

Abstract

Decarboxylative cross-coupling of alkyl carboxylic acids with vinyl halides has been accomplished through the synergistic merger of photoredox and nickel catalysis. This new methodology has been successfully applied to a variety of α-oxy and α-amino acids, as well as simple hydrocarbon-substituted acids. Diverse vinyl iodides and bromides give rise to vinylation products in high efficiency under mild, operationally simple reaction conditions.
Arguably one of the most important developments in synthetic chemistry has been the advent of transition-metal-catalyzed cross-coupling reactions.1,2 Such processes have had a profound impact on almost all areas of chemical synthesis, stemming from their ability to form C–C, C–N, and C–O bonds in a highly predictable and chemoselective fashion. At the present time, the majority of transition-metal-mediated C–C couplings rely on the use of nucleophilic substrates that are prefunctionalized with organometallic traceless activation groups (TAGs, e.g., boronic acids, stannanes, zincates, and Grignards). However, an ever-increasing impetus to improve the versatility, cost, and operational utility of transition-metal-based methods has led to the development of elegant protocols that employ organic, native functionality as activation handles for complex fragment coupling reactions.3
One complementary approach to the use of organometallic TAGs has been the implementation of simple carboxylic acids, an organic activation group that is widely available from abundant biomass feedstocks, generally inexpensive, and compatible with multistep reaction sequences in native or latent form (e.g., one step from esters, amides, and olefins).3b Indeed, since the pioneering work of Gooßen et al.,4 the decarboxylative cross-coupling between carboxylic acids and aryl halides has found application in the construction of Csp2–Csp2 and Csp–Csp2 bonds.5 More elusive, however, is the successful implementation of alkyl carboxylic acids for the production of Csp3–Csp2 bonds in complex fragment couplings. This deficiency can be readily appreciated given the diminished reactivity of Csp3 carboxylic acids toward decarboxylative transmetalation in the presence of commonly employed metal catalysts. Indeed, most examples to date of decarboxylative Csp3–Csp2 bond formations rely on the use of activated substrates, such as electron-deficient benzylic,6 cyclohexadienyl,7 α-cyano,8 or β-ester carboxylic acids.9,10 Clearly, a modern strategy enabling simple alkyl carboxylic acids to function broadly as generic nucleophiles in conventional cross-coupling reactions would be of substantial utility to synthetic chemists operating within both academic and industrial settings.
In recent years, synergistic or dual catalysis has come to the fore as a valuable mechanistic paradigm for the invention of novel chemical transformations that are currently not possible via the action of a single catalyst.11 Successful application of synergistic catalysis typically requires the simultaneous activation and engagement of two distinct coupling partners with two separate catalysts (wherein each catalyst independently operates on a respective substrate). Recently, our laboratory introduced a new dual catalysis platform that allows the decarboxylative coupling of Csp3 carboxylic acids with aryl halides under the combined action of visible-light photoredox catalysis and Ni catalysis.12−16 Importantly, this new synergistic protocol represents a general approach toward the coupling of alkyl, α-amino, and α-oxy acids with a wide range of aryl and heteroaryl halides (eq 1).17
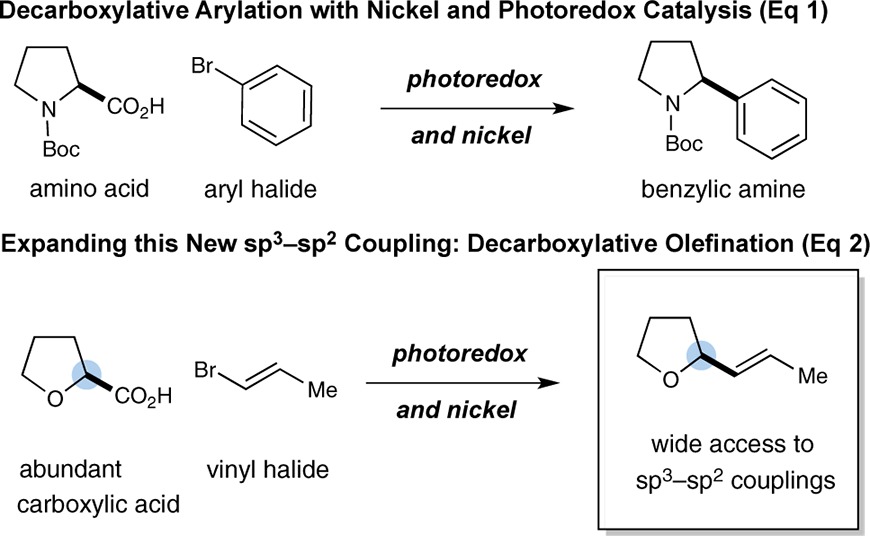 |
1 |
In an effort to demonstrate the wide-ranging applications of this new photoredox–nickel dual catalysis activation mode, we recently sought to explore the feasibility of a decarboxylative coupling between vinyl halides with a broad array of commercial Csp3 carboxylic acids. We recognized that the successful realization of these ideals would enable a new C–C bond-forming reaction that would allow direct access to complex alkyl vinyl, allylic amino, and allylic oxy products in only one step from simple, abundant, and inexpensive starting materials (eq 2). Perhaps most important, to our knowledge this transformation has not previously been accomplished in a generic format.
