi: 10.1002/ijc.2910040406. [DOI] [PubMed] [Google Scholar]
- 6.Geser A, Lenoir GM, Anvret M, et al. Epstein-Barr virus markers in a series of Burkitt's lymphomas from the West Nile District, Uganda. Eur J Cancer Clin Oncol. 1983;19:1393–1404. doi: 10.1016/0277-5379(93)90009-t. [DOI] [PubMed] [Google Scholar]
- 7.Geser A, de The G, Lenoir G, et al. Final case reporting from the Ugandan prospective study of the relationship between EBV and Burkitt's lymphoma. Int J Cancer. 1982;29:397–400. doi: 10.1002/ijc.2910290406. [DOI] [PubMed] [Google Scholar]
- 8.Zur Hausen H, Schulte-Holthausen H, Klein G, et al. EBV DNA in biopsies of Burkitt tumours and anaplastic carcinomas of the nasopharynx. Nature. 1970;228:1056–1058. doi: 10.1038/2281056a0. [DOI] [PubMed] [Google Scholar]
- 9.Lindahl T, Klein G, Reedman BM, et al. Relationship between Epstein-Barr virus (EBV) DNA and the EBV-determined nuclear antigen (EBNA) in Burkitt lymphoma biopsies and other lymphoproliferative malignancies. Int J Cancer. 1974;13:764–772. doi: 10.1002/ijc.2910130605. [DOI] [PubMed] [Google Scholar]
- 10.Reedman BM, Klein G. Cellular localization of an Epstein-Barr virus (EBV)-associated complement-fixing antigen in producer and non-producer lymphoblastoid cell lines. Int J Cancer. 1973;11:499–520. doi: 10.1002/ijc.2910110302. [DOI] [PubMed] [Google Scholar]
- 11.Henle W, Diehl V, Kohn G, et al. Herpes-type virus and chromo-some marker in normal leukocytes after growth with irradiated Burkitt cells. Science. 1967;157:1064–1065. doi: 10.1126/science.157.3792.1064. [DOI] [PubMed] [Google Scholar]
- 12.Pope JH, Horne MK, Scott W. Transformation of foetal human leukocytes in vitro by filtrates of a human leukaemic cell line containing herpes-like virus. Int J Cancer. 1968;3:857–866. doi: 10.1002/ijc.2910030619. [DOI] [PubMed] [Google Scholar]
- 13.IARC/WHO Epstein-Barr Virus and Kaposi's Sarcoma Herpesvirus/Human Herpesvirus 8. 1997/01/01. IARC. 1997 [PMC free article] [PubMed] [Google Scholar]
- 14.Delecluse HJ, Hammerschmidt W. The genetic approach to the Epstein-Barr virus: from basic virology to gene therapy. Mol Pathol. 2000;53:270–279. doi: 10.1136/mp.53.5.270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kieff E, Rickinson AB. Epstein-Barr virus and its replication. In: Knipe DM, Howley PM, editors. Fields Virology. 5th. Philadelphia, USA: Lippincott, Williams & Wilkins; 2007. pp. 2603–2654. [Google Scholar]
- 16.Masucci MG, Torsteindottir S, Colombani J, et al. Down-regulation of class I HLA antigens and of the Epstein-Barr virus-encoded latent membrane protein in Burkitt lymphoma lines. Proc Natl Acad Sci U S A. 1987;84:4567–4571. doi: 10.1073/pnas.84.13.4567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rowe DT, Rowe M, Evan GI, et al. Restricted expression of EBV latent genes and T-lymphocyte-detected membrane antigen in Burkitt's lymphoma cells. EMBO J. 1986;5:2599–2607. doi: 10.1002/j.1460-2075.1986.tb04540.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rowe M, Rowe DT, Gregory CD, et al. Differences in B cell growth phenotype reflect novel patterns of Epstein-Barr virus latent gene expression in Burkitt's lymphoma cells. EMBO J. 1987;6:2743–2751. doi: 10.1002/j.1460-2075.1987.tb02568.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Amoroso R, Fitzsimmons L, Thomas WA, et al. Quantitative studies of Epstein-Barr virus-encoded microRNAs provide novel insights into their regulation. J Virol. 2011;85:996–1010. doi: 10.1128/JVI.01528-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Burkitt D. A “tumour safari” in East and Central Africa. Br J Cancer. 1962;16:379–386. doi: 10.1038/bjc.1962.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Burkitt D. A children's cancer dependent on climatic factors. Nature. 1962;194:232–234. doi: 10.1038/194232a0. [DOI] [PubMed] [Google Scholar]
- 22.Dalldorf G, Linsell CA, Barnhart FE, et al. An epidemiologic approach to the lymphomas of African children and Burkitt's sacroma of the jaws. Perspect Biol Med. 1964;7:435–449. doi: 10.1353/pbm.1964.0023. [DOI] [PubMed] [Google Scholar]
- 23.Kafuko GW, Burkitt DP. Burkitt's lymphoma and malaria. Int J Cancer. 1970;6:1–9. doi: 10.1002/ijc.2910060102. [DOI] [PubMed] [Google Scholar]
- 24.Geser A, Brubaker G, Draper CC. Effect of a malaria suppression program on the incidence of African Burkitt's lymphoma. Am J Epidemiol. 1989;129:740–752. doi: 10.1093/oxfordjournals.aje.a115189. [DOI] [PubMed] [Google Scholar]
- 25.Carpenter LM, Newton R, Casabonne D, et al. Antibodies against malaria and Epstein-Barr virus in childhood Burkitt lymphoma: a case-control study in Uganda. Int J Cancer. 2008;122:1319–1323. doi: 10.1002/ijc.23254. [DOI] [PubMed] [Google Scholar]
