

Abstract
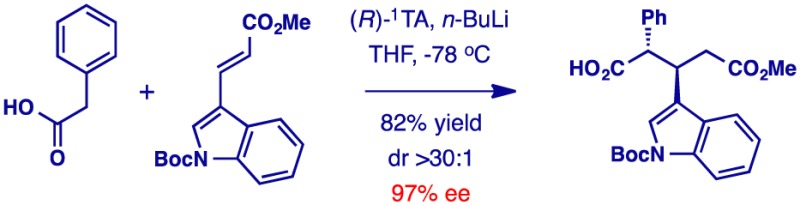
Michael addition is a premier synthetic method for carbon–carbon and carbon–heteroatom bond formation. Using chiral dilithium amides as traceless auxiliaries, we report the direct enantioselective Michael addition of carboxylic acids. A free carboxyl group in the product provides versatility for further functionalization, and the chiral reagent can be readily recovered by extraction with aqueous acid. The method has been applied in the enantioselective total synthesis of the purported structure of pulveraven B.
Enantioselective Michael addition of lithium enolates is a process of significant utility. From simple precursors, this reaction has the potential to generate multiple consecutive stereocenters that are difficult to access by other methods. There has been considerable progress in methodology that has tapped into this potential,1,2 including several methods based on covalent chiral auxiliaries.3,4 Asymmetric transformations with lithium enolates derived from carboxylic acids are performed predominantly with covalently attached chiral auxiliaries. These reactions provide a broad arsenal of methods for enantioselective synthesis and are indispensable in the synthesis of many complex natural products and pharmaceuticals.5 We recently reported that high enantioselectivities could be achieved in the direct one-step alkylation of arylacetic acids using chiral lithium amides.6 In this process, the chiral C2-symmetric dilithium amide functions as a chiral auxiliary within a mixed enediolate–dilithium amide aggregate formed in situ,7 thus bypassing the additional steps required to attach and remove covalently bound chiral auxiliaries. X-ray crystallography, 6Li, 13C, and 15N NMR spectroscopy, and DFT calculations pointed to the structure of the aggregate depicted in Scheme 1.7,8
Scheme 1.
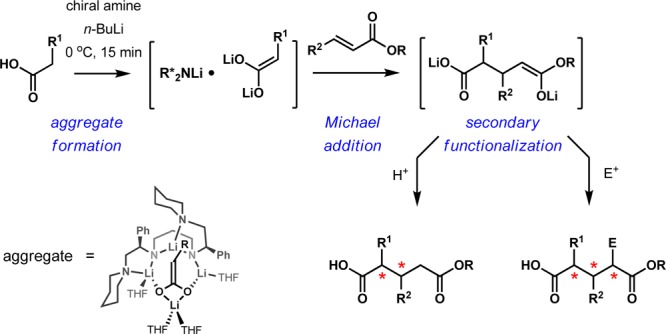
We describe herein a direct enantioselective Michael addition of carboxylic acids via enediolates mediated by chiral lithium amides (Scheme 1).9−11 The reaction occurs with high stereocontrol in both the relative and absolute sense, further highlighting the utility of chiral lithium amides as traceless auxiliaries for asymmetric synthesis.
