

Abstract
The discovery that ten-eleven translocation (TET) proteins are α-ketoglutarate-dependent dioxygenases involved in the conversion of 5-methylcytosines (5-mC) to 5-hydroxymethylcytosine (5-hmC), 5-formylcytosine and 5-carboxycytosine has revealed new pathways in the cytosine methylation and demethylation process. The description of inactivating mutations in TET2 suggests that cellular transformation is in part caused by the deregulation of this 5-mC conversion. The direct and indirect deregulation of methylation control through mutations in DNA methyltransferase and isocitrate dehydrogenase (IDH) genes, respectively, along with the importance of cytosine methylation in the control of normal and malignant cellular differentiation have provided a conceptual framework for understanding the early steps in cancer development. Here, we review recent advances in our understanding of the cytosine methylation cycle and its implication in cellular transformation, with an emphasis on TET enzymes and 5-hmC. Ongoing clinical trials targeting the activity of mutated IDH enzymes provide a proof of principle that DNA methylation is targetable, and will trigger further therapeutic applications aimed at controlling both early and late stages of cancer development.
Introduction
DNA methylation on carbon 5 of cytosine is one of the best-studied epigenetic marks in mammals and is known to play crucial roles in cellular processes, including gene expression regulation and control of differentiation. However, variations in DNA methylation appear during normal differentiation and aging, and may contribute to tumorigenesis. The processes of DNA methylation and demethylation as well as enzymes involved in these epigenetic mechanisms have been outlined recently but still need further characterization. Concomitantly, direct and indirect deregulation of methylation control has been uncovered in human malignancies from both hematopoietic and non-hematopoietic origins.
Ten-eleven translocation (TET) enzymes are 2-oxoglutarate, oxygen- and iron-dependent dioxygenases able to catalyze the oxidation of 5-methylcytosine (5-mC) into 5-hydroxymethylcytosine (5-hmC) [1,2]. They have been identified as key players in cytosine demethylation and in the control of cellular differentiation and transformation. Acquired point mutations and deletion events targeting TET genes are frequently observed in human cancers. These mutations affect TET2 and to some extent TET3 and result in partial or total inactivation of the gene. Metabolic perturbations resulting from mutations in genes encoding isocitrate dehydrogenase (IDH), fumarate hydratase (FH) or succinate dehydrogenase (SDH) also inhibit the TET enzymes and, in turn, DNA demethylation. Deregulation of DNA methylation may also be achieved directly through mutations in genes encoding DNA methyltransferase (DNMT) [3,4]. We are now starting to understand the control of TET protein activity, their DNA targeting, and their crosstalk with other epigenetic marks. For example, several proteins that interact with TET proteins (such as O-linked β-D-N acetylglucosamine transferase (OGT)) and with methylated and oxidized cytosines have been identified, highlighting their function in the regulation of chromatin structure. Following the implementation of specific detection methods, much has recently been learned regarding the quantity and location of the oxidized cytosine forms, mainly in embryonic stem (ES) cells, and we are now on the verge of a more complete understanding of their functions.
In this review, we discuss the established and emerging roles of TET enzymes and their functions in cytosine demethylation, with an emphasis on methylcytosine and its oxidized forms in normal tissues. We assess the roles of TET enzymes in hematological cancers and solid tumors, focusing on mutations involved in TET inactivation. Finally, we discuss the potential translational applications.
The cytosine methylation cycle
5-mC results from the transfer of a methyl group to cytosine within a CpG dinucleotide, mediated by DNMT enzymes encoded by five genes. DNMT1 is mainly responsible for the maintenance of genomic DNA methylation patterns (that is, after DNA replication), whereas DNMT2 (or tRNA cytosine-5-methyltransferase) is an RNA methyltransferase. DNMT3A and DNMT3B are mainly responsible for de novo DNA methylation [5]. However, all three enzymes may contribute to both maintenance and de novo DNA methylation [6]. The catalytically inactive DNMT3L interacts with these enzymes and the histone 3 tail to stimulate DNA methylation [7]. Furthermore, DNMT3A has recently been identified to be involved in crosstalk with epigenetic marks independently of DNMT3L [8].
Although DNA methylation has long been recognized, and cytosine methylation by DNMT3A and DNMT3B has been shown to be reversible in vitro [9], the mechanism of DNA demethylation was unclear until the functional analyses of the TET family proteins [1,2]. Due to its poor recognition of 5-hmC, which results from TET activity, DNMT1 is not able to perform the methylation of the neo-synthetized DNA strand (maintenance methylation). So the methylation information is lost in dividing cells, in a so-called passive manner (Figure 1). The three enzymes of the TET family (TET1, TET2 and TET3) are able to further oxidize 5-hmC into 5-formylcytosine (5-fC) and then 5-carboxycytosine (5-caC) [10,11]. Thymidine DNA glycosylase (TDG) is then able to remove 5-fC and 5-caC, triggering base-excision repair (BER) activity and the reintroduction of unmethylated cytosine [11-13]. The existence of decarboxylases that convert 5-caC to unmethylated cytosine is hypothetical. It has been suggested that the deamination of 5-hmC into 5-hydroxymethyluracil (5-hmU) occurs via activation-induced deaminase (AID) and apolipoprotein B mRNA editing enzyme (APOBEC), followed by TDG and BER mechanisms [14]. However, this remains controversial because 5-hmU residues may also originate from TET-mediated oxidation of thymine [15]. In addition, the activity of recombinant AID decreases with the size of the cytosine C5 electron cloud and does not show any activity on 5-hmC in vitro [16,17]. Indeed, AID exhibits its strongest activity against unmodified cytosine. Thymine resulting from deamination of 5-mC is not easily recognized by DNA repair machinery and is considered mutagenic. These branches of the cycle need to be further investigated in a cell- and tissue-dependent context. Regardless, TET proteins as well as several other proteins (Table 1) are essential players in the demethylation of 5-mC.
Figure 1.