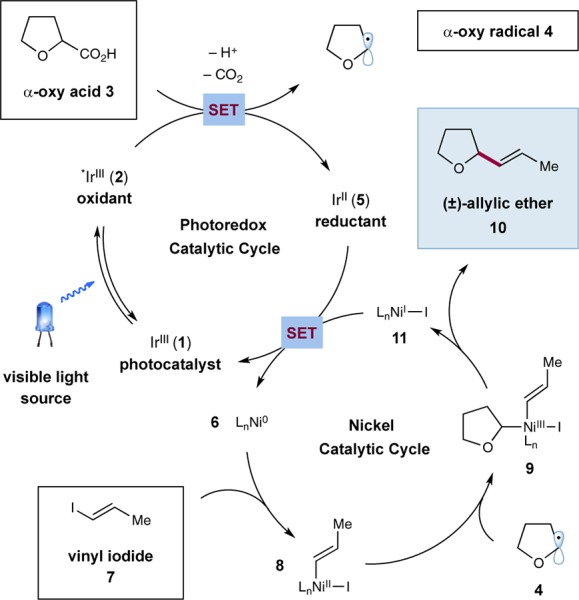
As shown in Scheme 1, our proposed olefination mechanism begins with photoexcitation of iridium(III) photocatalyst Ir[dF(CF3)ppy]2(dtbbpy)PF6 [dF(CF3)ppy = 2-(2,4-difluorophenyl)-5-(trifluoromethyl)pyridine, dtbbpy = 4,4′-di-tert-butyl-2,2′-bipyridine] (1) to produce the long-lived (τ = 2.3 μs)18 excited-state IrIII species 2. This photoexcited complex is a strong oxidant (E1/2red[*IrIII/IrII] = +1.21 V vs SCE in MeCN)18 and should undergo a thermodynamically favorable single electron transfer (SET) with the carboxylate formed by deprotonation of α-oxy acid 3 (e.g., for THF-2-CO2Cs, E1/2 = +1.08 V vs SCE in MeCN).19 This results in the formation of a carboxyl radical, which are known to rapidly undergo CO2-extrusion,20 to generate α-oxy radical 4 along with reduced IrII species 5. Concurrent with this photoredox mechanism, the Ni catalytic cycle will initiate with oxidative addition of LnNi0 species 6 into vinyl iodide 7 to generate vinyl NiII intermediate 8. Interception of α-oxy radical 4 by 8 would then generate organometallic NiIII adduct 9, which upon reductive elimination would deliver allylic ether product 10 and NiI species 11. Completion of the two catalytic cycles is then achieved by reduction of NiI species 11 (E1/2red[NiII/Ni0] = −1.2 V vs SCE in DMF)21 by the reduced state of the photocatalyst 5 (E1/2[IrIII/IrII] = −1.37 V vs SCE in MeCN)18 to regenerate photocatalyst 1 and Ni0 catalyst 6.
Scheme 1. Mechanism of Decarboxylative Olefination.