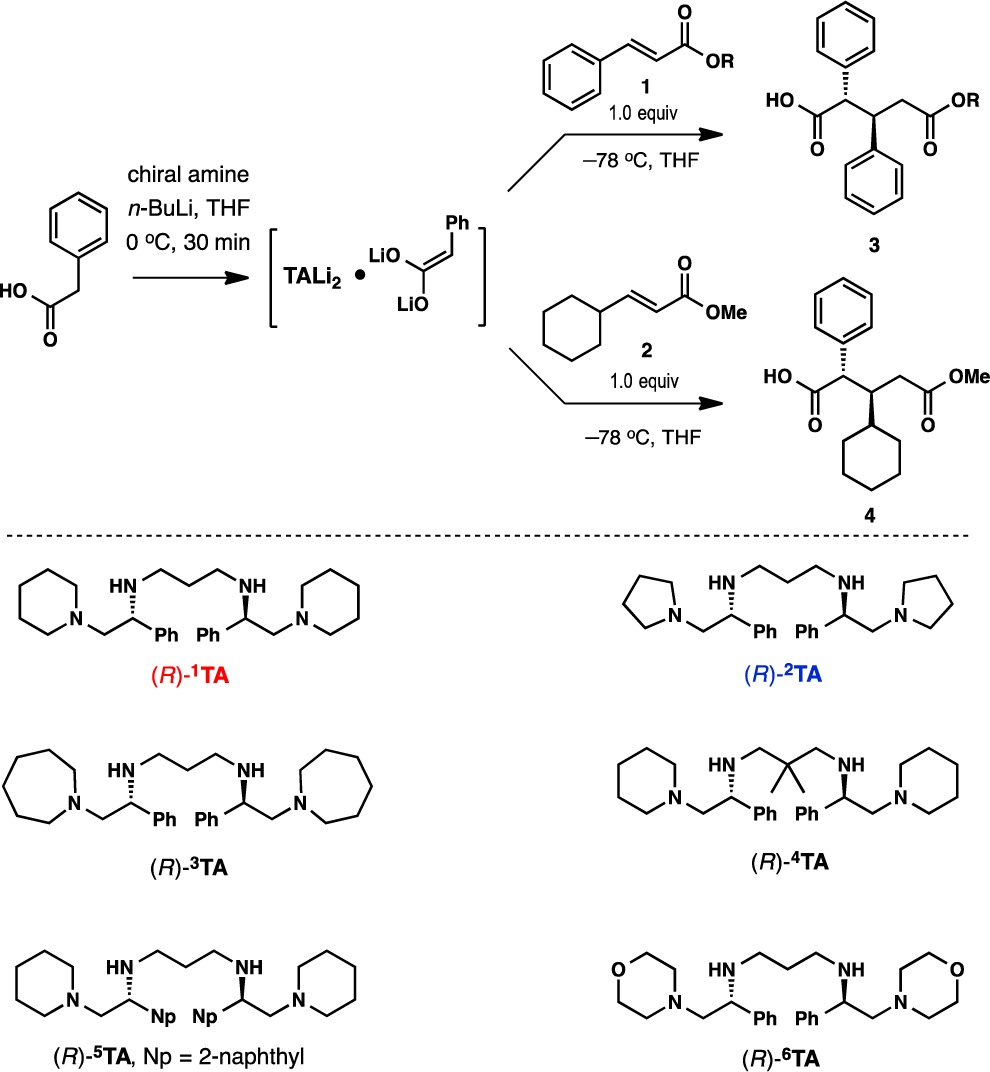
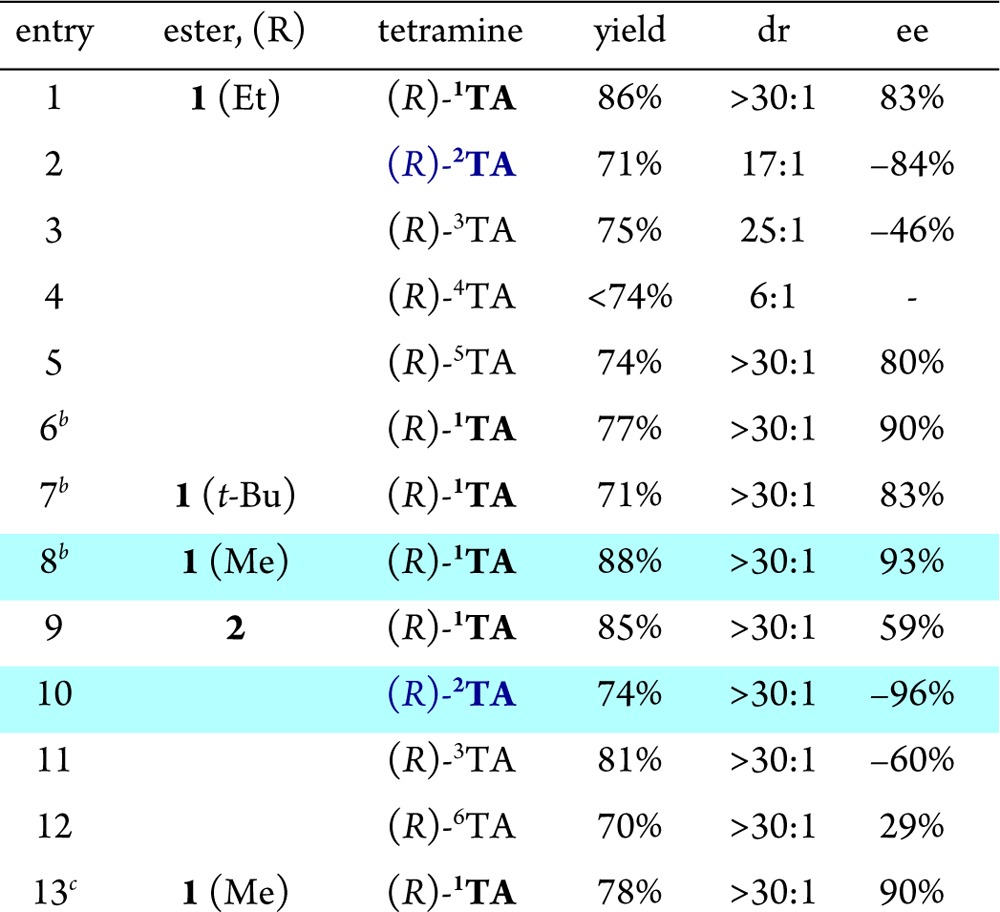
The addition of phenylacetic acid to alkyl cinnamates and methyl (E)-3-cyclohexylacrylate in the presence of dilithium amides from chiral C2-symmetric tetramines 1TA–6TA was investigated first (Table 1).12 From the onset, the reactions were characterized by facile, highly anti-selective 1,4-additions with high conversions at −78 °C.13 Piperidine-based tetramine (R)-1TA,14 which was previously shown to be highly effective in asymmetric alkylations of arylacetic acids, again proved to be optimal (88% yield, >30:1 dr, 83% ee; entry 1). One of the most striking observations is that a seemingly minor adjustment in the structure of the base, i.e., replacing the piperidine unit with pyrrolidine as in (R)-2TA, resulted in reversal of the enantiomeric preference, giving the opposite enantiomer ent-3 as the major product with 84% ee and 17:1 dr (entry 2). The previously reported alkylation reaction showed the same sense of enantiofacial preferences for these two bases.6 The azepine base also displayed a reversal of selectivity, although at a lower level (46% ee; entry 3). Introduction of the gem-dimethyl substitution on the propylene bridge in (R)-4TA gave a diminished yield of the product with lower diastereoselectivity (entry 4). Replacing the phenyl group with 2-naphthyl ((R)-5TA) had a minor impact on the course of the reaction.
Table 1. Chiral Lithium Amides for the Enantioselective Conjugate Addition of PhCH2CO2H to Esters 1 and 2a.
The ee values were measured using chiral HPLC analysis of methyl esters after derivatization with Me3SiCHN2; all results are corrected to bases with the R configuration.
Addition at −90 °C.
26 mmol scale, 5.97 g of 3; (R)-1TA was recovered in 99% yield.
Variations in the ester group revealed that the enantioselectivity was higher for methyl cinnamate (1, R = Me) than for ethyl and tert-butyl cinnamate. Under optimal conditions, the addition product was formed in 88% yield with 93% ee as a single diastereomer (Table 1, entry 8). A larger-scale experiment was performed at −78 °C on 26 mmol scale, affording 5.97 g of 3 (R = CH3) in 78% yield with 90% ee (entry 13). The product was isolated as a single diastereomer by recrystallization, and the reagent (R)-1TA was recovered by extraction in 99% yield.
