

Abstract
Background
T cells have the capacity to eliminate tumors but the signaling pathways by which they do so are incompletely understood. T cell priming requires activation of the transcription factors AP-1, NFAT and NF-κB downstream of the TCR, but whether activation of T cell-NF-κB in vivo is required for tumor control has not been addressed. In humans and mice with progressively growing tumors, the activity of T cell-intrinsic NF-κB is often reduced. However, it is not clear if this is causal for an inability to reject transformed cells, or if it is a consequence of tumor growth. T cell-NF-κB is important for T cell survival and effector differentiation and plays an important role in enabling T cells to reject cardiac and islet allografts, suggesting the possibility that it may also be required for tumor elimination. In this study, we tested whether normal T cell-NF-κB activation is necessary for the rejection of tumors whose growth is normally controlled by the immune system.
Methods
Mice with genetically impaired T cell-NF-κB activity were subcutaneously injected with MC57-SIY tumor cells. Tumor growth was measured over time, and the anti-tumor immune response was evaluated using flow cytometry and cytokine detection assays.
Results
Mice with impaired T cell-NF-κB activity were unable to reject tumors that were otherwise eliminated by wildtype mice, despite equal accumulation of tumor-reactive T cells. In addition, specific impairment of NF-κB signaling downstream of the TCR was sufficient to prevent tumor rejection. Tumor antigen-specific T cell-IFN-γ and TNF-α production, as well as cytotoxic ability, were all reduced in mice with impaired T cell-NF-κB, suggesting an important role for this transcription factor in the effector differentiation of tumor-specific effector T cells.
Conclusions
Our results have identified the NF-κB pathway as an important signaling axis in T cells, required for the elimination of growing tumors in vivo. Maintaining or enhancing T cell-NF-κB activity may be a promising avenue for anti-tumor immunotherapy.
Electronic supplementary material
The online version of this article (doi:10.1186/s40425-014-0045-x) contains supplementary material, which is available to authorized users.
Keywords: T cell, NF-κB, Tumor rejection, Priming, Effector function, Cytokine production, Cytotoxicity
Background
T cells specific for tumor antigens can play an important role in the elimination of growing tumors [1]. However, the signaling pathways elicited in T cells following tumor antigen-dependent engagement of the TCR that are required for tumor rejection are not fully understood. TCR signaling includes the activation and nuclear translocation of several transcription factors, such as nuclear factor of activated T cells (NFAT), activator protein-1 (AP-1) and nuclear factor-κ-light chain enhancer of activated B cells (NF-κB) [2]. However, ablation of NFAT1 from T cells results in improved rather than impaired tumor control, underscoring the importance of NFAT1 in T cell anergy [3]. Nevertheless, while targeted disruption of NFAT1 or NFAT2 genes separately did not prevent T cell activation and IL-2 production, expression of a dominant negative NFAT in T cells did [4], suggesting that complete elimination of NFAT activity may prevent tumor control in vivo, although this has not been tested directly. Similarly, mice with genetic ablation selectively in T cells of c-Fos, an AP-1 subunit, displayed better, rather than worse, tumor control while mice with transgenic expression of c-Fos in T cells conversely experienced accelerated tumor growth [5]. In this case, the immunosuppressive effect of c-Fos was due to the induction in T cells of the negative regulator programmed death-1 (PD-1), a direct target of AP-1. As with NFAT, complete ablation of the AP-1 subunits might be expected to prevent tumor rejection. The functional role of the third main transcription factor of T cell activation, NF-κB, in tumor control remains to be determined.
Factors produced by tumors can directly or indirectly inhibit TCR-induced NF-κB activation, as shown with T cells isolated from patients with renal cell carcinoma [6], non-small cell lung cancer [7], or exposed to ascites fluid from an ovarian cancer patient [8]. Similarly, mice with growing tumors can harbor reduced NF-κB activity in peripheral T cells [9,10]. However, whether a reduction in T cell-NF-κB activity can be a cause for why T cells fail to control tumor growth and not just a consequence of tumor expansion, or whether other transcription factors can compensate in vivo for deficient NF-κB activity, remains to be tested. Understanding the signaling pathways that contribute to tumor rejection when it is successful may help design therapies to promote tumor elimination when it is not spontaneously achieved.
The transcription factor NF-κB comprises a family of proteins that include DNA binders (p50, p52) and DNA transactivators (RelA, RelB and c-Rel) [11]. In the absence of a stimulus, heterodimers of these subunits are retained in the cytoplasm by inhibitors of NF-κB (IκB). TCR activation results in the phosphorylation of the lipid raft-associated CAspase Recruitment domain Membrane-Associated guanylate kinase protein 1 (CARMA1) [12]. Phosphorylated CARMA1 associates with the protein B cell lymphoma 10 (Bcl-10), which acts as a scaffold for the mucosa-associated lymphoid tissue lymphoma translocation gene-1 (MALT1). The complex formed by CARMA1, Bcl-10, and MALT1 induces the activation of the IκB kinase complex IKK (IKKα, IKKβ and NEMO), which then phosphorylates IκB, an event that targets IκB for K48 ubiquitination and degradation by the 26S proteasome. This uncovers a nuclear localization domain within NF-κB dimers that enables them to translocate into the nucleus and initiate gene transcription.
Several genetic mouse models of NF-κB impairment in T cells have been generated, including the transgenic expression selectively in T cells of a mutated form of IκBα that cannot be degraded (IκBα∆N-Tg mice) [13], the conditional deletion of IKKβ (CD4-cre x IKKβfl/fl mice) [14] and the elimination of CARMA1 expression (CARMA1-KO mice) [15-17]. T cells from the first 2 strains have impaired NF-κB activation not only downstream of the TCR, but also of other receptors that activate NF-κB in T cells, such as tumor necrosis factor receptor (TNFR) family members and Toll-like receptor (TLR) family members. In contrast, TCR-dependent but not TNFR- or TLR-dependent NF-κB signaling is absent in CARMA1-KO T cells. Using these mice, our group and others have shown that T cell-NF-κB plays a role in the proliferation and survival of T cells. Because of its requirement in cell-cycle progression, T cell-NF-κB is important for Th1 and Th17 differentiation; however, if proliferation is rescued, Th1 differentiation can proceed whereas T cell-NF-κB controls Th17 differentiation at an additional downstream checkpoint, by enabling accessibility of the IL-17 locus [18-22]. Whereas T cell NF-κB is required for the thymic development of natural Tregs [23-27], and c-Rel can play a modest role in the differentiation of peripherally induced Tregs (iTregs) [25-27], T cell-NF-κB can antagonize iTreg differentiation when strongly induced at high antigen doses [28]. In vivo, the role of T cell-NF-κB has proven complex. For instance, its importance in the rejection of transplanted organs was dependent on the tissue origin of the graft. While IκBα∆N-Tg and CARMA1-KO mice failed to reject fully mismatched cardiac allografts and displayed delayed rejection of islet allografts, they successfully rejected skin allografts with only slightly delayed kinetics [21,29,30]. Given these pleiotropic and complex functions of T cell-NF-κB, its role in tumor control cannot be easily anticipated.
