

Abstract
Tau hyperphosphorylation is a main cause of neuronal loss in Alzheimer's disease, which can be caused by many factors, including oxidative stress. The multifunctional protein p62, which exists in neurofibrillary tangles and causes aggregation of hyperphosphorylated tau, not only serves as a receptor in selective autophagy, but also regulates oxidative stress. However, whether p62 participates in oxidative stress-induced tau hyperphosphorylation remains unclear. In this study, we produced an Alzheimer's disease rat model by injecting β-amyloid protein into the hippocampus and β-galactose intraperitoneally. Hematoxylin-eosin staining was used for morphological analysis of brain tissue, and western blotting, immunohistochemistry and reverse transcription-PCR were employed to study p62 and autophagy related proteins, antioxidant defense system kelch-like ECH-associated protein 1-NF-E2-related factor 2 related proteins and hyperphosphorylated tau, respectively. The number of neurons in the brain decreased in Alzheimer's disease rats, and the autophagy related proteins Atg12-Atg5, microtubule-associated protein 1 light chain 3-phosphatidylethanolamine and Beclin1 increased significantly, while p62 expression reduced. Expression of kelch-like ECH-associated protein 1 increased, NF-E2-related factor 2 protein and the downstream gene products of glutamate cysteine ligase catalytic subunit and glutamate cysteine ligase modulatory subunit decreased, and hyperphosphorylated tau increased. These findings demonstrate that autophagy levels increased and p62 levels decreased in the brains of Alzheimer's disease rats. Moreover, the anti-oxidative capability of the NF-E2-related factor 2-antioxidant response element pathway was decreased, which may be the cause of tau hyperphosphorylation in Alzheimer's disease brain tissue and the subsequent structural and functional damage to neurons.
Keywords: Alzheimer's disease, autophagy, p62, NF-E2-related factor 2, tau hyperphosphorylation, neural regeneration
Abbreviation:
AD, Alzheimer's disease; NFT, neurofibrillary tangles; Keap1, kelch-like ECH-associated protein 1; Nrf2, NF-E2-related factor 2; Atg, autophagy related genes; LC3, microtubule-associated protein 1 light chain 3; GCLC, glutamate cysteine ligase catalytic subunit; GCLM, glutamate cysteine ligase modulatory subunit; ARE, antioxidant response element
INTRODUCTION
Relative to its high oxygen consumption, the antioxidant capacity of cerebral tissue is low. Given that neuronal cell membranes contain large amounts of unsaturated fatty acids, reactive oxygen species can cause neuronal degeneration when they accumulate[1]. Studies have confirmed that autophagy is activated when levels of reactive oxygen species increase in cells[2], and application of antioxidants such as N-acetyl-L-cysteine can reduce autophagy[2,3], which plays a vital role in maintaining neuronal homeostasis[4,5]. Autophagy is closely linked to oxidative stress, both of which contribute to damage of brain tissue in Alzheimer's disease (AD). p62 participates in selective autophagy as a receptor and helps to remove damaged proteins and organelles by mediating lysosomes to maintain the cell environment[6,7]. Neurofibrillary tangles (NFT) caused by aggregation of hyperphosphorylated tau is a main characteristic of AD[8,9]. Some studies have shown that p62 exists in NFT[10,11], which indicates that p62 levels may correlate with AD onset. p62 can also regulate oxidative stress and apoptosis[12]. p62 can influence the hydrolysis of Kelch-like ECH-associated protein 1 (Keap1) and affect its half-life in cells[13,14,15]. In vitro experiments have shown that increased expression of p62 can affect the function of Cul3-Rbx1-E3 ubiquitin ligases by isolating Keap1. However, when mutant p62 is expressed in cells, the levels of Keap1 are not affected[16]. Therefore, autophagy and p62 may influence the Keap1-NF-E2-related factor 2 (Nrf2) system and affect oxidative stress tolerance of brain tissue. Many factors can cause tau hyperphosphorylation in AD brain tissue[17]. For example, the activation of glycogen synthase kinase-3, cyclin-dependent kinase-5, and mitogen-activated protein kinases, or the suppression of protein phosphatase 2A and 2B can cause tau hyperphosphorylation.
Oxidative stress has been shown to also promote tau hyperphosphorylation[18]. Evidence exists that treatment of neuroblastoma cells with a low dose of buthionine sulfoximine to increase oxidative stress can induce the depletion of glutathione and increase the expression of hyperphosphorylated tau[19]. However, the reason for oxidative stress-induced tau hyperphosphorylation is unclear.
In this study, we used an AD rat model to monitor autophagy and the expression levels of p62. In addition, the role of the Keap1-Nrf2-ARE pathway and tau hyperphosphorylation, and morphological changes in brain tissue were investigated to monitor the pathogenesis of AD.
RESULTS
Quantitative analysis of experimental animals
Thirty-two male Wistar rats were randomly divided into three groups: control (n = 10; without any interference), saline group (n = 10; subjected to bilateral hippocampal injection and intraperitoneal injection of saline), and AD model group (n = 12; subjected to Aβ25-35 bilateral hippocampal injection and β-galactose intraperitoneal injection). Some rats did not survive past model preparation for unknown reasons. Therefore, a total of nine rats from the control group, eight rats from the saline group, and eight rats from the AD model group were included in the final analysis.
