

Abstract
We treated detonator-explosion-induced craniocerebral injury in rabbits with hyperbaric oxygen 1-24 hours post-injury. Expression of the apoptosis-regulating protein cytochrome c, the pro-apoptotic protein Bax and the apoptosis marker caspase-3 in the tissues surrounding the area of injury was significantly reduced, while that of the anti-apoptotic protein Bcl-2 was significantly increased. Our findings indicate that the curative effects of early hyperbaric oxygen on cortical cell apoptosis is associated with suppression of cytochrome c release from mitochondria. This mechanism underlies the observed reduction in Bax expression and upregulation of Bcl-2 expression.
Keywords: hyperbaric oxygen, craniocerebral injury, caspase-3, cytochrome c, Bax, Bcl-2, neural regeneration
Abbreviation:
HBOT, hyperbaric oxygen therapy; PBS, phosphate buffered saline
INTRODUCTION
The mitochondrion is a eukaryotic cell organelle that plays a critical role in cell apoptosis[1]. Indeed, the mitochondrion-mediated pathway is one of two pathways of apoptosis, the other being death receptor-mediated[2,3]. Early signal transduction in apoptosis mainly occurs via the mitochondrion pathway, in which, the mitochondrion acts as a receptor and amplifier of death signals in cells[4,5]. Cytochrome c is distributed in the mitochondrial inner and outer membranes[6] where it functions as an important cell regulator[7]. Its release from mitochondria is regarded as an early critical event of apoptosis[8,9,10,11]. In the presence of ischemia or hypoxia injury in brain tissues, changes in mitochondrial morphology, membrane potential and function result in elevated cytochrome c levels in the cytoplasm and reduced concentrations in the mitochondria[12,13], which finally triggers the caspase-induced apoptotic pathway[14,15]. In apoptosis related gene families, bcl-2 and bax may be involved in this process[16,17,18]. Previous studies found that hyperbaric oxygen therapy (HBOT) can maintain the mitochondrial membrane potential such that cell apoptosis can be reduced in rats with cortical injury[19,20]. In addition, early HBOT resulted in inhibitory effects on brain cell autophagy and apoptosis in an animal model of cerebral edema induced by explosive injury[21,22,23]. Although clinical HBOT also achieves curative effects after craniocerebral injury, the precise mechanism behind this remains unknown. We therefore carried out HBOT after brain injury and determined changes in caspase-3, cytochrome c, Bax and Bcl-2 levels in brain tissues of rabbits.
RESULTS
Quantitative analysis of experimental animals
A total of 150 New Zealand rabbits were randomly assigned to control (n = 10), model (n = 70) and HBOT (n = 70) groups. A blast-induced brain injury was established in the model and HBOT groups, and the HBOT group received HBOT treatment 1 hour after injury. Control and model groups received air at normal pressure instead of HBOT. Animals with blast-induced apnea were subjected to respiratory tract nursing or cardio-pulmonary resuscitation if necessary. Vital signs gradually stabilized and the animals regained consciousness 3–4 hours later, but they were depressed with a low appetite. They were housed in the laboratory animal center, but five died 3 days post-injury. A total of 155 rabbits were analyzed following supplementation.
Early HBOT suppressed cytochrome c expression in brain tissues around the injury site
Immunohistochemical detection revealed little cytochrome c expression in the cerebral cortex of control rabbits. Significant cytochrome c expression was detected in the brain tissues around the injury site 1 hour after the blast injury. This gradually increased with time, peaked at 12 hours, then gradually decreased and had restored to normal levels by day 3 (P > 0.05). Cytochrome c expression in the brain tissues around the injury site was significantly reduced in the early stages (1–24 hours post-injury) of HBOT (P < 0.05), but remained higher than control group (P < 0.05) then restored to normal levels by day 3 (P > 0.05; Figure 1).
Figure 1.