Similar selectivity trends were observed with methyl (E)-3-cyclohexylacrylate (2) (Table 1, entries 9–12). In this case, the pyrrolidine base (R)-2TA proved to be significantly more effective, giving adduct 4 in 74% isolated yield with 96% ee as a single anti diastereomer (cf. entries 9 and 10). The absolute and relative configurations of addition products 3 and 4 were established using X-ray crystallographic analysis as well as correlation with known compounds.15
Guided by the initial experiments, we selected the piperidine base (R)-1TA and the pyrrolidine base (R)-2TA for further investigations. The scope of Michael acceptors was examined first (Table 2). In all of the examples, ∼1:1 stoichiometry between the Michael donor and acceptor was applied. Many aryl and heteroaryl substituents were suitable for the reaction using the piperidine base (R)-1TA. The diastereoselectivity was uniformly high for all of the α,β-unsaturated esters (>20:1), and the enantioselectivity was in the range of 69–97% ee, with the highest value observed for products 5a and 5f bearing 2-methoxyphenyl and 3-indolyl groups, respectively. The 3-nitroaryl substituent (5c) afforded a much lower yield of the addition product, likely because of the high reactivity of the substrate; however, high enantioselectivity was maintained. In our screening efforts, we placed an emphasis on heteroaryl substituents because of their relevance to medicinal chemistry (5d–i). Although the enantioselectivity was somewhat reduced, it was in the practical range of 70–97% ee for a variety of groups, including N-methyl-2-pyrrolyl (5d, 86% ee), 2-benzofuryl (5e, 73% ee), N-Boc-3-indolyl (5f, 97% ee), 2-furyl (5g, 85% ee), 3,5-dimethyl-4-isoxazolyl (5h, 86% ee), and 2-thiazolyl (5i), which gave the lowest ee of 69%. Notably, the potentially sensitive N-Boc group in 5f was compatible with the reaction conditions; in fact, this was one of the best-performing reactions.
Table 2. Scope of α,β-Unsaturated Estersc.