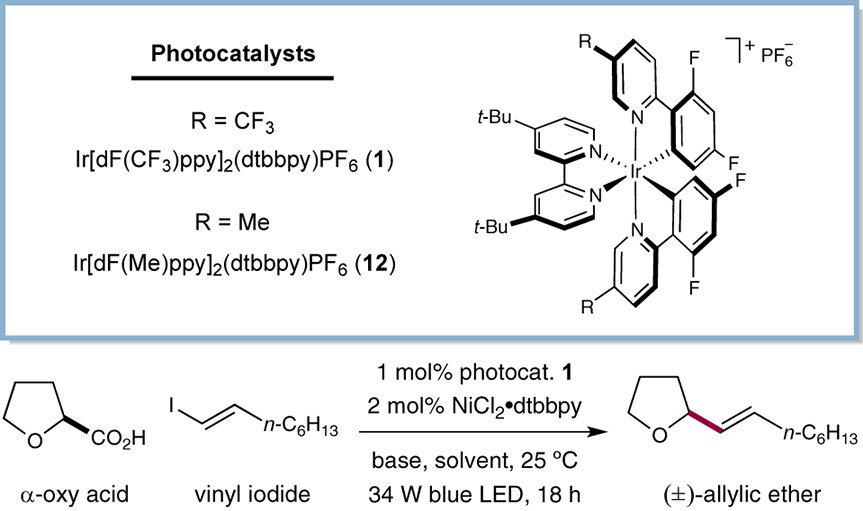
Initial investigations into the proposed decarboxylative cross-coupling focused on the reaction of tetrahydrofuran-2-carboxylic acid with (E)-1-iodo-1-octene (Table 1). We were delighted to find that irradiation of the carboxylic acid and vinyl iodide in the presence of photocatalyst 1, NiCl2·glyme (10 mol%), dtbbpy (10 mol%), and Cs2CO3 provided the desired allylic ether product in excellent yield (entry 1, 83% yield). Control experiments highlighted the essential roles of the photocatalyst, Ni catalyst, base, and light in this transformation (entries 2–5, <5% yield). While the vinylation product was obtained in high yield under the conditions described in entry 1, we recognized early on that the implementation of high dilution and Ni loadings would not be operationally optimal. However, conducting the same experiment with only 2 mol% NiCl2·glyme and at 0.1 M in DMF led to a significant reduction in reaction efficiency (entry 6, 22% yield). An evaluation of solvents revealed that DMSO provided a significant improvement in yield (entry 7, 52% yield), and changing the base from insoluble Cs2CO3 to 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) also proved to be beneficial (entry 8, 61% yield). Finally, the identification of Ir[dF(Me)ppy]2(dtbbpy)PF6 [dF(Me)ppy = 2-(2,4-difluorophenyl)-5-methylpyridine] (12) as the optimal photocatalyst provided a significant enhancement in yield, furnishing the desired cross-coupled product in 92% yield (entry 9) while retaining higher reaction concentrations and low catalyst loadings.22 Time studies have revealed that the use of photocatalyst 1 under similar reaction molarities (0.1 M) leads to catalyst decomposition prior to full conversion of the starting materials, resulting in diminished yields (entry 8, 61% yield). We believe the dramatic enhancement in efficiency in changing to photocatalyst 12 from 1 (entry 9, 92% yield) correlates to the improved stability of catalyst 12 toward radical-mediated decomposition under concentrated conditions.23
Table 1. Optimization of the Decarboxylative Olefinationa.
| entry | conditions | base | solvent | yield |
|---|---|---|---|---|
| 1b | 10 mol% NiCl2·dtbbpy | Cs2CO3 | DMF | 83% |
| 2b | no photocatalyst | Cs2CO3 | DMF | 0% |
| 3b | no NiCl2·dtbbpy | Cs2CO3 | DMF | <5% |
| 4b | no base | – | DMF | 0% |
| 5b | no light | Cs2CO3 | DMF | 0% |
| 6c | as shown | Cs2CO3 | DMF | 22% |
| 7c | as shown | Cs2CO3 | DMSO | 52% |
| 8c | as shown | DBU | DMSO | 61% |
| 9c | 1 mol% photocat. 12 | DBU | DMSO | 92% |
Reactions performed using (E)-1-iodo-1-octene (0.1 mmol) and tetrahydrofuran-2-carboxylic acid (1.7 equiv), dtbbpy = 4,4′-di-tert-butyl-2,2′-bipyridine. Yields determined by 1H NMR analysis using an internal standard.
Reactions performed at 0.025 M for 72 h.
Reactions performed at 0.1 M.
