

Abstract
Objective
The discovery of novel disease-modifying drugs for osteoarthritis (OA) is limited by the lack of adequate genetically-defined cartilage tissues for application in high-throughput screening systems. We addressed this need by synthesizing cartilage from induced pluripotent stem cells (iPSCs) to establish and validate an in vitro model of OA.
Methods
iPSC-derived or native mouse cartilage samples were treated with the cytokine interleukin-1α (IL-1α) for 3 days to model the inflammatory environment of OA. Biochemical content, mechanical properties, and gene expression of the resulting tissues were assayed. In addition, the inflammatory and catabolic environment of the media was assessed. To establish high-throughput capability, we utilized a 96-well plate format and conducted a screen of previously identified candidate OA drugs. Glycosaminoglycan release into the media was used as the primary output for screening.
Results
Treatment of iPSC-derived or native cartilage with IL-1α induced characteristic features of OA in a rapid and dose-dependent manner. In addition to the loss of glycosaminoglycans and tissue mechanical properties, IL-1α treatment induced expression of matrix metalloproteinases and increased production of the inflammatory mediators nitric oxide and prostaglandin E2. In the high-throughput screen validation, all candidate OA therapeutics provided some benefit, but only the NF-κB inhibitor SC-514 effectively reduced cartilage loss in response to IL-1α.
Conclusions
This work demonstrates the utility of iPSCs for studying cartilage pathology, and provides a platform for identifying novel, patient-specific therapeutics that prevent cartilage degradation and modify the course of OA development.
Introduction
Osteoarthritis (OA) is a significant health and economic burden, and the impact of the disease is predicted to rise due to an aging population (1). Currently, management of OA focuses on lifestyle modifications and the use of nutraceuticals, anti-inflammatory drugs, and viscosupplementation to limit pain (2). Because these treatments are unable to prevent disease progression, many patients advance to the endpoint of total joint replacement (2). While many pharmaceutical agents are under investigation, none have been able to demonstrate sufficient clinical efficacy to gain regulatory approval based on disease modification (3). The development of novel disease-modifying osteoarthritis drugs (DMOADs) would be greatly enhanced by the ability to efficiently screen candidate molecules for protection against OA. In this study, we recapitulate key characteristics of OA in engineered cartilage and validate the potential to use this system for identification of promising candidate drugs.
OA is characterized by progressive joint failure that involves multiple tissues, particularly the irreversible degradation of articular cartilage (4). Cartilage degradation results from an imbalance in the homeostasis of two key matrix components that endow the tissue with its mechanical properties—glycosaminoglycans (GAGs) and type II collagen (5). The pathogenesis of OA and the loss of cartilage homeostasis is dependent in part on the action of inflammatory cytokines such as interleukin-1 (IL-1) (6, 7) that also mediate the production of pro-inflammatory mediators [i.e., nitric oxide (NO) and prostaglandin E2 (PGE2)] and matrix degrading enzymes. These catabolic enzymes include matrix metalloproteinases (MMPs) that disrupt collagen fibers (8, 9) and members of the A disintegrin and metalloproteinase with thrombospondin motifs (ADAMTS) family that degrade aggrecan and release GAGs (10, 11).
The loss of matrix components leads to a decrease in the stiffness of the tissue and susceptibility to further degradation (12, 13). Focusing drug discovery efforts on blocking pathways that cause early cartilage loss has been proposed as a promising approach due to the challenges of reversing the disease after significant degradation has occurred (14). Proposed targets for reducing inflammation in OA include inhibiting intracellular signaling through the nuclear factor kappa-B (NF-κB) pathway (15), or blocking cyclooxygenase-2 (COX-2) enzyme activity (16). Other options for halting early cartilage degradation may include inhibiting catabolic enzyme activity (17, 18), or providing cytokines with anti-inflammatory activity, such as interleukin-4 (IL-4) (19).
The use of high-throughput drug screening approaches for DMOAD discovery is currently limited by the lack of a source for abundant cartilage tissue from a single genetic background. Investigators have therefore utilized monolayer culture systems despite the importance of cell-matrix interactions to cartilage function (5). Because primary chondrocytes dedifferentiate with passage in culture (20), DMOAD screening has typically been performed with cell lines (21, 22) or adult stem cells (23–25) that can be expanded to sufficient quantities while maintaining differentiation potential. Screens for mediators of chondrogenic differentiation have provided valuable candidate compounds and insights into chondrogenesis (23), but this approach does not necessarily identify therapeutics that target the catabolic pathways present during OA. An abundant supply of cartilage tissue would allow for an alternative approach of screening for compounds that modulate tissue degradation in response to a catabolic stimulus.
Induced pluripotent stem cells (iPSCs) are an attractive source for high-throughput drug screening due to their capacity for nearly unlimited self-renewal and differentiation in a genetically-defined context (26). In previous work, we utilized a rigorous, multi-stage chondrogenic differentiation protocol to generate engineered cartilage tissue from murine iPSCs (27). In this study, we used IL-1α to recapitulate characteristic features of OA in iPSC-derived cartilage and established a method to screen for therapeutics that prevent cartilage loss (Fig. 1). We confirmed our approach with native murine hip cartilage and validated the system using previously identified candidate DMOADs.
Figure 1. Overview of the multistep approach used to develop an in vitro model of osteoarthritis (OA) using cartilage derived from murine iPSCs.

iPSCs were first differentiated in micromasses and then purified based on a collagen type II-GFP reporter. Purified cells were expanded and then cultured as pellets to produce a robust cartilaginous matrix. As a model of OA, iPSC-derived cartilage was cultured with interleukin-1α to create a reproducible degenerative response. Native mouse cartilage served as a control to ensure that the breakdown of iPSC cartilage was consistent with an OA phenotype. After validating the model, pellet formation, culture, and analyses were adapted to a 96-well plate system so that candidate OA therapeutics could be screened efficiently. CM: chondrogenic medium, BMP-4: bone morphogenetic protein 4, bFGF: basic fibroblastic growth factor, TGF-β3: transforming growth factor beta 3, dex: dexamethasone.