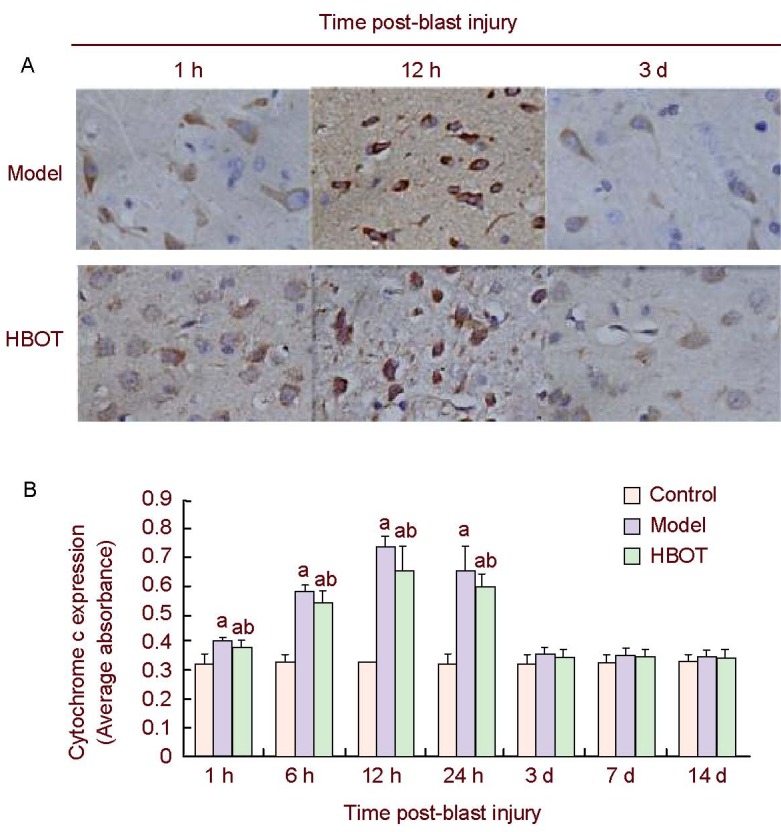
Cytochrome c expression in rabbit brain tissues around injury site.
(A) Immunohistochemical staining of rabbit brain tissues around injury site showed that cytochrome c was expressed in the cytoplasm, appearing as brown-yellow particles; the background was stained light yellow or remained unstained (× 400).
(B) Histogram of cytochrome c expression in rabbit brain tissues around injury site. High absorbance value represents high level of cytochrome c expression. Data were expressed as mean ± SD. Intergroup comparison was conducted using the Student-Newman-Keuls method. aP < 0.05, vs. control group; bP < 0.05, vs. model group.
HBOT: Hyperbaric oxygen treatment; h: hour; d: day.
Early HBOT suppressed Bax expression in rabbit brain tissues around the injury site
Immunohistochemistry showed little Bax expression in the cerebral cortex of control group rabbits. Bax expression was detected in the brain tissues around the injury site 1 hour post-blast injury, gradually increased over time, peaked at 12 hours, gradually decreased and was restored to normal levels by day 14 (P > 0.05). Bax expression in the brain tissues around the injury site was reduced in the early stage (1–24 hours post-injury) of HBOT (P < 0.05) but remained higher than the control group (P < 0.05), and was restored to normal levels by day 7 (P > 0.05; Figure 2).
Figure 2.
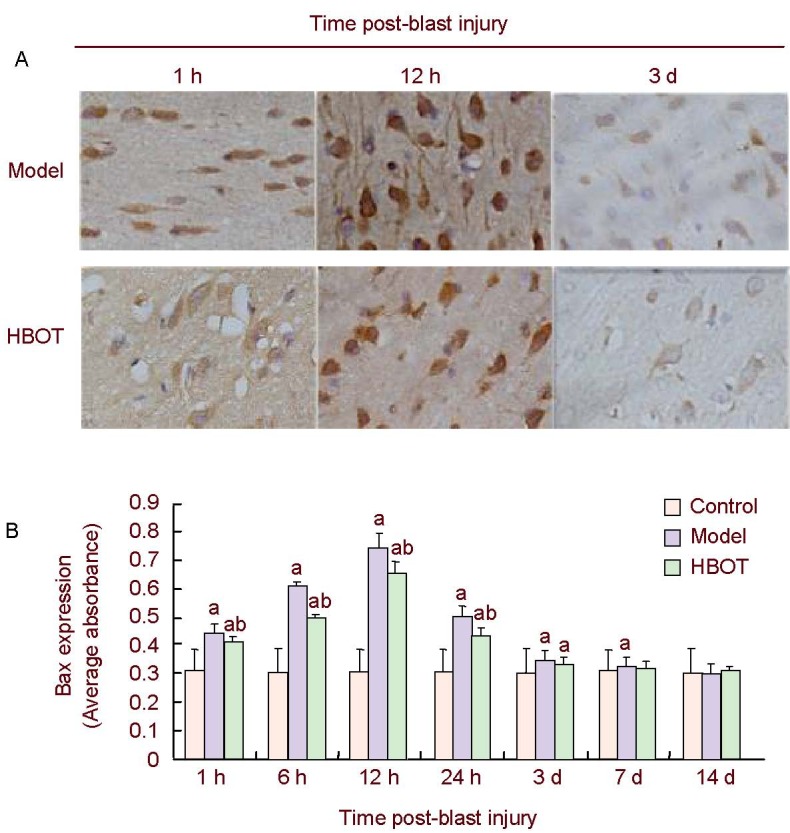
Bax expression in rabbit brain tissues around the injury site.