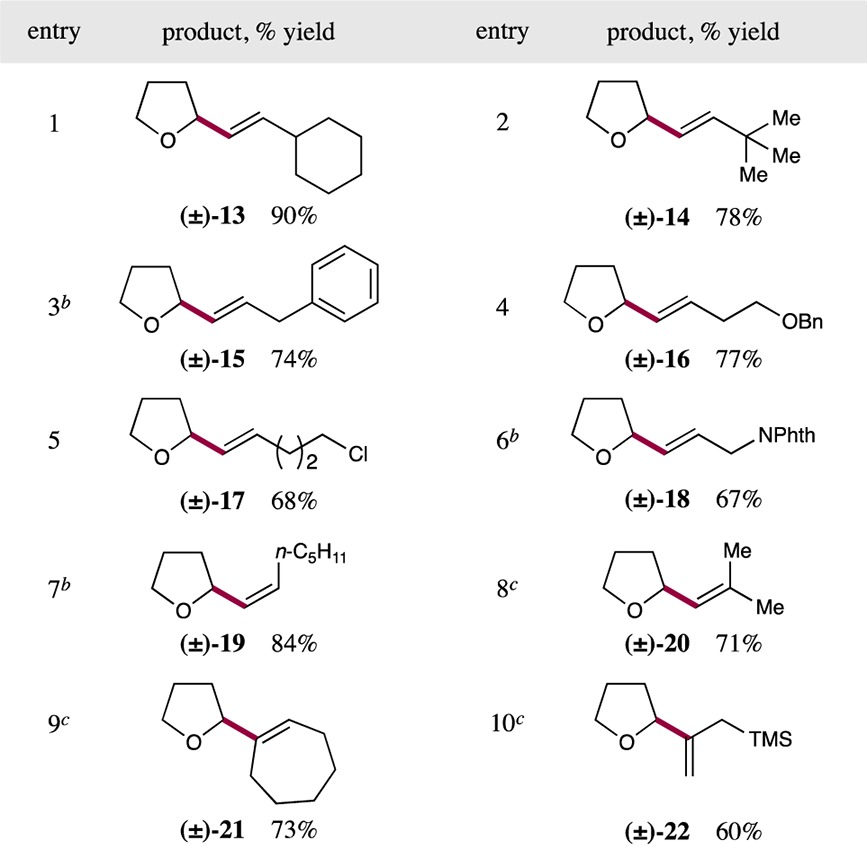
With the optimal dual catalysis conditions in hand, we next sought to examine the scope of the vinyl halide component in this new Csp3–Csp2 bond-forming protocol. As shown in Table 2, a range of structurally diverse vinyl halides undergo efficient cross-coupling with tetrahydrofuran-2-carboxylic acid (as the representative nucleophile). These include β-alkyl-substituted (E)-vinyl iodides, which provide cross-coupled products in high yield regardless of the size of the β-substituent (entries 1 and 2, 90% and 78% yield, respectively). Vinyl iodides possessing various functional groups, including aromatic rings, benzyl ethers, alkyl chlorides, and phthalimidoyl-protected amines also delivered the corresponding allyl ether products in good yield (entries 3–6, 67–77% yield). In addition to (E)-vinyl iodides, those with (Z)-geometry could be readily incorporated to generate cis-olefin containing products in excellent yield and with no observable geometrical isomerization (entry 7, 84% yield).24 Notably, for vinyl halides possessing α-substitution or β,β-disubstitution, the use of the corresponding vinyl bromide provided higher yields of the cross-coupled adduct. Under these conditions, vinyl bromides allowed the generation of trisubstituted olefin products 20 and 21 with excellent efficiency (entries 8 and 9, 71% and 73% yield, respectively). Finally, the synthesis of allyl silane 22 demonstrates the ability of this decarboxylative cross-coupling protocol to generate olefin products containing synthetically useful functional handles that might be readily exploited in subsequent transformations (entry 10, 60% yield). It is important to note that throughout all of our investigations into this new vinylation protocol, the stereochemical information present in the vinyl halides was translated directly to the olefin-bearing products, with no observed erosion in E:Z ratio.24
Table 2. Decarboxylative Olefination: Vinyl Halide Scopea.