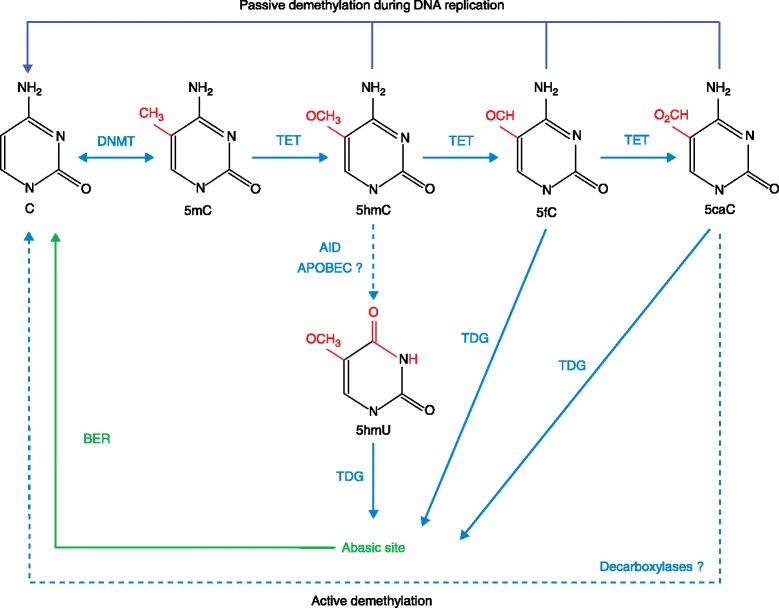
Regulation of DNA methylation and demethylation. DNA demethylation can occur spontaneously via the DNMT enzymes that methylated the nucleotide cytosine (5-methylcytosine, 5-mC) originally. A passive replication-dependent mechanism of DNA methylation is also possible. Several active demethylation pathways have been postulated. TET family proteins catalyze the oxidation of 5-mC into 5-hydroxymethylcytosine (5-hmC) and can further oxidize 5-hmC to 5-formylcytosine (5-fC) and 5-carboxycytosine (5-caC). 5-hmC recognition and transformation into 5-hydroxymethyluracyl (5-hmU) by activation-induced deaminase (AID) to facilitate repair by DNA glycosylase and the base-excision repair (BER) pathway is still controversial. These latter activities are also thought to process 5-fC and 5-caC into unmodified cytosine . The decarboxylases involved in this process are still to be identified. APOBEC, apolipoprotein B mRNA editing enzyme; DNMT, DNA methyltransferase; T, thymine; TDG, thymine DNA glycosylase; TET, ten-eleven translocation.
Table 1.
Functions and expression of human and murine proteins involved in the cytosine methylation/demethylation cycle
| Proteins | Functions | Expression levels during development and in embryonic stem cells | Expression levels in adult tissues | Expression levels in adult hematopoiesis |
|---|---|---|---|---|
| TET1 | Oxidization of 5-mC | High in mES cells, low in oocytes and zygotes [2,30,31] | Variable expression [56] | Low [83] |
| TET2 | Oxidization of 5-mC | High in mES cells, low in oocytes and zygotes [2,30,31] | Widely expressed [56] | High [83] |
| TET3 | Oxidization of 5-mC | Low in mES cells, high in oocytes and zygotes [2,30,31] | Limited expression in colon, stomach, adrenal glands and peripheral blood cells [56] | Low [83] |
| DNMT1 | Methylation maintenance during DNA replication | High in early embryonic stages [129] | Ubiquitous [130,131] | Uniform but low in neutrophils [132] |
| DNMT3A | De novo methylation | High in early embryonic stages [129] | Ubiquitous [130,131] | Uniform but high in T lymphocytes and neutrophils [132] |
| DNMT3B | De novo methylation | High in later embryonic stages and differentiated cells [129] | Low levels except testis, thyroid and bone marrow [130,131] | Low expression except in human CD34+ cells [132] |
| AID | Cytidine deamination | High in immature B cells from fetal bone marrow and liver [133] | High in lymph nodes and moderated in spleen and bone marrow [134] | Mainly in activated mature B cells [134] |
| TDG | Glycosylation and deamination | Ubiquitous from ED 7.5 to 13.5 in mouse, high in central and peripheral nervous system, thymus, lung, liver, kidney, adrenal glands and intestine at ED 14.5 [135] | Mouse aorta [98] | Not reported |
| IDH1 | Isocitrate decarboxylation of citric acid cycle | Not reported | Cytoplasm [92,103] | Not reported |
| IDH2 | Isocitrate decarboxylation of citric acid cycle | Not reported | Mitochondria [92,103] | Not reported |
| FH | Hydration of fumarate of citric acid cycle | Not reported | Mitochondria [136] | Mature erythrocytes [136] |
| SDH | Oxidation of succinate of citric acid cycle | Not reported | Mitochondria [136] | Not reported |
References listed in this table are based on mouse model studies, except for [56,103,83,130-133]. 5-mC, 5-methylcytosine; AID, activation-induced deaminase; DNMT, DNA methyltransferase; ED, embryonic day; FH, fumarate hydratase; IDH, isocitrate dehydrogenase; mES, murine embryonic stem; SDH, succinate dehydrogenase; TDG, thymine DNA glycosylase; TET, ten-eleven translocation.
DNA methylation distribution and localization
CpGs represent between 1% and 4% of genomic mammalian DNA and approximately 75% of them are methylated. Most CpGs are located in repetitive DNA elements, indicating that cytosine methylation is used as a defense mechanism against transposons and other parasitic elements to maintain the function and stability of the genome [18]. CpGs are also concentrated in CpG islands, which are mainly unmethylated and are associated with DNA accessibility and gene transcription. These CpG islands are usually found close to gene promoters, and their methylation status is strongly correlated with the transcription state of the genes nearby. Methylation can also be identified within gene bodies. It preferentially occurs in a CxG context (where x can be T, A or C) in ES cells and this intragenic methylation is mainly associated with highly expressed genes [19]. Promoter and gene body methylation are being extensively investigated to elucidate specific mechanisms and factors responsible for gene expression modulation. Recently, DNMT3B was reported to be involved in the remethylation of gene body-associated genes following treatment of a colon cancer cell line with DNMT inhibitors [20].
DNA hydroxymethylation distribution and localization
5-hmC was first identified in 1952 in bacteriophage T4 [21], and again 20 years later in the mammalian genome, in which it was found to constitute 0% to 17% of the total number of cytosine bases of brain-extracted DNA in mouse, rat and frog [22]. More recently, 5-hmC was estimated to constitute 0.6% of nucleotides in Purkinje cells, 0.2% in granule cells [1] and 0.03% in mouse ES cells [2]. However, the classical analyses of bisulfite-treated DNA do not discriminate between 5-mC and 5-hmC. Discrepancies among published studies may be due to different methodologies and analytical processes [23-26]. These studies nevertheless provide a general picture of the genome-wide distribution of modified cytosines in ES cells and other tissues.