- 26.Mutalima N, Molyneux E, Jaffe H, et al. Associations between Burkitt lymphoma among children in Malawi and infection with HIV, EBV and malaria: results from a case-control study. PLoS One. 2008;3:e2505. doi: 10.1371/journal.pone.0002505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rochford R, Cannon MJ, Moormann AM. Endemic Burkitt's lymphoma: a polymicrobial disease? Nat Rev Microbiol. 2005;3:182–187. doi: 10.1038/nrmicro1089. [DOI] [PubMed] [Google Scholar]
- 28.Manolov G, Manolova Y. Marker band in one chromosome 14 from Burkitt lymphomas. Nature. 1972;237:33–34. doi: 10.1038/237033a0. [DOI] [PubMed] [Google Scholar]
- 29.Dalla-Favera R, Bregni M, Erikson J, et al. Human c-myc oncogene is located on the region of chromosome 8 that is translocated in Burkitt lymphoma cells. Proc Natl Acad Sci U S A. 1982;79:7824–7827. doi: 10.1073/pnas.79.24.7824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Taub R, Kirsch I, Morton C, et al. Translocation of the c-myc gene into the immunoglobulin heavy chain locus in human Burkitt lymphoma and murine plasmacytoma cells. Proc Natl Acad Sci U S A. 1982;79:7837–7841. doi: 10.1073/pnas.79.24.7837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.O'Conor GT, Rappaport H, Smith EB. Childhood lymphoma resembling “Burkitt Tumor” in the United States. Cancer. 1965;18:411–417. doi: 10.1002/1097-0142(196504)18:4<411::aid-cncr2820180403>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- 32.O'Conor GT, Davies JN. Malignant tumors in African children. With special reference to malignant lymphoma. J Pediatr. 1960;56:526–535. doi: 10.1016/s0022-3476(60)80369-1. [DOI] [PubMed] [Google Scholar]
- 33.O'Conor GT. Significant aspects of childhood lymphoma in Africa. Cancer Res. 1963;23:1514–1518. [PubMed] [Google Scholar]
- 34.Leoncini L, Raphaël M, Stein H, et al. Burkitt lymphoma. In: Swedlow SH, Campo E, Harris NL, et al., editors. WHO classification of tumours of haematopoietic and lymphoid tissues. 4th. Lyon, France: International Agency for Research on Cancer; 2008. [Google Scholar]
- 35.Mbulaiteye SM, Pullarkat ST, Nathwani BN, et al. Epstein-Barr virus patterns in US Burkitt lymphoma tumors from the SEER residual tissue repository during 1979-2009. APMIS. 2014;122:5–15. doi: 10.1111/apm.12078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Magrath I. The pathogenesis of Burkitt's lymphoma. Adv Cancer Res. 1990;55:133–270. doi: 10.1016/s0065-230x(08)60470-4. [DOI] [PubMed] [Google Scholar]
- 37.Hummel M, Bentink S, Berger H, et al. A biologic definition of Burkitt's lymphoma from transcriptional and genomic profiling. N Engl J Med. 2006;354:2419–2430. doi: 10.1056/NEJMoa055351. [DOI] [PubMed] [Google Scholar]
- 38.Dave SS, Fu K, Wright GW, et al. Molecular diagnosis of Burkitt's lymphoma. N Engl J Med. 2006;354:2431–2442. doi: 10.1056/NEJMoa055759. [DOI] [PubMed] [Google Scholar]
- 39.Rickinson AB, Kieff E. Epstein-Barr virus. In: Knipe DM, Howley PM, editors. Fields Virology. 5th. Philadelphia, USA: Lippincott, Williams & Wilkins; 2007. pp. 2655–2700. [Google Scholar]
- 40.Rowe M, Kelly GL, Bell AI, et al. Burkitt's lymphoma: the Rosetta Stone deciphering Epstein-Barr virus biology. Semin Cancer Biol. 2009;19:377–388. doi: 10.1016/j.semcancer.2009.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Thorley-Lawson DA. Epstein-Barr virus: exploiting the immune system. Nature Rev Immunol. 2001;1:75–82. doi: 10.1038/35095584. [DOI] [PubMed] [Google Scholar]
- 42.Thorley-Lawson DA, Hawkins JB, Tracy SI, et al. The pathogenesis of Epstein-Barr virus persistent infection. Curr Opin Virol. 2013;3:227–232. doi: 10.1016/j.coviro.2013.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kurth J, Hansmann ML, Rajewsky K, et al. Epstein-Barr virus-infected B cells expanding in germinal centers of infectious mononucleosis patients do not participate in the germinal center reaction. Proc Natl Acad Sci U S A. 2003;100:4730–4735. doi: 10.1073/pnas.2627966100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kurth J, Spieker T, Wustrow J, et al. EBV-infected B cells in infectious mononucleosis: viral strategies for spreading in the B cell compartment and establishing latency. Immunity. 2000;13:485–495. doi: 10.1016/s1074-7613(00)00048-0. [DOI] [PubMed] [Google Scholar]