Morphological changes in the AD rat brain (Figure 1)
Figure 1.
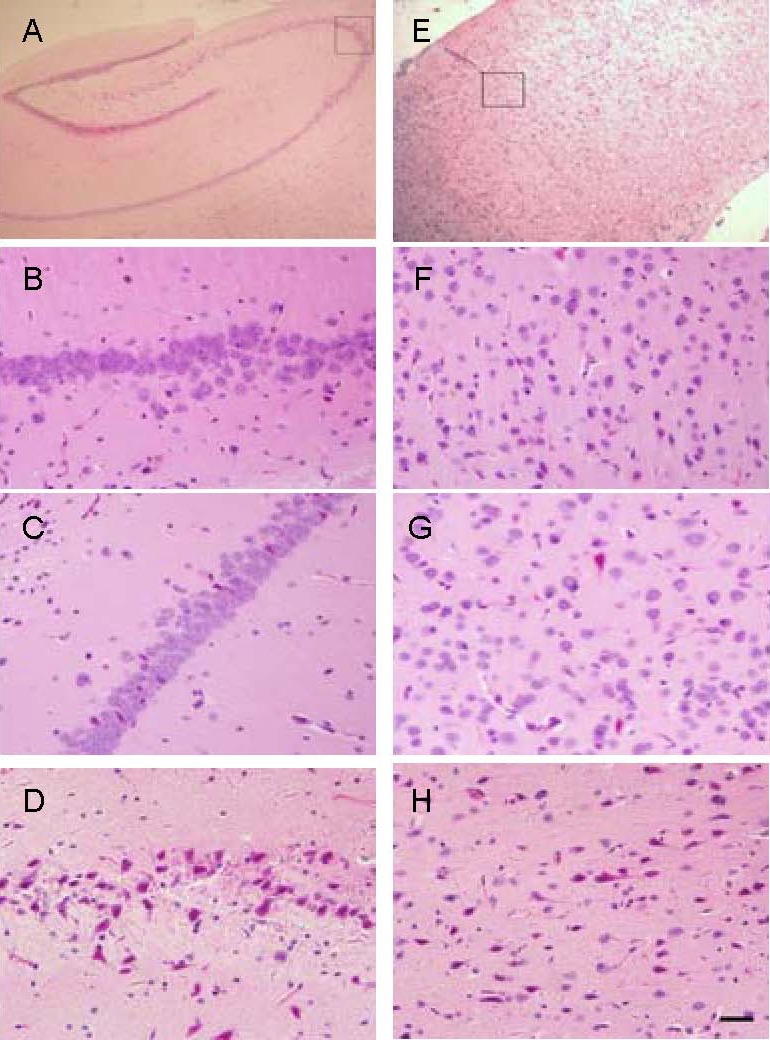
Morphological changes in the hippocampal CA3 area and forebrain (hematoxylin-eosin staining). Scale bar: 20 μm.
(A) Hippocampal CA3 area indicated in the square.
(B–D) The hippocampus in the control, saline, and Alzheimer's disease (AD) model groups, respectively. (B, C) Neurons were arranged closely, had a circular shape, nuclear membrane was clear in the control and saline groups; (D) the neurons had irregular shapes and the nuclear membrane was indistinct in the AD model group.
(E) Forebrain cortex indicated in the square.
(F–H) The forebrain cortex in the control, saline, and AD model groups, respectively. (F, G) Neurons had a circular shape, the nuclear membrane was clear in the control and saline groups; (H) neurons had irregular shapes and the nuclear membrane was indistinct in the AD model group.
Hematoxylin-eosin staining results (Figure 1) showed that the number of neurons in the hippocampal CA3 area in the AD model group (D) decreased, and that neurons had an irregular shape and showed signs of pyknosis and hyperchromasia.
The border between the nucleus and cytoplasm was indistinct, neuronal layering was reduced, the number of neurons in the forebrain cortex in the AD model group was reduced, and pyknosis and hyperchromasia were observed.
Expression of the autophagy-related proteins autophagy-related genes (Atg12)- Atg5, microtubule-associated protein 1 light chain 3 (LC3) and beclin1, and p62 protein in the AD rat cerebral cortex and hippocampus (Figure 2)
Figure 2.
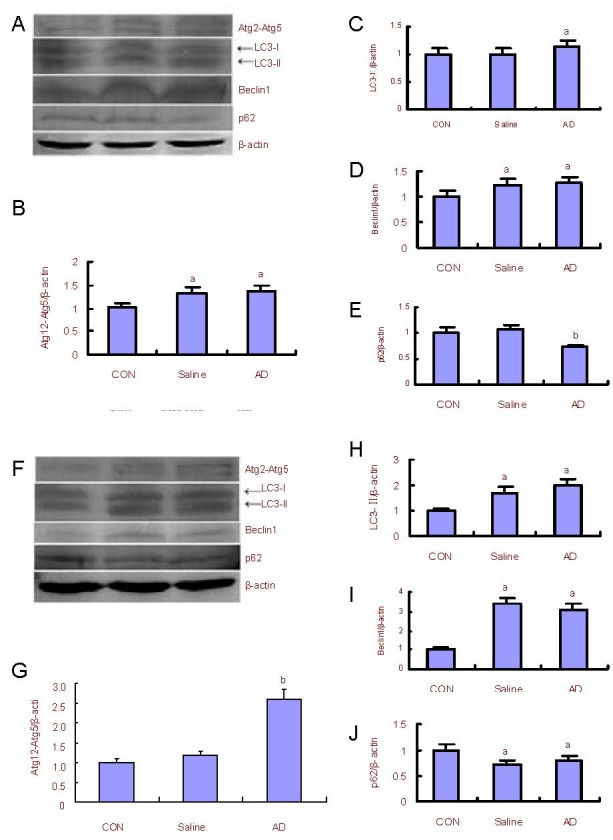
Changes in autophagy-associated proteins and p62 expression in the rat cerebral cortex and hippocampus.