Materials and Methods
Induced pluripotent stem cells (iPSC) culture and differentiation
iPSCs were derived, cultured, and differentiated towards the chondrogenic lineage as previously described (27). Briefly, tail fibroblasts from adult C57BL/6 mice were reprogrammed to pluripotency with a single doxycycline-inducible lentiviral vector that expressed mouse cDNAs for Oct4 (Pou5f1), Sox2, Klf4, and c-Myc (28). Undifferentiated iPSCs were maintained on mitomycin C-treated mouse embryonic feeder (MEFs, Millipore) in iPSC media containing DMEM-HG (Gibco), 20% lot-selected fetal bovine serum (Atlanta Biologicals), 100 nM MEM nonessential amino acids (NEAA, Gibco), 55 μm β-mercaptoethanol (2-me, Gibco), 25 ng/ml gentamicin (Gibco), and 1,000 U/ml mouse leukemia inhibitory factor (Millipore). Nucleofection (Amaxa) of a construct with the type II collagen promoter driving GFP (29) generated clones with stable integration that were further selected for specific GFP expression upon chondrogenic differentiation.
Initial chondrogenic differentiation was carried out in high density micromass culture for 15 days while cultured in serum-free chondrogenic medium containing DMEM-HG, NEAA, 2-me, ITS+ premix (BD), and penicillin-streptomycin, 50 μg/ml L-ascorbic acid 2-phosphate, and 40 μg/ml L-proline. For days 3–5 only, 50 ng/ml mBMP-4 (R&D Systems) and 100 nM dexamethasone (Sigma) were added to the medium. Micromasses were then digested and single cells were sorted on the basis of positive GFP expression using flow cytometry. GFP-positive cells were expanded for two passages in chondrogenic medium with the addition of 10% FBS and 4 ng/ml basic fibroblast growth factor (Roche). Cells were then pelleted by centrifugation using either 250,000 cells in 15 ml centrifuge tubes or 125,000 cells in 96 well round-bottom plates. Serum-free chondrogenic medium with the addition of 10 ng/ml TGF-β3 (R&D Systems) and 100 nM dexamethasone was replaced every 2–3 days for 3 weeks to generate iPSC-derived engineered cartilage (“iPSC cartilage”). iPSC cartilage derivation and inflammatory degradation was repeated twice, and each experimental group was comprised of at least 6 pellets.
Mouse hip cartilage isolation
Based on prior reports characterizing the inflammatory degradation of mouse cartilage (30, 31), a femoral head cartilage explant model was used. Briefly, hips from 12 day old C57BL/6 mice were disarticulated to expose the femoral head and the cartilage layer was separated from underlying bone with the use of thin forceps. Cartilage explants were cultured for two days in serum-free chondrogenic medium to allow for culture equilibration before use in experiments as “native cartilage”. A total of 6 femoral heads were isolated for each experimental group, representing 6 distinct mice.
Treatment with interleukin-1α (IL-1α)
For iPSC cartilage, IL-1α treatment was initiated following 21 days of chondrogenic pellet culture. IL-1α treatment of native cartilage was begun after 2 days of equilibration in media. For both cartilages, treatment with recombinant mouse IL-1α (R&D Systems) was conducted in serum-free chondrogenic medium. Dose-response experiments with iPSC cartilage were conducted using control, 10 pg/ml, 100 pg/ml, or 1 ng/ml doses. Since the treatment of native articular cartilage with IL-1 has been conducted regularly in the literature (32, 33), only the control and 1 ng/ml doses were used for native mouse cartilage samples. For gene expression studies in iPSC cartilage, IL-1 was given for 24 hrs. All other IL-1α treatments, in both iPSC and native cartilage, were conducted for 3 days.
Delivery of candidate therapeutics
Five candidate therapeutics were selected to be screened in this iPSC-based OA model because they have previously been shown to reduce aspects of inflammatory degradation in articular cartilage explants (16–19) or synovial explants (15). iPSC cartilage pellets were formed in a 96 well format and treated with each of the candidate molecules using concentrations from the literature: IL-4 (30 ng/ml, R&D Systems) (19), tissue inhibitor of metalloproteinase-3 (TIMP-3, 1 μg/ml, R&D Systems) (17), NS-398 (50 μM, Cayman Chemical) (16), SC-514 (50 μM, Cayman Chemical) (15), and GM-6001 (10 μM, EMD Biosciences) (18). Thirty minutes after addition of the candidate molecules, IL-1α was added to each pellet at a final concentration of 1 ng/ml in serum-free chondrogenic medium and cultured for three days. Control pellets were treated without IL-1 or with IL-1 plus DMSO (0.01%, Sigma) as a carrier control for the small molecules (NS-398, SC-514, and GM-6001). After 3 days, media and samples were harvested for analysis.
RNA isolation and PCR
To gain an understanding of the gene expression prior to analyzing the protein levels at day 3, all qPCR analyses were conducted at day 1. Following 24 hours of culture with or without IL-1α, RNA was isolated from iPSC cartilage using liquid nitrogen pulverization to disrupt the tissue, TRIzol (Invitrogen) and a glycogen carrier (Invitrogen) to extract the RNA. Reverse-transcription was performed with SuperScript VILO (Life Technologies) to generate cDNA. PCR was performed using Power SYBR Green Master Mix (Applied Biosystems) on StepOne Plus real time cycler (Applied Biosystems). Analysis was performed with the −2ΔΔCt method using 18s as a housekeeping gene. A sample size of n = 4 was used for all qPCR analyses. The gene specific primers used for qPCR were: chemokine C-C motif ligand 2 (Ccl2) F-GGCTCAGCCAGATGCAGTTAA, R-CCTACTCATTGGGATCATCTTGCT; Interleukin-6 (Il6) F-GAGGATACCACTCCCAACAGACC, R-AAGTGCATCATCGTTGTTCATACA; Adamts4 F-GACCTTCCGTGAAGAGCAGTGT, R-CCTGGCAGGTGAGTTTGCAT; Adamts5 F-GCCCACCCAATGGTAAATCTTT, R-TGACTCCTTTTGCATCAGACTGA; Mmp9 F-CGAACTTCGACACTGACAAGAAGT, R-GCACGCTGGAATGATCTAAGC; Mmp13 F-GGGCTCTGAATGGTTATGACATTC, R-AGCGCTCAGTCTCTTCACCTCTT; Aggrecan (Acan) F-GCATGAGAGAGGCGAATGGA, R-CTGATCTCGTAGCGATCTTTCTTCT; Collagen IIα1 (Col2a1) F-TCCAGATGACTTTCCTCCGTCTA, R-AGGTAGGCGATGCTGTTCTTACA; and 18s rRNA F-CGGCTACCACATCCAAGGAA, R-GGGCCTCGAAAGAGTCCTGT.