- 45.Roughan JE, Torgbor C, Thorley-Lawson DA. Germinal center B cells latently infected with Epstein-Barr virus proliferate extensively but do not increase in number. J Virol. 2010;84:1158–1168. doi: 10.1128/JVI.01780-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Joseph AM, Babcock GJ, Thorley-Lawson DA. Cells expressing the Epstein-Barr virus growth program are present in and restricted to the naive B-cell subset of healthy tonsils. J Virol. 2000;74:9964–9971. doi: 10.1128/jvi.74.21.9964-9971.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Niedobitek G, Agathanggelou A, Herbst H, et al. Epstein-Barr virus (EBV) infection in infectious mononucleosis: virus latency, replication and phenotype of EBV-infected cells. J Pathol. 1997;182:151–159. doi: 10.1002/(SICI)1096-9896(199706)182:2<151::AID-PATH824>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 48.Babcock GJ, Decker LL, Volk M, et al. EBV persistence in memory B cells in vivo. Immunity. 1998;9:395–404. doi: 10.1016/s1074-7613(00)80622-6. [DOI] [PubMed] [Google Scholar]
- 49.Babcock GJ, Hochberg D, Thorley-Lawson AD. The expression pattern of Epstein-Barr virus latent genes in vivo is dependent upon the differentiation stage of the infected B cell. Immunity. 2000;13:497–506. doi: 10.1016/s1074-7613(00)00049-2. [DOI] [PubMed] [Google Scholar]
- 50.Hislop AD, Taylor GS, Sauce D, et al. Cellular responses to viral infection in humans: lessons from Epstein-Barr virus. Annu Rev Immunol. 2007;25:587–617. doi: 10.1146/annurev.immunol.25.022106.141553. [DOI] [PubMed] [Google Scholar]
- 51.Heath E, Begue-Pastor N, Chaganti S, et al. Epstein-Barr virus infection of naive B cells in vitro frequently selects clones with mutated immunoglobulin genotypes: implications for virus biology. PLoS Pathog. 2012;8:e1002697. doi: 10.1371/journal.ppat.1002697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Roughan JE, Thorley-Lawson DA. The intersection of Epstein-Barr virus with the germinal center. J Virol. 2009;83:3968–3976. doi: 10.1128/JVI.02609-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kuppers R. Mechanisms of B-cell lymphoma pathogenesis. Nat Rev Cancer. 2005;5:251–262. doi: 10.1038/nrc1589. [DOI] [PubMed] [Google Scholar]
- 54.Gregory CD, Kirchgens C, Edwards CF, et al. Epstein-Barr virus-transformed human precursor B cell lines: altered growth phenotype of lines with germ-line or rearranged but nonexpressed heavy chain genes. Eur J Immunol. 1987;17:1199–1207. doi: 10.1002/eji.1830170818. [DOI] [PubMed] [Google Scholar]
- 55.Mancao C, Altmann M, Jungnickel B, et al. Rescue of “crippled” germinal center B cells from apoptosis by Epstein-Barr virus. Blood. 2005;106:4339–4344. doi: 10.1182/blood-2005-06-2341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bechtel D, Kurth J, Unkel C, et al. Transformation of BCR-deficient germinal-center B cells by EBV supports a major role of the virus in the pathogenesis of Hodgkin and posttransplantation lymphomas. Blood. 2005;106:4345–4350. doi: 10.1182/blood-2005-06-2342. [DOI] [PubMed] [Google Scholar]
- 57.Chaganti S, Bell AI, Pastor NB, et al. Epstein-Barr virus infection in vitro can rescue germinal center B cells with inactivated immunoglobulin genes. Blood. 2005;106:4249–4252. doi: 10.1182/blood-2005-06-2327. [DOI] [PubMed] [Google Scholar]
- 58.Brauninger A, Spieker T, Mottok A, et al. Epstein-Barr virus (EBV)-positive lymphoproliferations in post-transplant patients show immunoglobulin V gene mutation patterns suggesting interference of EBV with normal B cell differentiation processes. Eur J Immunol. 2003;33:1593–1602. doi: 10.1002/eji.200323765. [DOI] [PubMed] [Google Scholar]
- 59.Capello D, Cerri M, Muti G, et al. Molecular histogenesis of posttransplantation lymphoproliferative disorders. Blood. 2003;102:3775–3785. doi: 10.1182/blood-2003-05-1683. [DOI] [PubMed] [Google Scholar]
- 60.Timms JM, Bell A, Flavell JR, et al. Target cells of Epstein-Barr-virus (EBV)-positive post-transplant lymphoproliferative disease: similarities to EBV-positive Hodgkin's lymphoma. Lancet. 2003;361:217–223. doi: 10.1016/S0140-6736(03)12271-4. [DOI] [PubMed] [Google Scholar]
- 61.Caldwell RG, Wilson JB, Anderson SJ, et al. Epstein-Barr virus LMP2A drives B cell development and survival in the absence of normal B cell receptor signals. Immunity. 1998;9:405–411. doi: 10.1016/s1074-7613(00)80623-8. [DOI] [PubMed] [Google Scholar]
- 62.Casola S, Otipoby KL, Alimzhanov M, et al. B cell receptor signal strength determines B cell fate. Nat Immunol. 2004;5:317–327. doi: 10.1038/ni1036. [DOI] [PubMed] [Google Scholar]
- 63.Mancao C, Hammerschmidt W. Epstein-Barr virus latent membrane protein 2A is a B-cell receptor mimic and essential for B-cell survival. Blood. 2007;110:3715–3721. doi: 10.1182/blood-2007-05-090142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Brauninger A, Schmitz R, Bechtel D, et al. Molecular biology of Hodgkin's and Reed/Sternberg cells in Hodgkin's lymphoma. Int J Cancer. 2006;118:1853–1861. doi: 10.1002/ijc.21716. [DOI] [PubMed] [Google Scholar]
