

Abstract
Genomic clustering of genes in a pathway is commonly found in prokaryotes due to transcriptional operons, but these are not present in most eukaryotes. Yet, there might be clustering to a lesser extent of pathway members in eukaryotic genomes, that assist coregulation of a set of functionally cooperating genes. We analyzed five sequenced eukaryotic genomes for clustering of genes assigned to the same pathway in the KEGG database. Between 98% and 30% of the analyzed pathways in a genome were found to exhibit significantly higher clustering levels than expected by chance. In descending order by the level of clustering, the genomes studied were Saccharomyces cerevisiae, Homo sapiens, Caenorhabditis elegans, Arabidopsis thaliana, and Drosophila melanogaster. Surprisingly, there is not much agreement between genomes in terms of which pathways are most clustered. Only seven of 69 pathways found in all species were significantly clustered in all five of them. This species-specific pattern of pathway clustering may reflect adaptations or evolutionary events unique to a particular lineage. We note that although operons are common in C. elegans, only 58% of the pathways showed significant clustering, which is less than in human. Virtually all pathways in S. cerevisiae showed significant clustering.
Cotranscription of genes in operons, which is the norm in prokaryotes, has not been observed in most eukaryotes. The known exceptions are nematodes and trypanosomes, but these operons are different from prokaryotic operons in terms of both mechanism and gene content (Blumenthal 1998). Genes in prokaryotic operons are frequently found to be functionally related and involved in the same pathway (e.g., Lawrence 1997; Overbeek et al. 1999).
There are however numerous examples in prokaryotes of functionally related nonhomologous genes that are found in close proximity in the genome even when not part of an operon. Overbeek et al. (1999) identified clusters that are conserved between bacterial genomes of functionally related genes. Kolesov et al. (2001) developed a method called SNAP (similarity-neighborhood approach) that uses orthology relations between genomes together with neighbor relations within a genome to infer cycles of functionally related genes. A correlation between gene proximity and function was also observed by Yanai et al. (2002), who discovered that pairs of genes that are adjacent in multiple genomes are often functionally related, also in genomes where they are not adjacent.
Lathe et al. (2000) determined that certain pairs of genes were found adjacent to each other in a broad range of bacterial species. Although rearrangements of gene order occurs regularly in bacteria, gene neighborhoods tend to be somewhat conserved. A set of functionally related genes maintained in close proximity in the genome was termed ‘uber-operon.’ Ribosomal genes in bacteria were found to be organized in uber-operons. Each ribosomal gene had other ribosomal genes as neighbors in all 15 genomes studied, although the complement of genes found in the uber-operons in each genome were not necessarily the same as those found in other genomes. Two other functionally related groups of genes were found in uber-operons as well. Lathe et al. (2000) showed that uber-operons can be used to correctly predict function of ‘hypothetical’ proteins.
Operons that have been detected in eukaryotes differ from those in prokaryotes in that eukaryotic polycistronic mRNAs are not translatable directly as in prokaryotes (Kozak 1999). Polycistronic mRNAs have been detected in Trypanosomes (Johnson et al. 1987) and C. elegans (Spieth et al. 1993) but are processed into monocistronic mRNA before translation occurs. Zorio et al. (1994) estimated that about 25% of C. elegans genes are organized in operons. They have observed trans-splicing to two leader sequences in the C. elegans genome. Blumenthal (1998) found that genes in operons are frequently due to tandem duplication and are similar at the sequence level as well as being involved in similar functions. However, genes have also been found in C. elegans operons that are not similar at the sequence level but are known to be functionally related (Blumenthal 1998). Although polycistronic transcription units have yet to be found in yeast, Zhang and Smith (1998) found functionally related and adjacent genes. Such gene pairs are generally controlled by the same promoter region, located between the two genes on opposite strands. In addition, Kruglyak and Tang (2000) ascertained that there is a much higher correlation of expression patterns for adjacent genes in yeast than for randomly selected genes.