(A) Immunohistochemical staining of Bax expression in rabbit brain tissues around the injury site showed that Bax was mainly expressed in the nuclear membrane, and occasionally in the endoplasmic reticulum and mitochondrial outer membrane, appearing as brown-yellow particles (× 400).
(B) Histogram of Bax expression in rabbit brain tissues around the injury site. High absorbance value represents high level of Bax expression. Data were expressed as mean ± SD. Intergroup comparison was conducted using the Student-Newman-Keuls method. aP < 0.05, vs. control group; bP < 0.05, vs. model group.
HBOT: Hyperbaric oxygen treatment; h: hour; d: day.
Early HBOT suppressed Bcl-2 expression in rabbit brain tissues around the injury site
Immunohistochemistry showed little Bcl-2 expression in the cerebral cortex of control group rabbits. By contrast, Bcl-2 expression was detected in the brain tissues around the injury site 1 hour post-blast injury, gradually increasing over time and peaking at 6 hours. Levels then gradually decreased and were restored to normal by day 7 (P > 0.05). Bcl-2 expression in the brain tissues around the injury site was significantly increased in the early stage (1–24 hours post-bast injury) of HBOT compared with the model group (P < 0.05; Figure 3).
Figure 3.

Bcl-2 expression in rabbit brain tissues around the injury site.
(A) Immunohistochemical staining of Bcl-2 expression in rabbit brain tissues around the injury site showed that Bcl-2 was mainly expressed in the nuclear membrane, and was occasionally observed in the endoplasmic reticulum and mitochondrial outer membrane, appearing as brown-yellow particles (× 400).
(B) Histogram of Bcl-2 expression in rabbit brain tissues around the injury site. High absorbance value represents high level of Bcl-2 expression. Data were expressed as mean ± SD. Intergroup comparison was conducted using the Student-Newman-Keuls method. aP < 0.05, vs. control group; bP < 0.05, vs. model group.
HBOT: Hyperbaric oxygen treatment; h: hour; d: day.
Early HBOT suppressed caspase-3 expression in rabbit brain tissues around the injury site
Caspase-3 activation is a marker of apoptosis in its irreversible stage[24], and, as such, is used as a major marker for apoptosis. Immunohistochemistry showed little caspase-3 expression in the cerebral cortex of control group rabbits. However, caspase-3 expression was detected in the brain tissues around the injury site 1 hour after the blast. Levels then gradually increased, peaked at 24 hours, gradually decreased and were restored to normal by day 14 (P > 0.05). Caspase-3 expression in brain tissues around the injury site was decreased in the early stage (1-24 hours post-blast injury) of HBOT (P < 0.05) but remained higher than the control group (P < 0.05), and was restored to normal levels by day 14 (P > 0.05; Figure 4).
Figure 4.

Caspase-3 expression in rabbit brain tissues around the injury site.
(A) Immunohistochemical staining of caspase-3 expression in rabbit brain tissues around the injury site showed that caspase-3 was mainly expressed in the cytoplasm, appearing as brown-yellow particles (× 400).
(B) Histogram of caspase-3 expression in rabbit brain tissues around the injury site. High absorbance value represents high level of caspase-3 expression. Data were expressed as mean ± SD. Intergroup comparison was conducted using the Student-Newman-Keuls method. aP < 0.05, vs. control group; bP < 0.05, vs. model group.
HBOT: Hyperbaric oxygen treatment; h: hour; d: day.
DISCUSSION
In the present study, cytochrome c expression was detected in the brain tissues surrounding the injury site 1 hour post-blast injury. This suggests that the blast induced ischemia and hypoxia in the brain tissues, leading to the production of active oxygen and increased intracellular Ca2+ concentrations. In turn, this would open the mitochondrial permeability transition pore and alter mitochondrial permeability. As a result, the mitochondrial membrane potential would be reduced, affecting normal respiratory chain function and leading to cytochrome c release into the cytoplasm[25].
