

Abstract
Motivation: The model bacterium Escherichia coli is among the best studied prokaryotes, yet nearly half of its proteins are still of unknown biological function. This is despite a wealth of available large-scale physical and genetic interaction data. To address this, we extended the GeneMANIA function prediction web application developed for model eukaryotes to support E.coli.
Results: We integrated 48 distinct E.coli functional interaction datasets and used the GeneMANIA algorithm to produce thousands of novel functional predictions and prioritize genes for further functional assays. Our analysis achieved cross-validation performance comparable to that reported for eukaryotic model organisms, and revealed new functions for previously uncharacterized genes in specific bioprocesses, including components required for cell adhesion, iron–sulphur complex assembly and ribosome biogenesis. The GeneMANIA approach for network-based function prediction provides an innovative new tool for probing mechanisms underlying bacterial bioprocesses.
Contact: gary.bader@utoronto.ca; mohan.babu@uregina.ca
Supplementary information: Supplementary data are available at Bioinformatics online.
1 INTRODUCTION
As the primary model organism for microbial biology, Escherichia coli has been studied for decades using countless large- and small-scale biochemical assays of gene function. More recently, the physical (protein–protein) and functional (gene–gene or epistatic) relationships between E.coli genes have been extensively studied by our group (Hu et al., 2009; Babu et al., 2011a, 2014; Rajagopala et al., 2014) and others (Arifuzzaman et al., 2006; Typas et al., 2008) in the hopes of understanding the entire complement of biological pathways in a prokaryotic organism. These studies have revealed much of the physical and functional organization of the E.coli bacterial proteome. However, as many of the low-throughput studies were particularly concerned with specific, smaller groups of genes, and the larger scale studies were conducted using methodologies that inherently enrich for certain physical (i.e. transient versus more stable protein interactions) or genetic interactions, defining a single pathway level map of E.coli function can be problematic. Complicating matters further is the inherent difficulty in querying, navigating, and visualizing such complex biological networks in a meaningful way as each study only identifies part of the map and is idiosyncratically biased. Thus, despite rapid progress, we are far from understanding the biological roles and functional relationships of the 4247 E.coli genes from an integrated ‘systems’ perspective. As ∼ 45% (1925 of 4247) of this organism’s genome (i.e. K-12 W3110) still remains functionally unannotated, methods more sensitive at interpreting existing data appear warranted.
Underlying this disconnect between the volume of data available and the lack of annotation is a paucity of user friendly tools for the accurate and automatic inference of a gene’s function. While many gene function prediction systems based on functional interaction networks exist (Alexeyenko and Sonnhammer, 2009), few are readily available for prokaryotes [e.g. eNet (Hu et al., 2009); EcID (Andres Leon et al., 2009); STRING (Franceschini et al., 2013)], and none consider the breadth of evidence supporting functional interactions available today, such as phenomics and epistatic interactions, which have only recently become available.
Here, we extend the GeneMANIA resource to support E.coli (Mostafavi et al., 2008; Zuberi et al., 2013) for gene function prediction. We validate novel predictions supporting a role in iron (Fe) - sulphur (S) cluster binding, cell adhesion and ribosomal protein degradation and biogenesis for more than half a dozen uncharacterized (orphaned) E.coli genes. An online implementation of GeneMANIA including all E.coli biological networks used to generate our predictions has been made publically available (www.genemania.org), and we have also created a stand-alone program and plugin for the Cytoscape network visualization environment (Shannon et al., 2003; Montojo et al., 2010). We find that integration of these E.coli datasets into a single unified network using GeneMANIA furthers our understanding of how bacterial components are connected in complexes and pathways, and enables functional prediction of previously uncharacterized or under-characterized bacterial gene products.
2 METHODS
2.1 E.coli (K-12) genomes and biological networks
Since Gene Expression Omnibus (GEO) datasets (see Supplementary Methods for details), protein domains, coexpression and all experimental interactions were generated in the K-12 genomes of W3110 or MG1655 (which are highly similar), for gene function prediction, we merged the gene identifiers from both these genomes, generating a non-redundant dataset of 4455 genes (excluding insertion sequence elements). In total, 48 biological networks from various literature sources were compiled for function prediction, which are currently displayed on the GeneMANIA.