It thus seems that in most eukaryotes, the transcription factor machinery is sufficient for ensuring coregulation, and that colocalization in the genome is not a general requirement. Consequently, there should be no selection for lining up coregulated genes directly next to each other in the genome. However, there could exist some degree of selection for keeping coregulated genes in the same region of a chromosome, for instance to make them available to transcription more efficiently as a group. This is supported by the fact that coregulated human genes are often linked functionally in “synexpression” groups (Niehrs and Pollet 1999). In addition, genes which are coexpressed may be found clustered in eukaryotes. Highly expressed genes in humans have been found to be colocalized in the genome (Caron et al. 2001). Spellman and Rubin (2002) determined that about 20% of Drosophilia genes fall into clusters of coexpressed groups.
We wanted to use the currently available eukaryotic genome sequences to investigate the levels of clustering within pathways. The research presented here is based on genes in metabolic pathways as defined in the Kyoto Encyclopedia for Genes and Genomes (KEGG), with missing enzymes filled in by homology. The species included in this study were: Homo sapiens, Caenorhabditis elegans, Drosophila melanogaster, Arabidopsis thaliana, and Saccharomyces cerevisiae. We analyzed which pathways are significantly clustered in each species, and whether the clustered pathways are the same in different species. The clustering level was measured by calculating the overall degree of gene colocalization compared to random. The ‘clusters’ are thus not necessarily compact, but may correspond to rather large regions with high concentrations of pathway members, although nonmembers may also be present in such regions. In addition we propose a procedure to delineate actual clusters of genes, but do not prove those here. The clustering statistics and other auxiliary information can be found at ftp://ftp.cgb.ki.se/pub/data/pathwayclusters.
RESULTS
The pathways used in this study are the metabolic pathways in KEGG that include genes from at least one of the five species investigated here. Pathways that were incomplete in a genome were “repaired” by searching for homologs of the missing genes known in other species, using a stringent cutoff. We collected data for 105 different pathways in total. The number of pathways with data in each individual genome ranged from 79–98 (see Table 1).
Table 1.
Pathways Analyzed and Percentage Showing Significant Clustering in Unmerged and Merged Data Sets
| Organism | # Pathways analyzed | # Genes | % Significant unmerged data | % Significant merged data | % In random data |
| H. sapiens | 98 | 975 | 78% | 65% | 11% |
| C. elegans | 86 | 516 | 74% | 58% | 11% |
| D. melanogaster | 85 | 484 | 50% | 30% | 12% |
| A. thaliana | 79 | 318 | 60% | 43% | 11% |
| S. cerevisiae | 89 | 682 | 100% | 98% | 10% |
The percent significant refers to pathways in which the score is more than 3* (3rd quartile − median) + median. The same analysis was carried out on randomized pathways where genes were picked randomly from all genes, using the merged data.
To determine whether genes in a pathway are found in closer proximity than expected by chance, we developed a formula for calculating a clustering score. Such a formula needs to fulfill a set of criteria: (1) The score should increase monotonously with increased proximity, (2) the score between genes on different chromosomes needs to be defined and to be the same as the average score between two randomly placed genes on an average chromosome, and (3) the score should be normalized for genome and pathway size to allow comparative studies. To estimate the significance of the observed average clustering score of a pathway in a genome, it was compared to the distribution of 200 iterations of placing the same number of genes on randomly picked known gene positions. Friedman and Hughes (2001) used a similar approach for determining whether observed gene patterns are expected by chance.
A potential source of overestimating the amount of clustering is the fact that genes frequently undergo tandem duplications. In our analysis, such tandem pairs would be assigned to the same pathway if the duplication was recent, yet would not necessarily reflect functional coupling. Both genes may have the same function, or one gene may not have any function (pseudogene). Such cases of recent tandem duplication can be omitted from the analysis by counting any stretch of adjacent genes with the same EC number and greater than 60% identity as one gene. We call this mode of analysis “merged”, and present both merged and unmerged results. The merged results should be seen as a conservative estimate, as some neighbors may be truly distinct genes in a pathway.