Cytochrome c expression peaked at 12 hours post-bast injury, possibly because the mitochondrial permeability transition pore was opened further and the ATP substrate in the mitochondrial matrix was transferred to the cytoplasm, resulting in the collapse of the membrane potential, respiratory chain decoupling, ATP synthesis reduction and outer membrane rupture. Cytochrome c levels were similar to those of the control group at 3 days post-blast injury, this indicates that its release occurred soon after blast injury. Indeed, cytochrome c was released into the cytoplasm prior to the peak in caspase-3 levels, which is consistent with its role in inducing caspase activation and cell apoptosis[19,20,26,27,28,29]. Both cytochrome c and caspase-3 expression in the brain tissues around the injury site was significantly reduced following HBOT, indicating that HBOT suppresses apoptosis[21].
Factors that stimulate apoptosis can induce Bax to form a dimer and bind with the mitochondrial membrane to promote cytochrome c release[30], while Bcl-2 stably binds the surface of the mitochondrial membrane at its C-end transmembrane domain and inhibits cytochrome c mitochondrial release[31,32]. In the present study, Bax and Bcl-2 expression was detected 1 hour post-blast injury, and Bcl-2 expression peaked earlier than that of Bax. It is likely that Bcl-2 protein expression was enhanced post-blast injury in response to stress to protect neurons. However, over time, increasing apoptosis stimulating factors enhanced Bax expression, leading to apoptosis. After HBOT administration, Bax expression was reduced while Bcl-2 expression increased, suggesting that HBOT suppresses Bax but increases Bcl-2 in the early post-injury stage. Early HBOT may inhibit cell apoptosis through increasing Bcl-2 expression, reducing Bax expression, inhibiting cytochrome c release from mitochondria and suppressing caspase activation.
In summary, HBOT administered in the early stage post-injury can block mitochondrial cytochrome c release into the cytoplasm to some extent and block the mitochondrial pathway to suppress cell apoptosis. This mechanism is associated with its effects of inhibiting Bax expression and increasing Bcl-2 expression.
MATERIALS AND METHODS
Design
A randomized, controlled, animal experiment.
Time and setting
The study was performed at the Animal Experimental Center, Daping Hospital, Third Military Medical University of Chinese PLA, China between February and October 2009. Immunohistochemistry was conducted in the Laboratory of Immunohistochemistry, Anhui Medical University, China between February and July 2010.
Materials
A total of 150 healthy, adult New Zealand rabbits, males and females, weighing 2.0–2.5 kg, were provided by the Animal Experimental Center, Daping Hospital (license No. SYXK (army) 2007-017). They were housed at 26 ± 1.5°C with a humidity of 60%. Animal experimental procedures were performed in accordance with the Guidance Suggestions for the Care and Use of Laboratory Animals, issued by the Ministry of Science and Technology of China[33].
Methods
Establishment of blast brain injury model
Rabbits in model and HBOT groups were placed under a self-made explosion frame. A paper detonator (Chongqing 845 Factory, China) equivalent to 600 mg TriNitro Toluene (TNT) was fixed 6.5 cm above the rabbits’ brains, 1.5 cm to the median, 2 cm anterior to the line of both ears to induce an explosion. The explosion pressure was detected using a 113A31 sensor (PCB, Buffalo, NY, USA) and a Wavebook/516A data acquisition system[34] (Japan; supplementary Video 1 online).
HBOT
Rabbits in the HBOT group underwent HBOT in a transparent oxygen animal experimental chamber, and were exposed to 2 atmosphere absolute, equal to 2.03 MPa. The rabbits were compressed with pure oxygen for 5 minutes, boosting pressure for 30 minutes, stabilizing pressure for 1 hour and reducing pressure for 30 minutes. This was performed twice a day, once in the morning and once in the afternoon, for 2 hours each time. Control and model group rabbits were exposed to air of normal pressure.
Preparation of brain tissue paraffin sections
Ten rabbits from each group were selected 1, 6, 12, and 24 hours, and 3, 7 and 14 days post-blast injury, anesthetized and sacrificed (supplementary Figure 1 online). Brain tissues surrounding the injury site (0.5 cm from the margin of injury, approximately 3.0 mm3) were harvested, fixed in 4% paraformaldehyde, dehydrated, cleared, immersed in paraffin, embedded, and sectioned (4 μm thick).