The distribution of 5-hmC differs in several organs and tissues in mouse [27] and human [28]. The 5-hmC content also varies during development and cell differentiation [29]. For example, pluripotency correlates with high levels of 5-hmC, as observed in the inner cell mass, in multipotent adult stem cells as well as in progenitor cells. Embryonic or induced pluripotent stem cells also show a high 5-hmC level. Among differentiated cells, neuronal cells retain a high 5-hmC content [30,31]. In the blastocyst stage, erasure of DNA methylation and hydroxymethylation marks is followed by their re-establishment by TET proteins and subsequent de novo methylation by DNMT3A and DNMT3B. In the totipotent zygote, the paternal pronucleus shows high levels of 5-hmC [32,33] caused by genome-wide hydroxylation of 5-mC [34,35], in contrast to the low level of 5-hmC in the maternal pronucleus. This phenomenon is linked to TET3 translocation from the cytoplasm to the paternal pronucleus immediately after fertilization [36]. In addition, the maternal factor PGC7 protects 5-mC from Tet3-mediated conversion to 5-hmC by binding to maternal chromatin containing dimethylated histone H3 lysine 9 [37]. The maternal genome undergoes progressive 5-mC demethylation upon cell division. Genome-wide mapping of 5-hmC with specific antibodies or chemical labeling has enabled the design of 5-hmC distribution maps in mouse and human ES cells, as well as in neurological tissues. These studies have revealed that the 5-hmC mark is not uniformly distributed in the genome and is abundant in gene-rich euchromatin regions, particularly at promoters, exons and transcription start sites of genes expressed at low levels [38]. 5-hmC is mainly enriched in low CpG content regions and in bivalent gene promoters, characterized by both transcriptional permissive trimethylated histone H3 lysine 4 (H3K4me3) and repressive trimethylated histone H3 lysine 27 (H3K27me3) marks. Furthermore, TET2-mutated diffuse large B-cell lymphomas have been associated with a hypermethylation signature on gene promoters identified as bivalent in human ES cells [39]. More recently, 5-hmC was identified in intergenic regions in human ES cells. More specifically, 5-hmC was found in regions comprising cis-regulatory elements, such as active enhancers, with co-localization of the histone modification marks monomethylated histone H3 lysine 4 (H3K4me1) and acetylated histone H3 lysine 27 (H3K27ac), and transcription factor binding sites for pluripotency factors, such as OCT4 and NANOG, or the insulator binding protein CTCF [40,41].
Function of oxidized cytosine forms
TET proteins participate in the regulation of gene transcription through the controlled generation of 5-hmC, 5-fC and 5-caC and their subsequent recognition by protein complexes involved in modulating chromatin structure or DNA repair [42-46].
Promoter methylation is associated with the repression of gene expression in somatic cells. It is not clear yet whether specific readers of methylated DNA, such as methyl-CpG binding protein 2 (MeCP2), or methyl-binding domain (MBD) proteins are recruited to the methylated DNA and prevent the binding of transcription factors, or if they participate directly in the establishment of compact chromatin and gene repression. MBD1, MBD2 and MBD4 preferentially bind methylated DNA, in contrast to MBD3, MBD5 and MBD6 that prefer to bind to non-methylated DNA [47], although MBD5 and MBD6 associate with heterochromatin [48]. MBD4, possessing a DNA glycosylase domain, is also involved in BER following deamination events and is able to interact with mismatch repair proteins [49,50]. Methyl-CpG binding proteins were thought to be unable to bind 5-hmC [51] until recently [43], although the ability of MBD3 to specifically bind 5-hmC [45] is still controversial [44]. The DNA damage control proteins UHRF1 and UHRF2 also have 5-mC binding ability through their SET- and RING-associated domains. Additionally, UHRF1 is able to bind hemimethylated DNA and recruit DNMT1 [52,53]; it has recently been proposed that it may also be able to bind both methylated and hydroxymethylated DNA [42]. However, these 5-mC and 5-hmC readers are rarely found to be mutated in cancer (Table 2).
Table 2.
Somatic mutations affecting TET genes in cancer
| Genes | Mutations in solid tumors | Mutations in hematological malignancies |
|---|---|---|
| TET genes | ||
| TET1 | Rare in endometrioid carcinomaa, colorectal carcinomas [105,137], lung [106,138,139] and bladder cancer [140] | Rare in AML [141] and CLL [142] |
| TET2 | Rare in endometrioid carcinomaa, colorectal carcinomas [105,137], melanoma and lung cancer [106,138,139] | Frequent in various cancers (see Table 3) |
| TET3 | Rare in endometrioid carcinoma and colorectal carcinomas [105,137] | Rare in CLL [142], PTCL [90] and T-ALL [143] |
| Epigenetic regulators of TET genes | ||
| Methyltransferases | ||
| DNMT1 | Rare in endometrioid carcinomaa, colorectal carcinomas [105,137], and lung cancer [106,138,139] | Rare in AML [141] |
| DNMT3A | Rare in endometrioid carcinomaa, colorectal carcinomas [105,137], lung cancer [106,138,139] and 2% in non-small cell lung cancer [144] | AML [128], MDS [145] and T-cell lymphomas [90,91] |
| Deaminase and glycosylase | ||
| AID | Rare in glioblastoma and medulloblastoma [146], endometrioid carcinomaa, colon cancer [105,137] and lung cancer [106,138,139] | T-ALL [147] |
| TDG | Rare in endometrioid carcinomaa, rare in glioblastomaa, colon cancer [104,136], lung [106,138,139] and thyroid cancer [148] | Not reported |
| Histone crosstalk regulators | ||
| OGT | Rare in endometrioid carcinomaa, colorectal carcinomas [105,137], lung cancer [106,138,139] and breast cancer [115,149] | DLBCL [150] and CLL [142] |
| IDAX | Rare in breast cancera, glioblastomaa, endometrioid carcinomaa, kidneya, colon cancer [105,137], lung cancer [106,138,139] and 1.2% in mouth and pharynx carcinoma [117] | Not reported |
| 5-mC and 5-hmC readers | ||
| MBD1 | Rare in endometrioid carcinomaa, colorectal carcinomas [105,137], lung cancer [106,138,139], breast cancer [149] and melanoma [109] | Rare in ALL [151] |
| MBD4 | Rare in endometrioid carcinomaa, colorectal carcinomas [105,137], lung cancer [106,138,139], breast cancer [115] and melanoma [109] | Rare in AML [141] |
| UHRF1 | Rare in endometrioid carcinomaa, colorectal carcinomas [105,137] and lung cancer [106,138,139] | Rare in B-ALL [152] |
| Other genes affecting TET functions | ||
| Metabolic enzymes | ||
| IDH1 | Rare in paragangliomas [153], frequent in chondrosarcomas [154], thyroid [155,156], prostate [157] and central nervous system cancers [102,158,159] | Frequent in AML [88], MDS [160], DLBCL [161] and B-ALL [162] |
| IDH2 | Rare in endometrioid carcinomaa and colorectal carcinomas [105,137], frequent in chondrosarcomas [154] and central nervous system cancers [102,158,159] | Frequent in AML [88], MDS [160] and AITL [89] |
| FH | Renal cell carcinoma [163] and paragangliomas [104] | Not reported |
| SDH | Renal cell carcinoma [164] and paragangliomas [104] | Not reported |
aCOSMIC database. Some mutations listed in this table have not been confirmed as somatic mutations. 5-mC, 5-methylcytosine; 5-hmC, 5-hydroxymethylcytosine; AID, activation-induced deaminase; AML, acute myeloid leukemia; AITL, angioimmunoblastic T-cell lymphoma; B-ALL, B-cell acute lymphoblastic leukemia; CLL, chronic lymphocytic leukemia; DLBCL, diffuse large B-cell lymphoma; DNMT, DNA methyltransferase; FH, fumarate hydratase; IDAX, Inhibition of the Dvl and Axin complex; IDH, isocitrate dehydrogenase; MBD, methyl-binding domain; MDS, myelodysplastic syndrome; OGT, O-linked β-D-N acetylglucosamine transferase; PTCL, peripheral T-cell lymphoma; SDH, succinate dehydrogenase; T-ALL, T-cell acute lymphoblastic leukemia; TDG, thymine DNA glycosylase; TET, ten-eleven translocation; UHRF, ubiquitin-like with PHD and ring finger domains.