We next turned our attention to examining the scope of the carboxylic acid component. As revealed in Table 3, we were delighted to find that a broad range of substrates bearing Csp3 carboxylates readily participate as coupling partners in this CO2-extrusion/vinylation protocol. For example, five- and six-membered cyclic α-oxy carboxylic acids readily undergo cross-coupling to form the allyl ethers in excellent yields (23 and 24, 92% and 74% yield, respectively). Pleasingly, a highly functionalized ribose-derived cyclic α-oxy acid was also found to be a competent coupling partner, providing 25 in excellent yield and diastereoselectivity (89% yield, 18:1 dr). This result clearly demonstrates the potential utility of this new coupling protocol for late-stage and orthogonal nucleoside functionalization (an area of significant interest among pharmaceutical agencies developing antiviral agents).25 Decarboxylative olefination could also be extended to acyclic primary and secondary α-oxy acids, including benzyl-protected lactic and glycolic acids, generating the allylic ether products in high yields (26 and 27, 78% and 77% yield, respectively). Furthermore, an erythronic acid derivative was readily functionalized to generate olefin 28 in high yield albeit with low diastereoselectivity (79% yield, 1.4:1.0 dr).
Table 3. Decarboxylative Olefination: Carboxylic Acid Scopea.
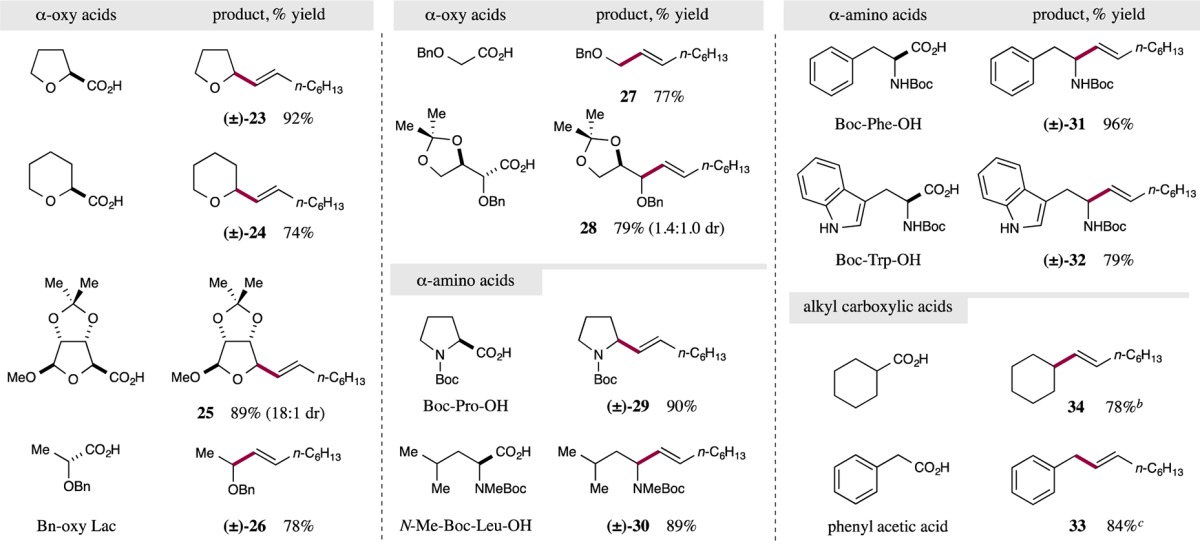
Reactions performed using the optimized conditions from Table 1 with 0.5 mmol (E)-1-iodo-1-octene or (E)-1-bromo-1-octene. Yields are of isolated products. Ratios of diastereomers determined by 1H NMR analysis. For detailed experimental procedures, see SI.
Primary alkyl carboxylic acids provided encouraging levels of efficiency, see SI. for experimental results.
Good yields were obtained for phenyl acetic acid derivatives p-OMe (96% yield) and m-Cl (68% yield). For experimental procedures, see SI.
In addition to α-oxy acids, α-amino acids readily undergo decarboxylative vinylation. A broad array of cyclic and acyclic N-tert-butoxycarbonyl (Boc)-protected amino acids were found to be highly efficient coupling partners, generating synthetically versatile allylic amine products in excellent yields (29–32, 79–96% yield) from biomass derivatives.