Western blot analysis (A) and quantification of the expression of Atg12-Atg5, LC3-II, beclin1 and p62 in the cerebral cortex of the saline and AD model group as compared with the control (CON) group (adjusted as 1.0) (B–E).
Western blot analysis (F) and quantification of the expression of Atg12-Atg5, LC3-II, beclin1, and p62 in the hippocampus as compared with the control group (adjusted as 1.0) (G–J).
β-actin was used as an internal standard. The assay was performed three times and data were expressed as mean ± SD. aP < 0.05, bP < 0.01, vs. the control group (one-way analysis of variance followed by Dunnett's t-test).
Atg: Autophagy-related genes; LC3: microtubule-associated protein 1 light chain 3; AD: Alzheimer's disease.
Western blotting results (Figure 2) showed that compared with the control group, expression levels of the Atg 12-Atg5, LC3 and beclin1 significantly increased (P < 0.05 or P < 0.01), and p62 expression significantly decreased (P < 0.01 or P < 0.05) in the cerebral cortex and hippocampus of the AD rat model.
Expression of Keap1 and Nrf2 in the AD rat cerebral cortex and hippocampus
Immunohistochemical staining results (Figure 3) showed that the expression of Keap1 in the cerebral cortex and hippocampus of AD model rats increased significantly when compared with control rats (P < 0.05).
Figure 3.
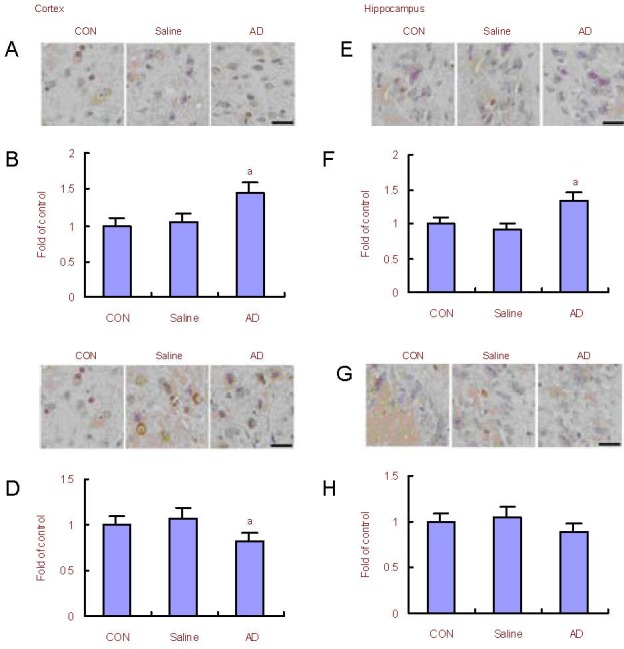
Changes in Keap1 and Nrf2 expression in the rat cerebral cortex and hippocampus.
(A, B) Immunohistochemical and quantitative analysis for the expression of Keap1 in the cerebral cortex of Alzheimer's disease (AD) model rats. The expression of Keap1 in AD model rats was increased compared with the control (CON) group.
(C, D) Immunohistochemical and quantitative analysis for the expression of Nrf2 in the cerebral cortex of AD model rats. The expression of Nrf2 in AD model rats increased compared with control rats.
(E, F) Immunohistochemical and quantitative analysis for the expression of Keap1 in the hippocampus of AD model rats. The expression of Keap1 in AD model rats increased compared with control rats.
(G, H) Immunohistochemical and quantitative analysis for the expression of Nrf2 in the hippocampus of AD model rats. The expression of Nrf2 in AD model rats remained unchanged compared with control rats.
Scale bars: 20 μm. aP < 0.05, vs. control rats (one-way analysis of variance followed by Dunnett's t-test). The assay was performed three times and data were expressed as mean ± SD.
Nrf2: NF-E2-related factor 2; Keap1: kelch-like ECH-associated protein 1.
The expression of Nrf2 in the cerebral cortex of AD model rats decreased significantly compared with control rats (P < 0.05). The expression of Nrf2 protein in the hippocampus remained unchanged (P > 0.05).
Expression of glutamate cysteine ligase catalytic subunit (GCLC) and glutamate cysteine ligase modulatory subunit (GCLM) mRNA in the AD rat cerebral cortex and hippocampus
Reverse transcription-PCR results (Figure 4) showed that mRNA transcription of GCLC and GCLM subunits reduced in the cerebral cortex (P < 0.05 or P < 0.01). There was no significant difference in mRNA transcription of GCLC and GCLM subunits in the hippocampus between the AD model and control groups.