Immunohistochemistry for caspase-3, cytochrome c, Bax and Bcl-2 expression in brain tissues surrounding the injury site
The sections were fixed in paraformaldehyde for 15 minutes, washed with phosphate buffered saline (PBS), for 3 × 3 minutes, mixed with 3% H2O2 and left at room temperature for 10 minutes. They were then washed again with PBS for 3 × 3 minutes, mixed with normal goat serum blocking solution at room temperature for 30 minutes, and incubated with rabbit anti-caspase-3, cytochrome c, Bax and Bcl-2 polyclonal antibody (1:200; Boster, Wuhan, China) overnight at 4°C. The sections were rewarmed at room temperature for 10 minutes, washed with PBS for 3 × 5 minutes, then incubated with 100 μL goat anti-rabbit IgG (1:100; Boster) per section at 37°C for 30 minutes. They were again washed with PBS for 3 × 5 minutes, treated with 100 μL streptavidin-biotin complex solution (Boster) per section, washed with PBS for 3 × 5 minutes, and colorized with diaminobenzidine for 5–10 minutes. Sections were then washed with tap water, counterstained with hematoxylin for 1 minute, differentiated with hydrochloric acid and ethanol for about 10 seconds, washed with tap water for 10–15 minutes, dehydrated, cleared, mounted, dried, and observed by light microscope (Nikon, Tokyo, Japan). Ten fields of view from each section (400 × magnification) were randomly selected and the mean absorbance value was calculated using the JEDA 801D morphologic image analysis system (Molecular Devices, Sunnyvale, CA, USA) and MetaMorph software (Molecular Devices)[35].
Statistical analysis
Data were analyzed using SPSS 17.0 software (SPSS, Chicago, IL, USA) and expressed as mean ± SD. Differences at different time points between groups were compared using one-way analysis of variance, and paired comparisons were performed by the Student-Newman-Keuls method. A value of P < 0.05 was considered statistically significant.
Acknowledgments:
We thank the Experiment Center of the Third Military Medical University of Chinese PLA for model establishment, and the Basic Teaching and Research Division, Anhui Medical University for technical support. We also thank the staff at the Department of Neurosurgery, the 105 Hospital of Chinese PLA for their help in study design and manuscript writing.
Footnotes
Funding: This study was supported by the Eleventh-Five Major Subject of Nanjing Military Area Command (Functional MRI of HBOT for acute severe traumatic brain injury), No. 06Z19; and the Military Medical Science and Technology Innovation Foundation in 2009 (Clinical study of CTP and NRS in traumatic SAH patients), No. 09Z009.
Conflicts of interest: None declared.
Ethical approval: This study received permission from the Animal Ethics Committee of Third Military Medical University, China.
Supplementary information: Supplementary data associated with this article can be found in the online version, by visiting http://www.nrronline.org.
(Edited by Lu CR, Yao CY/Su LL/Song LP)
REFERENCES
- [1].An WW, Wang MW, Tashiro S, et al. Norcantharidin induces human melanoma A375-S2 cell apoptosis through mitochondrial and caspase pathways. J Korean Med Sci. 2004;19(4):560–566. doi: 10.3346/jkms.2004.19.4.560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Huang J, Wu L, Tashiro S, et al. Reactive oxygen species mediate oridonin-induced HepG2 apoptosis through p53, MAPK, and mitochondrial signaling pathways. J Pharmacol Sci. 2008;107(4):370–379. doi: 10.1254/jphs.08044fp. [DOI] [PubMed] [Google Scholar]
- [3].Cheng HY, Hsieh MT, Wu CR, et al. Schizandrin protects primary cultures of rat cortical cells from glutamate-induced excitotoxicity. J Pharmacol Sci. 2008;107(1):21–31. doi: 10.1254/jphs.fp0072394. [DOI] [PubMed] [Google Scholar]