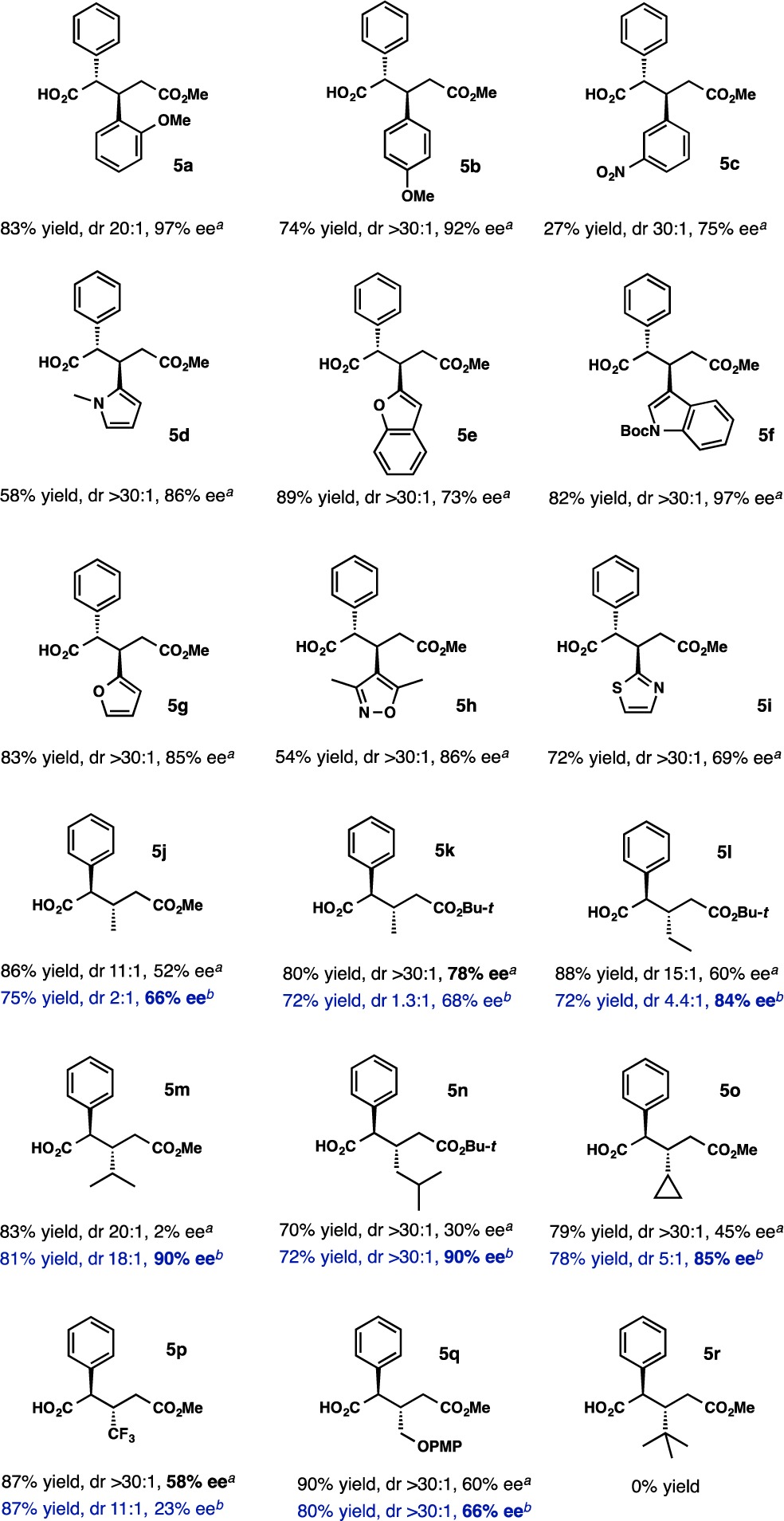
(R)-1TA as the base.
(R)-2TA as the base.
All results are normalized to bases with the R configuration; ee values were determined by HPLC analyses of methyl esters.
On the basis of the preliminary studies, aliphatic α,β-unsaturated esters were studied with both chiral bases, (R)-1TA and (R)-2TA (Table 2). With crotonates (5j, 5k), we found that the best enantioselectivity of 78% was obtained with the tert-butyl ester using the original piperidine base (R)-1TA. For all of the other substrates except for methyl 4,4,4-trifluorocrotonate (5p, 58% ee), the enantioselectivity was superior with (R)-2TA. Ethyl (5l), isopropyl (5m), isobutyl (5n), cyclopropyl (5o), and (4-methoxyphenyloxy)methyl (5q) substituents were effectively introduced. The enantioselectivities were in the range of 85–90%, except for 5q, which was isolated with 66% ee. Under the conditions studied, no reactivity was observed with sterically demanding methyl (E)-4,4-dimethylpent-2-enoate (cf. 5r in Table 2).