- 65.Mosialos G, Birkenbach M, Yalamanchili R, et al. The Epstein-Barr virus transforming protein LMP1 engages signaling proteins for the Tumor Necrosis Factor receptor family. Cell. 1995;80:389–399. doi: 10.1016/0092-8674(95)90489-1. [DOI] [PubMed] [Google Scholar]
- 66.Floettmann JE, Rowe M. Epstein-Barr virus latent membrane protein-1 (LMP1) C-terminus activation region-2 (CTAR2) maps to the far C-terminus and requires oligomersation for NF-kB activation. Oncogene. 1997;15:1851–1858. doi: 10.1038/sj.onc.1201359. [DOI] [PubMed] [Google Scholar]
- 67.Gires O, Zimber-Strobl U, Gonnella R, et al. Latent membrane protein 1 of Epstein-Barr virus mimics a constitutively active receptor molecule. EMBO J. 1997;16:6131–6140. doi: 10.1093/emboj/16.20.6131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Henderson S, Rowe M, Gregory C, et al. Induction of bcl-2 expression by Epstein-Barr virus latent membrane protein 1 protects infected B cells from programmed cell death. Cell. 1991;65:1107–1115. doi: 10.1016/0092-8674(91)90007-l. [DOI] [PubMed] [Google Scholar]
- 69.Cader FZ, Vockerodt M, Bose S, et al. The EBV oncogene LMP1 protects lymphoma cells from cell death through the collagen-mediated activation of DDR1. Blood. 2013;122:4237–4245. doi: 10.1182/blood-2013-04-499004. [DOI] [PubMed] [Google Scholar]
- 70.Kamranvar SA, Gruhne B, Szeles A, et al. Epstein-Barr virus promotes genomic instability in Burkitt's lymphoma. Oncogene. 2007;26:5115–5123. doi: 10.1038/sj.onc.1210324. [DOI] [PubMed] [Google Scholar]
- 71.Gualandi G, Giselico L, Carloni M, et al. Enhancement of genetic instability in human B cells by Epstein-Barr virus latent infection. Mutagenesis. 2001;16:203–208. doi: 10.1093/mutage/16.3.203. [DOI] [PubMed] [Google Scholar]
- 72.Lacoste S, Wiechec E, Dos Santos Silva AG, et al. Chromosomal rearrangements after ex vivo Epstein-Barr virus (EBV) infection of human B cells. Oncogene. 2010;29:503–515. doi: 10.1038/onc.2009.359. [DOI] [PubMed] [Google Scholar]
- 73.Kamranvar SA, Chen X, Masucci MG. Telomere dysfunction and activation of alternative lengthening of telomeres in B-lymphocytes infected by Epstein-Barr virus. Oncogene. 2013;32:5522–5530. doi: 10.1038/onc.2013.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kuhn-Hallek I, Sage DR, Stein L, et al. Expression of recombination activating genes (RAG-1 and RAG-2) in Epstein-Barr virus-bearing B cells. Blood. 1995;85:1289–1299. [PubMed] [Google Scholar]
- 75.Epeldegui M, Hung YP, McQuay A, et al. Infection of human B cells with Epstein-Barr virus results in the expression of somatic hypermutation-inducing molecules and in the accrual of oncogene mutations. Mol Immunol. 2007;44:934–942. doi: 10.1016/j.molimm.2006.03.018. [DOI] [PubMed] [Google Scholar]
- 76.Dalldorf G. Lymphomas of African children with different forms or environmental influences. JAMA. 1962;181:1026–1028. doi: 10.1001/jama.1962.03050380004002. [DOI] [PubMed] [Google Scholar]
- 77.Ziegler JL, Beckstead JA, Volberding PA, et al. Non-Hodgkin's lymphoma in 90 homosexual men. Relation to generalized lymphadenopathy and the acquired immunodeficiency syndrome. N Engl J Med. 1984;311:565–570. doi: 10.1056/NEJM198408303110904. [DOI] [PubMed] [Google Scholar]
- 78.Ziegler JL, Drew WL, Miner RC, et al. Outbreak of Burkitt's-like lymphoma in homosexual men. Lancet. 1982;2:631–633. doi: 10.1016/s0140-6736(82)92740-4. [DOI] [PubMed] [Google Scholar]
- 79.Wiggill TM, Mantina H, Willem P, et al. Changing pattern of lymphoma subgroups at a tertiary academic complex in a high-prevalence HIV setting: a South African perspective. J Acquir Immune Defic Syndr. 2011;56:460–466. doi: 10.1097/QAI.0b013e31820bb06a. [DOI] [PubMed] [Google Scholar]
- 80.O'Conor GT. Persistent immunologic stimulation as a factor in oncogenesis, with special reference to Burkitt's tumor. Am J Med. 1970;48:279–285. doi: 10.1016/0002-9343(70)90057-4. [DOI] [PubMed] [Google Scholar]
- 81.Donati D, Mok B, Chene A, et al. Increased B cell survival and preferential activation of the memory compartment by a malaria polyclonal B cell activator. J Immunol. 2006;177:3035–3044. doi: 10.4049/jimmunol.177.5.3035. [DOI] [PubMed] [Google Scholar]
- 82.Rasti N, Falk KI, Donati D, et al. Circulating Epstein-Barr virus in children living in malaria-endemic areas. Scand J Immunol. 2005;61:461–465. doi: 10.1111/j.1365-3083.2005.01589.x. [DOI] [PubMed] [Google Scholar]
- 83.Moss DJ, Burrows SR, Castelino DJ, et al. A comparison of Epstein-Barr virus-specific T-cell immunity in malaria-endemic and -nonendemic regions of Papua New Guinea. Int J Cancer. 1983;31:727–732. doi: 10.1002/ijc.2910310609. [DOI] [PubMed] [Google Scholar]