In the current study, we investigated whether normal activation of NF-κB in T cells is required for the rejection of tumors that are normally controlled by wildtype mice. Because IκBα∆N-Tg mice have defects in thymic selection resulting in reduced numbers of thymic and peripheral T cells perhaps due to the early expression of the transgene driven by the Lck promoter, CD4-cre x IKKβfl/fl and CARMA1-KO mice were employed to address this question, as they have closer to normal numbers of CD4+ and CD8+ conventional T cells. We found that mice with impaired T cell-NF-κB activity were unable to reject tumors that were otherwise eliminated by wildtype mice, and specific impairment of NF-κB signaling downstream of the TCR was sufficient to preclude anti-tumor responses. Reduced T cell-NF-κB activity did not reduce expansion of tumor-specific T cells, but resulted in decreased production of tumor antigen-specific IFN-γ and TNF-α, as well as diminished cytotoxicity against tumor antigen-expressing cells in vivo. Thus, T cell-NF-κB is required for the successful elimination of tumors. Our data identify this signaling pathway as a potential therapeutic target to enhance anti-tumor immunity in cancer patients.
Results
Rejection of MC57-SIY is dependent on T cell-IKKβ
To determine whether T cell-NF-κB signaling was required for tumor rejection, we examined the ability of T cell-NF-κB-impaired mice to reject a transplantable tumor. To this end, we utilized CD4-cre x IKKβfl/fl mice, the T cells from which lack the kinase IKKβ that is required for the phosphorylation of IκB and consequent nuclear translocation of NF-κB molecules. In these mice, cre-mediated deletion of IKKβ occurs at the double positive stage of thymocyte development, resulting in absence of IKKβ from all peripheral CD4+ and CD8+ αβT cells. CD4-cre x IKKβfl/fl mice have been previously described [14,31], and their T cells displayed normal survival and activation by polyclonal stimuli in vitro, expanded efficiently in response to superantigen administration in vivo, but were deficient in recall responses, help for germinal center formation, and lymphopenia-induced homeostatic proliferation [31,32]. CD4-cre x IKKβfl/fl mice and littermate controls (CD4-cre x IKKβ+/fl) were subcutaneously injected with 106 MC57-SIY tumor cells, a mouse fibrosarcoma cell line engineered to express the model peptide antigen SIYRYYGL (SIY). This transplantable tumor is spontaneously rejected by wildtype mice [33]. While littermate controls successfully rejected the MC57-SIY tumors, CD4-cre x IKKβfl/fl mice did not (Figure 1a). To determine if this was due to an inability of CD4-cre x IKKβfl/fl mice to mount any kind of immune response when antigens were present in a cutaneous location, we examined whether they could reject skin grafts. CD4-cre x IKKβfl/fl mice (C57BL/6 background, H-2b) successfully rejected a fully mismatched BALB/c (H-2d) skin allograft, albeit with slower kinetics than wildtype mice (Figure 1b). Therefore, T cell-IKKβ was necessary for elimination of MC57-SIY tumors but was not required for the complete destruction of skin allografts.
Figure 1.
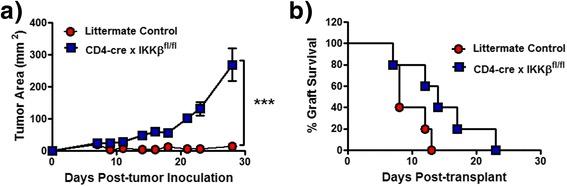
IKKβ expression in T cells is required for MC57-SIY tumor rejection. a) One million MC57-SIY tumor cells were subcutaneously injected into CD4-cre x IKKβfl/fl mice, and tumor growth was measured over time. b) Fully mismatched skin from wildtype BALB/c mice (H-2d) was transplanted into CD4-cre x IKKβfl/fl mice (H-2b). Results are representative of at least 2 experiments. ***p < 0.001.
Initial T cell priming is unimpaired in CD4-cre x IKKβfl/fl mice
Lack of tumor rejection in CD4-cre x IKKβfl/fl mice may depend on impaired survival, proliferation, differentiation, migration or effector function of tumor-reactive T cells in vivo. To investigate the fate of anti-tumor T cells, we used fluorescently labeled pentamers of the MHC class I Kb molecule coupled to the SIY peptide to identify SIY-reactive T cells. Mice inoculated with MC57-SIY tumor cells were sacrificed on day 7, which was determined to be the height of the anti-tumor response [33], and splenocytes were analyzed by flow cytometry. Frequencies (Figure 2a) and absolute numbers (Figure 2b) of SIY-specific T cells were similar between CD4-cre x IKKβfl/fl mice and littermate controls, suggesting similar expansion and survival of SIY-specific CD8+ T cells following tumor exposure regardless of the presence or absence of IKKβ.
Figure 2.
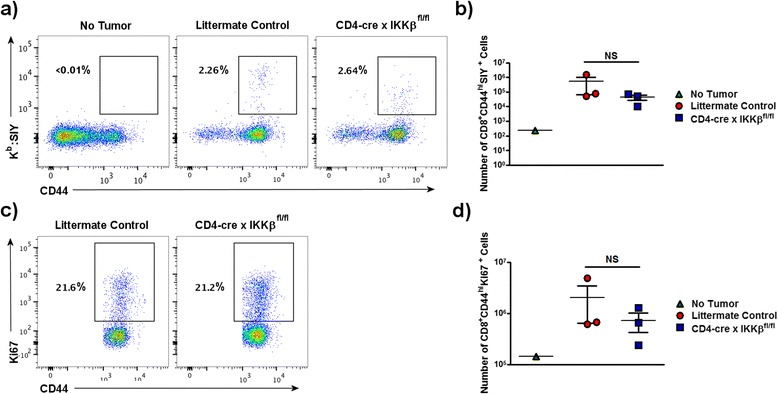
Normal T cell priming in CD4-cre x IKKβ fl/fl mice. CD4-cre x IKKβfl/fl mice were subcutaneously injected with 106 MC57-SIY tumor cells, were sacrificed on day 7 and splenocytes were prepared for flow cytometry. Graphical representation (a and c) and absolute numbers (b and d) of SYI:Kb+ tumor-specific CD8+ T cells (a and b) and of Ki67+ proliferating CD8+CD44hi cells (c and d). Results are representative of at least 2 independent experiments. NS = not significant.
Because SIY is a model antigen, we also compared the proliferation of all activated CD8+ T cells that should also contain T cells reactive to non-SIY tumor antigens as well as tumor non-specific T cells. Splenocytes isolated 7 days following tumor inoculation were gated on CD8+CD44hi populations and expression of Ki67, a nuclear protein induced during the active phases of the cell cycle, was assessed. As shown in Figures 2c and 2d, the proportion and total number of proliferating CD8+ T cells increased similarly following tumor injection in control and CD4-cre x IKKβfl/fl T cells, indicating that the initial T cell priming and cell survival following tumor implantation occurred normally despite lack of T cell-IKKβ.
T cell-IKKβ is required for anti-tumor effector function
To determine if a differentiation step downstream of T cell proliferation was affected by lack of IKKβ, IFN-γ production upon tumor antigen re-challenge in vitro was measured by ELISpot in splenocytes harvested 7 days post-tumor injection. Fewer CD4-cre x IKKβfl/fl than wildtype splenocytes secreted IFN-γ upon restimulation with irradiated MC57-SIY tumor cells (Figure 3a). Additionally, the production of IFN-γ from CD4-cre x IKKβfl/fl mice was reduced on a per-cell basis compared to littermate controls, as assessed by mean ELISpot size (Figure 3b).
Figure 3.