2.2 Validation
GeneMANIA performance was evaluated by 5-fold cross-validation on each Gene Ontology (GO) annotation category (GO gene sets were downloaded from go_daily-termdb.obo-xml.gz; dated 2013-12-03). In each instance, true examples were withheld proteins with the corresponding annotation, and negative examples were all other proteins. Cross-validation and area under the ROC (receiver operating characteristic) curve (AUC) was computed using the ‘Network Assessor’ component of the GeneMANIA command line tool (Montojo et al., 2010). To gauge the contribution of each feature to overall prediction performance, networks were withheld and average AUC and error rate estimated across all GO annotations.
2.3 Fe-S cluster, ribosome and adhesion function prediction
The query gene derived subnetwork corresponding to the selected process was generated using the GeneMANIA plugin (Montojo et al., 2010) for Cytoscape (Shannon et al., 2003), available from the Cytoscape App Store (Lotia et al., 2013). Relative network weightings were determined using the GeneMANIA web interface.
2.4 Bacterial strains and media
Strains used were either the wild-type E.coli K-12 BW25113 or single gene deletion mutant strains marked with a kanamycin resistance marker from the Keio knockout library (Baba et al., 2006) for validation experiments. Mutant strains were streaked onto Luria-Bertani (LB) agar plates supplemented with 30 µg/ml kanamycin and incubated at 37°C overnight to obtain single colonies.
2.5 Growth curves
Overnight cultures of wild-type and mutant strains prepared from single colonies were inoculated in LB supplemented with either no antibiotic, 6 µg/ml streptomycin, 0.5 µg/ml tetracycline, 750 µM reduced l-glutathione, 250 µM 2, 2′-dipyridyl or a combination, as indicated, at an OD600 of ∼0.01. Concentrations of additives were based on analogous recently conducted experiments (Wong et al., 2014). Cultures were grown in Bioscreen C (Growth curves) honeycomb 100-well plates in 200 µl volumes at 37°C and the turbidity of the cultures was measured using the wide-band filter (450–580 nm) at 15-min intervals.
2.6 Degradation of ribosomal proteins, ribosomal profiles and translation fidelity
The reaction mixture used for degradation assays contained 1.2 µM ClpP, 3.9 µM substrate and an ATP regeneration system (13 units/ml of creatine kinase and 16 mM creatine phosphate) in buffer containing 25 mM HEPES (pH 7.5), 5 mM MgCl2, 5 mM KCl, 0.03% (w/v) Tween 20 and 10% glycerol. Components were incubated at 37°C for 3 min before adding 1.0 µM ClpX to start the reaction. At given time points, aliquots were withdrawn and mixed with 4× Laemmli buffer to stop the reaction. Proteins were then resolved on SDS-PAGE gels and visualized by Coomassie staining.
The S30 crude extracts were loaded on linear sucrose density gradients as described (Jiang et al., 2007), with 40% sucrose used as cushion and ribosomes or other complex subunits isolated by high-speed ultracentrifugation at 4°C for 16 h as previously described (Campbell and Brown, 2008). Translational fidelity of mutant strains expressed via reported expression plasmids was evaluated as previously described (Thompson et al., 2002; Hu et al., 2009; Babu et al., 2011b).
2.7 Biofilm assay
The biofilm assay was performed as previously described (O'Toole et al., 1999), with minor modifications. Briefly, 5 µl of overnight E.coli cultures grown in LB at 32°C was added to sterile 96-well polystyrene dish containing 100 µl of fresh LB medium supplemented with 0.45% glucose. Culture dish was incubated overnight ( ∼18 h) at 32°C, and the biofilm was stained with 0.5% crystal violet for 5 min. Excess crystal violet was washed off with sterile water. An ethanol–acetone mixture (80:20) was added to the wells to release the dye, and the biofilm that adhered to the surface of the well was imaged using a Canon digital camera. Biofilm formation was assessed by the intensity of residual coloration.
3 RESULTS AND DISCUSSION
3.1 Networks supporting function prediction
We collected five types of E.coli GeneMANIA functional interaction networks: physical interactions, transcript coexpression, genetic interactions, shared protein domains (SPDs) and ‘other’ networks inferred from genomic context and chemogenomic (i.e. phenomic) profiles (Supplementary Table S1). Specifically, we included 48 experimentally derived E.coli biological networks, from our group and others that span these evidence types. While the physical interaction networks were unweighted, all remaining network types were weighted by profile correlation, wherein higher correlations are more indicative of shared function (Mostafavi et al., 2008). To evaluate the extent to which these lines of evidence types overlapped, we counted the number of parallel edges (i.e. edges from different networks connected to the same node pairs) in each supporting network (to a maximum of 50) for each gene in the E.coli genome. More than 57 000 edges were supported by several networks, while the majority (>800 000) of edges existed in only a single network, or were only weakly supported (i.e. with low edge weight; Supplementary Fig. S1). Notably, coexpression contributed most (>500 000) of these edges due to the large number of experiments included (33), followed by networks derived from genetic screens (>145 000).