Table 1 shows the number of pathways found to be significantly clustered in each genome. We used three times the distance between the 3rd quartile and the median of the random cluster score distribution as the threshold for significance. This approach was used because it makes no assumptions about the shape of the distribution. As seen in Figures 1 and 2, a more stringent significance criterion would have produced essentially the same result, because most significantly clustered pathways are far above the cutoff. To verify that the result is not expected by chance, we randomized the gene content of each pathway such that the gene number was kept constant but the gene locations were picked randomly from all genes in the organism. Between 10% and 12% of the randomized pathways reached our significance cutoff, whereas the observed fractions of significantly clustered pathways ranged between 30% and 98% (both using merged data).
Figure 1.

Clustering results for four pathways, illustrating different levels in clustering score observed among different genomes, pathways, and merged/unmerged data. Pathway 00361 is significantly clustered in all species, whereas the other pathways show varying degrees of clustering. In S. cerevisiae, all but two pathways showed significant clustering (using merged data). Note that the scale of the clustering score is logarithmic.
Figure 2.

Distribution of pathway clustering scores (using merged data). The x-axis represents how much higher from the median the score is in terms of difference between the median and the 3rd quartile. Also shown is the score distribution from randomly chosen pathways (from all organisms) in which the genes were picked randomly from all genes in the particular organism.
The number of pathways with significant clustering is consistently lower using merged data. In yeast there is only a 2% difference, whereas in human, worm, and Arabidopsis the drop is more substantial, yet no more than about one-quarter of the original number. In fly we see the largest effect, with 40% of the clustered pathways dropping to an insignificant clustering score in the merged data set. Differences were found between pathways as well. Data from a few selected pathways are shown in detail in Figure 1. The distribution of clustering scores expressed as 3rd quartile-median ranges (3QMRs) for all pathways and genomes is shown in Figure 2. For yeast, human, and worm, the bulk of the pathways fall in very significant regions.
Plots of actual gene locations in a pathway are shown in Figure 3, illustrating the difference between two pathways with the same number of genes, one with significant clustering and one without. In cases like this, it can be useful to define loose clusters of genes as groups that form a string with genomic distances less than a threshold value. We calculated such thresholds individually for each genome by counting distances for 1000 pairs of randomly placed genes on the same chromosome. These pairwise distances appeared to approximate a normal distribution, and we determined the average and standard deviation for a random pair of genes in each genome (Table 2). To define a loose cluster of genes, we used the average random distance minus three standard deviations as a threshold (99% significance level). Such loose clusters are shown for two pathways in Figure 3. It is clear that the clustering is a relatively diffuse large-scale phenomenon, and not immediately striking to the eye, but after closer inspection the significantly clustered pathway does contain many more nearby genes than the insignificant one. The degree of clustering measured as fraction of genes in loose clusters in significantly and insignificantly clustered pathways for the five species was: H. sapiens: 50% / 18%; D. melanogaster: 68% / 45%; C. elegans: 74% / 44%; A. thaliana: 52% / 44%; S. cerevisiae: 37% / 0%.
Figure 3.

Distribution of genes in pathways with and without significant clustering in A. thaliana. Pathway 00360 has a highly significant clustering score, whereas pathway 03020 does not. Merged data were used. The boxes represent putative loose gene clusters.
Table 2.
Average Distance of Random Pairs of Genes on a Chromosome
| Species | Average distance | SD |
| H. sapiens | 62.79 Mb | 14.1 Mb |
| C. elegans | 5.58 Mb | 0.89 Mb |
| D. melanogaster | 7.76 Mb | 1.39 Mb |
| A. thaliana | 8.81 Mb | 1.92 Mb |
| S. cerevisiae | 0.33 Mb | 0.077 Mb |
This information may be used to determine if genes deviate enough from the random distribution to be considered clustered. We used the average distance minus three standard deviations as significance cutoff for defining genes to be included in loose clusters in Figure 3.
Recent tandem duplications sometimes gave rise to extremely high clustering scores. In most cases when a pathway had an unmerged cluster score higher than 1000, this number dropped substantially after merging, although not necessarily to an insignificant level. Hence recent tandem duplication can only be partially responsible for the high levels of clustering observed.