Atomic Force Microscopy (AFM)
Mechanical testing to determine the cartilage elastic modulus was performed using AFM (MFP-3D, Asylum Research) as previously described (27). Briefly, iPSC-derived or native cartilage samples were harvested following three days of treatment and immobilized on glass slides using agarose. Samples were tested with a 25-μm-diameter polystyrene bead attached to a silicon nitride cantilever (Novascan Technologies). Indentations were performed on 8–10 sites of each sample and the force-indentation curves were fit to a modified Hertz model to calculate elastic modulus. A sample size of n = 6 was used for mechanical testing.
Biochemical analysis for DNA, glycosaminoglycans (GAGs), and collagen
iPSC cartilage and native cartilage samples were digested overnight with papain and analyzed for biochemical content as previously described (34). Briefly, double stranded DNA content was measured using the PicoGreen assay (Molecular Probes) and GAG content was detected using the 1,9-dimethylmethylene blue assay with a 525 nm wavelength. Collagen content was measured using a modified hydroxyproline assay (35). A sample size of n = 5 was used for all biochemical assays.
Histology
After three days of culture, iPSC cartilage and native cartilage samples were fixed in 4% paraformaldehyde and embedded in paraffin. Sections of 8 μm were stained with safranin-O/fast green/hematoxylin or picrosirius red. Immunohistochemistry for collagen type II was performed as previously described (27) using a primary antibody from the Developmental Studies Hybridoma Bank (II-II6B3, 1:1), a secondary antibody form Abcam (ab97021, 1:500), and the AEC Red Single chromogen (Invitrogen). Two samples per group were used for histological analysis.
Media analyses for GAGs, nitric oxide (NO), prostaglandin E2 (PGE2), and matrix metalloproteinase (MMP) activity
At the time of tissue harvest, the cell-conditioned media was collected and stored at −20°C in aliquots until analyzed for GAGs, NO and MMP activity as described previously (36). Briefly, GAGs were measured with the 1,9-dimethylmethylene blue as described for digested samples, NO was analyzed using the concentration of nitrate and nitrite with Greiss reagent, and MMP activity was measured using a quenched fluorogenic substrate (Dab-Gly-Pro-Leu-Gly-Met-Arg-Gly-Lys-Flu) after activation of latent MMPs with p-aminophenylmercuric acetate. For PGE2, the concentration was measured using a commercial ELISA (R&D Systems) (16). A sample size of n = 6 was used for all media analyses.
Statistical analysis
iPSC cartilage samples were analyzed independently using one-way ANOVA with Tukey’s HSD post-hoc test when applicable (α = 0.05). Native cartilage samples were analyzed with a T-test (α = 0.05).
Results
IL-1α induced changes in the gene expression of iPSC cartilage
Treatment of iPSC cartilage with IL-1α for 24 hours induced a rapid dose-dependent upregulation in the expression of six genes known to regulate the inflammatory and matrix-degrading features of OA (6) (Fig. 2). Upregulation of these genes at 24 hrs is consistent with prior studies that have examined IL-1 stimulated chondrocytes (7) and native cartilage (37). Increased expression of inflammatory and catabolic genes was statistically significant for all IL-1 doses tested. At the highest dose of 1 ng/ml, expression of soluble inflammatory mediators Ccl2 and Il6 were dramatically increased by 640- and 1700-fold, respectively. Expression of catabolic enzymes were also significantly upreguated under these conditions with the aggrecanases Adamts4 and 5 increasing 8- and 24-fold, respectively, and the MMPs Mmp9 and Mmp13 increasing 140- and 85-fold, respectively. Expression of chondrogenic genes Acan and Col2a1 were reduced in iPSC cartilage following treatment with 1 ng/ml of IL-1, but were unchanged at lower doses.
Figure 2. Gene expression analysis of iPSC-derived cartilage following a 24 hr treatment with IL-1α.

Expression of inflammatory (Ccl2, Il6) and catabolic (Adamts4, -5, Mmp9, -13) genes were all increased in a dose dependent manner. These increases were significant at all doses of IL-1α. Expression of chondrogenic genes (Acan, Col2a1) were decreased following treatment with 1 ng/ml of IL-1α, but unchanged at lower doses. A 1-way ANOVA was used with Tukey’s post-hoc test. Data are presented as mean ± SEM, and groups not connected by the same letter are significantly different from each other.
Loss of mechanical integrity
iPSC cartilage treated for 3 days with IL-1α showed a dose-dependent decrease in the elastic modulus of the tissue (Fig. 3a). Control samples possessed the highest modulus (19.31 ± 2.76 kPa), while samples subjected to 1 ng/mL IL-1α displayed the lowest modulus (6.05 ± 0.74 kPa). The elastic modulus of native hip cartilage was slightly higher than that of iPSC cartilage, and consistent with prior reports of mouse cartilage tested using AFM (38). A similar loss in structural integrity following IL-1α treatment was seen in native tissue, with the elastic modulus decreasing approximately 30% (Fig. 3a).
Figure 3. Mechanical properties and biochemical content of iPSC-derived and native cartilage following 3 days of IL-1α treatment.