The scope of carboxylic acids investigated as Michael donors with the piperidine base (R)-1TA is summarized in Table 3. Initially, variation of the position of the chlorine on the phenyl ring revealed that ortho substitution resulted in a drastic reduction of enantioselectivity. While 4- and 3-chlorophenylacetic acid afforded addition products with 93 and 86% ee, respectively, 33% ee was observed with 2-chlorophenylacetic acid. The selectivity could be substantially improved to 70% ee by using the alternative base (R)-2TA (6c). Again, the enantiomeric product was favored. Similar trends were observed for 2- and 3-naphthylacetic acid (6e, 6f). The former afforded the addition product with 87% ee, while the selectivity with the latter was 33% ee, which could be enhanced to −79% ee using (R)-2TA. 2-(Benzo[d]dioxol-5-yl)acetic acid afforded 6d with 89% ee.
Table 3. Scope of Carboxylic Acidsc.
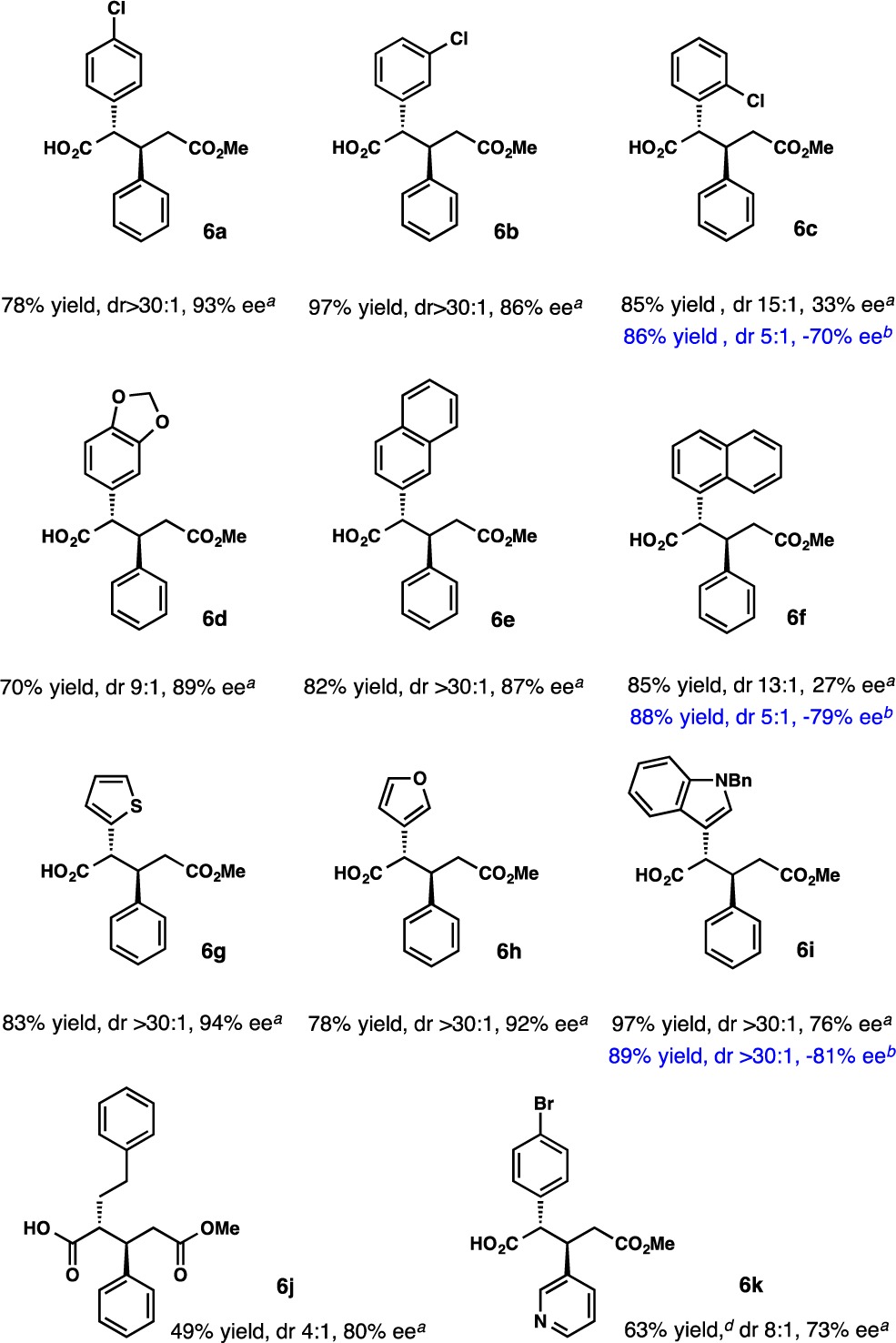
(R)-1TA as the base.
(R)-2TA as the base.
All results are normalized to bases with the R configuration; ee values were determined by HPLC analyses of methyl esters.