To what extent is clustering of a pathway conserved between different genomes? Although pathway data are inherently incomplete, we have enough coverage to analyze crude patterns of shared clustering. Table 3 shows the number of pathways shared among different genomes, divided into categories of the total number of genomes containing the pathway. Of the 69 pathways found in all five genomes, only seven were significantly clustered in all of them. (Using unmerged data, 18 of them were clustered.) A complete list of pathways including which species we have data for, number of genes in the pathway, and clustering scores can be found on the Web site table. Trusting the merged data more, we conclude that universally clustered pathways are relatively rare. The seven universally clustered pathways are: glycolysis, aminoacyl-tRNA biosynthesis, ATP synthase, DNA polymerase, hexachlorocyclohexane degradation, cyanoamino acid metabolism, and photosynthesis (which in KEGG includes ATP synthesis, explaining why it can be found in all organisms).
Table 3.
Distribution of Shared Pathway Clustering Between Genomes
| 5 | 4 | 3 | 2 | 1 | 0 | |
| 5 | 7 (18) | 11 (25) | 35 (21) | 11 (5) | 5 (0) | 0 (0) |
| 4 | 2 (2) | 2 (4) | 6 (6) | 3 (1) | 0 (0) | |
| 3 | 0 (0) | 3 (4) | 1 (0) | 0 (0) | ||
| 2 | 1 (1) | 5 (7) | 4 (2) | |||
| 1 | 6 (6) | 2 (2) |
Each row indicates the number of species for which pathway information could be collected. The columns indicate the number of species that show significant clustering of the pathway. Results for unmerged data sets are given within brackets. For example, there are 35 pathways that are significantly clustered (using merged data) in exactly three out of five genomes.
It may be expected that pathways with many genes exhibit higher clustering scores than pathways with few members. This should not have an effect when comparing to the randomized clustering scores, but could influence the relative comparison of pathways. A plot of gene number in the pathway versus its clustering score (Fig. 4) indicates that no clear correlation between them exists; there are pathways with high scores and few genes as well as pathways with low scores and many genes.
Figure 4.

Relationship between clustering score per gene and number of genes in the pathway (merged data).
DISCUSSION
Operons or clusters of genes which are linked functionally are found frequently in prokaryotes. Identification of operons and uber-operons has been documented in prokaryotes (Overbeek et al. 1999; Salgado et al. 2000; Kolesov et al. 2001; Tamames 2001). Salgado et al. (2000) found that they could predict genes which are in an operon by functional relation of closely placed genes in bacterial genomes and, in addition, found cases of genes which were not in an operon but were adjacent, transcribed in the same direction (a directon), and are functionally related. We have investigated genes involved in pathways in eukaryotes to determine whether similar observations can be detected here. In all genomes studied, we frequently found that genes in a pathway are in closer proximity than would be expected by chance. The genes that are ‘clustered’ are much more distant than those detected in prokaryotes; however there may still be an advantage for functionally related eukaryotic genes to be close in a genome even if separated by other genes. In some cases, clustering of functionally related genes appears to be due to recent tandem duplications. In a few cases, merging tandemly duplicated genes (with the same EC number) caused the clustering score to drop to insignificant levels, but generally similar clustering scores were found in both data sets. In other words, most genes in a pathway that are adjacent in the genome do not come from recent tandem duplications. Some of the observed clustering effect could stem from local duplications followed by functional divergence. However, given the generally high motility of genes in a genome, this effect can hardly explain the high levels of clustering observed; it seems instead that the clustering of pathway members must be a selected trait. In further support of this, there is no detectable correlation between sequence similarity and clustering score in our data (data not shown).
Lawrence (1999) proposed a selfish operon model, which suggests that genes are clustered in operons to benefit the genes and not the organism. In this regard, genes, especially in prokaryotes, may be transmitted by both vertical and horizontal transfer and remain as an intact functional unit. Glansdorff (1999) suggested that operons arose in thermophilic bacteria, where it is important for proteins to complex very quickly in order to become more stable, less toxic, or as a means of facilitating protein interactions. An argument against the selfish operon model is that operons are poorly conserved between different organisms, and perhaps operons are simply selected for to promote coregulation and efficient transcription and translation (Lathe et al. 2000).