- 84.Urban BC, Ferguson DJ, Pain A, et al. Plasmodium falciparum-infected erythrocytes modulate the maturation of dendritic cells. Nature. 1999;400:73–77. doi: 10.1038/21900. [DOI] [PubMed] [Google Scholar]
- 85.Whittle HC, Brown J, Marsh K, et al. T-cell control of Epstein-Barr virus-infected B cells is lost during P. falciparum malaria. Nature. 1984;312:449–450. doi: 10.1038/312449a0. [DOI] [PubMed] [Google Scholar]
- 86.Moormann AM, Chelimo K, Sumba PO, et al. Exposure to holoendemic malaria results in suppression of Epstein-Barr virus-specific T cell immunosurveillance in Kenyan children. J Infect Dis. 2007;195:799–808. doi: 10.1086/511984. [DOI] [PubMed] [Google Scholar]
- 87.Moormann AM, Chelimo K, Sumba OP, et al. Exposure to holoendemic malaria results in elevated Epstein-Barr virus loads in children. J Infect Dis. 2005;191:1233–1238. doi: 10.1086/428910. [DOI] [PubMed] [Google Scholar]
- 88.Njie R, Bell AI, Jia H, et al. The effects of acute malaria on Epstein-Barr virus (EBV) load and EBV-specific T cell immunity in Gambian children. J Infect Dis. 2009;199:31–38. doi: 10.1086/594373. [DOI] [PubMed] [Google Scholar]
- 89.Torgbor C, Awuah P, Deitsch K, et al. A multifactorial role for P. falciparum malaria in endemic Burkitt's lymphoma pathogenesis. PLoS Pathog. 2014;10:e1004170. doi: 10.1371/journal.ppat.1004170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Rowe M, Khanna R, Jacob CA, et al. Restoration of endogenous antigen processing in Burkitt's lymphoma cells by Epstein-Barr virus latent membrane protein-1: coordinate upregulation of peptide transporters and HLA-class I antigen expression. Eur J Immunol. 1995;25:1374–1384. doi: 10.1002/eji.1830250536. [DOI] [PubMed] [Google Scholar]
- 91.Levitskaya J, Sharipo A, Leonchiks A, et al. Inhibition of ubiquitin/proteasome-dependent protein degradation by the Gly-Ala repeat domain of the Epstein-Barr virus nuclear antigen 1. Proc Natl Acad Sci U S A. 1997;94:12616–12621. doi: 10.1073/pnas.94.23.12616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Shimizu N, Tanabe-Tochikura A, Kuroiwa Y, et al. Isolation of Epstein-Barr virus (EBV)-negative cell clones from the EBV-positive Burkitt's lymphoma (BL) line Akata: malignant phenotypes of BL cells are dependent on EBV. J Virol. 1994;68:6069–6073. doi: 10.1128/jvi.68.9.6069-6073.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Komano J, Sugiura M, Takada K. Epstein-Barr virus contributes to the malignant phenotype and to apoptosis resistance in Burkitt's lymphoma cell line Akata. J Virol. 1998;72:9150–9156. doi: 10.1128/jvi.72.11.9150-9156.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Ruf IK, Rhyne PW, Yang H, et al. Epstein-barr virus regulates c-MYC, apoptosis, and tumorigenicity in Burkitt lymphoma. Mol Cell Biol. 1999;19:1651–1660. doi: 10.1128/mcb.19.3.1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kennedy G, Komano J, Sugden B. Epstein-Barr virus provides a survival factor to Burkitt's lymphomas. Proc Natl Acad Sci U S A. 2003;100:14269–14274. doi: 10.1073/pnas.2336099100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Saridakis V, Sheng Y, Sarkari F, et al. Structure of the p53 binding domain of HAUSP/USP7 bound to Epstein-Barr nuclear antigen 1 implications for EBV-mediated immortalization. Mol Cell. 2005;18:25–36. doi: 10.1016/j.molcel.2005.02.029. [DOI] [PubMed] [Google Scholar]
- 97.Ruf IK, Lackey KA, Warudkar S, et al. Protection from interferon-induced apoptosis by Epstein-Barr virus small RNAs is not mediated by inhibition of PKR. J Virol. 2005;79:14562–14569. doi: 10.1128/JVI.79.23.14562-14569.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Ruf IK, Rhyne PW, Yang C, et al. Epstein-Barr virus small RNAs potentiate tumorigenicity of Burkitt lymphoma cells independently of an effect on apoptosis. J Virol. 2000;74:10223–10228. doi: 10.1128/jvi.74.21.10223-10228.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Komano J, Maruo S, Kurozumi K, et al. Oncogenic role of Epstein-Barr virus-encoded RNAs in Burkitt's lymphoma cell line Akata. J Virol. 1999;73:9827–9831. doi: 10.1128/jvi.73.12.9827-9831.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Vereide DT, Seto E, Chiu YF, et al. Epstein-Barr virus maintains lymphomas via its miRNAs. Oncogene. 2014;33:1258–1264. doi: 10.1038/onc.2013.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Kelly GL, Milner AE, Tierney RJ, et al. Epstein-Barr virus nuclear antigen 2 (EBNA2) gene deletion is consistently linked with EBNA3A, -3B, and -3C expression in Burkitt's lymphoma cells and with increased resistance to apoptosis. J Virol. 2005;79:10709–10717. doi: 10.1128/JVI.79.16.10709-10717.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Kelly G, Bell A, Rickinson A. Epstein-Barr virus-associated Burkitt lymphomagenesis selects for downregulation of the nuclear antigen EBNA2. Nat Med. 2002;8:1098–1104. doi: 10.1038/nm758. [DOI] [PubMed] [Google Scholar]