GeneMANIA uses the ‘guilt-by-association’ function prediction approach (Oliver, 2000), wherein a user provides a ‘seed list’ of known related genes that is then extended to include other genes that are predicted to share a similar function based on overlapping connection within the biological networks (Mostafavi et al., 2008). In the simplest mode of operation, the user needs only to enter in a gene or genes of interest. In generating functional predictions, the GeneMANIA algorithm is designed to automatically weight networks on the basis of relevance to the query set. This weighting is calculated for each query, so that network weights can vary based on user input. These assigned network weights are provided to the user, so that they may assess the relative predictive power of the biological evidence types and the basis for each prediction.
3.2 Cross-validation and performance of network categories
To assess the sensitivity and specificity of GeneMANIA in making functional predictions in E.coli, we used 5-fold cross-validation to measure its ability to correctly identify known functionally annotated E.coli genes given input genes possessing the same functional annotation. These results were compared across GeneMANIA score thresholds using AUC. The average error, measured as 1 - AUC, across all GO biological process, cellular component and molecular function annotations ranged from 0.10 for cellular component annotations to 0.15 for biological process annotations (Fig. 1a), which is comparable to that achieved with yeast Saccharomyces cerevisiae (Mostafavi et al., 2008). Moreover, comparison of function predictions derived from STRING to GeneMANIA predictions indicated that GeneMANIA achieved significantly better performance (Supplementary Table S2, see Supplementary Methods).
Fig. 1.
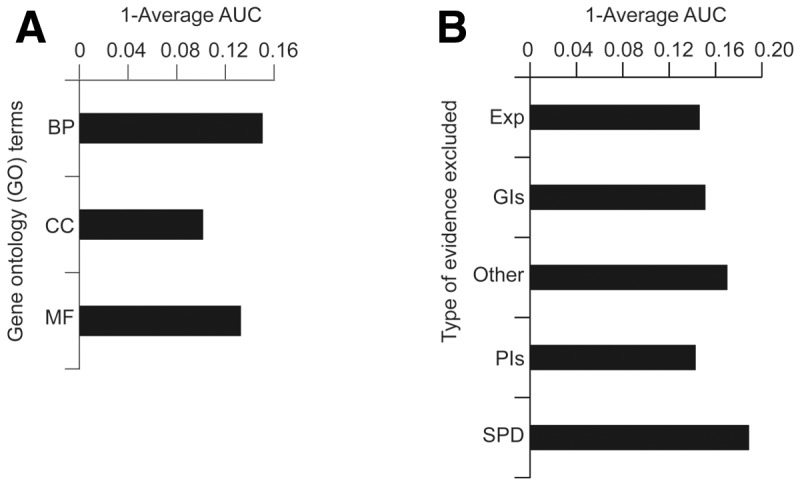
Median error rates of function prediction. (A) Average error rate of GeneMANIA function prediction estimated by 5-fold cross-validation for each GO biological process (BP), cellular component (CC) and molecular function (MF) annotation. (B) Effect of eliminating various categories of evidence on error. Exp, coexpression; GIs, genetic interactions; PIs, physical interactions and SPD, shared protein domains
Next, we evaluated the contribution of each input network type by individually excluding each type of network and re-evaluating overall performance (Fig. 1b) using the GeneMANIA network assessor function (Montojo, 2014). We find that while the SPD and other network types contribute substantially to the overall predictive accuracy of the program, some networks appeared to have little influence on average. However, this was not the case for all GO terms. For example, physical interactions contribute more substantial reductions in error for GO terms such as ribosome assembly (Supplementary Table S3). This underscores one of the principle benefits of the GeneMANIA algorithm: namely, its ability to adaptively weight evidence based on the input genes.