(A) IL-1 exposure produced a dose-dependant decrease in the elastic modulus of both iPSC and native cartilage. (B) The DNA content of iPSC cartilage decreased slightly following treatment with 1 ng/ml of IL-1α, while there was no significant change in the native cartilage. IL-1α doses above 100 pg/ml produced a decrease in the total sulfated GAG content (C) and the sulfated GAG/DNA (D) of iPSC-derived and native cartilage (E) The total collagen content of iPSC-derived cartilage decreased slightly with the 1 ng/ml dose, but there was no significant change in the total collagen normalized to DNA (F). A 1-way ANOVA was used with Tukey’s post-hoc test. Data are presented as mean ± SEM, and groups not connected by the same letter are significantly different from each other.
Changes in biochemical composition with IL-1α exposure
Native cartilage and iPSC cartilage were similar in biochemical content in the control samples, but native tissue explants contained slightly more cells (as assessed by DNA content), GAG, and total collagen (Fig. 3). IL-1 exposure at 1 ng/ml caused a decrease in the DNA content of iPSC cartilage, but no changes were seen in native tissue (Fig 3b). Treatment of iPSC cartilage with IL-1α caused a dose-dependent loss of total GAG content and GAG per DNA, with a significant decrease at 100 pg/ml and a loss of approximately half of the original GAG content with 1 ng/ml (Fig. 3c–d). Native cartilage also showed a significant 23% loss in GAG per DNA when treated with 1 ng/ml IL-1α (Fig. 3d). While the collagen content of iPSC cartilage did decrease following treatment with 1 ng/ml of IL-1 (Fig. 3e), the total collagen per cell was not altered in either tissue after 3 days of IL-1α treatment (Fig. 3f).
Histological analysis
Safranin-O staining of the samples mimicked the biochemical data and indicated a significant loss of sulfated GAGs following IL-1α treatment (Fig. 4). GAG staining was clearly decreased with a dose of 100 pg/ml and was almost completely eliminated with a dose of 1 ng/ml. Native cartilage also lost GAG staining in response to treatment with 1 ng/ml IL-1α, but it was mainly localized to the outermost articular cartilage layer. Picrosirius red staining also matched the biochemical results and indicated no change in the total collagen content of either tissue after exposure to IL-1 (Fig. 4). In contrast, collagen type II immunostaining did decrease in iPSC cartilage treated with 1 ng/ml of IL-1 (Fig. 4), indicating that the collagen may be degrading, even if it is not released from the tissue.
Figure 4. Histological staining of iPSC-derived and native mouse femoral cartilage following treatment with IL-1α.
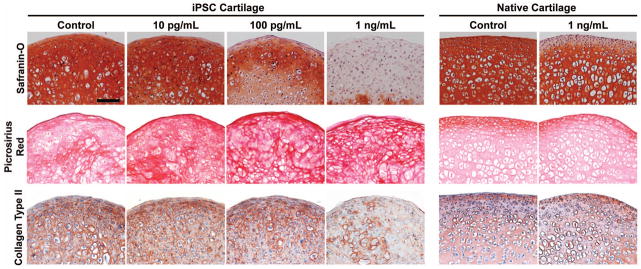
Safranin-O staining of iPSC and native cartilage, indicating a loss of GAG content from both tissues following IL-1α treatment at ≥100 pg/ml. Pircrosirius red staining indicated extensive collagen content in all samples, and no significant changes following cytokine treatment. While there was no change in total collagen content, a loss of collagen II immunostaining was seen, but only in iPSC cartilage treated with 1 ng/ml of IL-1. (Scale bar = 100 μm)
Degenerative environment in the media after IL-1α treatment
After three days of culture with IL-1α, media harvested from both iPSC cartilage and native cartilage samples contained high levels of sulfated GAGs that had been released from the tissue (Fig. 5a). In both tissues the GAG content in the media was nearly an exact inverse of the GAG/DNA values in the tissue. With a dose of 1 ng/ml, >60% of the total GAG in iPSC cartilage samples was found in the media. A likely contributor to this GAG loss was the dramatic and dose-dependent increase in specific MMP activity in the media. In both iPSC and native cartilage samples an IL-1 dose of 1 ng/ml induced a 300-fold increase in MMP activity within the media (Fig. 5b). IL-1α doses of 100 pg/ml or higher increased the release of nitric oxide from both native and iPSC cartilage (up to 5.8-fold), but the control levels of NO were higher in the native tissue samples (Fig. 5c). PGE2 release was also basally higher in native cartilage samples, but IL-1 treatment induced a larger increase in the release of this molecule from iPSC cartilage (up to 34-fold) (Fig. 5d).
Figure 5. Media analysis of iPSC-derived and native cartilage samples under inflammatory conditions.

(A) GAG release to the media increased in both tissues for IL-1α doses ≥100 pg/ml. (B) Specific MMP activity increased in a dose dependent manner at all concentrations. Concentration of nitric oxide (C) and PGE2 (C) increased following treatment with cytokine doses ≥100 pg/ml. A 1-way ANOVA was used with Tukey’s post-hoc test. Data are presented as mean ± SEM, and groups not connected by the same letter are significantly different from each other.
Screen of candidate therapeutics
In order to extend the capabilities of the in vitro model of OA to an effective drug screening system, we prioritized increasing the throughput capability of tissue formation. By distributing the cell suspension to 96-well round bottom plates instead of traditional 15 ml tubes for centrifugation, a large number of pellet cultures were simultaneously established. Pellets in this format retained homogenous staining for GAGs that were released upon treatment with 1 ng/ml IL-1α (Fig. 6a). Media analysis demonstrated that all of the candidate DMOADs provided some protective effect against IL-1 mediated degeneration of the iPSC cartilage. The NF-κB inhibitor SC-514 was effective at preventing approximately half of the GAG loss to the media, and IL-4 was also significantly protective (Fig. 6b). Safranin-O staining of the pellets confirmed these results and indicated increased GAG retention in these groups (Fig. 6a). Notably, the remaining molecules—TIMP-3, the COX-2 inhibitor NS-398, and broad spectrum MMP inhibitor GM-6001—did not significantly prevent IL-1α mediated GAG loss in this system.