Most importantly, simple hydrocarbon-substituted carboxylic acids that lack radical stabilizing α-heteroatoms can also be readily employed in this new coupling protocol. For example, phenylacetic acid proved to be a valuable coupling partner, presumably due to the formation of a stabilized benzyl radical intermediate, furnishing allyl benzene 33 in 84% yield. Last, cyclohexanecarboxylic acid readily undergoes efficient cross-coupling to provide 1-cyclohexyloct-1-ene (34) with excellent efficiency (78% yield). This result is of particular note as it demonstrates the potential value of this cross-coupling methodology to functionalize simple alkyl carboxylic acids, without the requirement of a radical-stabilizing α-substituent.26 Moreover, this result highlights the potential breadth of simple, hydrocarbon-based nucleophile partners that may be employed in this Csp3–Csp2 coupling.
 |
3 |
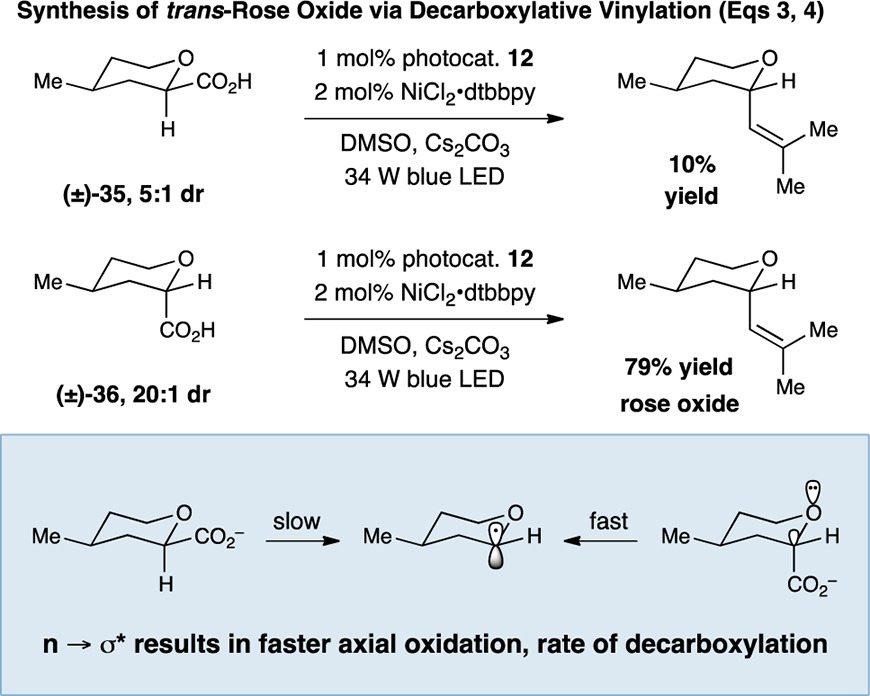
In order to further demonstrate the utility of this decarboxylative vinylation method, we applied it to the racemic synthesis of trans-rose oxide, a natural product and widely used fragrance (eq 3). As shown above, exposure of trans-tetrahydropyran carboxylic acid 36 to this photoredox–Ni coupling provided trans-rose oxide in high yield and with useful levels of diastereoselectivity. Intriguingly, however, when the corresponding cis-tetrahydropyran carboxylic acid 35 was employed, high coupling efficiencies were not observed, instead leading to recovery of 35 enriched in the cis-diastereomer. This result is rationalized by the change in bond strength and oxidation potential of the C–CO2 moiety as it exists in either the anomeric axial or the nonanomeric equatorial topography. We assume that the axial C–CO2 bond is weaker, and the rate of decarboxylation is faster (in competition with back electron transfer) due to the hyperconjugative stabilization by the ring oxygen lone pair in the case of the trans-tetrahydropyran carboxylic acid system.
Acknowledgments
Financial support was provided by NIHGMS (R01 GM103558-03) and kind gifts from Merck, AbbVie, Bristol-Myers Squibb, and Amgen.