In ES cells, the distributions of 5-fC and 5-caC resemble those of 5-hmC, with a preference for enhancers, and bivalent and silent promoters. Analyses of proteins interacting with cytosine-oxidized forms have identified glycosylase and DNA repair proteins interacting with 5-fC at a higher level compared with other cytosine forms, suggesting that 5-fC may trigger repair-associated removal [44].
TET proteins
TET1 was first identified as a rare fusion partner of the mixed lineage leukemia gene, resulting from the chromosomal translocation t(10;11)(q22;23) in acute leukemia [2,54-57]. The difference between TET proteins relies on their structure (Figure 2) but also on their distinct expression patterns: TET2 is more highly expressed in the hematopoietic system than TET3 and TET1. It is currently thought that the common and main function of TET proteins is to establish or maintain protective boundaries to prevent unwanted methylation of non-methylated regions [58]. Each TET protein may also have specific functions: for example, TET1 oxidizes 5-mC to 5-hmC, and TET2 and TET3 stimulate the removal of 5-hmC [59]. In ES cells, TET2 may preferentially act on gene bodies, and TET1 at promoters [60]. The role of TET-mediated cytosine oxidation at distal enhancers is currently being thoroughly investigated. Super enhancers (enhancer clusters) that produce enhancer-transcribed RNAs in mouse ES cells have recently been associated with H3K27ac, TET1 and a decrease in DNA methylation level at pluripotency-dedicated loci [61]. Also, a specific role for TET2 in the control of enhancer activity has been suggested in the context of murine ES cell differentiation [62]. This mechanism remains to be investigated in the context of cancer, and more specifically in hematological disorders.
Figure 2.
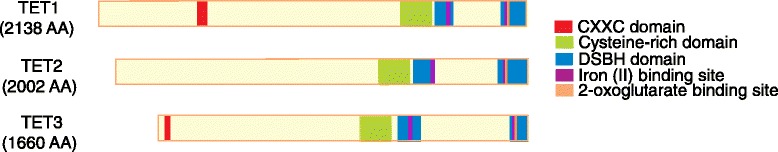
Primary structure and function of human TET proteins. All TET proteins present a double-stranded β helix (DSBH), a cysteine-rich domain, and one 2-oxoglutarate and three iron (II) binding sites in the carboxyl terminus, which constitute their dioxygenase catalytic domain. An amino-terminal CXXC zinc finger domain is only identified in TET1 and TET3, allowing these enzymes to bind DNA directly to CpG. Recently, the CXXC4 gene (also named inhibition of the Dvl and Axin complex, IDAX), located upstream of TET2 on chromosome 4, has been reported to tether TET2 to DNA through a physical interaction [65]. AA, amino acid; TET, ten-eleven translocation.
Interaction with other proteins
The stability and activity of TET proteins are regulated in several ways. Vitamin C has been reported as a cofactor that enhances the activity of TET enzymes [63,64]. The Dvl-binding protein inhibition of the Dvl and Axin complex (IDAX) can recruit TET2 to unmethylated DNA via the CXXC domain, and at the same time is able to induce its proteolytic degradation by caspase activation [65]. Other proteins interact with TET proteins, such as early B-cell factor 1 [66], or modulate their subcellular localization, such as AID [67], but it is not yet clear whether they affect TET stability and function. This is also the case for OGT, which can associate with TET proteins [68-70] but appears to differently affect the three proteins. For instance, OGT has been described to trigger the export of TET3 from the nucleus and thus impair its activity [71]. A better understanding of multiple TET functions will arise from the identification of TET partners in normal and cancerous cellular contexts.
Crosstalk with other epigenetic mechanisms
In addition to transcriptional regulation through the readers of 5-hmC, 5-fC and 5-caC, another level of transcriptional regulation mediated by TETs comes from the interplay between DNA and histone modifiers. TET1 has been shown to interact with histone deacetylases through the transcriptional corepressor SIN3 transcription regulator family member A, thereby promoting transcriptional repression [72]. TET proteins can recruit OGT enzymes to chromatin, which catalyzes the addition of O-linked β-D-N acetylglucosamine to serine and threonine within histones and other proteins. TET proteins also interact indirectly with the complex proteins associated with SET1 (COMPASS) complex, which is responsible for mono-, di- and trimethylation of histone 3 lysine 4 and is associated with active transcription. This interaction occurs through the OGT-mediated glycosylation of the COMPASS subunit host cell factor 1. The COMPASS complex of proteins is involved in the regulation of master genes, such as HOX, during development, balanced by the action of the polycomb repressive complex (PRC), which catalyzes the repressive mark H3K27me3. In addition, TET1 shares target genes with PRC2 in ES cells [73]. In conclusion, TET proteins also serve as platforms for other epigenetic activities [74].
Other TET functions
The TET family is conserved during evolution. Drosophila, for example, has one homologous gene, whose function remains undetermined because of the particular DNA methylation pattern of flies [75]. Additional TET functions might be uncovered in the future, and a recent report indicates that mammalian TET proteins may catalyze the formation of 5-hydroxymethylcytidine in vitro, suggesting a role in RNA modification [76]. Recently, TET triple knockout mouse ES cells were generated using the CRISPR/Cas9 system, suggesting a novel function of these proteins in telomere length regulation [77]. Indeed, triple knockout ES cells have an increased telomere length associated with a higher frequency of telomere-sister chromatid exchange. Although TET proteins seem to be involved in telomere shortening, their precise roles need to be further investigated in the context of both normal and cancerous cells.