3.3 Predicting gene function
To identify functions for E.coli genes, we input genes by GO annotation (assuming at least 10 genes per GO term) to GeneMANIA using the command line tools available at http://pages.genemania.org/tools/. The top 100 results were retrieved for each GO term (Supplementary Table S4). To confirm the value of some of the higher ranking predictions, we focused experimental effort on functionally uncharacterized proteins that were predicted to have a novel function. For example, Fe–S cluster biogenesis proteins were found to be associated with three uncharacterized proteins: YnfH, YhgA and YdhZ (Fig. 2a). This association was based largely on SPDs, coexpression and other sources of evidence including large-scale phenomics data (Nichols et al., 2011) (Fig. 2a). Since a large number of genes that confer aminoglycoside sensitivity are involved in Fe–S cluster biogenesis and aerobic respiration (Kohanski et al., 2007; Babu et al., 2014; Wong et al., 2014), we tested the sensitivity of ynfH, yhgA and ydhZ single mutants to sublethal concentrations of streptomycin antibiotic. Both wild-type and single mutant deletion strains exhibited similar growth curves in the absence of antibiotic or in the presence of a non-aminoglycoside (tetracycline) drug (Fig. 2a). However, at a sublethal dosage of the aminoglycoside streptomycin, wild-type cells reached a lower density of cells in stationary phase compared with the mutant cells. Addition of the iron chelator 2,2'-dipyridyl (DP) or the essential antioxidant, glutathione (GSH) to the streptomycin containing medium relieved the growth reduction in wild-type cells to a level comparable to single mutants (Fig. 2a), indicating the participation of YnfH, YhgA and YdhZ in an Fe–S cluster-related processes.
Fig. 2.

Novel factors involved in Fe-S assembly and ribosome biogenesis. (A) Subnetwork of non-essential proteins of unknown function (pink) connecting the components of Fe-S (blue) cluster binding (i) based on SPD, Exp and other large-scale (ii) network sources. Growth profiles of wild type (WT) and mutant strains in the absence or presence of sublethal concentration of indicated antibiotics, iron chelator and antioxidant (iii). Tetracycline is used as control. Each data point represents the mean ± SD (error bars) of three independent replicates (see Supplementary Table S5) (Color version of this figure is available at Bioinformatics online.) (B) Subnetwork of ClpP (pink) is connected with the ribosome factors (blue) (i) based on Exp and other genomic sources (ii). SDS-PAGE gels (iii) showing the degradation of ribosomal (r) S7 protein over time (h) after the addition of ClpX to the mixture containing ClpP, casein kinase (CK), ribosomal S7 protein and ATP regeneration system (left), whereas no S7 degradation was observed in the absence of ATP and ClpP over time (right, negative controls). Molecular masses (kDa) of marker proteins (M) by SDS-PAGE are shown
Another example of novel assigned function was the implication of the gene clpP in ribosome biogenesis, a prediction driven largely by strong coexpression. Consistent with this prediction, the ClpP, a serine protease that forms an active degradation complex with ClpX ATPase was found to degrade the ribosomal subunit S7 in the presence of ATP (Fig. 2b), but the mechanism by which the ClpXP protease recognizes the ribosomal S7 for degradation (Flynn et al., 2003) is not yet known.
Similarly, based on SPDs, coexpression and other data sources (e.g. phenomics and genomic context), a ribosomal link was predicted between a previously uncharacterized protein, YihD, and the methyltransferase factors (TrmA, RsmJ) responsible for methylation of 16 S rRNA and tRNAs. To evaluate this possible connection, we tested the ribosome profile of yihD deficient strains. In contrast to wild type, yihD mutant had an elevated amount of free 30 S and 50 S ribosomal subunits, and a concomitant decrease in 70 S formation (Supplementary Fig. S2a). Consistent with this result, yihD mutant also exhibited significantly higher read-through of amber (UAG) and opal (UGA) stop codon alleles, and +1 and −1 frameshift mutations in a β-galactosidase reporter system (O'Connor, et al., 1992). Collectively, these results support our prediction of ClpP and YihD involvement in ribosome biogenesis.
Finally, SPDs and correlated transcript expression profiles connected the unannotated proteins YdeT, YdhQ, YhjY with components involved in bacterial adhesion and biofilm formation. To evaluate this link, we measured mutant strains harboring deletions in each of these genes for their ability to form a biofilm in vitro (see Methods). In contrast to wild-type cells, all three single mutants displayed surface attachment through biofilm formation (Supplementary Fig. S2b) as has been previously noted for other biofilm-associated mutants (Ma and Wood, 2009). Notably, only one of the eight experimentally tested function predictions from GeneMANIA were ranked highly by the STRING-derived function predictor (Supplementary Table S6, see Supplementary Methods).