Figure 4.
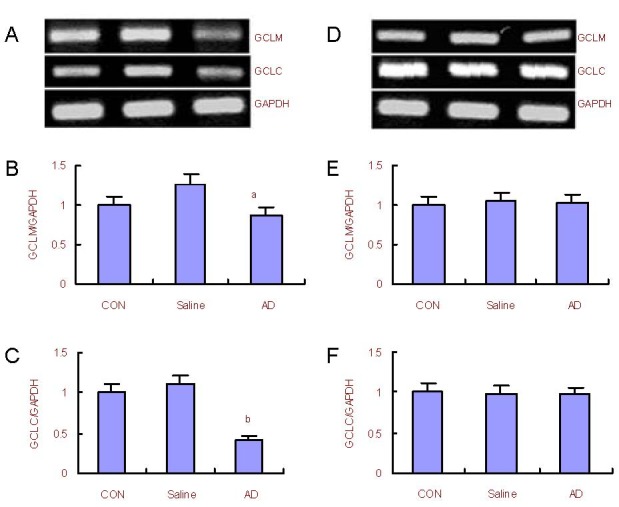
Changes in glutamate cysteine ligase catalytic subunit (GCLC) and glutamate cysteine ligase modulatory subunit (GCLM) mRNA expression in the rat cerebral cortex and hippocampus.
(A) RT-PCR analysis for mRNA expression of GCLM and GCLC in the cerebral cortex; (B, C) quantification of GCLM and GCLC mRNA expression in the cerebral cortex compared with the control (CON) group (adjusted as 1.0).
(D) Reverse transcription-PCR analysis for mRNA expression of GCLM and GCLC in the hippocampus; (E, F) quantification of GCLM and GCLC mRNA expression in the hippocampus compared with the control group (adjusted as 1.0).
GAPDH was used as an internal standard. aP < 0.05, bP < 0.01, vs. control group (one-way analysis of variance followed by Dunnett's t-test). The assay was performed three times and data were expressed as mean ± SD.
Expression of tau and hyperphosphorylated total tau in the AD rat cerebral cortex and hippocampus
Western blot analysis results (Figure 5) showed that the expression of total tau in the hippocampus and cerebral cortex of AD model rats was reduced (P < 0.05), and that expression of hyperphosphorylated tau increased in the hippocampus and cerebral cortex of AD model rats when compared with control rats (P < 0.05).
Figure 5.
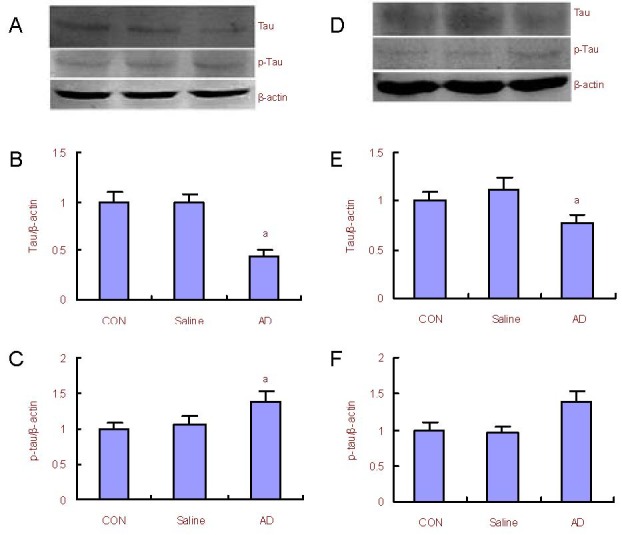
Changes in tau and p-tau protein expression in the rat cerebral cortex and hippocampus.
(A) Western blot analysis for the expression of total tau and p-tau in the cerebral cortex; (B, C) quantification of tau and p-tau protein expression in the cerebral cortex compared with the control (CON) group (adjusted as 1.0).
(D) Western blot analysis for the expression of total tau and p-tau protein in the hippocampus; (E, F) quantification of tau and p-tau protein expression in the hippocampus compared with the control group (adjusted as 1.0).
β-actin was used as an internal standard. aP < 0.05, vs. the control group (one-way analysis of variance followed by Dunnett's t-test). The assay was performed three times and data were expressed as mean ± SD.
DISCUSSION
We monitored autophagic cell death and the expression levels of p62, a multi-domain protein associated with autophagy, to investigate the role of the Keap1-Nrf2-ARE pathway and tau hyperphosphorylation, and further studied the morphological changes in the brain of AD rats. Results from this study showed that autophagy levels increased, expression of p62 decreased, and that the antioxidant efficiency of the Nrf2-ARE pathway decreased. Hyperphosphorylated tau was increased, which was predominantly manifested by morphological changes in brain tissue from AD rats.