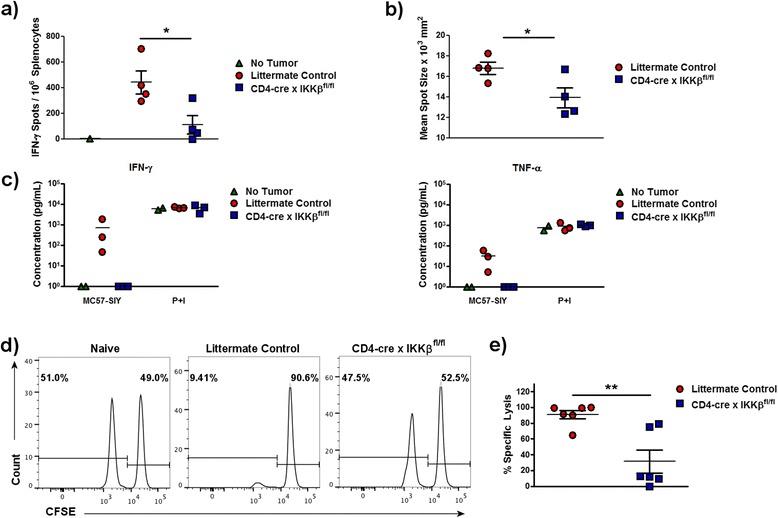
T cell-IKKβ activity is required for anti-tumor effector function. CD4-cre x IKKβfl/fl and littermate control mice were subcutaneously injected with 106 MC57-SIY tumor cells and sacrificed 7 days later. a) Splenocytes were restimulated in vitro with γ-irradiated MC57-SIY tumor cells, and frequency of tumor-specific IFN-γ-secreting cells was determined by ELISpot. b) Mean spot size (from a) was used to evaluate amount of IFN-γ secretion on a per-cell basis. Results are representative of 2 experiments. c) Quantification of soluble IFN-γ and TNF-α from CD8+ splenocytes restimulated in vitro with γ-irradiated MC57-SIY tumor cells or PMA + ionomycin, as assessed by cytokine bead array. d) Mice bearing MC57-SIY tumors for 7 days were injected with a 1:1 ratio of CFSE-labeled cells loaded with (CFSElow) or without (CFSEhigh) SIY peptide. Eighteen hours later, mice were sacrificed and the presence of the target cells was assessed by flow cytometry. Results are representative of at least 2 experiments. e) Specific lysis of SIY-specific cells was calculated using the ratio of the transferred populations as described in Materials and Methods. Results combine 2 independent experiments. *p < 0.05, **p < 0.01.
Because cells other than T cells can produce IFN-γ, and to determine if CD8+ T cells specifically had impaired cytokine production in CD4-cre x IKKβfl/fl tumor-bearing mice, CD8+ T cells were enriched from the spleen on day 7 post-tumor implantation using magnetic bead-based negative selection, and restimulated with irradiated tumor cells or PMA + ionomycin. Cytokine secretion was assessed using a cytokine bead array. CD8+ T cells from CD4-cre x IKKβfl/fl mice had dramatically reduced IFN-γ and TNF-α production in response to tumor cell re-stimulation (Figure 3c). Surprisingly, these cells retained the ability to produce IFN-γ and TNF-α in response to PMA and ionomycin (Figure 3c and see Additional file 1: Figure S1), a combination of stimuli that bypasses the TCR and also stimulates the diversity of CD8+ T cells present in the culture. These findings suggest either that activated T cells were specifically unresponsive to tumor antigens upon re-challenge, or that they had failed to differentiate into IFN-γ- or TNF-α-producing cells in response to the tumor in vivo.
To further examine the extent of the functional deficiency in IKKβ-deficient tumor-specific T cells, we performed an in vivo cytotoxicity assay. CD4-cre x IKKβfl/fl mice and control littermates were injected with MC57-SIY tumor cells, and on day 7 post-inoculation, a 1:1 ratio of CFSE-labeled T cell-depleted splenocytes loaded with SIY tumor antigen (exposed to low CFSE concentration) versus left unloaded (exposed to high CFSE concentration) was transferred intravenously into each mouse. Mice were sacrificed 18 hours later and specific target lysis was calculated using the ratio of the two transferred populations evaluated by flow cytometry (Figure 3d). Compared to wildtype littermate controls, CD4-cre x IKKβfl/fl tumor-bearing mice demonstrated a very significant reduction in specific lysis (Figure 3e). Taken together, these findings suggest that, while other factors may contribute to the survival and proliferation of T cells, IKKβ expression is required to enable effector function during the anti-tumor immune response.
Rejection of the MC57-SIY tumor is dependent on NF-κB signaling downstream of the T cell receptor
NF-κB in T cells can be activated by IKKβ downstream of several receptors, including TLRs, members of the TNFR superfamily, and the TCR. To assess whether T cell-IKKβ requirement for tumor rejection was due to TLR-dependent NF-κB activation as TLRs may detect damage-associated molecular patterns released during tumorigenesis, tumor rejection in MyD88-KO mice was analyzed. MyD88 is an adaptor downstream of most TLRs and necessary for TLRs to activate NF-κB [34]. MC57-SIY tumor cells were injected into MyD88-KO mice and wildtype controls both on the C57BL/6 background, and tumor growth was measured over time. Both MyD88-KO and wildtype mice rejected the tumors (Figure 4a), indicating that TLR-dependent NF-κB signaling is not required for the rejection of MC57-SIY tumors.
Figure 4.
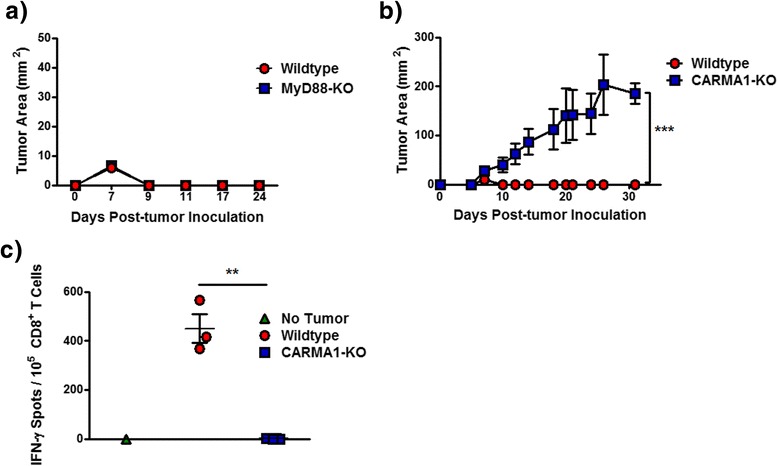
Rejection of the MC57-SIY tumor is dependent on CARMA1 but not MyD88 expression. One million MC57-SIY tumor cells were subcutaneously injected into either MyD88-KO (a) or CARMA1-KO (b) mice, and tumor growth was measured over time. c) Wildtype and CARMA1-KO mice were subcutaneously injected with 106 MC57-SIY tumor cells and sacrificed 7 days later. Enriched splenic CD8+ T cells were restimulated with γ-irradiated MC57-SIY cells and an ELISpot was used to measure the frequency of cells secreting IFN-γ. The experiment was repeated 3 times with 3–5 mice per group/experiment. **p < 0.01, ***p < 0.001.
To address whether NF-κB signaling specifically downstream of the TCR was required for tumor rejection, tumor growth was analyzed in CARMA1-KO mice that lack the key adaptor connecting the TCR to the NF-κB activating machinery. Whereas wildtype mice successfully rejected the tumors, CARMA1-KO mice failed to prevent growth of MC57-SIY tumors (Figure 4b). As observed for CD4-cre x IKKβfl/fl mice, this was not due to a complete failure of CARMA1-KO mice to mount any immune response in vivo, as we have previously shown that they successfully reject fully allogeneic BALB/c skin grafts [21]. Similarly to CD4-cre x IKKβfl/fl mice, enriched CD8+ splenic T cells from tumor-bearing CARMA1-KO mice failed to produce IFN-γ in response to re-stimulation with irradiated tumor cells (Figure 4c), indicating a failure to mount a successful anti-tumor effector response. Taken together, these findings indicate that NF-κB activation downstream of the TCR is required for the elimination of MC57-SIY tumors.