TET and cancer
Here, we discuss the role of TET proteins in cancer focusing on TET2 mutations and activity impairment, first in hematopoietic malignances and then in solid tumors.
TET in hematopoietic malignancies
TET mutations
Inactivation of TET2 by genomic deletions or mutations has been reported in a wide range of adult hematological malignancies, including acute myeloid leukemia (AML), myelodysplastic syndrome (MDS) and myeloproliferative neoplasms (MPN) [78-80], as well as in lymphoid malignancies [39,81] (Table 3). In myeloid malignancies, TET2 mutations are associated with a decrease in 5-hmC levels and an increase in 5-mC levels with respect to TET2-wild-type samples [82-84]. Many TET2 acquired missense mutations have been described. Mutations that target the evolutionarily conserved catalytic domain of the protein are predicted to impair its function. Other missense mutations, occurring, for example, in the amino-terminal part of the protein, may also affect its function in an as yet uncharacterized manner. TET2 mutations are observed on only one of the two gene copies, indicating that partial inactivation of the protein may contribute to cellular transformation [78]. There are marked differences between the three TET genes in terms of their expression levels. TET2, for example, has a higher expression level in hematological cells than TET1 or TET3. TET3 expression levels are higher than TET1 levels in hematopoietic progenitor cells. Mutations in TET3 have also been described but are much less frequent, probably because of its lower expression in hematopoiesis. Regarding TET1, most of the currently described mutations are missense mutations, whose functional consequences have not been established.
Table 3.
Prevalence of TET1 , TET2 and TET3 mutations in hematological malignancies and solid tumors
| Cancer | TET1 mutation prevalence (%) | TET2 mutation prevalence (%) | TET3 mutation prevalence (%) |
|---|---|---|---|
| Myeloid malignancies | |||
| MDS | Not reported | 6-26 [78,79,165-169] | Not reported |
| MDS/MPN | |||
| CMML (adult) | Not reported | 20-58 [78,169-174] | Not reported |
| MPD | |||
| PV | Not reported | 6-16 [78,169,170] | Not reported |
| ET | Not reported | 4-5 [78,169,170] | Not reported |
| MF | Not reported | 2-17 [78,169,170] | Not reported |
| CML | Not reported | 2-4 [175,176] | Not reported |
| AML | |||
| De novo (adult) | Rare [141] | 12-27 [169,170,177-181] | Not reported |
| De novo (pediatric) | Not reported | 2-4 [182,183] | Not reported |
| Secondary AML | Not reported | 17-32 [80,85,184,185] | Not reported |
| Mastocytosis | Not reported | 20-29 [87,186] | Not reported |
| BPDCN | Not reported | 25-54 [187-189] | Not reported |
| Lymphoid malignancies | |||
| B-cell lymphoma | |||
| DLBCL | Not reported | 6-12 [39,81] | Not reported |
| MCL | Not reported | 0-4 [81,190] | Not reported |
| Follicular lymphoma | Not reported | 2 [81] | Not reported |
| CLL | Rare [142] | Not reported | Rare [142] |
| T cell lymphoma | |||
| AITL | Not reported | 33-83 [81,86,91,191,192] | Not reported |
| PTCL and PTCL, NOS | Not reported | 20-49 [81,86,91,191,192] | Rare [90] |
| T-ALL | Not reported | Not reported | Rare [143] |
| Solid tumors from | |||
| Endometrium | 9* | 7* | 4* |
| Breast | Rare [106] | Rare [115] | Rare* |
| Central nervous system | Rare [193] | Rare* | Rare* |
| Kidney | Rare* | Rare [113] | Rare* |
| Large intestine | 7 [105,137] | 4 [105,137] | 5 [105,137] |
| Liver | Rare [194] | Rare* | Rare [195] |
| Lung | 5 [106,114,138] | 2 [115,138,139] | Rare [138,139] |
| Ovary | Rare* | Rare* | Rare [112] |
| Pancreas | Rare [196,197] | Rare* | Rare* |
| Prostate | Rare [106,198] | Rare [198,199] | Rare [199] |
| Skin | Rare [109] | 1 [109] | Rare [116] |
| Stomach | 4 [200,201] | Not reported | Rare* |
| Urinary tract | 4 [140] | 4* | Rare* |
AITL, angioimmunoblastic T cell lymphoma; AML, acute myeloid leukemia; BPDCN, blastic plasmacytoid dendritic cell neoplasm; CLL, chronic lymphocytic leukemia; CML, chronic myeloid leukemia; CMML, chronic myelomonocytic leukemia; DLBCL, diffuse large B cell lymphoma; ET, essential thrombocytosis; FL, follicular lymphoma; MCL, mantle cell lymphoma; MDS, myelodysplastic syndrome; MF, myelofribrosis; MDS/MPN, myelodysplastic syndrome/myeloproliferative neoplasm; MPD, myeloproliferative disorder; PV, polycythemia vera; PTCL, peripheral T cell lymphoma; PTCL,NOS, peripheral T cell lymphoma not otherwise specified; T-ALL, T-cell acute lymphoblastic leukemia; TET, Ten eleven translocation. *COSMIC database. Some mutations listed in this table have not been confirmed as somatic mutations.
Associations with other mutations
Mouse and human studies have shown that the loss of TET2 endows cells with a growth advantage over wild-type cells, but does not lead to full transformation. Although this is not always the case, TET2 mutation frequently occurs before the JAK2V617F mutation in the development of MPN [78,85], suggesting that TET2 mutation may occur very early in cancer development. TET2 mutations also occur in early progenitors in MDS. Acquired TET2 mutations are also observed in lymphoma, both B- and T-cell types, and particularly in angioimmunoblastic T-cell lymphoma (AITL). In both T- and B-cell lymphomas, TET2 mutations have been identified in multipotent progenitors [86] that are able to participate in both myeloid and lymphoid differentiation. Together, these observations indicate that TET2 loss predisposes but does not trigger cellular transformation. The tumor phenotype depends on cooperating mutations, such as JAK2 or KIT mutations for MPN [87].
In AML, TET2 mutations occur with other major mutations, particularly internal tandem duplication of FLT3, as well as mutations in RAS, NPM1 and DNMT3A. Mutations in TET2, IDH1 and IDH2 are, however, mutually exclusive [88]. The situation is markedly different in AITL. Here, TET2 mutations are closely associated with DNMT3A mutations [86] and, even more intriguing, do occur together with IDH2 mutations [89-91].