First, the expression of the autophagy-related proteins Atg12-Atg5, LC3-II and beclin1 increased in AD model rats. Beclin1 can combine with other proteins to form a complex composed of the autophagosome membrane. The ubiquitin-like linking proteins Atg12-Atg5 and LC3-II play a role in extension and closure of the autophagosome membrane. LC3-II connects to the inner and outer membrane of the autophagosome, and participates in the complete formation of the autophagosome. Therefore, LC3-II can be seen as an autophagy marker protein[20,21]. Thus, increased expression of beclin1, Atg12-Atg5, and LC3-II in AD brain tissue suggests enhancement of autophagy. Our result is different from previous studies, which may be due to our analysis time. We analyzed autophagy-related proteins at an early stage (after 2-3 months of AD model preparation), while previous studies found that autophagy levels decreased at later stages. However, it is possible that the increased expression of Beclin1 and LC3-II in the saline group may be due to the mechanical stimulation of injection during the process of model establishment.
Second, the expression of p62 in AD model rats decreased. p62 is linked to LC3 of the autophagosome by the LC3-interacting region (LIR) and is degraded by the autophagy-lysosome pathway. Thus, expression levels of p62 are related to autophagy. It can be postulated from the above results that elevated autophagy can induce a decrease in p62 in AD rats. p62 may protect cells in other ways, such as modulation of the oxidative stress related signaling pathway[22]. Therefore, we investigated the influence of decreased p62 on the Keap1-Nrf-ARE system in brain tissue from AD rats.
Results from this study showed that Keap1 increased in the hippocampus and cortex of AD model rats. Nrf2 decreased in the cortex of AD model rats, but remained unchanged in the hippocampus. Keap1 is the receptor of Nrf2. Nrf2 can be cleared by the 26S proteasome after being ubiquitinated by Keap1 through the cullin-3 pathway[23]. This suggests that increased Keap1 can decrease Nrf2.
Nrf2 regulates the promoter of the antioxidant response element (ARE), coding a biphasic detoxifying enzyme and peroxiredoxins, which includes genes for [NAD(P)H: NQO1], glutathione synthetase, and g-glutamylcysteine synthetase (g-GCS)[24]. Cells eliminate reactive oxygen species by increasing the expression of these proteins. To study the effect of Nrf2 on downstream genes, the mRNA expression of the glutamate cysteine ligase (GCL) subunits GCLC and GCLM were monitored. GCL is the rate-limiting enzyme of glutathione biosynthesis.
RT-PCR results showed that the transcription of GCLC and GCLM mRNA was reduced in the cortex, but remained unchanged in the hippocampus. The expression of GCLC and GCLM mRNA was consistent with Nrf2 expression.
Our results showed that the expression of Keap1 can be enhanced, which is induced by p62 degradation. Keap1 promotes the elimination of Nrf2, and further influences downstream mRNA expression of GCLC and GCLM after Nrf2 enters to the nucleolus. These results indicate that the decrease of p62 can weaken the anti-oxidative capacity in the brain of AD model rats. To investigate the effect of anti-oxidative capacity deficiency, the expression of tau hyperphosphorylation was monitored. In addition, morphological measurements were performed to monitor changes in neurons and other structures.
Results from this study demonstrated that the expression of total tau protein in the hippocampus and cortex of AD model rats was reduced, and that expression of phosphorylated tau was increased.
Morphological analysis showed that the structure of neurons, neuropil, and mitochondria changed predominantly in the hippocampal CA3 region and cortex of AD model rats.
In conclusion, the present results reveal that elevated autophagy may induce the excessive degradation of p62, and further influence the anti-oxidative capability of the Nrf2-ARE pathway, which is one of the causes for tau hyperphosphorylation in the AD brain and the subsequent structural and functional damage to neurons.
MATERIALS AND METHODS
Design
A randomized controlled animal experiment.
Time and setting
The experiment was performed in the State Key Laboratory of Pathological Physiology, Norman Bethune College of Medicine, Jilin University, China during December 2009-December 2011.
Materials
Male Wistar rats, weighing 250–300 g, were purchased from the Laboratory Animal Center of Norman Bethune College of Medicine, Jilin University, China (license No. SCXK (Ji) 2007-0003). The animals were raised at room temperature under a 12 hour light/dark illumination cycle and were fed freely. This study was performed with the approval from the Animal Care and Ethics Committee of Jilin University in China. All experiments were performed according to the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Methods
AD model preparation
The AD rat model was prepared according to a previously described method[25,26]. Aβ25-35 peptide (1 mg) was solubilized in 100 μL sterile physiological saline at 10 μg/μL. After parafilm sealing, the solution was put in a 37°C chamber for 96 hours to facilitate aggregation. Rats were deeply anesthetized by intraperitoneal injection with 2% (w/v) pentobarbital sodium (45 mg/kg). The animals were immobilized on a stereotaxic apparatus (KO-PF company, NY, USA), and a bilateral hippocampal injection was performed with position coordinates according to the Rat Brain Stereotaxic Atlas[27]. The skull was drilled at 3.5 mm behind bregma, 2.0 mm beside the midline with an incisor hook, 3.3 mm below the interaural line, and the dura mater was exposed. The microinjector was slowly inserted vertically to a subdural depth of 2.7 mm. In the AD model group, 2 μL Aβ25-35 (Sigma, St. Louis, MO, USA) was slowly injected into the bilateral hippocampi, while the saline group was injected once with 2 μL normal saline. The needle was kept in position for 15 minutes to ensure that the solution was fully dispersed. The AD model group was injected with 1.5 mL β-galactosidase (Ding Guo Biological Technology Co., Ltd., Beijing, China) by intraperitoneal injection, and the saline group animals were injected with 1.5 mL normal saline by intraperitoneal injection every day. The same procedure was carried out for 20 days. Control group animals received no injection. After 3 days of Aβ25-35 injection, the spatial memory of animals was tested and evaluated using the Morris water maze test (Chinese Academy of Medical Sciences, Beijing, China). On the first day of the Morris water maze test, rats were acclimatized to the new environment. The water platform was removed to allow the rats to swim freely for 120 seconds. Rats were randomly placed in the water during the training phase, facing the wall, and forced to look for the platform that was set under the water. The time was recorded when the rat found the platform within 120 seconds (latency). If the rat could not find the platform within 120 seconds, it was guided to the platform and allowed to stay there for 60 seconds. The latency was recorded as 120 seconds. There was a significant difference in the time taken to find the platform between the AD model group and control group (P < 0.05), indicating that AD model establishment was successful.