Discussion
NF-κB is activated in many cancer types and has also been associated with angiogenesis, tumor progression and metastasis [35]. For instance, we have shown that the development of acute T cell lymphoplastic leukemia driven by Notch-1 in mice depends on the ability of Notch to activate the NF-κB pathway [36]. Recently, NF-κB in parenchymal tumor cells was also shown to promote T cell recruitment in mice and humans [37]. However, the role of NF-κB within non-transformed T cells in their ability to control growth of parenchymal cancers had not been previously investigated. Our data show that both CD4-cre x IKKβfl/fl and CARMA1-KO mice fail to reject tumors that are otherwise eliminated by wildtype mice. Although the results point to the importance of T cell-NF-κB in tumor rejection, we cannot rule out that other signaling pathways downstream of IKKβ or CARMA1 may play a role in addition to, or instead of, NF-κB. Indeed, IKKβ has been shown to phosphorylate other substrates than IκB, including the Forkhead transcription factor FOXO3a, the mRNA destabilizer 14-3-3b, the insulin receptor substrate 1 and the docking protein 1 [38]. Similarly, CARMA1 has been shown to activate JNK2 in addition to NF-κB [39]. However, preliminary results indicate that IκBα∆N-Tg mice, in which T cells express a transgenic super-repressor form of IκBα, also have a defect in controlling MC57-SIY tumor growth (data not shown). The convergence of this same phenotype in 3 different genetic models, the common intersecting point being T cell-NF-κB impairment, strongly supports the hypothesis that normal activation of NF-κB in T cells is essential for tumor control in vivo. Moreover, other signaling pathways downstream of the TCR, such as NFAT or AP-1, cannot compensate for the absence of T cell-NF-κB. The tumor-controlling role of T cell-NF-κB suggests that augmenting the signaling of NF-κB may facilitate tumor rejection.
Although T cell-NF-κB has been implicated by us and by others in the survival and proliferation of conventional T cells in other model systems, our current results indicate that tumor-specific CD4-cre x IKKβfl/fl CD8+ T cells were able to expand and accumulate in tumor-bearing mice. Thus, other signaling pathways may be sufficient to support T cell proliferation and expansion in the absence of IKKβ in vivo. For instance, IKKα and IKKβ can substitute for one another for thymocyte development [40]. In addition, the lack of a defect in IKKβ-deficient T cell survival and proliferation in this tumor model allowed us to probe the role of NF-κB downstream of these early T cell priming events and identify an essential role for IKKβ in the acquisition of IFN-γ and TNF-α production by tumor-specific CD8+ T cells as well as of cytotoxicity in vivo. This observation is consistent with reports indicating that an NF-κB-binding site controls granzyme B transcription and that the c-Rel subunit of NF-κB (but not the p50 subunit) can bind to enhancer sites on the IFN-γ gene [41,42]. This impaired effector response is likely one of the mechanisms by which CD4-cre x IKKβfl/fl mice fail to reject MC57-SIY tumors, as T cell-IFN-γ and cytotoxicity are functions known to play important roles in the rejection of various growing tumors [43-45]. The fact that CD8+ T cells from CD4-cre x IKKβfl/fl tumor-bearing mice could produce IFN-γ in response to PMA/ionomycin suggests that tumor-specific IKKβ-deficient T cells may be anergic or hyporesponsive despite their initial proliferation, as anergic T cells have defects in Ras signaling which can be functionally rescued by bypassing early steps of T cell activation [46]. Alternatively, PMA/ionomycin may be eliciting IFN-γ from previously differentiated non tumor-reactive T cells, suggesting that IKKβ may be dispensable for effector differentiation in certain settings. It is conceivable that exposure of mice prior to tumor inoculation to environmental antigens in the presence of particular signals such as from pattern-recognition receptors, costimulatory molecules, or inflammatory cytokines may have enabled differentiation of a subset of T cells in the absence of IKKβ. Of note, while the expansion of CD4-cre x IKKβfl/fl CD8+ T cells was preserved in our studies, these cells displayed reduced binding to Kb:SIY fluorescent pentamers (see Figure 2a), perhaps reflecting loss of high avidity clones. Indeed, varying TCR:ligand affinity in TCRβ mutant mice has been correlated with differential NF-κB activation and distinct CD8+ T cell fate decisions [47]. Thus, tumor immune responses in mice with impaired T cell-NF-κB may be limited because of deletion or lack of expansion of clones with high affinity for tumor antigens.
T cell-NF-κB is differently required for the elimination of distinct tissues in vivo, as it is essential for the rejection of cardiac but not skin allografts, although the tempo of skin rejection is slower than in control mice [29,48]. The fate of MC57-SIY tumors appears to segregate with that of cardiac rather than skin allografts, despite the tumors being positioned in a similar location as the skin graft bed. The underlying mechanisms for the differential role of T cell-NF-κB in the rejection of the various tissues are incompletely understood, but several factors likely play a role in providing sufficient signal strength to enable T cell activation despite reduced NF-κB. These may include differences in antigen load, site and quality of initial T cell priming and types of APCs present in the different tissues. For instance, we have previously shown that skin Langerhans cells but not splenic dendritic cells can activate NF-κB-impaired T cells and that transfer of donor-matched Langerhans cells is sufficient to drive rejection of cardiac allografts in T cell-NF-κB-impaired mice [49]. Whether absence of tumor control in mice with impaired T cell-NF-κB is due to these tumors lacking potent immune-stimulatory APCs remains to be demonstrated, but is consistent with the reduced T cell effector function observed in these mice.
NF-κB in T cells can be activated downstream of several surface receptors, including the TCR, most TLRs, and all TNFR family members [50]. The fact that CARMA1-KO but not MyD88-KO mice fail to reject MC57-SIY tumors suggest that TCR-NF-κB but not TLR-NF-κB plays a role in tumor control. TNFR family members are mostly expressed on activated rather than naïve T cells and include HVEM, CD27, CD30, OX40 and 4-1BB [51]. Engagement of these receptors by their respective soluble or membrane-bound ligands serves to costimulate activated T cells and results in TRAF-dependent activation of NF-κB [51-53], which is prevented by lack of expression of IKKβ but not of CARMA1. Whether NF-κB activity downstream of TNFR family members also plays a role in T cell-mediated spontaneous control of tumor growth remains to be investigated. Agonistic antibodies to 4-1BB, OX40 and CD27 that are immune-stimulatory are being tested as potential immunotherapies for cancer patients and the efficacy of anti-4-1BB pre-clinically has been shown to depend on TRAF2-mediated NF-κB activation [54].
Our data suggest that T cell-NF-κB is essential for the spontaneous rejection of transplantable tumors. It remains to be tested if it also plays a role in tumor surveillance and elimination of nascent autochthonous tumors. CD4-cre x IKKβfl/fl or CARMA1-KO mice do not appear to develop spontaneous tumors as they age, but this may be because the animals are kept in specific pathogen-free facilities and are not exposed to oncogeneic viruses or environmental carcinogens which may be necessary to drive tumor initiation [55]. Whether inhibition of T cell-NF-κB accelerates the development of spontaneous genetic or carcinogen-induced tumors, or prevents the efficacy of current immunotherapies such as blockade of the CTLA-4/B7 or PD-1/PDL1 pathways will be of interest to study in the future. In particular, we have previously shown that CTLA-4 and PD-1 inhibit TCR-mediated NF-κB activity [56,57] such that their blockade in vivo may conceivably function through enhancement of NF-κB signaling.