- 103.Kelly GL, Long HM, Stylianou J, et al. An Epstein-Barr virus anti-apoptotic protein constitutively expressed in transformed cells and implicated in Burkitt lymphomagenesis: the Wp/BHRF1 link. PLoS Pathog. 2009;5:e1000341. doi: 10.1371/journal.ppat.1000341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Kelly GL, Stylianou J, Rasaiyaah J, et al. Different patterns of Epstein-Barr virus latency in endemic Burkitt lymphoma (BL) lead to distinct variants within the BL-associated gene expression signature. J Virol. 2013;87:2882–2894. doi: 10.1128/JVI.03003-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Staege MS, Lee SP, Frisan T, et al. MYC overexpression imposes a nonimmunogenic phenotype on Epstein-Barr virus-infected B cells. Proc Natl Acad Sci U S A. 2002;99:4550–4555. doi: 10.1073/pnas.072495599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Anderton E, Yee J, Smith P, et al. Two Epstein-Barr virus (EBV) oncoproteins cooperate to repress expression of the proapoptotic tumour-suppressor Bim: clues to the pathogenesis of Burkitt's lymphoma. Oncogene. 2008;27:421–433. doi: 10.1038/sj.onc.1210668. [DOI] [PubMed] [Google Scholar]
- 107.Feederle R, Linnstaedt SD, Bannert H, et al. A viral microRNA cluster strongly potentiates the transforming properties of a human herpesvirus. PLoS Pathog. 2011;7:e1001294. doi: 10.1371/journal.ppat.1001294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Pajic A, Staege MS, Dudziak D, et al. Antagonistic effects of c-myc and Epstein-Barr virus latent genes on the phenotype of human B cells. Int J Cancer. 2001;93:810–816. doi: 10.1002/ijc.1404. [DOI] [PubMed] [Google Scholar]
- 109.Bellan C, Lazzi S, Hummel M, et al. Immunoglobulin gene analysis reveals 2 distinct cells of origin for EBV-positive and EBV-negative Burkitt lymphomas. Blood. 2005;106:1031–1036. doi: 10.1182/blood-2005-01-0168. [DOI] [PubMed] [Google Scholar]
- 110.Pelicci PG, Knowles DM, 2nd, Magrath I, et al. Chromosomal breakpoints and structural alterations of the c-myc locus differ in endemic and sporadic forms of Burkitt lymphoma. Proc Natl Acad Sci U S A. 1986;83:2984–2988. doi: 10.1073/pnas.83.9.2984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Guikema JE, de Boer C, Haralambieva E, et al. IGH switch breakpoints in Burkitt lymphoma: exclusive involvement of noncanonical class switch recombination. Genes Chromosomes Cancer. 2006;45:808–819. doi: 10.1002/gcc.20345. [DOI] [PubMed] [Google Scholar]
- 112.Shiramizu B, Barriga F, Neequaye J, et al. Patterns of chro-mosomal breakpoint locations in Burkitt's lymphoma: relevance to geography and Epstein-Barr virus association. Blood. 1991;77:1516–1526. [PubMed] [Google Scholar]
- 113.Schrader A, Bentink S, Spang R, et al. High Myc activity is an independent negative prognostic factor for diffuse large B cell lymphomas. Int J Cancer. 2012;131:E348–E361. doi: 10.1002/ijc.26423. [DOI] [PubMed] [Google Scholar]
- 114.Polack A, Hortnagel K, Pajic A, et al. c-myc activation renders proliferation of Epstein-Barr virus (EBV)-transformed cells independent of EBV nuclear antigen 2 and latent membrane protein 1. Proc Natl Acade Sci U S A. 1996;93:10411–10416. doi: 10.1073/pnas.93.19.10411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Janz S, Muller J, Shaughnessy J, et al. Detection of recombinations between c-myc and immunoglobulin switch alpha in murine plasma cell tumors and preneoplastic lesions by polymerase chain reaction. Proc Natl Acad Sci U S A. 1993;90:7361–7365. doi: 10.1073/pnas.90.15.7361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Muller JR, Janz S, Goedert JJ, et al. Persistence of immunoglobulin heavy chain/c-myc recombination-positive lymphocyte clones in the blood of human immunodeficiency virus-infected homosexual men. Proc Natl Acad Sci U S A. 1995;92:6577–6581. doi: 10.1073/pnas.92.14.6577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Klein G. Dysregulation of lymphocyte proliferation by chromosomal translocations and sequential genetic changes. BioEssays. 2000;22:414–422. doi: 10.1002/(SICI)1521-1878(200005)22:5<414::AID-BIES3>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 118.Lin CY, Loven J, Rahl PB, et al. Transcriptional amplification in tumor cells with elevated c-Myc. Cell. 2012;151:56–67. doi: 10.1016/j.cell.2012.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.McCarthy N. Tumorigenesis: megaphone MYC. Nat Rev Cancer. 2012;12:733. doi: 10.1038/nrc3384. [DOI] [PubMed] [Google Scholar]