Hematoxylin-eosin staining of neurons in the hippocampal CA3 region and forebrain
Rats were deeply anesthetized intraperitoneally with 10% (w/v) chloral hydrate (35 mg/100 g) and perfused transcardially with normal saline at 4°C followed by 10% (v/v) neutral buffered formalin (pH 7.4). The brain tissues were immediately resected and coronal sections of the brain were fixed in 10% (v/v) neutral buffered formalin at 4°C for 24 hours. The brains were processed for routine paraffin embedding. Serial coronal sections (4 μL) of the hippocampal CA3 region and forebrain were cut and mounted onto poly-L-lysine coated slides, deparaffinized with xylene, rehydrated through graded ethanol, and stained with hematoxylin-eosin. Finally, brain cell morphology was observed by light microscopy (Olympus, Tokyo, Japan).
Immunohistochemical analysis of p62, Keap1 and Nrf2 in the cortex and hippocampus
Brain coronal sections were used for immunohistochemistry. Immunohistochemistry was performed using the ultra-sensitive SP immunohistochemistry kit (Maixin, Fuzhou, China). Briefly, sections were deparaffinized with xylene, rehydrated through graded ethanol and dewaxed. Subsequently, antigen retrieval was performed by heating the sections in 10 mM citric acid buffer (pH 6.0). Endogenous peroxidase was blocked with 50 μL hydrogen peroxide (reagent A) for 10 minutes and 50 μL blocking serum (reagent B) for 60 minutes at room temperature. Sections were incubated with 1:50 or 1:100 rabbit polyclonal antibodies against p62, Keap1 and Nrf2 (Santa Cruz Biotechnology, CA, USA) overnight at 4°C, and then with biotinylated goat anti-rabbit secondary antibody (reagent C) for 20 minutes at room temperature. The sections were then incubated for 20 minutes with avidin and biotinylated horseradish peroxidase (reagent D). Immunoreactivity was visualized using the ABC kit (Maixin, Fuzhou, China) with diaminobenzidine. Selected sections were counterstained with hematoxylin and were analyzed under a BX51 Olympus light microscope (Image-Pro Plus 6.0, Olympus, Tokyo, Japan). The number of cells positive for Keap1 and Nrf2 in the cortex and hippocampus in each group was counted using the methods described by Neese et al[28].
Western blot analysis of Atg12, LC3-II, beclin1, P62, tau, and p-tau in the cortex and hippocampus
Frozen cortex and hippocampus from different groups were homogenized, and total protein was extracted. Rat brain homogenate was prepared from 100 mg frozen cortical and hippocampal tissue in 0.5 mL homogenization buffer (20 mM Tris HCl pH 7.5, 0.25 M sucrose, 2 mM EDTA, 1 μg/mL phenylmethanesulfonyl fluoride (PMSF), 1 μg/mL leupeptin, 1 μg/mL aprotinin, 1 μg/mL pepstatin). The homogenates were centrifuged for 10 minutes at 1 200 × g. The supernatants were centrifuged for 5 minutes at 12 000 × g for total protein. Protein concentration was determined using a protein assay kit (Bio-Rad, Hercules, CA, USA). Western blotting was performed using standard techniques. Lysate proteins (35 μg) were separated by 12% or 15% (w/v) sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred onto nitrocellulose transfer membranes (Millipore, Bedford, MA, USA). Membranes were blocked in PBS-Tween 20 buffer containing 10 mM Tris-HCl, 5% (w/v) non-fat dry milk, 100 mM NaCl, and 0.1% (v/v) Tween 20 (pH 7.6) for 1 hour at room temperature, and incubated with primary antibodies overnight at 4°C. The primary antibodies were: mouse monoclonal antibody against β-actin (as an internal control, sc-47778; 1:200), rabbit polyclonal antibody against Atg12 (sc-68884; 1:200), rabbit polyclonal antibody against MAP LC3β (sc-28266; 1:200), rabbit polyclonal antibody against p62 (sc-25575; 1:200), beclin1 (sc-48341; 1:200), tau (Tau5, sc-58860; 1:200) and p-tau (sc-101817; 1:200)(Santa Cruz Biotechnology). Membranes were then incubated with horseradish peroxidase-conjugated goat anti-rabbit secondary antibody (1:2 000)(Thermo, Waltham, MA, USA) at a 1:2 000 dilution for 1 hour at room temperature. Immunoreactive bands were visualized with diaminobenzidine (Sigma). Protein levels were quantified using Quantity One software (Bio-Rad) by measuring the band intensity (area × absorbance) in each group and normalizing it to the internal control.