Conclusions
T cell-NF-κB plays an important role in tumor control, indicating that reduced T cell-NF-κB observed in tumor-bearing hosts can be a cause of tumor progression and that other transcription factors cannot compensate in vivo for deficient NF-κB activity in T cells. These data suggest that enhancing this axis either genetically or with immunotherapies such as agonistic antibodies against TNFR family members are important therapeutic considerations. Based on our findings, we propose that the maintenance of NF-κB activity in T cells could facilitate tumor rejection by supporting T cell effector function, specifically pro-inflammatory cytokine secretion and specific cytotoxicity. It will be important to design such therapies to target only T cells, as enhancing NF-κB in cancer cells could contribute to tumor progression.
Methods
Animal models
C57BL/6 and BALB/c mice were obtained from Harlan Laboratories (Indianapolis, IN). CD4-cre mice, purchased from Jackson Laboratories [017336; B6.Cg-Tg(Cd4-cre)1Cwi/BfluJ], express the cre recombinase under the control of the CD4 promoter and have been previously described [58,59]. IKKβfl/fl mice, which express a loxP-flanked IKKβ locus and have been previously described [60], were generously provided by Dr. Michael Karin (University of California San Diego, San Diego, CA). CD4-cre mice were crossed with IKKβfl/fl mice in house (CD4-cre x IKKβfl/fl mice). CARMA1-KO mice were originally generated on the 129 background [16], were a gift from Dr. Daniel Littman (New York University, New York, NY) and were backcrossed for 6–10 generations onto the C57BL/6 background at the University of Chicago specific pathogen-free (SPF) barrier facility. MyD88-KO mice are deficient in the adaptor molecule MyD88 in all tissues [61] and were a gift from Dr. Alexander Chervonsky (University of Chicago, Chicago, IL). All mice were on the C57BL/6 background and bred at the University of Chicago SPF facility in agreement with our Institutional Animal Care and Use Committee (IACUC) and according to the National Institutes of Health guidelines for animal use.
Tumor cell lines
The MC57 methylcholanthrene-induced fibrosarcoma cell line was provided by Dr. Hans Schreiber (University of Chicago, Chicago, IL). MC57-SIY was engineered to express the model antigen SIYRYYGL, which can be recognized by CD8+ T cells in the context of H2-Kb [33].
Tumor challenge and measurement
Tumor cells were washed and resuspended in PBS at a concentration of 107 cells/mL. A volume of 0.1 mL (106 tumor cells) was injected subcutaneously into the right flank of each mouse. Tumors were measured with calipers by a single investigator, and tumor size was calculated as the product of the greatest tumor diameter length and its perpendicular width.
Skin transplantation
The tail skin of euthanized BALB/c (H-2d) donor mice was cleaned with 70% ethanol. 1×1 cm segments of skin were removed with sterile scissors and attached onto a Petri dish maintained at 4°C, where they were kept moist with a sterile gauze soaked in 0.9% NaCl. C57BL/6 (H-2b) recipient mice were anesthetized with ketamine/xylazine. The surgical site was shaved and disinfected with povidone-iodine. Using sterile curved scissors, an area of flank skin was removed. The donor skin graft was placed onto the prepared graft bed, and secured with two Bandaids. After 7 days, the adhesive Bandaids were removed. Graft survival was assessed daily and rejection was defined as greater than 80% of the skin becoming necrotic.
Splenocyte isolation
Spleens were homogenized in hypotonic ammonium chloride potassium (ACK) lysis buffer for 5 minutes to lyse red blood cells. Cells were resuspended in Dulbecco’s Modified Eagle Medium (DMEM, Invitrogen) supplemented with 5% fetal bovine serum (FBS; HyClone, Logan, UT), penicillin (100 U/mL), streptomycin (100 μg/mL), HEPES buffer (50 μM), β-mercaptoethanol (50 μM), and non-essential amino acids (1% final volume) (complete DMEM). Cells were filtered through cell strainers, centrifuged, and resuspended in complete DMEM. Live cells were counted with the Countess Automated Cell Counter (Invitrogen), using the trypan blue exclusion method.
T cell enrichment
CD8+ T cells were enriched by negative selection using magnetic beads according to the instructions of the manufacturer (Stem Cell, Vancouver, Canada). Purity of T cells was verified in each experiment to be equal or superior to 85%.
Flow cytometry and pentamer staining
Flow cytometric analyses were performed on single-cell suspensions stained in FACS buffer (PBS, 1% BSA, and 0.01% NaN3). Cells were then washed, resuspended in FACS buffer, and immediately analyzed by flow cytometry. Biotinylated pentamers of Kb complexed to SIY peptide (ProImmune, Oxford, U.K.) were used according to the manufacturer’s protocol and coupled to streptavidin-PE. Cells were labeled with PE-, PE-Cy7-, APC-, APC-Cy7-, or FITC- conjugated antibodies. The antibodies used targeted murine CD4, CD8α or CD8β, B220, or CD44. These antibodies were obtained from BD Biosciences (San Jose, CA), eBioscience (San Diego, CA), or Biolegend (San Diego, CA). In certain experiments, cells were stained with 7-AAD obtained from Invitrogen (Carlsbad, CA). For proliferation assays, cells were permeabilized, fixed and stained with APC-conjugated Ki67 according to the manufacturer’s instructions (BD Biosciences). Samples were acquired using Accuri, FACSCanto, or LSR Fortessa (BD Biosciences) flow cytometers. Data were analyzed using FlowJo software (TreeStar).
ELISpot assay
The mouse IFN-γELISpot assay was conducted using the BD Bioscience (San Jose, CA) kit according to the manufacturer’s protocol. ELISpot plates were coated with anti-mouse IFN-γ antibody and stored overnight at 4°C. Plates were then washed and blocked with DMEM supplemented with 10% FBS for 2 hours at room temperature. Splenocytes (106 cells/well) or enriched CD8+ T cells (2 × 105 cells/well) were plated. Stimulation was performed with irradiated MC57-SIY tumor cells (20,000 rad) or PMA (50 ng/ml) and ionomycin (0.5 μg/ml) as a positive control. Plates were stored at 37°C in a 7.5% CO2 incubator overnight, washed, and coated with detection antibody for 2 hours at room temperature. They were again washed and coated with avidin-peroxidase for 1 hour at room temperature. Plates were washed and developed by addition of 3-amino-9-ethylcarbazole (AEC) substrate. Developed plates were dried overnight, read using an ImmunoSpot Series 3 Analyzer, and analyzed with ImmunoSpot software.
Cytokine bead array
The mouse cytokine bead array was conducted using the Bio-Rad Laboratories Bio-Plex Pro™ Mouse Cytokine Th17 Panel A 6-Plex Group (Hercules, CA) kit according to the manufacturer’s protocol. Mice were challenged for 7 days with 106 MC57-SIY tumor cells. CD8+ T cells were then isolated and restimulated with irradiated tumor cells or PMA + ionomycin. Supernatants were collected, and cytokine concentration was assessed. In addition, supernatants from PMA + ionomycin-stimulated samples were serially diluted and analyzed for IFN-γ content by ELISA to ensure that oversaturation of the assay was not masking differences between the samples.
Cytotoxicity assay
Naïve syngeneic mice were sacrificed, and single-cell suspensions of whole splenocytes were prepared. T cells were depleted from splenocytes by negative selection for CD4+ and CD8+ cells using magnetic beads according to the instructions of the manufacturer (Stem Cell, Vancouver, Canada). Half of the remaining splenocytes were incubated with SIY peptide at a concentration of 1 μM at 37°C for one hour. All cells were washed. Peptide-loaded cells were stained with 0.5 μM CFSE, and remaining splenocytes were stained with 5 μM CFSE. Cells were counted using the Accuri flow cytometer, and equal populations of cells were intravenously transferred into tumor-bearing mice one week post-tumor inoculation. The day after transfer, tumor-bearing mice were sacrificed, and populations of transferred cells were analyzed by flow cytometry. Specific lysis was calculated using the formula, where .