- [4].Maruyama J, Naguro I, Takeda K, et al. Stress-activated MAP kinase cascades in cellular senescence. Curr Med Chem. 2009;16(10):1229–1235. doi: 10.2174/092986709787846613. [DOI] [PubMed] [Google Scholar]
- [5].Aguzzi A, Maggioni D, Nicolini G, et al. MAP kinase modulation in squamous cell carcinoma of the oral cavity. Anticancer Res. 2009;29(1):303–308. [PubMed] [Google Scholar]
- [6].Tamilselvan J, Jayaraman G, Sivarajan K, et al. Age-dependent upregulation of p53 and cytochrome c release and susceptibility to apoptosis in skeletal muscle fiber of aged rats: role of carnitine and lipoic acid. Free Radic Biol Med. 2007;43(12):1656–1669. doi: 10.1016/j.freeradbiomed.2007.08.028. [DOI] [PubMed] [Google Scholar]
- [7].Nakatsuka H, Ohta S, Tanaka J, et al. Cytochrome c release from mitochondria to the cytosol was suppressed in the ischemia-tolerance-induced hippocampal CA1 region after 5-min forebrain ischemia in gerbils. Neurosci Lett. 2000;278(1-2):53–56. doi: 10.1016/s0304-3940(99)00894-0. [DOI] [PubMed] [Google Scholar]
- [8].Budd SL, Tenneti L, Lishnak T, et al. Mitochondrial and extramitochondrial apoptotic signaling pathways in cerebrocortical neurons. Proc Natl Acad Sci U S A. 2000;97(11):6161–6166. doi: 10.1073/pnas.100121097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Green DR, Reed JC. Mitochondria and apoptosis. Science. 1998;281(5381):1309–1312. doi: 10.1126/science.281.5381.1309. [DOI] [PubMed] [Google Scholar]
- [10].Heusch G, Boengler K, Schulz R. Cardioprotection: nitric oxide, protein kinases, and mitochondria. Circulation. 2008;118(19):1915–1919. doi: 10.1161/CIRCULATIONAHA.108.805242. [DOI] [PubMed] [Google Scholar]
- [11].Nakatsuka H, Ohta S, Tanaka J, et al. Cytochrome c release from mitochondria to the cytosol was suppressed in the ischemia-tolerance-induced hippocampal CA1 region after 5-min forebrain ischemia in gerbils. Neurosci Lett. 2000;278(1-2):53–56. doi: 10.1016/s0304-3940(99)00894-0. [DOI] [PubMed] [Google Scholar]
- [12].Kluck RM, Bossy-Wetzel E, Green DR, et al. The release of cytochrome c from mitochondria: a primary site for Bcl-2 regulation of apoptosis. Science. 1997;275(5303):1132–1136. doi: 10.1126/science.275.5303.1132. [DOI] [PubMed] [Google Scholar]
- [13].Kroemer G, Galluzzi L, Brenner C. Mitochondrial membrane permeabilization in cell death. Physiol Rev. 2007;87(1):99–163. doi: 10.1152/physrev.00013.2006. [DOI] [PubMed] [Google Scholar]
- [14].Li P, Nijhawan D, Budihardjo I, et al. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell. 1997;91(4):479–489. doi: 10.1016/s0092-8674(00)80434-1. [DOI] [PubMed] [Google Scholar]
- [15].Jeong SY, Seol DW. The role of mitochondria in apoptosis. BMB Rep. 2008;41(1):11–22. doi: 10.5483/bmbrep.2008.41.1.011. [DOI] [PubMed] [Google Scholar]
- [16].Sareen D, van Ginkel PR, Takach JC, et al. Mitochondria as the primary target of resveratrol-induced apoptosis in human retinoblastoma cells. Invest Ophthalmol Vis Sci. 2006;47(9):3708–3716. doi: 10.1167/iovs.06-0119. [DOI] [PubMed] [Google Scholar]
- [17].Dlugosz PJ, Billen LP, Annis MG, et al. Bcl-2 changes conformation to inhibit Bax oligomerization. EMBO J. 2006;25(11):2287–2296. doi: 10.1038/sj.emboj.7601126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Skommer J, Wlodkowic D, Deptala A. Larger than life: Mitochondria and the Bcl-2 family. Leuk Res. 2007;31(3):277–286. doi: 10.1016/j.leukres.2006.06.027. [DOI] [PubMed] [Google Scholar]
- [19].Soustiel JF, Palzur E, Vlodavsky E, et al. The effect of oxygenation level on cerebral post-traumatic apoptotsis is modulated by the 18-kDa translocator protein (also known as peripheral-type benzodiazepine receptor) in a rat model of cortical contusion. Neuropathol Appl Neurobiol. 2008;34(4):412–423. doi: 10.1111/j.1365-2990.2007.00906.x. [DOI] [PubMed] [Google Scholar]