Figure 6. Screen of candidate therapeutics using a high-throughput format.

(A) Safranin-O staining of pellets indicates a loss of GAG from the tissue following IL-1α treatment. Only treatment with SC-514 provided significant preservation of GAG content in the pellets (scale = 500 μm). (B) GAG release to the media was significantly reduced by SC-514 and IL-4. (C) MMP activity in the samples was completely inhibited by GM-6001 treatment, but also largely decreased by SC-514. Nitric oxide (D) and PGE2 (E) concentrations in the media were mildly reduced by most of the candidate therapeutics, with the greatest decreases after NS-398 and SC-514 treatment. A 1-way ANOVA was used with Tukey’s post-hoc test. Data are presented as mean ± SEM, and the pound indicates that the treatment statistically significant compared to the IL-1α only control.
Importantly, all of the candidate DMOADs were effective on their specific targets and mimicked their prior effects on native tissue, even if they did not completely protect the tissue from GAG release. For example, the MMP inhibitor GM-6001 abolished MMP activity (Fig. 6c) and the COX-2 inhibitor NS-398 inhibited both nitric oxide (Fig. 6d) and PGE2 production (Fig. 6e). These results are consistent with prior reports that have examined the treatment of native cartilage explants with GM-6001 (18) or NS-398 (16). SC-514 was the only molecule that provided a broad protective effect, reducing GAG loss, MMP production, nitric oxide production, and PGE2 production (Fig. 6b–e). GAG release to the media appeared to be the best measure of an effective therapeutic in this model.
Discussion
The goal of this work was to use iPSCs to develop a scalable and high-throughput system for identifying novel candidate drugs that may inhibit cartilage degradation in vitro. Our findings show that iPSC-derived cartilage responds to IL-1 with changes that are characteristic of osteoarthritic cartilage. Our approach has several important features needed for an effective DMOAD screening platform: 1) scalability and control of the cells’ genetic background through the use of iPSC-derived cartilage, 2) versatility due to a defined OA stimulus that could be tailored to match particular joint environments, and 3) sensitivity and high-throughput screening through media analysis of critical cartilage components and secreted mediators.
There has been much excitement about the use of iPSC-based “disease in a dish” models to investigate pathological mechanisms and screen for compounds that restore normal function (26). One strategy is to derive iPSCs from individuals with genetic defects and focus on any phenotypic differences to control iPSCs after differentiation to the cell type of interest (39, 40). However, phenotypic differences may not be readily apparent for late onset diseases or with the use of iPSCs derived from individuals without a strong predisposing genetic mutation. In these cases, further maturation in a tissue engineering context and treatment with disease-initiating stimuli may be necessary to obtain a robust phenotype useful for drug screening (41, 42). A critical step in this approach is to validate that the phenotype obtained in the in vitro model is relevant to disease pathology.
Treatment of iPSC cartilage with IL-1α recapitulated OA-like changes at the level of gene expression, matrix content, and tissue properties. Even a low dose of IL-1α was sufficient to upregulate the expression of catabolic genes at 24 hours, which is consistent with prior reports using native cartilage (7, 37). The upregulation of both ADAMTS members is indicative of a robust response, as Adamts4 but not Adamts5 has been implicated in the inflammatory response of cartilage explants, but only Adamts5 knockout mice are protected from OA (10). The 300-fold increase in MMP activity measured after 3 days indicates that the gene expression changes at 24 hrs ultimately resulted in degeneration of the iPSC cartilage. Treatment with IL-1α for 72 hours caused extensive loss of GAGs and a decrease in type II collagen staining, while the total amount of collagen was unaltered. This finding is consistent with cartilage explant studies and the natural course of OA, where GAG loss is indicative of early pathology and the loss of collagen is seen at late stages of disease (43).
In addition to cellular and matrix changes, OA is characterized by biomechanical changes in the articular cartilage that result in tissue dysfunction (12). IL-1α treatment of native and iPSC-derived cartilage resulted in a dose-dependent decrease in the elastic modulus of the tissue. Interestingly, the decrease in compressive modulus was more dramatic than the loss of GAG from the tissue, and was also detectable at lower doses of IL-1. These results suggest that structural and mechanical changes may precede matrix loss in this system, particularly near the surface of the tissue. The sensitivity of mechanical testing for detecting small degenerative changes also indicates that it could be a valuable output measure for DMOAD screening. High-throughput compressive testing of cartilage constructs was recently demonstrated in 48-well plates (44), and should be adaptable to increased throughput drug screening systems.
Both iPSC and native cartilage showed degradation in response to 1 ng/ml of IL-1α, but the loss of biochemical composition and biomechanical properties was more dramatic in the iPSC-derived tissue. Prior reports have also indicated that engineered cartilage is more sensitive to IL-1 treatment than native cartilage (33). This may relate to the lower GAG and collagen content present in the engineered tissue at the time of IL-1α treatment, as the maturity of cartilage constructs is known to mediate the response to cytokine stimulation (33, 45). In addition to allowing comparisons between iPSC and native cartilage, the use of murine cells also facilitates follow-up studies with established mouse models of OA to confirm the ability of the in vitro model to predict in vivo efficacy. Using this same drug screening system with human iPSCs may generate different drug candidates and could help investigators recognize differences in mouse and human OA for interpretation of pre-clinical data in drug development (26).
The previously identified candidate DMOADs employed to validate the system were chosen based on their ability to inhibit pathways related to inflammatory-mediated cartilage degradation in native tissue (15–19). Additionally, the molecules targeted different downstream mediators of IL-1 signaling in order to gain insight into which of the pleiotropic IL-1 effects are critical for matrix degradation. In this preliminary screen, the therapeutic molecules were administered prior to the OA stimulus in an effort to provide a best case scenario for the compounds to demonstrate efficacy. Even under these ideal conditions, only some of these agents were able to prevent GAG release into the media. These results provide support that our system is sensitive enough to find potential DMOADs and specific enough to avoid treatments that alter OA pathways but do not inhibit matrix degradation. The most effective molecule in our system, SC-514, inhibits the IκB kinase 2 of the NF-κB pathway and was previously shown to reduce PGE2, IL-6, and IL-8 production in stimulated synovial fibroblasts (15). The NF-κB pathway affects matrix production and chondrocyte differentiation in addition to inflammatory mediators (46), providing one possible explanation for the efficacy in this model as compared to the other inhibitors tested. In the future, molecules that show a protective effect, such as SC-514, will need to be examined during long-term culture in this model system to ensure that they can maintain their efficacy over time. While only a limited number of compounds were tested here as proof-of-concept for our model system, these results suggest that future screens for novel compounds using this system could benefit from focusing on pro-inflammatory mechanisms such as the NF-κB pathway.