TET and IDH mutations
IDH mutant proteins can inhibit TET2 activity. The IDH genes encode enzymes of the citric acid cycle that convert isocitrate into α-ketoglutarate (αKG) in a nicotinamide adenine dinucleotide phosphate-dependent manner. A variety of human cancers, including AML [92-94], show recurrent missense mutations in IDH1 and IDH2 that endow the mutant protein with the ability to synthesize 2-hydroxyglutarate (2HG) from αKG (Table 2). 2HG is a competitive inhibitor of αKG and may inhibit all αKG-dependent dioxygenases, including EGLN prolyl hydroxylases, Jumanji C histone demethylases and TET proteins. In AML, TET2 and IDH mutations are mutually exclusive, suggesting that they target the same pathway [84]. Consistent with this, TET2- and IDH-mutated primary AML samples show comparable DNA methylation profiles [84,95].
Other examples of TET2 activity targeting in myeloid malignancies
A recent report indicates that mutations in the WT1 gene are exclusive from TET2, IDH1 and IDH2 mutations and impair TET2 activity in human AML. The WT1 gene encodes a zinc finger transcription factor and is mutated in approximately 8% of patients. Similar to patients with mutations in IDH1, IDH2 and TET2, samples from patients with WT1-mutated primary AML show decreased 5-hmC levels and changes in 5-hmC localization. This study indicates the involvement of WT1 in the regulation of hydroxymethylation and provides an example of TET2 function impairment without TET2 mutations [96].
Mouse models have shown that microRNAs (miRNAs) miR26a and miR29a are able to regulate TET expression by targeting their 3’ untranslated regions (UTRs) [97,98]. Other miRNAs, such as miR125b, miR29b, miR29c, miR101 and miR7, have also been implicated in TET regulation using a 3’ UTR human and mouse reporter screen [99]. Recently, miR22 has been shown to be responsible for the downregulation of all three TET genes [100]. Indeed, conditional expression of miR22 in a transgenic mouse model led to reduced levels of 5-hmC, amplification of the hematopoietic stem/progenitor compartment, and development of hematopoietic malignancies. miR22 is highly expressed in more than half of adult MDS and AML samples, providing another example that TET2 activity can be knocked down in the absence of a somatic mutation.
5-hmC and TET in solid tumors
Deregulation of cytosine hydroxymethylation by TET activity
Abnormal patterns of cytosine methylation have been observed in some solid tumors, including melanoma. The melanoma epigenome widely lacks 5-hmC, in association with tumor progression and downregulation of the TET family genes [101]. However, somatic TET mutations are exceedingly rare in this cancer, suggesting that another mechanism is affecting TET activity. Considering that TET enzymes are dependent on αKG, alteration in genes participating in its production may contribute to the inhibition of TET activity. Accordingly, IDH1 or IDH2 mutations are described in 10% of melanomas. These data support a role for deregulation of DNA methylation control during tumor progression rather than during the initial phases.
IDH mutations were first observed in human gliomas [102]. The IDH-mutated samples exhibited a hypermethylation phenotype, due to the inactivation of TET proteins by 2HG [103]. In paragangliomas, inactivating mutations in the SDHx and FH genes, encoding citric acid cycle enzymes (Table 2), result in the accumulation of succinate or fumarate, respectively, and competitive inhibition of αKG-dependent dioxygenases, similar to 2HG [104]. SDH mutations induce a hypermethylation phenotype compared to tumors with wild-type SDH, and are associated with transcriptional silencing. This argues for a driver role for demethylation deregulation in the development of these tumors.
TET mutations
TET mutations are rare in solid tumors [105-117]. In many instances, acquired mutations are missense mutations whose functional consequences on TET protein activity are uncertain. A survey of TET2 mutations in the COSMIC database showed more deleterious mutations in hematological malignancies than in solid tumors (29.8% versus 7.3% for frameshift mutations and 28.1% versus 10.3% for nonsense mutations). Conversely, there are fewer potentially benign mutations in hematological malignancies than in solid tumors (0.25% versus 17.6% in solid tumors for coding-silent mutations and 26.5% versus 63.1% for missense mutations). The dominant expression of TET2 (with respect to TET1 and TET3) in hematopoiesis results in a strong effect of TET2 deficiency on 5-hmC levels. Aside from the potential specific functions of TET2, because expression of the three TET genes is equivalent in other tissues, the consequences of TET2 deficiency on global cytosine (hydroxy)methylation is expected to be less important than in hematopoietic tissues. IDH, SDH and FH mutations, which result in the inhibition of virtually all αKG-dependent dioxygenases, including all three TET proteins, would therefore more strongly impact DNA methylation control than a single TET gene mutation.
Implications for disease
Studies of TET2 deficiencies in tumor development have revealed the importance of DNA methylation in cellular processes as well as in the progressive development of adult type hematological malignancies.
In terms of potential clinical applicability, it appears difficult to specifically and directly target these TET dioxygenases for cancer treatment because they are inactivated in cancer. Indeed, recent efforts have focused on indirect correction of TET function and 5-hmC deregulation in cancer.
TET inactivation induces a methylation imbalance, including hypermethylation of tumor suppressor genes in malignant clones. These genes may be targeted by hypomethylating agents already used in clinical studies, such as 5-azacitidine and decitabine [118,119]. The global hypomethylation effect of these drugs, which remains nonspecific, seems to be accompanied by local hypermethylation, whose long-term consequences are unknown [20]. The molecular mechanisms of action of these drugs need to be further investigated, and extensive clinical trials are needed to prove their efficacy and to identify biomarkers of clinical responses.
In IDH1- or IDH2-mutated cancers, the oncometabolite 2HG acts as a biomarker of compromised enzyme activity [120]. This led to the development of IDH2 inhibitors, now tested in clinical trials [121]. Similarly, FH and SDH inhibitors could be developed to prevent the overall effect of metabolic TET inactivation in cancer. The activities of TET as well as DNMT enzymes are regulated, in part, by the concentrations of their required cofactors. Thus, the metabolic state of the cell is an antitumor target, by preventing the activity of the mutated protein but also by manipulating agonist or antagonist functions. In addition to the detection of TET2 mutations that pre-date full-blown malignancies, recent studies have highlighted preleukemic phases in AML that are associated with mutations in other genes affecting DNA methylation, such as DNMT3A, IDH1 and IDH2 [122], and in genes involved in chromatin structure, such as SMC1A (structural maintenance of chromosome 1A) [123]. These observations suggest that manipulating the control of chromatin structure may be efficient for the treatment of both early and late phases of disease.