Reverse transcription-PCR of GCLC, GCLM in the cortex and hippocampus
Frozen rat cerebral cortex and hippocampus were homogenized. Total RNA was isolated from the cerebral cortex and hippocampus using Trizol reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's protocol. cDNAs were generated through reverse-transcription polymerase chain reaction with the SuperScript preamplification system (Promega, Madison, MI, USA). The primer sequences are as follows: GCLC (sense: 5’-GTC TTC AGG TGA CAT TCC AAG C-3’, antisense: 5’-TGT TCT TCA GGG GCT CCA GTC-3’) (213 bp); GCLM (sense: 5’-CTG CTA AAC TGT TCA TTG TAG G-3’, antisense: 5’-CTA TTG GGT TTT ACC TGT G-3’)(280 bp); GAPDH (glyceraldehyde-3-phosphatedehydrogenase) (sense: 5’-GGG TGA TGC TGG TGC TGA GTA TGT-3’, antisense: 5’-AAG AAT GGG AGT TGC TGT TGA AGT-3’) (619 bp). All primers were synthesized by Sangon Biotech Co., Ltd. (Shanghai, China). GAPDH was used as an endogenous control for quantifying mRNA. The PCR products were electrophoresed on a 1% (w/v) agarose gel containing ethidium bromide (Sigma), visualized with a Tanon-1600 gel image processing system (Tanon, Shanghai, China) and analyzed by a GIS 1D gel image system software (Tanon).
Statistical analysis
All data were presented as mean ± SD and analyzed using SPSS 16.0 (SPSS, Chicago, IL, USA) and Origin 8.0 (OriginLab Corp, Northampton, MA, USA). One-way analysis of variance followed by Dunnett's t-test was used for data analysis among groups. A P value less than 0.05 was considered statistically significant for all analyses.
Footnotes
Conflicts of interest: None declared.
Ethical approval: All protocols for animal studies were approved by the Animal Care and Ethics Committee of Jilin University in China.
(Edited by Zhao B, Zou LY/Song LP)
REFERENCES
- [1].Clark TA, Lee HP, Rolston RK, et al. Oxidative stress and its implications for future treatments and management of Alzheimer disease. Int J Biomed Sci. 2010;6(3):225–227. [PMC free article] [PubMed] [Google Scholar]
- [2].Scherz-Shouval R, Shvets E, Fass E, et al. Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. EMBO J. 2007;26(7):1749–1760. doi: 10.1038/sj.emboj.7601623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Underwood BR, Imarisio S, Fleming A, et al. Antioxidants can inhibit basal autophagy and enhance neurodegeneration in models of polyglutamine disease. Hum Mol Genet. 2010;19(17):3413–3429. doi: 10.1093/hmg/ddq253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Hara T, Nakamura K, Matsui M, et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature. 2006;441(7095):885–889. doi: 10.1038/nature04724. [DOI] [PubMed] [Google Scholar]
- [5].Komatsu M, Waguri S, Koike M, et al. Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell. 2007;131:1149–1163. doi: 10.1016/j.cell.2007.10.035. [DOI] [PubMed] [Google Scholar]
- [6].Bjørkøy G, Lamark T, Brech A, et al. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J Cell Biol. 2005;171(4):603–614. doi: 10.1083/jcb.200507002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Ichimura Y, Kumanomidou T, Sou YS, et al. Structural basis for sorting mechanism of p62 in selective autophagy. J Biol Chem. 2008;283(33):22847–22857. doi: 10.1074/jbc.M802182200. [DOI] [PubMed] [Google Scholar]
- [8].Alonso AC, Li B, Grundke-Iqbal I, et al. Mechanism of tau-induced neurodegeneration in Alzheimer disease and related tauopathies. Curr Alzheimer Res. 2008;5(4):375–384. doi: 10.2174/156720508785132307. [DOI] [PubMed] [Google Scholar]
- [9].Reynolds MR, Berry RW, Binder LI. Nitration in neurodegeneration: deciphering the “Hows” “nYs”. Biochemistry. 2007;46:7325–7336. doi: 10.1021/bi700430y. [DOI] [PubMed] [Google Scholar]
- [10].Ramesh Babu J, Lamar Seibenhener M, Peng J, et al. Genetic inactivation of p62 leads to accumulation of hyperphosphorylated tau and neurodegeneration. J Neurochem. 2008;106(1):107–120. doi: 10.1111/j.1471-4159.2008.05340.x. [DOI] [PubMed] [Google Scholar]
- [11].Kuusisto E, Salminen A, Alafuzoff I. Early accumulation of p62 in neurofibrillary tangles in Alzheimer's disease: possible role in tangle formation. Neuropathol Appl Neurobiol. 2002;28(3):228–237. doi: 10.1046/j.1365-2990.2002.00394.x. [DOI] [PubMed] [Google Scholar]