Prokaryotic genomes tend to be small and do not need to be as tightly packed as eukaryotic chromosomes. The eukaryotic genome requires extensive compaction of DNA to fit into the nucleus. Packing is accomplished by wrapping the DNA around histones and other proteins. When genes are transcribed, a great deal of unpacking of regions of DNA is required. The region of DNA which is unfolded contains a number of genes, and keeping functionally related genes near, even if not adjacent, may ease the burden of unpacking of DNA in the cell for transcription. Evidence of such clusters of coexpressed genes have been detected in human, fly, and worm. These clusters contain 10–30 genes in fly and 2–5 in worm (Roy et al. 2002; Spellman and Rubin 2002).
Eukaryotic operons have been detected in various forms. In C. elegans, the splice leader for the second gene in polycistronic transcription units has only been detected in the genus Caenorhabditis and not in other closely related genera (Spieth et al. 1993). This may indicate that polycistronic transcription units are a relatively novel feature in this genome and may have been selected for in recent evolution. It does not appear to increase the amount of genomic clustering of functionally linked genes, as C. elegans pathways are not more clustered than in human.
Huynen et al. (2001) found that gene pairs are less conserved in eukaryotes than prokaryotes, where a gene pair is defined as two adjacent genes. In eukaryotes, conserved gene order was not found at a higher rate than in randomized genomes when looking solely at adjacent genes. Evidence obtained in the present study indicates that clustering of genes may not be limited to adjacent genes, and that gene order conservation studies should be expanded to include larger areas than the immediate neighborhood.
In the present study the question remains as to why clustering of genes in pathways may occur at a higher level in certain eukaryotes and less in others. Specifically, why is clustering observed at a very high level in S. cerevisiae and at a low level in D. melanogaster? A study by Ranz et al. (2001) demonstrated that the Drosophila genome shows a higher rate of chromosomal evolution than other eukaryotes. This may contribute to more dispersion of gene clusters than in other eukaryotes. Similar analysis of additional eukaryotic genomes as they become available may shed more light on questions raised by our research.
METHODS
Genomic Data
Collections of all known and predicted proteins for each species in this study were taken directly from the genome sequencing sites in February 2001 (Protein databases). The species included and databases from which information was collected are: H. sapiens (www.ensemble.org), C. elegans (www.wormbase.org), D. melanogaster (www.flybase.org), A. thaliana (www.arabidopsis.org), and S. cerevisiae (genome-www.stanford.edu/Saccharomyces). For all proteins in each genome, we stored the protein identifier, the genomic location, and the sequence. We also created separate files of gene names and genomic locations to facilitate search of genomic locations (Locations tables).
Pathway Data
Pathways were initially based on the metabolic pathways defined in the KEGG (http://www.genome.ad.jp/kegg/kegg/html) for each organism included in our analysis. Each step in a pathway is identified by an EC number, which in turn is linked to genes in the individual organisms. Because KEGG is not complete with all current genomic data, we searched for missing genes in each pathway. This was accomplished by using genes from each species known to be involved in each step of each pathway and by using BLAST (Altschul et al. 1990) to search for homologs in our collections of proteins from the other species involved in this study. BLAST results were run through MSPcrunch (Sonnhammer and Durbin 1994) with a cutoff of 60% Identity. The 60% cutoff was chosen after analyzing rates of false-positive and false-negative assignments of EC numbers at different levels of identity in BLAST matches. As shown in Figure 5, a cutoff of 60% removes nearly all matches (99.4%) between genes with different EC numbers across the genomes studied. Many matches between genes with the same EC number fall below this cutoff, but the bulk of these are distantly related genes that by chance have the same function. We did not use ortholog finding programs to identify enzymes in other species belonging to an EC group, as many enzymes which perform the same function in different species are not always detectable as orthologs. Sequences that were previously not included and had matches above the cutoff to a sequence in another species with the “missing” EC number were assigned this EC number. This EC number was kept associated with the sequence in further analyses of “repaired” pathways. The fractions of genes in a pathway that came from such repair assignments ranged from 20% to 46%.
Figure 5.

Comparison of percent identity in BLAST matches of enzymes with the same EC number to those with different EC numbers, across all organisms and pathways in this study. Less than 0.6% of all pairs with different EC numbers had an identity above 60%.