Supporting Information Available
Experimental procedures and spectral data. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- van Santen R. A. In Catalysis: From Principles to Applications, 1st ed.; Beller M., Renken A., van Santen R. A., Eds.; Wiley-VCH: Weinheim, 2012; p 3. [Google Scholar]
- For selected reviews, see:; a Metal-Catalyzed Cross-Coupling Reactions, 2nd ed.; de Meijere A., Diederich F., Eds.; Wiley-VCH: Weinheim, 2004. [Google Scholar]; b Seechurn C. C. C. J.; Kitching M. O.; Colacot T. J.; Snieckus V. Angew. Chem., Int. Ed. 2012, 51, 5062. [DOI] [PubMed] [Google Scholar]
- a Godula K.; Sames D. Science 2006, 312, 67. [DOI] [PubMed] [Google Scholar]; b Baudoin O. Angew. Chem., Int. Ed. 2007, 46, 1373. [DOI] [PubMed] [Google Scholar]; c Chen X.; Engle K. M.; Wang D.-H.; Yu J.-Q. Angew. Chem., Int. Ed. 2009, 48, 5094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gooßen L. J.; Deng G.; Levy L. M. Science 2006, 313, 662. [DOI] [PubMed] [Google Scholar]
- For selected reviews, see:; a Rodríguez N.; Goossen L. J. Chem. Soc. Rev. 2011, 40, 5030. [DOI] [PubMed] [Google Scholar]; b Gooßen L. J.; Gooßen K. In Topics in Organometallic Chemistry, Vol. 44; Gooßen L. J., Ed.; Springer-Verlag: Berlin, 2013; p 121. [Google Scholar]; c Park K.; Lee S. RSC Adv. 2013, 3, 14165. [Google Scholar]
- a Shang R.; Yang Z.-W.; Wang Y.; Zhang S.-L.; Liu L. J. Am. Chem. Soc. 2010, 132, 14391. [DOI] [PubMed] [Google Scholar]; b Shang R.; Huang Z.; Chu L.; Fu Y.; Liu L. Org. Lett. 2011, 13, 4240. [DOI] [PubMed] [Google Scholar]; c Xu Z.; Wang Q.; Zhu J. Angew. Chem., Int. Ed. 2013, 52, 3272. [DOI] [PubMed] [Google Scholar]
- Chou C.-M.; Chatterjee I.; Studer A. Angew. Chem., Int. Ed. 2011, 50, 8614. [DOI] [PubMed] [Google Scholar]
- a Yeung P. Y.; Chung K. H.; Kwong F. Y. Org. Lett. 2011, 13, 2912. [DOI] [PubMed] [Google Scholar]; b Shang R.; Ji D.-S.; Chu L.; Fu Y.; Liu L. Angew. Chem., Int. Ed. 2011, 50, 4470. [DOI] [PubMed] [Google Scholar]
- Feng Y.-S.; Wu W.; Xu Z.-Q.; Li Y.; Li M.; Xu H.-J. Tetrahedron 2012, 68, 2113. [Google Scholar]
- Additional reports on decarboxylative functionalization of aliphatic acids:; a Bi H.-P.; Chen W.-W.; Liang Y.-M.; Li C.-J. Org. Lett. 2009, 11, 3246. [DOI] [PubMed] [Google Scholar]; b Zhang C.; Seidel D. J. Am. Chem. Soc. 2010, 132, 1798. [DOI] [PubMed] [Google Scholar]; c Bi H.-P.; Qingfeng T.; Guan M.; Chen W.-W.; Liang Y.-M.; Yao X.; Li C.-J. J. Org. Chem. 2010, 75, 783. [DOI] [PubMed] [Google Scholar]; d Wang Y.; Zhang L.; Yang Y.; Zhang P.; Du Z.; Wang C. J. Am. Chem. Soc. 2013, 135, 18048. [DOI] [PubMed] [Google Scholar]; e Yin F.; Wang Z.; Li Z.; Li C. J. Am. Chem. Soc. 2012, 134, 10401. [DOI] [PubMed] [Google Scholar]; f Wang Z.; Zhu L.; Yin F.; Su Z.; Li Z.; Li C. J. Am. Chem. Soc. 2012, 134, 4258. [DOI] [PubMed] [Google Scholar]; g Liu X.; Wang Z.; Cheng X.; Li C. J. Am. Chem. Soc. 2012, 134, 14330. [DOI] [PubMed] [Google Scholar]