The versatility of this iPSC-based screening system suggests that it can easily be modified to enhance its utility for particular applications. For example, while IL-1α was used in this work, a different OA stimulus such as fibronectin fragments (47) or other inflammatory cytokines such as tumor necrosis factor alpha or oncostatin M (48) may induce different features of OA. Furthermore, as these cells can be readily encapsulated in artificial matrices (27), the system also has the potential for use under conditions of injurious mechanical loading (49). The use of iPSCs, which can be derived from any individual, allows for genotype-specific drug screens that could be used for personalized medicine approaches or to provide insight into the genetic variability of disease progression (41). Alternatively, genome editing of iPSCs can probe the specific effects of genetic variation without confounding patient differences and therefore investigate the interaction between genetic and environmental contributors to OA (50). The ability to study different mechanisms and pathways that are potentially involved in OA using a single model system will hopefully expedite the discovery of effective DMOADs.
Acknowledgments
We thank Dr. William Horton of Portland Shriners Research Center for providing the Col2-GFP construct. We would also like to thank Nancy Martin of the Flow Cytometry Shared Resource at Duke University and Shannon K. O’Connor for their technical assistance. Funding from National Institutes of Health Grants AR50245, AR48852, AG15768, AG46927 and AR48182; the AO Foundation; the AOSpine Foundation; the Arthritis Foundation; and the Flight Attendant Medical Research Institute. Training was supported by a National Science Foundation Graduate Research Fellowship (B.O.D.); NIH T32AI007217 (V.P.W.); and Arthritis Foundation Postdoctoral Fellowships (J.S.-A. and V.P.W.).
Footnotes
Author contributions
All authors were involved in drafting the article or revising it critically for important intellectual content, and all authors approved the final version to be published. Dr. Guilak had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Study conception and design: Willard, Diekman, Christoforou, Leong, Guilak
Acquisition of data: Willard, Diekman, Sanchez-Adams
Analysis and interpretation of data: Willard, Diekman, Sanchez-Adams, Guilak
The authors declare no competing financial interests.
References
- 1.Lawrence RC, Felson DT, Helmick CG, Arnold LM, Choi H, Deyo RA, et al. Estimates of the prevalence of arthritis and other rheumatic conditions in the United States. Part II. Arthritis Rheum. 2008;58(1):26–35. doi: 10.1002/art.23176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhang W, Moskowitz RW, Nuki G, Abramson S, Altman RD, Arden N, et al. OARSI recommendations for the management of hip and knee osteoarthritis, Part II: OARSI evidence-based, expert consensus guidelines. Osteoarthritis and cartilage/OARS, Osteoarthritis Research Society. 2008;16(2):137–62. doi: 10.1016/j.joca.2007.12.013. [DOI] [PubMed] [Google Scholar]
- 3.Hunter DJ. Pharmacologic therapy for osteoarthritis--the era of disease modification. Nature reviews Rheumatology. 2011;7(1):13–22. doi: 10.1038/nrrheum.2010.178. [DOI] [PubMed] [Google Scholar]
- 4.Bijlsma JW, Berenbaum F, Lafeber FP. Osteoarthritis: an update with relevance for clinical practice. Lancet. 2011;377(9783):2115–26. doi: 10.1016/S0140-6736(11)60243-2. [DOI] [PubMed] [Google Scholar]
- 5.Goldring MB, Marcu KB. Cartilage homeostasis in health and rheumatic diseases. Arthritis research & therapy. 2009;11(3):224. doi: 10.1186/ar2592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kapoor M, Martel-Pelletier J, Lajeunesse D, Pelletier JP, Fahmi H. Role of proinflammatory cytokines in the pathophysiology of osteoarthritis. Nature reviews Rheumatology. 2011;7(1):33–42. doi: 10.1038/nrrheum.2010.196. [DOI] [PubMed] [Google Scholar]
- 7.Sandell LJ, Xing X, Franz C, Davies S, Chang LW, Patra D. Exuberant expression of chemokine genes by adult human articular chondrocytes in response to IL-1beta. Osteoarthritis and cartilage/OARS, Osteoarthritis Research Society. 2008;16(12):1560–71. doi: 10.1016/j.joca.2008.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hollander AP, Heathfield TF, Webber C, Iwata Y, Bourne R, Rorabeck C, et al. Increased damage to type II collagen in osteoarthritic articular cartilage detected by a new immunoassay. J Clin Invest. 1994;93(4):1722–32. doi: 10.1172/JCI117156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Poole AR, Nelson F, Dahlberg L, Tchetina E, Kobayashi M, Yasuda T, et al. Proteolysis of the collagen fibril in osteoarthritis. Biochemical Society symposium. 2003;(70):115–23. doi: 10.1042/bss0700115. [DOI] [PubMed] [Google Scholar]
- 10.Bondeson J, Wainwright S, Hughes C, Caterson B. The regulation of the ADAMTS4 and ADAMTS5 aggrecanases in osteoarthritis: a review. Clinical and experimental rheumatology. 2008;26(1):139–45. [PubMed] [Google Scholar]
- 11.Sandy JD, Flannery CR, Neame PJ, Lohmander LS. The structure of aggrecan fragments in human synovial fluid. Evidence for the involvement in osteoarthritis of a novel proteinase which cleaves the Glu 373-Ala 374 bond of the interglobular domain. J Clin Invest. 1992;89(5):1512–6. doi: 10.1172/JCI115742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Setton LA, Elliott DM, Mow VC. Altered mechanics of cartilage with osteoarthritis: human osteoarthritis and an experimental model of joint degeneration. Osteoarthritis and cartilage/OARS, Osteoarthritis Research Society. 1999;7(1):2–14. doi: 10.1053/joca.1998.0170. [DOI] [PubMed] [Google Scholar]