For the dimethyl ester.
The heteroarylacetic acids 2-thiophene- and 3-furanacetic acid afforded 6g and 6h with high enantioselectivity (Table 3). Although N-Boc-3-indoleacetic acid proved to be a poor substrate in the addition reaction, N-benzyl-3-indoleacetic acid afforded 6i in a very high yield with 76% ee. Improved selectivity was observed with (R)-2TA as the base, which again afforded the product with the opposite sense of enantioinduction.
Although a systematic study of aliphatic carboxylic acids as Michael donors was beyond the scope of this contribution, addition of 4-phenylbutyric acid was an important initial advance in this direction (6j; Table 3). In contrast to our previous work on alkylation reactions, a rather high ee of 80% (dr 4:1) was observed with (R)-1TA as the base. Studies of enediolates from aliphatic acids continue and will be reported in due course. Variation in both coupling partners using more functionalized substrates is tolerated (6k).
The versatility of the Michael addition methodology is illustrated by the applications depicted in Scheme 2. The first application capitalized on the reactivity of the initially formed enolate by exploiting it in a secondary alkylation reaction with iodomethane in one pot. This process enabled the formation of three consecutive tertiary stereocenters with excellent stereocontrol in 74% yield (Scheme 2a). The free carboxyl group is an exceptionally convenient surrogate for an amino group via the Curtius rearrangement transform. In the event, treatment of acid 7 with diphenylphosphoryl azide followed by benzyl alcohol delivered γ-amino acid derivative 8 in 53% unoptimized yield.
Scheme 2.
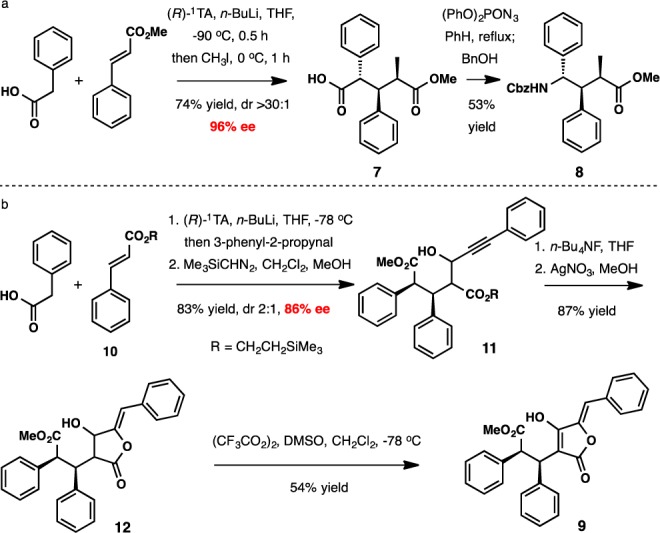
The second application was the enantioselective synthesis of the purported structure of pulveraven B (9) (Scheme 2b), reported as a constituent of the edible mushroom Pulveroboletus ravenelii in 2003.16 It displayed selective inhibition of carcinogen-induced pre-neoplastic lesion formation in mouse mammary organ culture with IC50 = 0.8 μM. The potency was reduced 10-fold for its epimer pulveraven A.
In the synthesis described herein, the initial Michael adduct was subjected in situ to aldol coupling with 3-phenyl-2-propynal, affording a 2:1 mixture of aldol products 11 with 86% ee for both diastereomers (83% yield). After cleavage of the trimethylsilylethyl ester with n-Bu4NF, a Ag-catalyzed cyclization afforded γ-lactone 12.17 Oxidation delivered the tetronic acid with a structure proposed for pulveraven B. However, the optical rotation and NMR spectral data did not match those reported for the natural product.16
In conclusion, we have developed a method for direct enantio- and diastereoselective conjugate addition of carboxylic acids to α,β-unsaturated esters. The stereoselectivity is imparted by a chiral lithium amide–enediolate aggregate formed from chiral C2-symmetric Koga-type tetramines. The study revealed intriguing and unexpected patterns of stereoselectivty that are the subject of current mechanistic investigations.18 Multiple selective bond formations were illustrated (1) by a highly stereoselective one-pot alkylation with iodomethane and (2) by an aldol coupling within the context of the enantioselective total synthesis of pulveraven B, which revealed that its structure appears to be misassigned.
Acknowledgments
We thank Drs. Beaver and Bio of Amgen for the donation of >3 kg of tetramine (R)-1TA. This work was supported by NIH (NIGMS, R01-077379). J.A.E. was supported by the NSF Graduate Research Fellowship Program (DGE 1144085). J.J.J. was supported by the UCSB Dean’s Graduate Mentor Fellowship.