Statistical analyses
Comparisons of means were performed with GraphPad Prism (GraphPad Software) using the two-way ANOVA where appropriate with Bonferroni’s correction for multiple comparisons. Where appropriate, comparisons of means were performed with InStat software using the t-test. Differences were considered significant for p values <0.05.
Acknowledgements
The authors would like to acknowledge Michelle Miller for help with data interpretation, statistical analysis, and revision of the manuscript. We would also like to thank Drs. Michael Karin (UC San Diego), Alexander Chervonsky (U of Chicago) and Daniel Littman (NYU) for donating mice that were essential to the completion of this study. TFG was supported by RO1 CA127475 and a Team Science Award from the Melanoma Research Alliance. CE was supported by a T32 post-doctoral fellowship from the University of Chicago Committee on Cancer Biology and from Bolsa SFRH/BPD/80353/2011 from the Portuguese Fundaçao para a Ciencia e Tecnologia. MLA was supported by a pilot grant from the University of Chicago’s Comprehensive Cancer Center and from NIH/NIAID RO1 AI052352. This work was also supported by Cancer Center Support Grant #CA014599.
Additional file
{kind=link}
Stimulation with PMA + ionomycin elicits similar IFN-γ production from control and IKKβ-deficient CD8+ T cells. Supernatants from Figure 3c were serially diluted and the concentration of IFN-γ was measured by ELISA.
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
SEB participated in the design of the study and interpretation of data, carried out tumor growth experiments and functional assays, and performed statistical analyses. YW carried out the skin transplant experiments. LLM participated in the design and discussion of the experiments. TFG, CE and MA conceived the study, participated in its design and coordination, and edited the manuscript. All authors read and approved the final manuscript.
Authors’ information
Cesar Evaristo and Maria-Luisa Alegre are co-last authors
Contributor Information
Sarah E Barnes, Email: sebarnes@uchicago.edu.
Ying Wang, Email: yingwang@medicine.bsd.uchicago.edu.
Luqiu Chen, Email: lchen@medicine.bsd.uchicago.edu.
Luciana L Molinero, Email: molinero.luciana@gene.com.
Thomas F Gajewski, Email: tgawjewsk@medicine.bsd.uchicago.edu.
Cesar Evaristo, Email: cesar.evaristo@gmail.com.
Maria-Luisa Alegre, Email: malegre@medicine.bsd.uchicago.edu.
References
- 1.Coulie PG, Van den Eynde BJ, van der Bruggen P, Boon T. Tumour antigens recognized by T lymphocytes: at the core of cancer immunotherapy. Nat Rev Cancer. 2014;14(2):135–146. doi: 10.1038/nrc3670. [DOI] [PubMed] [Google Scholar]
- 2.Chakraborty AK, Weiss A. Insights into the initiation of TCR signaling. Nat Immunol. 2014;15(9):798–807. doi: 10.1038/ni.2940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Abe BT, Shin DS, Mocholi E, Macian F. NFAT1 supports tumor-induced anergy of CD4(+) T cells. Cancer Res. 2012;72(18):4642–4651. doi: 10.1158/0008-5472.CAN-11-3775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chow CW, Rincon M, Davis RJ. Requirement for transcription factor NFAT in interleukin-2 expression. Mol Cell Biol. 1999;19(3):2300–2307. doi: 10.1128/mcb.19.3.2300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Xiao G, Deng A, Liu H, Ge G, Liu X. Activator protein 1 suppresses antitumor T-cell function via the induction of programmed death 1. Proc Natl Acad Sci U S A. 2012;109(38):15419–15424. doi: 10.1073/pnas.1206370109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Uzzo RG, Rayman P, Kolenko V, Clark PE, Cathcart MK, Bloom T, Novick AC, Bukowski RM, Hamilton T, Finke JH. Renal cell carcinoma-derived gangliosides suppress nuclear factor-kappaB activation in T cells. J Clin Invest. 1999;104(6):769–776. doi: 10.1172/JCI6775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Broderick L, Brooks SP, Takita H, Baer AN, Bernstein JM, Bankert RB. IL-12 reverses anergy to T cell receptor triggering in human lung tumor-associated memory T cells. Clin Immunol. 2006;118(2–3):159–169. doi: 10.1016/j.clim.2005.09.008. [DOI] [PubMed] [Google Scholar]
- 8.Simpson-Abelson MR, Loyall JL, Lehman HK, Barnas JL, Minderman H, O’Loughlin KL, Wallace PK, George TC, Peng P, Kelleher RJ, Jr, Odunsi K, Bankert RB. Human ovarian tumor ascites fluids rapidly and reversibly inhibit T cell receptor-induced NF-kappaB and NFAT signaling in tumor-associated T cells. Cancer Immun. 2013;13:14. [PMC free article] [PubMed] [Google Scholar]
- 9.Correa MR, Ochoa AC, Ghosh P, Mizoguchi H, Harvey L, Longo DL. Sequential development of structural and functional alterations in T cells from tumor-bearing mice. J Immunol. 1997;158(11):5292–5296. [PubMed] [Google Scholar]
- 10.Ghosh P, Sica A, Young HA, Ye J, Franco JL, Wiltrout RH, Longo DL, Rice NR, Komschlies KL. Alterations in NF kappa B/Rel family proteins in splenic T-cells from tumor-bearing mice and reversal following therapy. Cancer Res. 1994;54(11):2969–2972. [PubMed] [Google Scholar]
- 11.Bonizzi G, Karin M. The two NF-kappaB activation pathways and their role in innate and adaptive immunity. Trends Immunol. 2004;25(6):280–288. doi: 10.1016/j.it.2004.03.008. [DOI] [PubMed] [Google Scholar]
- 12.Roche MI, Ramadas RA, Medoff BD. The role of CARMA1 in T cells. Crit Rev Immunol. 2013;33(3):219–243. doi: 10.1615/CritRevImmunol.2013007056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Boothby MR, Mora AL, Scherer DC, Brockman JA, Ballard DW. Perturbation of the T lymphocyte lineage in transgenic mice expressing a constitutive repressor of nuclear factor (NF)-kappaB. J Exp Med. 1997;185:1897–1907. doi: 10.1084/jem.185.11.1897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schmidt-Supprian M, Courtois G, Tian J, Coyle AJ, Israel A, Rajewsky K, Pasparakis M. Mature T cells depend on signaling through the IKK complex. Immunity. 2003;19(3):377–389. doi: 10.1016/S1074-7613(03)00237-1. [DOI] [PubMed] [Google Scholar]
- 15.Jun JE, Wilson LE, Vinuesa CG, Lesage S, Blery M, Miosge LA, Cook MC, Kucharska EM, Hara H, Penninger JM, Domashenz H, Hong NA, Glynne RJK, Nelms KA, Goodnow CC. Identifying the MAGUK protein Carma-1 as a central regulator of humoral immune responses and atopy by genome-wide mouse mutagenesis. Immunity. 2003;18(6):751–762. doi: 10.1016/S1074-7613(03)00141-9. [DOI] [PubMed] [Google Scholar]