- Allen A. E.; MacMillan D. W. C. Chem. Sci. 2011, 3, 633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Zuo Z.; MacMillan D. W. C. J. Am. Chem. Soc. 2014, 136, 5257. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Chu L.; Ohta C.; Zuo Z.; MacMillan D. W. C. J. Am. Chem. Soc. 2014, 136, 10886. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Noble A.; MacMillan D. W. C. J. Am. Chem. Soc. 2014, 136, 11602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For recent reviews on photoredox catalysis, see:; a Tucker J. W.; Stephenson C. R. J. J. Org. Chem. 2012, 77, 1617. [DOI] [PubMed] [Google Scholar]; b Prier C. K.; Rankic D. A.; MacMillan D. W. C. Chem. Rev. 2013, 113, 5322. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Xie J.; Jin H.; Xu P.; Zhu C. Tetrahedron Lett. 2014, 55, 36. [Google Scholar]; d Schultz D. M.; Yoon T. P. Science 2014, 343, 985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For reports on the successful merger of photoredox and transition-metal catalysis, see:; a Osawa M.; Nagai H.; Akita M. Dalton Trans. 2007, 827. [DOI] [PubMed] [Google Scholar]; b Kalyani D.; McMurtrey K. B.; Neufeldt S. R.; Sanford M. S. J. Am. Chem. Soc. 2011, 133, 18566. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Ye Y.; Sanford M. S. J. Am. Chem. Soc. 2012, 134, 9034. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Rueping M.; Koenigs R. M.; Poscharny K.; Fabry D. C.; Leonori D.; Vila C. Chem.—Eur. J. 2012, 18, 5170. [DOI] [PubMed] [Google Scholar]; e Sahoo B.; Hopkinson M. N.; Glorius F. J. Am. Chem. Soc. 2013, 135, 5505. [DOI] [PubMed] [Google Scholar]; f Shu X. Z.; Zhang M.; He Y.; Frei H.; Toste F. D. J. Am. Chem. Soc. 2014, 136, 5844. [DOI] [PMC free article] [PubMed] [Google Scholar]; g Tellis J. C.; Primer D. N.; Molander G. A. Science 2014, 345, 433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Xie J.; Xu P.; Li H.; Xue Q.; Jin H.; Cheng Y.; Zhu C. Chem. Commun. 2013, 49, 5672. [DOI] [PubMed] [Google Scholar]; b Liu J.; Liu Q.; Yi H.; Qin C.; Bai R.; Qi X.; Lan Y.; Lei A. Angew. Chem., Int. Ed. 2014, 53, 502. [DOI] [PubMed] [Google Scholar]
- Pfister K. F.; Grunberg M. F.; Gooßen L. J. Adv. Synth. Catal. 2014, 356, 3302. [Google Scholar]
- Zuo Z.; Ahneman D. T.; Chu L.; Terrett J. A.; Doyle A. G.; MacMillan D. W. C. Science 2014, 345, 6195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowry M. S.; Goldsmith J. I.; Slinker J. D.; Rohl R.; Pascal R. A. Jr.; Malliaras G. G.; Bernhard S. Chem. Mater. 2005, 17, 5712. [Google Scholar]
- The reduction potential of cesium tetrahydrofuran-2-carboxylate was measured in acetonitrile following literature methods:; a Andrieux C. P.; Gonzalez F.; Saveant J. J. Electroanal. Chem. 2001, 498, 171. [Google Scholar]; b Galicia M.; Gonzalez F. J. J. Electrochem. Soc. 2002, 149, D46. [Google Scholar]; See Supporting Information for details.
- Bockman T. M.; Hubig S. M.; Kochi J. K. J. Org. Chem. 1997, 62, 2210. [DOI] [PubMed] [Google Scholar]
- Durandetti M.; Devaud M.; Perichon J. New J. Chem. 1996, 20, 659. [Google Scholar]
- Ladouceur S.; Fortin D.; Zysman-Colman E. Inorg. Chem. 2011, 50, 11514. [DOI] [PubMed] [Google Scholar]
- Devery J. J. III; Douglas J. J.; Nguyen J. D.; Cole K. P.; Flowers R. A. II; Stephenson C. R. J. Chem. Sci. 2015, 6, 537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erosion of E:Z ratio can be observed for β-substituted styrenes under photocatalytic conditions. For examples, see:; a Reference (11)c; b Singh K.; Staig S. J.; Weaver J. D. J. Am. Chem. Soc. 2014, 136, 5275. [DOI] [PubMed] [Google Scholar]
- De Clerq E. Nat. Rev. Drug Discovery 2002, 1, 13. [DOI] [PubMed] [Google Scholar]
- A primary carboxylic acid (5-phenylvaleric acid) provided the corresponding product in 11% yield. Tertiary alkyl carboxylic acids were not viable coupling partners.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.