Identification of Genomic Locations
Genomic locations were retrieved simply by searching the Location tables for all genes in a pathway and appending the chromosome and base-pair position information to the gene name and EC number. The analysis following was then done in two different ways. Gene locations as they were found were used, and in addition, genes with the same EC and which are at least 60% identical which are located adjacently, presumably from tandem duplication, were merged and counted as a single gene. “Adjacent” was defined as a pair with no intervening genes on either strand. All analysis following was performed with both the merged and unmerged files.
Calculation of Pathway Clustering Score
The clustering score was determined by a pairwise analysis of all genes in each pathway. For each pair we first determined whether the genes were on the same chromosome. The score for pairs of genes on the same chromosome was calculated with the following equation:
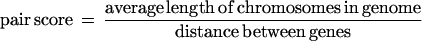 |
For genes on different chromosomes:
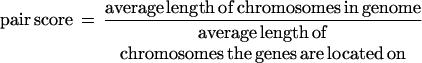 |
For genes on different chromosomes, the average length of the two chromosomes was taken as a substitute for a real distance that represents the maximum distance between genes on these chromosomes. We divided the average chromosome length by the pairwise distance to produce pair scores close to one for randomized locations and higher scores for close genes. The pathway clustering score is the sum of all pairwise scores divided by the number of genes in the pathway as normalization. Although there are alternative ways to calculate distances and clustering scores, the actual method should not matter when comparing to randomized data. We chose this method for reasons of convenience.
Comparison to Random
To determine whether the observed clustering score for each pathway could be found by chance, “randomized pathways” were generated for each pathway. Gene locations were randomly picked from all pathways in the Location tables, but matched the actual number of genes in each pathway and species. A clustering score was calculated with the identical method as the actual pathways. Two hundred iterations of random pathways were done to generate a distribution and determine the 1st and 3rd quartiles and the median.
Defining a Loose Cluster
We were able to group genes in each pathway into loose clusters by calculating a clustering cut-off distance. This was accomplished by extracting pairwise distances of neighboring genes on the same chromosome from the Location tables, and determining the average distance (see Table 2). All such distances generated approximately normal distributions, hence we used the average distance minus three standard deviations as a significant distance cut-off for loose gene clusters. A loose cluster was defined as a group of pathway members in which all member genes are closer than this cut-off. The corresponding genomic region may well contain other genes that are not considered members in the KEGG.
WEB SITE REFERENCES
www.genome.ad.jp/kegg/kegg/html; KEGG.
www.ensemble.org; ENSEMBL.
www.wormbase.org; WORMBASE.
www.flybase.org; FLYBASE.
www.arabidopsis.org; TAIR.
Acknowledgments
This work was supported by a grant from Pharmacia Corporation.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
E-MAIL erik.sonnhammer@cgb.ki.se; FAX +46 8 337983.
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.737703. Article published online before print in April 2003.
REFERENCES
- 1.Altschul S.F., Gish, W., Miller, W., Myers, E.W., and Lipman, D.J. 1990. Basic local alignment search tool. J. Mol. Biol. 215: 403-410. [DOI] [PubMed] [Google Scholar]
- 2.Blumenthal T. 1998. Gene clusters and polycistronic transcription in eukaryotes. BioEssays 20: 480-487. [DOI] [PubMed] [Google Scholar]
- 3.Caron H., van Schaik, B., van der Mee, M., Baas, F., Riggins, G., van Sluis, P., Hermus, M.C., van Asperen, R., Boon, K., Voute, P.A., et al. 2001. The human transcriptome map: Clustering of highly expressed genes in chromosomal domains. Science 291: 1289-1292. [DOI] [PubMed] [Google Scholar]