- [20].Palzur E, Zaaroor M, Vlodavsky E, et al. Neuroprotective effect of hyperbaric oxygen therapy in brain injury is mediated by preservation of mitochondrial membrane properties. Brain Res. 2008;1221:126–133. doi: 10.1016/j.brainres.2008.04.078. [DOI] [PubMed] [Google Scholar]
- [21].Tang H, Liu JC, Zhang YM, et al. Effect of hyperbaric oxygen on the changes of autophagy and apoptosis in cerebral cortex in rats with brain explosive injury. Zhonghua Shenjing Yixue Zazhi. 2010;9(10):1014–1017. [Google Scholar]
- [22].Sun WJ, Liu JC, Zhang YM, et al. Expression of AQP-4 and efficacy of hyperbaric oxygen therapy after blast-related brain injury in rabbits. Zhongguo Weiqinxi Waike Zazhi. 2011;16(3):136–138. [Google Scholar]
- [23].Zhang YM, QI ST, Huang GL, et al. Expression of Connexin-43 in brain tissue of rabbits with explosive brain injury. Zhongguo Weiqinxi Shenjing Waike Zazhi. 2010;15(6):279–281. [Google Scholar]
- [24].Huang J, Wu L, Tashiro S, et al. Reactive oxygen species mediate oridonin-induced HepG2 apoptosis through p53, MAPK, and mitochondrial signaling pathways. J Pharmacol Sci. 2008;107(4):370–379. doi: 10.1254/jphs.08044fp. [DOI] [PubMed] [Google Scholar]
- [25].Chandra D, Liu JW, Tang DG. Early mitochondrial activation and cytochrome c up-regulation during apoptosis. J Biol Chem. 2002;277(52):50842–50854. doi: 10.1074/jbc.M207622200. [DOI] [PubMed] [Google Scholar]
- [26].An WW, Wang MW, Tashiro S, et al. Norcantharidin induces human melanoma A375-S2 cell apoptosis through mitochondrial and caspase pathways. J Korean Med Sci. 2004;19(4):560–566. doi: 10.3346/jkms.2004.19.4.560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Cui N, Li S, Zhao X, et al. Expression of Bcl-2, Bax and Caspase-3 in nerve tissues of rats chronically exposed to 2,5-hexanedione. Neurochem Res. 2007;32(9):1566–1572. doi: 10.1007/s11064-007-9359-0. [DOI] [PubMed] [Google Scholar]
- [28].Das A, Guyton MK, Matzelle DD, et al. Time-dependent increases in protease activities for neuronal apoptosis in spinal cords of Lewis rats during development of acute experimental autoimmune encephalomyelitis. J Neurosci Res. 2008;86(13):2992–3001. doi: 10.1002/jnr.21737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].He A, Wang JA, Gui C, et al. Changes of mitochondrial pathway in hypoxia/reoxygenation induced cardiomyocytes apoptosis. Folia Histochem Cytobiol. 2007;45(4):397–400. [PubMed] [Google Scholar]
- [30].Wu YT, Tan HL, Huang Q, et al. Autophagy plays a protective role during zVAD-induced necrotic cell death. Autophagy. 2008;4(4):457–466. doi: 10.4161/auto.5662. [DOI] [PubMed] [Google Scholar]
- [31].Cartron PF, Priault M, Oliver L, et al. The N-terminal end of Bax contains a mitochondrial-targeting signal. J Biol Chem. 2003;278(13):11633–11641. doi: 10.1074/jbc.M208955200. [DOI] [PubMed] [Google Scholar]
- [32].Eliseev RA, Malecki J, Lester T, et al. Cyclophilin D interacts with Bcl2 and exerts an anti-apoptotic effect. J Biol Chem. 2009;284(15):9692–9699. doi: 10.1074/jbc.M808750200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].The Ministry of Science and Technology of the People's Republic of China. Guidance Suggestions for the Care and Use of Laboratory Animals. 2006-09-30 [Google Scholar]
- [34].Zhang YM, Liu JC, Yang YY, et al. Establishment of blast-related brain injury model in rabbits. Zhongguo Weiqinxi Shenjing Waike Zazhi. 2010;15(2):74–76. [Google Scholar]
- [35].Tong H, Chen GH, Liu RY, et al. Age-related learning and memory impairments in adult-onset hypothyroidism in Kunming mice. Physiol Behav. 2007;91(2-3):290–298. doi: 10.1016/j.physbeh.2007.03.008. [DOI] [PubMed] [Google Scholar]