- 13.Wilusz RE, Zauscher S, Guilak F. Micromechanical mapping of early osteoarthritic changes in the pericellular matrix of human articular cartilage. Osteoarthritis and cartilage/OARS, Osteoarthritis Research Society. 2013;21(12):1895–903. doi: 10.1016/j.joca.2013.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chevalier X, Eymard F, Richette P. Biologic agents in osteoarthritis: hopes and disappointments. Nature reviews Rheumatology. 2013;9(7):400–10. doi: 10.1038/nrrheum.2013.44. [DOI] [PubMed] [Google Scholar]
- 15.Kishore N, Sommers C, Mathialagan S, Guzova J, Yao M, Hauser S, et al. A selective IKK-2 inhibitor blocks NF-kappa B-dependent gene expression in interleukin-1 beta-stimulated synovial fibroblasts. The Journal of biological chemistry. 2003;278(35):32861–71. doi: 10.1074/jbc.M211439200. [DOI] [PubMed] [Google Scholar]
- 16.Fermor B, Weinberg JB, Pisetsky DS, Misukonis MA, Fink C, Guilak F. Induction of cyclooxygenase-2 by mechanical stress through a nitric oxide-regulated pathway. Osteoarthritis and cartilage/OARS, Osteoarthritis Research Society. 2002;10(10):792–8. doi: 10.1053/joca.2002.0832. [DOI] [PubMed] [Google Scholar]
- 17.Gendron C, Kashiwagi M, Hughes C, Caterson B, Nagase H. TIMP-3 inhibits aggrecanase-mediated glycosaminoglycan release from cartilage explants stimulated by catabolic factors. FEBS letters. 2003;555(3):431–6. doi: 10.1016/s0014-5793(03)01295-x. [DOI] [PubMed] [Google Scholar]
- 18.Charni-Ben Tabassi N, Desmarais S, Bay-Jensen AC, Delaisse JM, Percival MD, Garnero P. The type II collagen fragments Helix-II and CTX-II reveal different enzymatic pathways of human cartilage collagen degradation. Osteoarthritis and cartilage/OARS, Osteoarthritis Research Society. 2008;16(10):1183–91. doi: 10.1016/j.joca.2008.02.008. [DOI] [PubMed] [Google Scholar]
- 19.Chowdhury TT, Bader DL, Lee DA. Anti-inflammatory effects of IL-4 and dynamic compression in IL-1beta stimulated chondrocytes. Biochemical and biophysical research communications. 2006;339(1):241–7. doi: 10.1016/j.bbrc.2005.11.016. [DOI] [PubMed] [Google Scholar]
- 20.Dell’Accio F, De Bari C, Luyten FP. Molecular markers predictive of the capacity of expanded human articular chondrocytes to form stable cartilage in vivo. Arthritis Rheum. 2001;44(7):1608–19. doi: 10.1002/1529-0131(200107)44:7<1608::AID-ART284>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 21.Hojo H, Yano F, Ohba S, Igawa K, Nakajima K, Komiyama Y, et al. Identification of oxytetracycline as a chondrogenic compound using a cell-based screening system. Journal of bone and mineral metabolism. 2010;28(6):627–33. doi: 10.1007/s00774-010-0179-y. [DOI] [PubMed] [Google Scholar]
- 22.Yano F, Hojo H, Ohba S, Fukai A, Hosaka Y, Ikeda T, et al. A novel disease-modifying osteoarthritis drug candidate targeting Runx1. Annals of the rheumatic diseases. 2013;72(5):748–53. doi: 10.1136/annrheumdis-2012-201745. [DOI] [PubMed] [Google Scholar]
- 23.Johnson K, Zhu S, Tremblay MS, Payette JN, Wang J, Bouchez LC, et al. A stem cell-based approach to cartilage repair. Science. 2012;336(6082):717–21. doi: 10.1126/science.1215157. [DOI] [PubMed] [Google Scholar]
- 24.Yang SL, Harnish E, Leeuw T, Dietz U, Batchelder E, Wright PS, et al. Compound screening platform using human induced pluripotent stem cells to identify small molecules that promote chondrogenesis. Protein & cell. 2012 doi: 10.1007/s13238-012-2107-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Huang AH, Motlekar NA, Stein A, Diamond SL, Shore EM, Mauck RL. High-throughput screening for modulators of mesenchymal stem cell chondrogenesis. Annals of biomedical engineering. 2008;36(11):1909–21. doi: 10.1007/s10439-008-9562-4. [DOI] [PubMed] [Google Scholar]
- 26.Engle SJ, Puppala D. Integrating human pluripotent stem cells into drug development. Cell stem cell. 2013;12(6):669–77. doi: 10.1016/j.stem.2013.05.011. [DOI] [PubMed] [Google Scholar]
- 27.Diekman BO, Christoforou N, Willard VP, Sun H, Sanchez-Adams J, Leong KW, et al. Cartilage tissue engineering using differentiated and purified induced pluripotent stem cells. Proceedings of the National Academy of Sciences of the United States of America. 2012;109(47):19172–7. doi: 10.1073/pnas.1210422109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Carey BW, Markoulaki S, Hanna J, Saha K, Gao Q, Mitalipova M, et al. Reprogramming of murine and human somatic cells using a single polycistronic vector. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(1):157–62. doi: 10.1073/pnas.0811426106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Grant TD, Cho J, Ariail KS, Weksler NB, Smith RW, Horton WA. Col2-GFP reporter marks chondrocyte lineage and chondrogenesis during mouse skeletal development. Dev Dyn. 2000;218(2):394–400. doi: 10.1002/(SICI)1097-0177(200006)218:2<394::AID-DVDY12>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 30.Stanton H, Golub SB, Rogerson FM, Last K, Little CB, Fosang AJ. Investigating ADAMTS-mediated aggrecanolysis in mouse cartilage. Nature protocols. 2011;6(3):388–404. doi: 10.1038/nprot.2010.179. [DOI] [PubMed] [Google Scholar]