- [12].Duran A, Linares JF, Galvez AS, et al. The signaling adaptor p62 is an important NF-kappaB mediator in tumorigenesis. Cancer Cell. 2008;13(4):343–354. doi: 10.1016/j.ccr.2008.02.001. [DOI] [PubMed] [Google Scholar]
- [13].Pankiv S, Clausen TH, Lamark T, et al. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem. 2007;282(33):24131–24145. doi: 10.1074/jbc.M702824200. [DOI] [PubMed] [Google Scholar]
- [14].Komatsu M, Kurokawa H, Waguri S, et al. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat Cell Biol. 2010;12(3):213–223. doi: 10.1038/ncb2021. [DOI] [PubMed] [Google Scholar]
- [15].Copple IM, Lister A, Obeng AD, et al. Physical and functional interaction of sequestosome 1 with Keap1 regulates the Keap1-Nrf2 cell defense pathway. J Biol Chem. 2010;285(22):16782–16788. doi: 10.1074/jbc.M109.096545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Lau A, Wang XJ, Zhao F, et al. A noncanonical mechanism of Nrf2 activation by autophagy deficiency: direct interaction between Keap1 and p62. Mol Cell Biol. 2010;30(13):3275–3285. doi: 10.1128/MCB.00248-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Martin L, Latypova X, Terro F. Post-translational modifications of tau protein: implications for Alzheimer's disease. Neurochem Int. 2011;58(4):458–471. doi: 10.1016/j.neuint.2010.12.023. [DOI] [PubMed] [Google Scholar]
- [18].Costa AP, Tramontina AC, Biasibetti R, et al. Neuroglial alterations in rats submitted to the okadaic acid-induced model of dementia. Behav Brain Res. 2011;226(2):420–427. doi: 10.1016/j.bbr.2011.09.035. [DOI] [PubMed] [Google Scholar]
- [19].Su B, Wang XL, Lee HG, et al. Chronic oxidative stress causes increased tau phosphorylation in M17 neuroblastoma cells. Neurosci Lett. 2010;468(3):267–271. doi: 10.1016/j.neulet.2009.11.010. [DOI] [PubMed] [Google Scholar]
- [20].Hanada T, Noda NN, Satomi Y, et al. The Atg12-Atg5 conjugate has a novel E3-like activity for protein lipidation in autophagy. J Biol Chem. 2007;282(52):37298–37302. doi: 10.1074/jbc.C700195200. [DOI] [PubMed] [Google Scholar]
- [21].Suzuki K, Kubota Y, Sekito T, et al. Hierarchy of Atg proteins in pre-autophagosomal structure organization. Genes Cells. 2007;12:209–218. doi: 10.1111/j.1365-2443.2007.01050.x. [DOI] [PubMed] [Google Scholar]
- [22].Fujita KI, Maeda D, Xiao Q, et al. Nrf2-mediated induction of p62 controls Toll-like receptor-4-driven aggresome-like induced structure formation and autophagic degradation. Proc Natl Acad Sci U S A. 2011;108(4):1427–1432. doi: 10.1073/pnas.1014156108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Nguyen T, Sherratt PJ, Nioi P, et al. Nrf2 controls constitutive and inducible expression of ARE-driven genes through a dynamic pathway involving nucleocytoplasmic shuttling by Keap1. J Biol Chem. 2005;280:32485–32492. doi: 10.1074/jbc.M503074200. [DOI] [PubMed] [Google Scholar]
- [24].Ishii T, Itoh K, Takahashi S, et al. Transcription factor Nrf2 coordinately regulates a group of oxidative stress-inducible genes in macrophages. J Biol Chem. 2000;275(21):16023–16029. doi: 10.1074/jbc.275.21.16023. [DOI] [PubMed] [Google Scholar]
- [25].Pavia J, Alberch J, Alvarez I, et al. Repeated administration of beta amyboid (25-35) to rats decreases muscarinic receptor in cerebral cortex. Neurosci Lett. 2000;278(12):69–72. doi: 10.1016/s0304-3940(99)00900-3. [DOI] [PubMed] [Google Scholar]
- [26].Hua X, Lei M, Ding J, et al. Pathological and biochemical alterations of astrocytes in ovariectomized rats injected with D-galactose: a potential contribution to Alzheimer's disease processes. Exp Neurol. 2008;210(2):709–718. doi: 10.1016/j.expneurol.2008.01.009. [DOI] [PubMed] [Google Scholar]
- [27].Paxinos G, Watson CR, Emson PC. AChE-stained horizontal sections of the rat brain in stereotaxic coordinates. J Neurosci Methods. 1980;3(2):129–149. doi: 10.1016/0165-0270(80)90021-7. [DOI] [PubMed] [Google Scholar]
- [28].Neese SL, Sherill LK, Tan AA, et al. Vagus nerve stimulation may protect GABAergic neurons following traumatic brain injury in rats: An immunocytochemical study. Brain Res. 2007;1128:157–163. doi: 10.1016/j.brainres.2006.09.073. [DOI] [PMC free article] [PubMed] [Google Scholar]