Conclusions and future directions
DNA methylation patterns are markedly abnormal in malignant cells in comparison with normal tissues. Abnormal methylation has been postulated to inactivate tumor suppressor genes through cytosine methylation and to activate oncogenes through cytosine hydroxymethylation and demethylation (Figure 3). An unexpected number of oxidized cytosine forms have been uncovered, whose specific functions need to be investigated. Specific techniques allowing their thorough investigation at the nucleotide level are under development and will enable us to investigate the specific functions of these cytosines in normal cells. This is a requirement for understanding their roles in cellular transformation, because mutations detected in cancer can inactivate or impair DNA methylation (for example, DNMT3A mutations) or DNA demethylation (for example, TET2 or IDH mutations).
Figure 3.
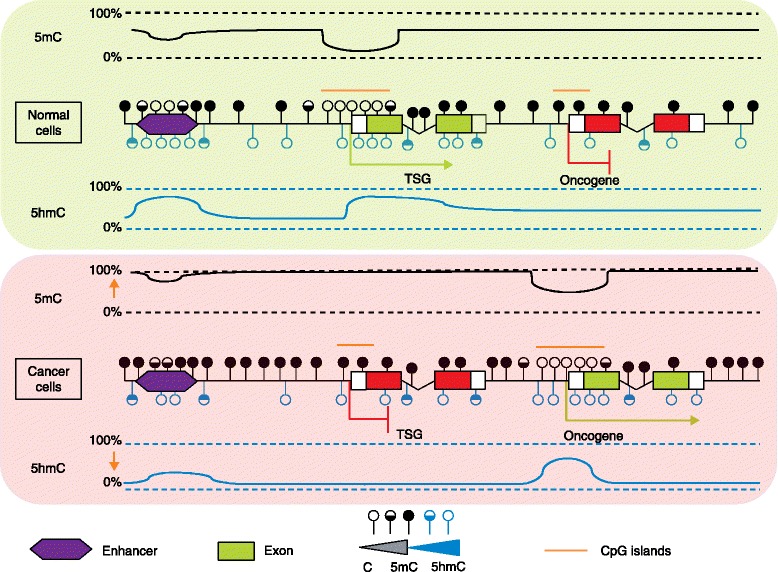
Schematic of methylation and hydroxymethylation patterns in normal and cancer cells. In normal cells, unmethylated cytosines are located in CpG islands and promoters of actively transcribed genes, whereas promoters of repressed genes are mainly composed of 5-methylcytosine (5-mC). 5-hydroxymethylcytosines (5-hmCs) are frequent in cis-regulatory elements such as enhancers, in low CpG content regions, and within gene bodies of transcribed genes. A global hypermethylation phenotype, with respect to normal tissue, is usually associated with tumoral transformation, including repressed tumor suppressor gene promoters. Hypomethylation can, however, be observed locally, in oncogene promoters, for example. Cancer cells present a global decrease of 5-hmC and local redistribution of this mark towards some oncogene promoters. C, cytosine; TSG, tumor suppressor gene.
Although cytosine methylation is associated with gene repression, the exact mechanisms leading from methylation to gene repression are still elusive, as are the crosstalk with other epigenetic modifications, the factors involved in chromatin modification and the regulation of their activities. DNA methylation and demethylation result from the regulation of different enzymatic activities, which compete with each other for DNA access.
This process is complex enough to appear stochastic, with a slow drift in methylation profiles that is tissue specific as well as age related. This drift leads to cellular heterogeneity and, with respect to methylation and gene repression, allows for cell selection. It is likely that TET2 deficiency increases cellular heterogeneity and facilitates the selection of fitter cells. We now require a complete understanding of the protein complexes involved in cytosine methylation and demethylation, including the exact role of each of the mammalian TET proteins and their regulatory signals, in order to target these processes.
Work with human samples and murine models has shown that TET2 deficiency does not trigger full-blown malignancies but predisposes to the development of tumors such as MPN, MDS and lymphoma. The different Tet2 knockout mouse models exhibit similar phenotypes but also present subtle differences that might be due to the loss of different regions of the gene. So far, it has not been possible to correlate clinical phenotypes with TET2 mutations (for example, regarding their precise location or heterozygosity). Other questions relate to the dependency of malignant cells on the initial TET2 mutation, and why some patients with TET2 mutations remain healthy while others develop a myeloid or a lymphoid malignancy [123-126]. Addressing such questions is important, not only with regard to mutations in genes involved in the control of DNA methylation (such as TET2 or DNMT3A), but also for mutation in genes controlling other functions that predate and may predispose to the development of adult malignancies [125-127].
Acknowledgements
Work in the laboratory was supported by grants from Institut National de la Santé Et de la Recherche Médicale (INSERM), Institut Gustave Roussy, Institut National du Cancer (INCa) - INCa-DGOS-INSERM 6043, Fondation pour la recherche Médicale (FRM) and Association Laurette Fugain. LS is supported by fellowships from Cancéropôle Ile de France and Fondation ARC. We thank Philippe Dessen for help with COSMIC data.
Abbreviations
- 2HG
2-hydroxyglutarate
- 5-caC
5-carboxycytosine
- 5-fC
5-formylcytosine
- 5-hmC
5-hydroxymethylcytosine
- 5-hmU
5-hydroxymethyluracil
- 5-mC
5-methylcytosine
- αKG
α-ketoglutarate
- AID
Activation-induced deaminase
- AITL
Angioimmunoblastic T-cell lymphoma
- AML
Acute myeloid leukemia
- BER
Base-excision repair
- COSMIC
Catalogue of somatic mutations in cancer
- DNMT
DNA methyltransferase
- ES
Embryonic stem
- FH
Fumarate hydratase
- IDH
Isocitrate dehydrogenase
- MBD
Methyl-binding domain
- MBP
Methyl-CpG binding
- MDS
Myelodysplastic syndrome
- miRNA
microRNA
- MPN
Myeloproliferative neoplasms
- OGT
O-linked β-D-N acetylglucosamine transferase
- PRC2
Polycomb repressive complex 2
- SDH
Succinate dehydrogenase
- TDG
Thymidine DNA glycosylase
- TET
Ten-eleven translocation
- UTR
Untranslated region
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
LS and OAB drafted the manuscript. All authors participated and agreed to the final version.
Contributor Information
Laurianne Scourzic, Email: laurianne.scourzic@inserm.fr.
Enguerran Mouly, Email: enguerran.mouly@inserm.fr.
Olivier A Bernard, Email: olivier.bernard@inserm.fr.