- 16.Egawa T, Albrecht B, Favier B, Sunshine MJ, Mirchandani K, O’Brien W, Thome M, Littman DR. Requirement for CARMA1 in antigen receptor-induced NF-kappa B activation and lymphocyte proliferation. Curr Biol. 2003;13(14):1252–1258. doi: 10.1016/S0960-9822(03)00491-3. [DOI] [PubMed] [Google Scholar]
- 17.Hara H, Wada T, Bakal C, Kozieradzki I, Suzuki S, Suzuki N, Nghiem M, Griffiths EK, Krawczyk C, Bauer B, D’Acquisto F, Ghosh S, Yeh WC, Baier G, Rottapel R, Penninger JM. The MAGUK family protein CARD11 is essential for lymphocyte activation. Immunity. 2003;18(6):763–775. doi: 10.1016/S1074-7613(03)00148-1. [DOI] [PubMed] [Google Scholar]
- 18.Aronica MA, Mora AL, Mitchell DB, Finn PW, Johnson JE, Sheller JR, Boothby MR. Preferential role for NF-kappa B/Rel signaling in the type 1 but not type 2 T cell-dependent immune response in vivo. J Immunol. 1999;163(9):5116–5124. [PubMed] [Google Scholar]
- 19.Mora A, Youn J, Keegan A, Boothby M. NF-kappa B/Rel participation in the lymphokine-dependent proliferation of T lymphoid cells. J Immunol. 2001;166(4):2218–2227. doi: 10.4049/jimmunol.166.4.2218. [DOI] [PubMed] [Google Scholar]
- 20.Mora AL, Corn RA, Stanic AK, Goenka S, Aronica M, Stanley S, Ballard DW, Joyce S, Boothby M. Antiapoptotic function of NF-kappaB in T lymphocytes is influenced by their differentiation status: roles of Fas, c-FLIP, and Bcl-xL. Cell Death Differ. 2003;10(9):1032–1044. doi: 10.1038/sj.cdd.4401257. [DOI] [PubMed] [Google Scholar]
- 21.Molinero LL, Cubre A, Mora-Solano C, Wang Y, Alegre ML. T cell receptor/CARMA1/NF-kappaB signaling controls T-helper (Th) 17 differentiation. Proc Natl Acad Sci U S A. 2012;109(45):18529–18534. doi: 10.1073/pnas.1204557109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stanic AK, Bezbradica JS, Park JJ, Matsuki N, Mora AL, Van Kaer L, Boothby MR, Joyce S. NF-kappaB controls cell fate specification, survival, and molecular differentiation of immunoregulatory natural T lymphocytes. J Immunol. 2004;172(4):2265–2273. doi: 10.4049/jimmunol.172.4.2265. [DOI] [PubMed] [Google Scholar]
- 23.Molinero LL, Yang J, Gajewski T, Abraham C, Farrar MA, Alegre ML. CARMA1 controls an early checkpoint in the thymic development of FoxP3+ regulatory T cells. J Immunol. 2009;182(11):6736–6743. doi: 10.4049/jimmunol.0900498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Barnes MJ, Krebs P, Harris N, Eidenschenk C, Gonzalez-Quintial R, Arnold CN, Crozat K, Sovath S, Moresco EM, Theofilopoulos AN, Beutler B, Hoebe K. Commitment to the Regulatory T Cell Lineage Requires CARMA1 in the Thymus but Not in the Periphery. PLoS Biol. 2009;7(3):e51. doi: 10.1371/journal.pbio.1000051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Isomura I, Palmer S, Grumont RJ, Bunting K, Hoyne G, Wilkinson N, Banerjee A, Proietto A, Gugasyan R, Wu L, McNally A, Steptoe RJ, Thomas R, Shannon MF, Gerondakis S. c-Rel is required for the development of thymic Foxp3+ CD4 regulatory T cells. J Exp Med. 2009;206(13):3001–3014. doi: 10.1084/jem.20091411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ruan Q, Kameswaran V, Tone Y, Li L, Liou HC, Greene MI, Tone M, Chen YH. Development of Foxp3(+) regulatory t cells is driven by the c-Rel enhanceosome. Immunity. 2009;31(6):932–940. doi: 10.1016/j.immuni.2009.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Visekruna A, Huber M, Hellhund A, Bothur E, Reinhard K, Bollig N, Schmidt N, Joeris T, Lohoff M, Steinhoff U. c-Rel is crucial for the induction of Foxp3(+) regulatory CD4(+) T cells but not T(H)17 cells. Eur J Immunol. 2010;40(3):671–676. doi: 10.1002/eji.200940260. [DOI] [PubMed] [Google Scholar]
- 28.Molinero LL, Miller ML, Evaristo C, Alegre ML. High TCR stimuli prevent induced regulatory T cell differentiation in a NF-kappaB-dependent manner. J Immunol. 2011;186(8):4609–4617. doi: 10.4049/jimmunol.1002361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhou P, Hwang KW, Palucki DA, Guo Z, Boothby M, Newell KA, Alegre ML. Impaired NF-kB activation in T cells permits tolerance to primary heart allografts and to secondary donor skin grafts. Amer J Transplant. 2003;3:139–147. doi: 10.1034/j.1600-6143.2003.00033.x. [DOI] [PubMed] [Google Scholar]
- 30.Porras DL, Wang Y, Zhou P, Molinero LL, Alegre ML. Role of T-cell-specific nuclear factor kappaB in islet allograft rejection. Transplantation. 2012;93(10):976–982. doi: 10.1097/TP.0b013e31824d11d7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schmidt-Supprian M, Tian J, Ji H, Terhorst C, Bhan AK, Grant EP, Pasparakis M, Casola S, Coyle AJ, Rajewsky K. I kappa B kinase 2 deficiency in T cells leads to defects in priming, B cell help, germinal center reactions, and homeostatic expansion. J Immunol. 2004;173(3):1612–1619. doi: 10.4049/jimmunol.173.3.1612. [DOI] [PubMed] [Google Scholar]
- 32.Silva A, Cornish G, Ley SC, Seddon B. NF-kappaB signaling mediates homeostatic maturation of new T cells. Proc Natl Acad Sci U S A. 2014;111(9):E846–E855. doi: 10.1073/pnas.1319397111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Spiotto MT, Yu P, Rowley DA, Nishimura MI, Meredith SC, Gajewski TF, Fu YX, Schreiber H. Increasing tumor antigen expression overcomes “ignorance” to solid tumors via crosspresentation by bone marrow-derived stromal cells. Immunity. 2002;17(6):737–747. doi: 10.1016/S1074-7613(02)00480-6. [DOI] [PubMed] [Google Scholar]
- 34.Kawai T, Akira S. Signaling to NF-kappaB by Toll-like receptors. Trends Mol Med. 2007;13(11):460–469. doi: 10.1016/j.molmed.2007.09.002. [DOI] [PubMed] [Google Scholar]
- 35.Prasad S, Ravindran J, Aggarwal BB. NF-kappaB and cancer: how intimate is this relationship. Mol Cell Biochem. 2010;336(1–2):25–37. doi: 10.1007/s11010-009-0267-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vilimas T, Mascarenhas J, Palomero T, Mandal M, Buonamici S, Meng F, Thompson B, Spaulding C, Macaroun S, Alegre ML, Kee BL, Ferrando A, Miele L, Aifantis I. Targeting the NF-kappaB signaling pathway in Notch1-induced T-cell leukemia. Nat Med. 2007;13(1):70–77. doi: 10.1038/nm1524. [DOI] [PubMed] [Google Scholar]
- 37.Hopewell EL, Zhao W, Fulp WJ, Bronk CC, Lopez AS, Massengill M, Antonia S, Celis E, Haura EB, Enkemann SA, Chen DT, Beg AA. Lung tumor NF-kappaB signaling promotes T cell-mediated immune surveillance. J Clin Invest. 2013;123(6):2509–2522. doi: 10.1172/JCI67250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schmid JA, Birbach A. IkappaB kinase beta (IKKbeta/IKK2/IKBKB)--a key molecule in signaling to the transcription factor NF-kappaB. Cytokine Growth Factor Rev. 2008;19(2):157–165. doi: 10.1016/j.cytogfr.2008.01.006. [DOI] [PubMed] [Google Scholar]