4 CONCLUSIONS
Until now, GeneMANIA has been limited to eukaryotes, where it has proven to be a powerful resource for probing gene function and revealing pairwise connections linking genes in yeast, fly, worm, human and other species (Zuberi et al., 2013). In this study, we have extended the predictive power of GeneMANIA to a leading model prokaryote, E.coli, an organism that has to date lacked comparable tools for functional interrogation that are simultaneously accurate, comprehensive (including the latest high-throughput data), and easy to use. This work combines the GeneMANIA algorithm with expansive networks of informative functional connections consisting of more than one million gene–gene associations based on physical interactions and shared genetic, domain, chemogenomics and coexpression profiles. This represents a rich resource, unparalleled in any other bacterial species to date, for further mechanistic characterization of both known and uncharacterized genes.
The GeneMANIA algorithm and supporting networks for E.coli and several model eukaryotes are made freely available via a user friendly GeneMANIA web interface (www.genemania.org) and as a plugin for the Cytoscape network visualization environment. This web-accessible resource facilitates exploration of functional inferences in hypothesis-driven follow-up studies aimed at elucidating mechanistic aspects associated with particular bioprocesses.
While the prediction performed by GeneMANIA provides a new method for leveraging functionally informative associations to explore bacterial gene function, the quality of function predictions, especially for loosely connected proteins, is expected to be improved over the coming years as new genomic resources, including protein and genetic interactions for the previously unexplored interactome and biological space become available. Nevertheless, just as we were able to identify novel functions for uncharacterized genes in Fe–S cluster binding, ribosome biogenesis and cell adhesion, we believe that this resource will enable additional functional discoveries in E.coli, and, through orthology mapping, in other experimentally and evolutionarily significant uncharacterized bacterial species (Supplementary Table S7, see Supplementary Methods), including opportunistic pathogens.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Gabe Musso (Harvard Medical School, Massachusetts, USA) for helpful discussions and comments. J.V., K.Z., H.R., R.A. and L.C. analyzed data. S.P. and A.K. compiled all data sources. A.Ga., V.D., B.S., E.L. and K.R. conducted experiments. Q.M. and G.B. shared GeneMANIA resources. J.V and M.B wrote the article, with input from A.G., A.E., Q.M., J.F.G., W.H. and G.B.
Funding: This work was supported by grants from Global Leadership Round in Genomics and Life Sciences to Q.M. and G.B., by National Resource for Network Biology (P41 GM103504) to G.B., Canadian Institutes of Health Research (CIHR) to W.A.H. (MOP-130374) and J.F.G. and A.E. (MOP-82852), and the National Sciences and Engineering Research Council of Canada to A.G. and M.B. (DG-20234). M.B. holds a CIHR New Investigator award. AGa was a recipient of a CIHR Vanier Graduate Scholarship. J.V. is supported by a Saskatchewan Postdoctoral Research Fellowship.
Conflict of interest: none declared.