- 31.Little CB, Mittaz L, Belluoccio D, Rogerson FM, Campbell IK, Meeker CT, et al. ADAMTS-1-knockout mice do not exhibit abnormalities in aggrecan turnover in vitro or in vivo. Arthritis Rheum. 2005;52(5):1461–72. doi: 10.1002/art.21022. [DOI] [PubMed] [Google Scholar]
- 32.McNulty AL, Rothfusz NE, Leddy HA, Guilak F. Synovial fluid concentrations and relative potency of interleukin-1 alpha and beta in cartilage and meniscus degradation. J Orthop Res. 2013;31(7):1039–45. doi: 10.1002/jor.22334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lima EG, Tan AR, Tai T, Bian L, Stoker AM, Ateshian GA, et al. Differences in interleukin-1 response between engineered and native cartilage. Tissue engineering Part A. 2008;14(10):1721–30. doi: 10.1089/ten.tea.2007.0347. [DOI] [PubMed] [Google Scholar]
- 34.Estes BT, Diekman BO, Gimble JM, Guilak F. Isolation of adipose-derived stem cells and their induction to a chondrogenic phenotype. Nature protocols. 2010;5(7):1294–311. doi: 10.1038/nprot.2010.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Detamore MS, Athanasiou KA. Effects of growth factors on temporomandibular joint disc cells. Archives of oral biology. 2004;49(7):577–83. doi: 10.1016/j.archoralbio.2004.01.015. [DOI] [PubMed] [Google Scholar]
- 36.McNulty AL, Miller MR, O’Connor SK, Guilak F. The effects of adipokines on cartilage and meniscus catabolism. Connective tissue research. 2011;52(6):523–33. doi: 10.3109/03008207.2011.597902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chan PS, Caron JP, Orth MW. Short-term gene expression changes in cartilage explants stimulated with interleukin beta plus glucosamine and chondroitin sulfate. J Rheumatol. 2006;33(7):1329–40. [PubMed] [Google Scholar]
- 38.Christensen SE, Coles JM, Zelenski NA, Furman BD, Leddy HA, Zauscher S, et al. Altered trabecular bone structure and delayed cartilage degeneration in the knees of collagen VI null mice. PLoS One. 2012;7(3):e33397. doi: 10.1371/journal.pone.0033397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lan F, Lee AS, Liang P, Sanchez-Freire V, Nguyen PK, Wang L, et al. Abnormal calcium handling properties underlie familial hypertrophic cardiomyopathy pathology in patient-specific induced pluripotent stem cells. Cell stem cell. 2013;12(1):101–13. doi: 10.1016/j.stem.2012.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Brennand KJ, Simone A, Jou J, Gelboin-Burkhart C, Tran N, Sangar S, et al. Modelling schizophrenia using human induced pluripotent stem cells. Nature. 2011;473(7346):221–5. doi: 10.1038/nature09915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cherry AB, Daley GQ. Reprogramming cellular identity for regenerative medicine. Cell. 2012;148(6):1110–22. doi: 10.1016/j.cell.2012.02.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bellin M, Marchetto MC, Gage FH, Mummery CL. Induced pluripotent stem cells: the new patient? Nature reviews Molecular cell biology. 2012 doi: 10.1038/nrm3448. [DOI] [PubMed] [Google Scholar]
- 43.Caterson B, Flannery CR, Hughes CE, Little CB. Mechanisms involved in cartilage proteoglycan catabolism. Matrix biology: journal of the International Society for Matrix Biology. 2000;19(4):333–44. doi: 10.1016/s0945-053x(00)00078-0. [DOI] [PubMed] [Google Scholar]
- 44.Mohanraj B, Hou C, Meloni GR, Cosgrove BD, Dodge GR, Mauck RL. A high throughput mechanical screening device for cartilage tissue engineering. Journal of biomechanics. 2013 doi: 10.1016/j.jbiomech.2013.10.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ousema PH, Moutos FT, Estes BT, Caplan AI, Lennon DP, Guilak F, et al. The inhibition by interleukin 1 of MSC chondrogenesis and the development of biomechanical properties in biomimetic 3D woven PCL scaffolds. Biomaterials. 2012 doi: 10.1016/j.biomaterials.2012.08.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Marcu KB, Otero M, Olivotto E, Borzi RM, Goldring MB. NF-kappaB signaling: multiple angles to target OA. Current drug targets. 2010;11(5):599–613. doi: 10.2174/138945010791011938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Forsyth CB, Pulai J, Loeser RF. Fibronectin fragments and blocking antibodies to alpha2beta1 and alpha5beta1 integrins stimulate mitogen-activated protein kinase signaling and increase collagenase 3 (matrix metalloproteinase 13) production by human articular chondrocytes. Arthritis Rheum. 2002;46(9):2368–76. doi: 10.1002/art.10502. [DOI] [PubMed] [Google Scholar]
- 48.Durigova M, Troeberg L, Nagase H, Roughley PJ, Mort JS. Involvement of ADAMTS5 and hyaluronidase in aggrecan degradation and release from OSM-stimulated cartilage. European cells & materials. 2011;21:31–45. doi: 10.22203/ecm.v021a03. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Stevens AL, Wishnok JS, White FM, Grodzinsky AJ, Tannenbaum SR. Mechanical injury and cytokines cause loss of cartilage integrity and upregulate proteins associated with catabolism, immunity, inflammation, and repair. Molecular & cellular proteomics: MCP. 2009;8(7):1475–89. doi: 10.1074/mcp.M800181-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Merkle FT, Eggan K. Modeling human disease with pluripotent stem cells: from genome association to function. Cell stem cell. 2013;12(6):656–68. doi: 10.1016/j.stem.2013.05.016. [DOI] [PubMed] [Google Scholar]