Supporting Information Available
Procedures and characterization data. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Examples of conjugate addition of lithium enolates in the presence of chiral additives:; a Yamamoto Y.; Suzuki H.; Yasuda Y.; Iida A.; Tomioka K. Tetrahedron Lett. 2008, 49, 4582. [Google Scholar]; b Yasuda K.; Shindo M.; Koga K. Tetrahedron Lett. 1996, 37, 6343. [Google Scholar]; Examples of mixed aggregates of lithium ester enolates:; c Juaristi E.; Beck A. K.; Hansen J.; Matt T.; Mukhopadhyay T.; Simson M.; Seebach D. Synthesis 1993, 1271. [Google Scholar]; d Duguet N.; Harrison-Marchand A.; Maddaluno J.; Tomioka K. Org. Lett. 2006, 8, 5745. [DOI] [PubMed] [Google Scholar]; e Lecachey B.; Duguet N.; Oulyadi H.; Fressigne C.; Harrison-Marchand A.; Yamamoto Y.; Tomioka K.; Maddaluno J. Org. Lett. 2009, 11, 1907. [DOI] [PubMed] [Google Scholar]
- Selected reviews of asymmetric conjugate additions:; a Mukherjee S.; Yang J. W.; Hoffmann S.; List B. Chem. Rev. 2007, 107, 5471. [DOI] [PubMed] [Google Scholar]; b Christoffers J.; Koripelly G.; Rosiak A.; Rössle M. Synthesis 2007, 1279. [Google Scholar]
- Early studies with chiral auxiliaries bound to the Michael donor or acceptor and applications in synthesis:; a Oppolzer W.; Pitteloud R.; Bernardinelli G.; Baettig K. Tetrahedron Lett. 1983, 24, 4975. [Google Scholar]; b Oppolzer W. Pure Appl. Chem. 1990, 62, 1241. [Google Scholar]; c Corey E. J.; Houpis I. N. J. Am. Chem. Soc. 1990, 112, 8997. [Google Scholar]; d Taber D. F.; Mack J. F.; Rheingold A. L.; Geib S. J. J. Org. Chem. 1989, 54, 3831. [Google Scholar]; e Jang D. P.; Chang J. W.; Uang B. J. Org. Lett. 2001, 3, 983. [PubMed] [Google Scholar]; f Holton R. A.; Williams A. D.; Kennedy R. M. J. Org. Chem. 1986, 51, 5480. [Google Scholar]; g Krafft M. E.; Kennedy R. M.; Holton R. A. Tetrahedron Lett. 1986, 27, 2087. [Google Scholar]; Also see:; h Jo S. Ph.D. Thesis, Florida State University, Tallahassee, FL, May 2008. [Google Scholar]
- a Enders D.; Teschner P.; Gröbner R.; Raabe G. Synthesis 1999, 237. [Google Scholar]; b Evans D. A.; Bilodeau M. T.; Somers T. C.; Clardy J.; Cherry D.; Kato Y. J. Org. Chem. 1991, 56, 5750. [Google Scholar]; c Smitrovich J. H.; Boice G. N.; Qu C.; DiMichele L.; Nelson T. D.; Huffman M. A.; Murry J.; McNamara J.; Reider P. J. Org. Lett. 2002, 4, 1963. [DOI] [PubMed] [Google Scholar]; d Smitrovich J. H.; DiMichele L.; Qu C.; Boice G. N.; Nelson T. D.; Huffman M. A.; Murry J. J. Org. Chem. 2004, 69, 1903. [DOI] [PubMed] [Google Scholar]; e Barluenga J.; Monserrat J. M.; Florez J.; Garcia-Granda S.; Martin E. Chem.—Eur. J. 1995, 1, 236. [Google Scholar]; f D’Angelo J.; Maddaluno J. J. Am. Chem. Soc. 1986, 108, 8112. [Google Scholar]
- a Evans D. A.; Helmchen G.; Rüping M. In Asymmetric Synthesis—The Essentials; Christmann M., Bräse S., Eds.; Wiley-VCH: Weinheim, 2007; p 3. [Google Scholar]; b Key Chiral Auxiliary Applications; Roose G., Ed.; Academic Press: Boston, 2014. [Google Scholar]
- Stivala C. E.; Zakarian A. J. Am. Chem. Soc. 2011, 133, 11936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Y.; Stivala C. E.; Wright A. M.; Hayton T.; Liang J.; Keresztes I.; Lobkovsky E.; Collum D. B.; Zakarian A. J. Am. Chem. Soc. 2013, 135, 16853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Su C.; Guang J.; Li W.; Wu K.; Hopson R.; Williard P. G. J. Am. Chem. Soc. 2014, 136, 11735. [DOI] [PubMed] [Google Scholar]; b Su C.; Hopson R.; Williard P. G. J. Am. Chem. Soc. 2014, 136, 3246. [DOI] [PubMed] [Google Scholar]; c Su C.; Hopson R.; Williard P. G. J. Am. Chem. Soc. 2013, 135, 14367. [DOI] [PubMed] [Google Scholar]; d Su C.; Hopson R.; Williard P. G. J. Org. Chem. 2013, 78, 7288. [DOI] [PubMed] [Google Scholar]; e Barozzino-Consiglio G.; Rouen M.; Oulyadi H.; Harrison-Marchand A.; Maddaluno J. Dalton Trans. 2014, 43, 14219. [DOI] [PubMed] [Google Scholar]