- 4.Friedman R. and Hughes, A.L. 2001. Gene duplication and the structure of eukaryotic genomes. Genome Res. 11: 373-381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Glansdorff N. 1999. On the origin of operons and their possible role in evolution toward thermophily. J. Mol. Evol. 49: 432-438. [DOI] [PubMed] [Google Scholar]
- 6.Huynen M.A., Snel, B., and Bork, P. 2001. Inversions and the dynamics of eukaryotic gene order. TRENDS in Genet. 17: 304-306. [DOI] [PubMed] [Google Scholar]
- 7.Johnson P.J., Kooter, J.M., and Borst, P. 1987. Inactivation of transcription by UV irradiation of T. brucei provides evidence for a multicistronic transcription unit including a VSG gene. Cell 51: 273-281. [DOI] [PubMed] [Google Scholar]
- 8.Kolesov G., Mewes, H.-W., and Frishman, D. 2001. SNAPing up functionally related genes based on context information: A colinearity-free approach. J. Mol. Biol. 311: 639-656. [DOI] [PubMed] [Google Scholar]
- 9.Kozak M. 1999. Initiation of translation in prokaryotes and eukaryotes. Gene 234: 187-208. [DOI] [PubMed] [Google Scholar]
- 10.Kruglyak S. and Tang, H. 2000. Regulation of adjacent yeast genes. Trends Genet. 16: 109-111. [DOI] [PubMed] [Google Scholar]
- 11.Lathe W.C., Snel, B., and Bork, P. 2000. Gene context conservation of a higher order than operons. Trends Biochem. Sci. 25: 474-479. [DOI] [PubMed] [Google Scholar]
- 12.Lawrence J.G. 1997. Selfish operons and speciation by gene transfer. Trends Microbiol. 5: 355-359. [DOI] [PubMed] [Google Scholar]
- 13.Lawrence J. 1999. Selfish operons: The evolutionary impact of gene clustering in prokaryotes and eukaryotes. Curr. Opin. Genet. Dev. 9: 642-648. [DOI] [PubMed] [Google Scholar]
- 14.Niehrs C. and Pollet, N. 1999. Synexpression groups in eukaryotes. Nature 402: 483-487. [DOI] [PubMed] [Google Scholar]
- 15.Overbeek R., Fonstein, M., D'Souza, M., Pusch, G.D., and Maltsev, N. 1999. The use of gene clusters to infer functional coupling. Proc. Natl. Acad. Sci. 96: 2896-2901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ranz J.M., Casals, F., and Ruiz, A. 2001. How malleable is the eukaryotic genome? Extreme rate of chromosomal rearrangement in the genus Drosophila. Genome Res. 11: 230-239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Roy P.J., Stuart, J.M., Lund, J., and Kim, S.K. 2002. Chromosomal clustering of muscle-expressed genes in Caenorhabditis elegans. Nature 418: 975-979. [DOI] [PubMed] [Google Scholar]
- 18.Salgado H., Moreno-Hagelsieb, G., Smith, T.F., and Collado-Vides, J. 2000. Operons in Escherichia coli: Genomic analyses and predictions. Proc. Natl. Acad. Sci. 97: 6652-6657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sonnhammer E.L. and Durbin, R. 1994. A workbench for large-scale sequence homology analysis. Comput. Appl. Biosci. 10: 301-307. [DOI] [PubMed] [Google Scholar]
- 20.Spellman P.T. and Rubin, G.M. 2002. Evidence for large domains of similarly expressed genes in the Drosophila genome. J. Biol. 1: 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Spieth J., Brooke, G., Kuesten, S., Lea, K., and Blumenthal, T. 1993. Operons in C. elegans: Polycistronic mRNA precursors are processed by trans-splicing of the SL2 to downstream coding regions. Cell 79: 521-532. [DOI] [PubMed] [Google Scholar]
- 22.Tamames J. 2001. Evolution of gene order conservation in prokaryotes. Genome Biol. 2: research0020.1-0020.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yanai I., Mellor, J.C., and DeLisis, C. 2002. Identifying functional links between genes using conserved chromosomal proximity. Trends Genet. 18: 176-179. [DOI] [PubMed] [Google Scholar]
- 24.Zhang X. and Smith, T.F. 1998. Yeast “Operons”. Microb. Comp. Genomics. 3: 133-140. [DOI] [PubMed] [Google Scholar]
- 25.Zorio D.A.R., Cheng, N.N., Blumenthal, T., and Spieth, J. 1994. Operons as a common form of chromosomal organization in C. elegans. Nature 372: 270-272. [DOI] [PubMed] [Google Scholar]