- 120.Nie Z, Hu G, Wei G, et al. c-Myc is a universal amplifier of expressed genes in lymphocytes and embryonic stem cells. Cell. 2012;151:68–79. doi: 10.1016/j.cell.2012.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Pelengaris S, Khan M, Evan G. c-MYC: more than just a matter of life and death. Nat Rev Cancer. 2002;2:764–776. doi: 10.1038/nrc904. [DOI] [PubMed] [Google Scholar]
- 122.Strasser A, Harris AW, Bath ML, et al. Novel primitive lymphoid tumours induced in transgenic mice by cooperation between myc and bcl-2. Nature. 1990;348:331–333. doi: 10.1038/348331a0. [DOI] [PubMed] [Google Scholar]
- 123.Michalak EM, Jansen ES, Happo L, et al. Puma and to a lesser extent Noxa are suppressors of Myc-induced lymphomagenesis. Cell Death Differ. 2009;16:684–696. doi: 10.1038/cdd.2008.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Happo L, Cragg MS, Phipson B, et al. Maximal killing of lymphoma cells by DNA damage-inducing therapy requires not only the p53 targets Puma and Noxa, but also Bim. Blood. 2010;116:5256–5267. doi: 10.1182/blood-2010-04-280818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Sander S, Calado DP, Srinivasan L, et al. Synergy between PI3K signaling and MYC in Burkitt lymphomagenesis. Cancer Cell. 2012;22:167–179. doi: 10.1016/j.ccr.2012.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Blyth K, Terry A, O'Hara M, et al. Synergy between a human c-myc transgene and p53 null genotype in murine thymic lymphomas: contrasting effects of homozygous and heterozygous p53 loss. Oncogene. 1995;10:1717–1723. [PubMed] [Google Scholar]
- 127.Jacobs JJ, Scheijen B, Voncken JW, et al. Bmi-1 collaborates with c-Myc in tumorigenesis by inhibiting c-Myc-induced apoptosis via INK4a/ARF. Genes Dev. 1999;13:2678–2690. doi: 10.1101/gad.13.20.2678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Eischen CM, Weber JD, Roussel MF, et al. Disruption of the ARF-Mdm2-p53 tumor suppressor pathway in Myc-induced lymphomagenesis. Genes Dev. 1999;13:2658–2669. doi: 10.1101/gad.13.20.2658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Drotar ME, Silva S, Barone E, et al. Epstein-Barr virus nuclear antigen-1 and Myc cooperate in lymphomagenesis. Int J Cancer. 2003;106:388–395. doi: 10.1002/ijc.11224. [DOI] [PubMed] [Google Scholar]
- 130.Farrell PJ, Allan GJ, Shanahan F, et al. p53 is frequently mutated in Burkitt's lymphoma cell lines. EMBO J. 1991;10:2879–2887. doi: 10.1002/j.1460-2075.1991.tb07837.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Gaidano G, Ballerini P, Gong JZ, et al. p53 mutations in human lymphoid malignancies: association with Burkitt lymphoma and chronic lymphocytic leukemia. Proc Natl Acad Sci U S A. 1991;88:5413–5417. doi: 10.1073/pnas.88.12.5413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Wiman KG, Magnusson KP, Ramqvist T, et al. Mutant p53 detected in a majority of Burkitt lymphoma cell lines by monoclonal antibody PAb240. Oncogene. 1991;6:1633–1639. [PubMed] [Google Scholar]
- 133.Bhatia KG, Gutierrez MI, Huppi K, et al. The pattern of p53 mutations in Burkitt's lymphoma differs from that of solid tumors. Cancer Res. 1992;52:4273–4276. [PubMed] [Google Scholar]
- 134.Preudhomme C, Dervite I, Wattel E, et al. Clinical significance of p53 mutations in newly diagnosed Burkitt's lymphoma and acute lymphoblastic leukemia: a report of 48 cases. J Clin Oncol. 1995;13:812–820. doi: 10.1200/JCO.1995.13.4.812. [DOI] [PubMed] [Google Scholar]
- 135.Lindstrom MS, Wiman KG. Role of genetic and epigenetic changes in Burkitt lymphoma. Semin Cancer Biol. 2002;12:381–387. doi: 10.1016/s1044-579x(02)00058-5. [DOI] [PubMed] [Google Scholar]
- 136.Schmitz R, Young RM, Ceribelli M, et al. Burkitt lymphoma pathogenesis and therapeutic targets from structural and functional genomics. Nature. 2012;490:116–120. doi: 10.1038/nature11378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Richter J, Schlesner M, Hoffmann S, et al. Recurrent mutation of the ID3 gene in Burkitt lymphoma identified by integrated genome, exome and transcriptome sequencing. Nat Genet. 2012;44:1316–1320. doi: 10.1038/ng.2469. [DOI] [PubMed] [Google Scholar]
- 138.Love C, Sun Z, Jima D, et al. The genetic landscape of mutations in Burkitt lymphoma. Nat Genet. 2012;44:1321–1325. doi: 10.1038/ng.2468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Johnston JM, Carroll WL. c-myc hypermutation in Burkitt's lymphoma. Leuk Lymphoma. 1992;8:431–439. doi: 10.3109/10428199209051025. [DOI] [PubMed] [Google Scholar]