- Recent reviews of asymmetric synthesis using chiral lithium amides:; a Simpkins N. S.; Weller M. D. Org. React. 2013, 79, 317. [Google Scholar]; b Harrison-Marchand A.; Maddaluno J.. Advances in the Chemistry of Chiral Lithium Amides. In Lithium Compounds in Organic Synthesis; Luisi R., Capriati V., Eds.; Wiley-VCH: Weinheim, 2014; p 463. [Google Scholar]
- Recent applications of chiral lithium amides in synthesis:; a He C.; Zhu C.; Wang B.; Ding H. Chem.—Eur. J. 2014, 20, 15053. [DOI] [PubMed] [Google Scholar]; b He C.; Zhu C.; Dai Z.; Tseng C.-C.; Ding H. Angew. Chem., Int. Ed. 2013, 52, 13256. [DOI] [PubMed] [Google Scholar]; c Araoz R.; Servent D.; Molgo J.; Iorga B.; Fruchart-Gaillard C.; Benoit E.; Gu Z.; Stivala C. E.; Zakarian A. J. Am. Chem. Soc. 2011, 133, 10499. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Stivala C. E.; Zakarian A. J. Am. Chem. Soc. 2008, 130, 3774. [DOI] [PubMed] [Google Scholar]; e Stivala C. E.; Gu Z.; Smith L. L.; Zakarian A. Org. Lett. 2012, 14, 804. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Alliot J.; Gravel E.; Pillon F.; Buisson D.-A.; Nicolas M.; Doris E. Chem. Commun. 2012, 48, 8111. [DOI] [PubMed] [Google Scholar]
- Alternative catalytic Michael addition of carboxylic acids via mixed anhydrides:Belmessieri D.; Morrill L. C.; Simal C.; Slawin A. M. Z.; Smith A. D. J. Am. Chem. Soc. 2011, 133, 2714. [DOI] [PubMed] [Google Scholar]
- Diamines such as compound i gave
low yields of the conjugate addition products, presumably because
of competitive oligomerization among other side reactions.
- No lithium amide addition to the Michael acceptor was observed. The reaction using lithium diisopropylamide as the base gave a 1.6:1 mixture of diastereomers.
- a Murakata M.; Nakajima M.; Koga K. J. Chem. Soc., Chem. Commun. 1990, 1657. [Google Scholar]; b Imai M.; Hagihara A.; Kawasaki H.; Manabe K.; Koga K. J. Am. Chem. Soc. 1994, 116, 8829. [Google Scholar]; For a kilogram-scale preparation, see:; c Frizzle M. J.; Caille S.; Marshall T. L.; McRae K.; Nadeau K.; Guo G.; Wu S.; Martinelli M. J.; Moniz G. A. Org. Process Res. Dev. 2007, 11, 215. [Google Scholar]; d Frizzle M. J.; Nani R. R.; Martinelli M. J.; Moniz G. A. Tetrahedron Lett. 2011, 52, 5613. [Google Scholar]
- See the Supporting Information for details. Also see ref (4d).
- a Duncan C. J. G.; Cuendet M.; Fronczek F. R.; Pezzuto J. M.; Mehta R. G.; Hamann M. T.; Ross S. A. J. Nat. Prod. 2003, 66, 103. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Quang D. N.; Hashimoto T.; Nukada M.; Yamamoto I.; Tanaka M.; Takaoka S.; Asakawa Y. Chem. Pharm. Bull. 2003, 51, 330. [DOI] [PubMed] [Google Scholar]
- a Jong T. T.; Williard P. G.; Porwoll J. P. J. Org. Chem. 1984, 49, 735. [Google Scholar]; b Tamura S.; Tonokawa M.; Murakami N. Tetrahedron Lett. 2010, 51, 3134. [Google Scholar]
- Stereochemical models for Michael addition of Li enolates:; a Oare D. A.; Heathcock C. J. Org. Chem. 1990, 55, 157. [Google Scholar]; b Kwan E. E.; Evans D. A. Org. Lett. 2010, 12, 5124. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Kwan E. E.; Scheerer J. R.; Evans D. A. J. Org. Chem. 2013, 78, 175. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.