REFERENCES
- Alexeyenko A, Sonnhammer EL. Global networks of functional coupling in eukaryotes from comprehensive data integration. Genome Res. 2009;19:1107–1116. doi: 10.1101/gr.087528.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andres Leon E, et al. EcID. A database for the inference of functional interactions in E. coli. Nucleic Acids Res. 2009;37:D629–D635. doi: 10.1093/nar/gkn853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arifuzzaman M, et al. Large-scale identification of protein-protein interaction of Escherichia coli K-12. Genome Res. 2006;16:686–691. doi: 10.1101/gr.4527806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baba T, et al. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol. Syst. Biol. 2006;2 doi: 10.1038/msb4100050. 2006.0008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babu M, et al. Ribosome-dependent ATPase interacts with conserved membrane protein in Escherichia coli to modulate protein synthesis and oxidative phosphorylation. PLoS One. 2011a;6:e18510. doi: 10.1371/journal.pone.0018510. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Babu M, et al. Genetic interaction maps in Escherichia coli reveal functional crosstalk among cell envelope biogenesis pathways. PLoS Genet. 2011b;7:e1002377. doi: 10.1371/journal.pgen.1002377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babu M, et al. Quantitative genome-wide genetic interaction screens reveal global epistatic relationships of protein complexes in Escherichia coli. PLoS Genet. 2014;10:e1004120. doi: 10.1371/journal.pgen.1004120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell TL, Brown ED. Genetic interaction screens with ordered overexpression and deletion clone sets implicate the Escherichia coli GTPase YjeQ in late ribosome biogenesis. J. Bacteriol. 2008;190:2537–2545. doi: 10.1128/JB.01744-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flynn JM, et al. Proteomic discovery of cellular substrates of the ClpXP protease reveals five classes of ClpX-recognition signals. Mol Cell. 2003;11:671–683. doi: 10.1016/s1097-2765(03)00060-1. [DOI] [PubMed] [Google Scholar]
- Franceschini A, et al. STRING v9.1: protein-protein interaction networks, with increased coverage and integration. Nucleic Acids Res. 2013;41:D808–815. doi: 10.1093/nar/gks1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu P, et al. Global functional atlas of Escherichia coli encompassing previously uncharacterized proteins. PLoS Biol. 2009;7:e1000096. doi: 10.1371/journal.pbio.1000096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang M, et al. Identification of novel Escherichia coli ribosome-associated proteins using isobaric tags and multidimensional protein identification techniques. J. Bacteriol. 2007;189:3434–3444. doi: 10.1128/JB.00090-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohanski MA, et al. A common mechanism of cellular death induced by bactericidal antibiotics. Cell. 2007;130:797–810. doi: 10.1016/j.cell.2007.06.049. [DOI] [PubMed] [Google Scholar]
- Lotia S, et al. Cytoscape app store. Bioinformatics. 2013;29:1350–1351. doi: 10.1093/bioinformatics/btt138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Q, Wood TK. OmpA influences Escherichia coli biofilm formation by repressing cellulose production through the CpxRA two-component system. Environ. Microbiol. 2009;11:2735–2746. doi: 10.1111/j.1462-2920.2009.02000.x. [DOI] [PubMed] [Google Scholar]
- Montojo J. Network assessor: an automated method for quantitative assessment of a network’s potential for gene function prediction. Frontiers Genet. 2014;5:123. doi: 10.3389/fgene.2014.00123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montojo J, et al. GeneMANIA Cytoscape plugin: fast gene function predictions on the desktop. Bioinformatics. 2010;26:2927–2928. doi: 10.1093/bioinformatics/btq562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mostafavi S, et al. GeneMANIA: a real-time multiple association network integration algorithm for predicting gene function. Genome Biol. 2008;9(Suppl 1):S4. doi: 10.1186/gb-2008-9-s1-s4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichols RJ, et al. Phenotypic landscape of a bacterial cell. Cell. 2011;144:143–156. doi: 10.1016/j.cell.2010.11.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Connor M, et al. A ribosomal ambiguity mutation in the 530 loop of E. coli 16S rRNA. Nucleic Acids Res. 1992;20:4221–4227. doi: 10.1093/nar/20.16.4221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Toole GA, et al. Genetic approaches to study of biofilms. Methods Enzymol. 1999;310:91–109. doi: 10.1016/s0076-6879(99)10008-9. [DOI] [PubMed] [Google Scholar]
- Oliver S. Guilt-by-association goes global. Nature. 2000;403:601–603. doi: 10.1038/35001165. [DOI] [PubMed] [Google Scholar]
- Rajagopala SV, et al. The binary protein-protein interaction landscape of Escherichia coli. Nat. Biotechnol. 2014;32:285–290. doi: 10.1038/nbt.2831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon P, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson J, et al. The protein synthesis inhibitors, oxazolidinones and chloramphenicol, cause extensive translational inaccuracy in vivo. J. Mol. Biol. 2002;322:273–279. doi: 10.1016/s0022-2836(02)00784-2. [DOI] [PubMed] [Google Scholar]
- Typas A, et al. High-throughput, quantitative analyses of genetic interactions in E. coli. Nat. Methods. 2008;5:781–787. doi: 10.1038/nmeth.1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong KS, et al. The MoxR ATPase RavA and its cofactor ViaA interact with the NADH:ubiquinone oxidoreductase I in Escherichia coli. PLoS One. 2014;9:e85529. doi: 10.1371/journal.pone.0085529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuberi K, et al. GeneMANIA prediction server 2013 update. Nucleic Acids Res. 2013;41:W115–W122. doi: 10.1093/nar/gkt533. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.