- 140.Cowling VH, Turner SA, Cole MD. Burkitt's lymphoma-associated c-Myc mutations converge on a dramatically altered target gene response and implicate Nol5a/Nop56 in oncogenesis. Oncogene. 2014;33:3519–3527. doi: 10.1038/onc.2013.338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Seitz V, Butzhammer P, Hirsch B, et al. Deep sequencing of MYC DNA-binding sites in Burkitt lymphoma. PLoS One. 2011;6:e26837. doi: 10.1371/journal.pone.0026837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Burkitt D. Long-term remissions following one and two-dose chemotherapy for African lymphoma. Cancer. 1967;20:756–759. doi: 10.1002/1097-0142(1967)20:5<756::aid-cncr2820200530>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- 143.Ferry JA. Burkitt's lymphoma: clinicopathologic features and differential diagnosis. Oncologist. 2006;11:375–383. doi: 10.1634/theoncologist.11-4-375. [DOI] [PubMed] [Google Scholar]
- 144.Oriol A, Ribera JM, Bergua J, et al. High-dose chemotherapy and immunotherapy in adult Burkitt lymphoma: comparison of results in human immunodeficiency virus-infected and noninfected patients. Cancer. 2008;113:117–125. doi: 10.1002/cncr.23522. [DOI] [PubMed] [Google Scholar]
- 145.Ngoma T, Adde M, Durosinmi M, et al. Treatment of Burkitt lymphoma in equatorial Africa using a simple three-drug combination followed by a salvage regimen for patients with persistent or recurrent disease. Br J Haematol. 2012;158:749–762. doi: 10.1111/j.1365-2141.2012.09236.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Magrath I. Towards curative therapy in Burkitt lymphoma: the role of early African studies in demonstrating the value of combination therapy and CNS prophylaxis. Adv Hematol. 2012;2012:130680. doi: 10.1155/2012/130680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Appelbaum FR, Deisseroth AB, Graw RG, Jr, et al. Prolonged complete remission following high dose chemotherapy of Burkitt's lymphoma in relapse. Cancer. 1978;41:1059–1063. doi: 10.1002/1097-0142(197803)41:3<1059::aid-cncr2820410339>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- 148.Thomas DA, Faderl S, O'Brien S, et al. Chemoimmunotherapy with hyper-CVAD plus rituximab for the treatment of adult Burkitt and Burkitt-type lymphoma or acute lymphoblastic leukemia. Cancer. 2006;106:1569–1580. doi: 10.1002/cncr.21776. [DOI] [PubMed] [Google Scholar]
- 149.Molyneux EM, Rochford R, Griffin B, et al. Burkitt's lymphoma. Lancet. 2012;379:1234–1244. doi: 10.1016/S0140-6736(11)61177-X. [DOI] [PubMed] [Google Scholar]
- 150.Vita M, Henriksson M. The Myc oncoprotein as a therapeutic target for human cancer. Semin Cancer Biol. 2006;16:318–330. doi: 10.1016/j.semcancer.2006.07.015. [DOI] [PubMed] [Google Scholar]
- 151.Mertz JA, Conery AR, Bryant BM, et al. Targeting MYC dependence in cancer by inhibiting BET bromodomains. Proc Natl Acad Sci U S A. 2011;108:16669–16674. doi: 10.1073/pnas.1108190108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Soucek L, Whitfield J, Martins CP, et al. Modelling Myc inhibition as a cancer therapy. Nature. 2008;455:679–683. doi: 10.1038/nature07260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Rodon J, Dienstmann R, Serra V, et al. Development of PI3K inhibitors: lessons learned from early clinical trials. Nat Rev Clin Oncol. 2013;10:143–153. doi: 10.1038/nrclinonc.2013.10. [DOI] [PubMed] [Google Scholar]
- 154.Kauffmann-Zeh A, Rodriguez-Viciana P, Ulrich E, et al. Suppression of c-Myc-induced apoptosis by Ras signalling through PI(3)K and PKB. Nature. 1997;385:544–548. doi: 10.1038/385544a0. [DOI] [PubMed] [Google Scholar]
- 155.Brennan P, Mehl AM, Jones M, et al. Phosphatidylinositol 3-kinase is essential for the proliferation of lymphoblastoid cells. Oncogene. 2002;21:1263–1271. doi: 10.1038/sj.onc.1205182. [DOI] [PubMed] [Google Scholar]
- 156.Kelly GL, Grabow S, Glaser SP, et al. Targeting of MCL-1 kills MYC-driven mouse and human lymphomas even when they bear mutations in p53. Genes Dev. 2014;28:58–70. doi: 10.1101/gad.232009.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Lessene G, Czabotar PE, Colman PM. BCL-2 family antagonists for cancer therapy. Nat Rev Drug Discov. 2008;7:989–1000. doi: 10.1038/nrd2658. [DOI] [PubMed] [Google Scholar]
- 158.Li N, Thompson S, Schultz DC, et al. Discovery of selective inhibitors against EBNA1 via high throughput in silico virtual screening. PLoS One. 2010;5:e10126. doi: 10.1371/journal.pone.0010126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Kelly GL, Rickinson AB. Burkitt lymphoma: revisiting the patho-genesis of a virus-associated malignancy. Hematol Am Soc Hematol Educ Program. 2007;2007:277–284. doi: 10.1182/asheducation-2007.1.277. [DOI] [PubMed] [Google Scholar]