- 39.Blonska M, Pappu BP, Matsumoto R, Li H, Su B, Wang D, Lin X. The CARMA1-Bcl10 signaling complex selectively regulates JNK2 kinase in the T cell receptor-signaling pathway. Immunity. 2007;26(1):55–66. doi: 10.1016/j.immuni.2006.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gerondakis S, Fulford TS, Messina NL, Grumont RJ. NF-kappaB control of T cell development. Nat Immunol. 2014;15(1):15–25. doi: 10.1038/ni.2785. [DOI] [PubMed] [Google Scholar]
- 41.Huang C, Bi E, Hu Y, Deng W, Tian Z, Dong C, Hu Y, Sun B. A novel NF-kappaB binding site controls human granzyme B gene transcription. J Immunol. 2006;176(7):4173–4181. doi: 10.4049/jimmunol.176.7.4173. [DOI] [PubMed] [Google Scholar]
- 42.Sica A, Tan TH, Rice N, Kretzschmar M, Ghosh P, Young HA. The c-rel protooncogene product c-Rel but not NF-kappa B binds to the intronic region of the human interferon-gamma gene at a site related to an interferon-stimulable response element. Proc Natl Acad Sci U S A. 1992;89(5):1740–1744. doi: 10.1073/pnas.89.5.1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kline J, Zhang L, Battaglia L, Cohen KS, Gajewski TF. Cellular and molecular requirements for rejection of B16 melanoma in the setting of regulatory T cell depletion and homeostatic proliferation. J Immunol. 2012;188(6):2630–2642. doi: 10.4049/jimmunol.1100845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mortenson ED, Park S, Jiang Z, Wang S, Fu YX. Effective anti-neu-initiated antitumor responses require the complex role of CD4+ T cells. Clin Cancer Res. 2013;19(6):1476–1486. doi: 10.1158/1078-0432.CCR-12-2522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Thiery J, Lieberman J. Perforin: a key pore-forming protein for immune control of viruses and cancer. Subcell Biochem. 2014;80:197–220. doi: 10.1007/978-94-017-8881-6_10. [DOI] [PubMed] [Google Scholar]
- 46.Fields PE, Gajewski TF, Fitch FW. Blocked Ras activation in anergic CD4+ T cells. Science. 1996;271:1276–1278. doi: 10.1126/science.271.5253.1276. [DOI] [PubMed] [Google Scholar]
- 47.Knudson KM, Hamilton SE, Daniels MA, Jameson SC, Teixeiro E. Cutting edge: The signals for the generation of T cell memory are qualitatively different depending on TCR ligand strength. J Immunol. 2013;191(12):5797–5801. doi: 10.4049/jimmunol.1300905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Finn PW, Stone JR, Boothby MR, Perkins DL. Inhibition of NF-kappaB-dependent T cell activation abrogates acute allograft rejection. J Immunol. 2001;167(10):5994–6001. doi: 10.4049/jimmunol.167.10.5994. [DOI] [PubMed] [Google Scholar]
- 49.Molinero L, Zhou P, Wang Y, Harlin H, Cosmano J, Yokota Y, Kee B, Abraham C, Alegre ML. Epidermal Langerhans cells play a major role in skin allograft rejection in mice with NF-kB-impaired T cells. Am J Transplant. 2008;8:21–31. doi: 10.1111/j.1600-6143.2007.02038.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Molinero LL, Alegre ML. Role of T cell-nuclear factor kappaB in transplantation. Transplant Rev. 2011;26(3):189–200. doi: 10.1016/j.trre.2011.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Alegre ML, Najafian N. Costimulatory molecules as targets for the induction of transplantation tolerance. Curr Mol Med. 2006;6(8):843–857. doi: 10.2174/156652406779010812. [DOI] [PubMed] [Google Scholar]
- 52.Arch RH, Thompson CB. 4-1BB and Ox40 are members of a tumor necrosis factor (TNF)-nerve growth factor receptor subfamily that bind TNF receptor-associated factors and activate nuclear factor kappaB. Mol Cell Biol. 1998;18(1):558–565. doi: 10.1128/mcb.18.1.558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Young Lee S, Yull Lee S, Kandala G, Liou M, Liou H, Choi Y. CD30/TNF receptor-associated factor interaction: NF-kB activation and binding specificity. Proc Natl Acad Sci USA. 1996;93(18):9699–9703. doi: 10.1073/pnas.93.18.9699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Martinez-Forero I, Azpilikueta A, Bolanos-Mateo E, Nistal-Villan E, Palazon A, Teijeira A, Perez-Chacon G, Morales-Kastresana A, Murillo O, Jure-Kunkel M, Zapata JM, Melero I. T cell costimulation with anti-CD137 monoclonal antibodies is mediated by K63-polyubiquitin-dependent signals from endosomes. J Immunol. 2013;190(12):6694–6706. doi: 10.4049/jimmunol.1203010. [DOI] [PubMed] [Google Scholar]
- 55.Dunn GP, Old LJ, Schreiber RD. The immunobiology of cancer immunosurveillance and immunoediting. Immunity. 2004;21(2):137–148. doi: 10.1016/j.immuni.2004.07.017. [DOI] [PubMed] [Google Scholar]
- 56.Harlin H, Hwang KW, Palucki DA, Kim O, Thompson CB, Boothby M, Alegre ML. CTLA-4 engagement regulates NF-kappaB activation in vivo. Eur J Immunol. 2002;32(8):2095–2104. doi: 10.1002/1521-4141(200208)32:8<2095::AID-IMMU2095>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 57.Qiao G, Li Z, Molinero L, Alegre ML, Ying H, Sun Z, Penninger JM, Zhang J. T-cell receptor-induced NF-kappaB activation is negatively regulated by E3 ubiquitin ligase Cbl-b. Mol Cell Biol. 2008;28(7):2470–2480. doi: 10.1128/MCB.01505-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lee PP, Fitzpatrick DR, Beard C, Jessup HK, Lehar S, Makar KW, Perez-Melgosa M, Sweetser MT, Schlissel MS, Nguyen S, Cherry SR, Tsai JK, Tucker SM, Weaver WM, Kelso A, Jaenisch R, Wilson CB. A critical role for Dnmt1 and DNA methylation in T cell development, function, and survival. Immunity. 2001;15(5):763–774. doi: 10.1016/S1074-7613(01)00227-8. [DOI] [PubMed] [Google Scholar]
- 59.Orban PC, Chui D, Marth JD. Tissue- and site-specific DNA recombination in transgenic mice. Proc Natl Acad Sci U S A. 1992;89(15):6861–6865. doi: 10.1073/pnas.89.15.6861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pasparakis M, Courtois G, Hafner M, Schmidt-Supprian M, Nenci A, Toksoy A, Krampert M, Goebeler M, Gillitzer R, Israel A, Krieg T, Rajewsky K, Haase I. TNF-mediated inflammatory skin disease in mice with epidermis-specific deletion of IKK2. Nature. 2002;417(6891):861–866. doi: 10.1038/nature00820. [DOI] [PubMed] [Google Scholar]
- 61.Adachi O, Kawai T, Takeda K, Matsumoto M, Tsutsui H, Sakagami M, Nakanishi K, Akira S. Targeted disruption of the MyD88 gene results in loss of IL-1- and IL-18-mediated function. Immunity. 1998;9(1):143–150. doi: 10.1016/S1074-7613(00)80596-8. [DOI] [PubMed] [Google Scholar]