References
- 1.Kriaucionis S, Heintz N. The nuclear DNA base 5-hydroxymethylcytosine is present in Purkinje neurons and the brain. Science. 2009;324:929–30. doi: 10.1126/science.1169786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tahiliani M, Koh KP, Shen Y, Pastor WA, Bandukwala H, Brudno Y, et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science. 2009;324:930–5. doi: 10.1126/science.1170116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Russler-Germain DA, Spencer DH, Young MA, Lamprecht TL, Miller CA, Fulton R, et al. The R882H DNMT3A mutation associated with AML dominantly inhibits wild-type DNMT3A by blocking its ability to form active tetramers. Cancer Cell. 2014;25:442–54. doi: 10.1016/j.ccr.2014.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Heyn H, Vidal E, Sayols S, Sanchez-Mut JV, Moran S, Medina I, et al. Whole-genome bisulfite DNA sequencing of a DNMT3B mutant patient. Epigenetics. 2012;7:542–50. doi: 10.4161/epi.20523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Okano M, Bell DW, Haber DA, Li E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell. 1999;99:247–57. doi: 10.1016/s0092-8674(00)81656-6. [DOI] [PubMed] [Google Scholar]
- 6.Arand J, Spieler D, Karius T, Branco MR, Meilinger D, Meissner A, et al. In vivo control of CpG and non-CpG DNA methylation by DNA methyltransferases. PLoS Genet. 2012;8:e1002750. doi: 10.1371/journal.pgen.1002750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ooi SK, Qiu C, Bernstein E, Li K, Jia D, Yang Z, et al. DNMT3L connects unmethylated lysine 4 of histone H3 to de novo methylation of DNA. Nature. 2007;448:714–7. doi: 10.1038/nature05987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Guo X, Wang L, Li J, Ding Z, Xiao J, Yin X, et al. Structural insight into autoinhibition and histone H3-induced activation of DNMT3A. Nature. 2014. doi:10.1038/nature13899 [DOI] [PubMed]
- 9.Chen CC, Wang KY, Shen CK. The mammalian de novo DNA methyltransferases DNMT3A and DNMT3B are also DNA 5-hydroxymethylcytosine dehydroxymethylases. J Biol Chem. 2012;287:33116–21. doi: 10.1074/jbc.C112.406975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ito S, Shen L, Dai Q, Wu SC, Collins LB, Swenberg JA, et al. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science. 2011;333:1300–3. doi: 10.1126/science.1210597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.He YF, Li BZ, Li Z, Liu P, Wang Y, Tang Q, et al. Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science. 2011;333:1303–7. doi: 10.1126/science.1210944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Maiti A, Drohat AC. Thymine DNA glycosylase can rapidly excise 5-formylcytosine and 5-carboxylcytosine: potential implications for active demethylation of CpG sites. J Biol Chem. 2011;286:35334–8. doi: 10.1074/jbc.C111.284620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang L, Lu X, Lu J, Liang H, Dai Q, Xu GL, et al. Thymine DNA glycosylase specifically recognizes 5-carboxylcytosine-modified DNA. Nat Chem Biol. 2012;8:328–30. doi: 10.1038/nchembio.914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Guo JU, Su Y, Zhong C, Ming GL, Song H. Hydroxylation of 5-methylcytosine by TET1 promotes active DNA demethylation in the adult brain. Cell. 2011;145:423–34. doi: 10.1016/j.cell.2011.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pfaffeneder T, Spada F, Wagner M, Brandmayr C, Laube SK, Eisen D, et al. Tet oxidizes thymine to 5-hydroxymethyluracil in mouse embryonic stem cell DNA. Nat Chem Biol. 2014;10:574–81. doi: 10.1038/nchembio.1532. [DOI] [PubMed] [Google Scholar]
- 16.Nabel CS, Jia H, Ye Y, Shen L, Goldschmidt HL, Stivers JT, et al. AID/APOBEC deaminases disfavor modified cytosines implicated in DNA demethylation. Nat Chem Biol. 2012;8:751–8. doi: 10.1038/nchembio.1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rangam G, Schmitz KM, Cobb AJ, Petersen-Mahrt SK. AID enzymatic activity is inversely proportional to the size of cytosine C5 orbital cloud. PLoS One. 2012;7:e43279. doi: 10.1371/journal.pone.0043279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ehrlich M, Gama-Sosa MA, Huang LH, Midgett RM, Kuo KC, McCune RA, et al. Amount and distribution of 5-methylcytosine in human DNA from different types of tissues of cells. Nucleic Acids Res. 1982;10:2709–21. doi: 10.1093/nar/10.8.2709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lister R, Pelizzola M, Dowen RH, Hawkins RD, Hon G, Tonti-Filippini J, et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature. 2009;462:315–22. doi: 10.1038/nature08514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yang X, Han H, De Carvalho DD, Lay FD, Jones PA, Liang G. Gene body methylation can alter gene expression and is a therapeutic target in cancer. Cancer Cell. 2014;26:577–90. doi: 10.1016/j.ccr.2014.07.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wyatt GR, Cohen SS. A new pyrimidine base from bacteriophage nucleic acids. Nature. 1952;170:1072–3. doi: 10.1038/1701072a0. [DOI] [PubMed] [Google Scholar]
- 22.Penn NW, Suwalski R, O'Riley C, Bojanowski K, Yura R. The presence of 5-hydroxymethylcytosine in animal deoxyribonucleic acid. Biochem J. 1972;126:781–90. doi: 10.1042/bj1260781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Booth MJ, Branco MR, Ficz G, Oxley D, Krueger F, Reik W, et al. Quantitative sequencing of 5-methylcytosine and 5-hydroxymethylcytosine at single-base resolution. Science. 2012;336:934–7. doi: 10.1126/science.1220671. [DOI] [PubMed] [Google Scholar]
- 24.Booth MJ, Marsico G, Bachman M, Beraldi D, Balasubramanian S. Quantitative sequencing of 5-formylcytosine in DNA at single-base resolution. Nat Chem. 2014;6:435–40. doi: 10.1038/nchem.1893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mohn F, Weber M, Schubeler D, Roloff TC. Methylated DNA immunoprecipitation (MeDIP) Methods Mol Biol. 2009;507:55–64. doi: 10.1007/978-1-59745-522-0_5. [DOI] [PubMed] [Google Scholar]
- 26.Flusberg BA, Webster DR, Lee JH, Travers KJ, Olivares EC, Clark TA, et al. Direct detection of DNA methylation during single-molecule, real-time sequencing. Nat Methods. 2010;7:461–5. doi: 10.1038/nmeth.1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Globisch D, Munzel M, Muller M, Michalakis S, Wagner M, Koch S, et al. Tissue distribution of 5-hydroxymethylcytosine and search for active demethylation intermediates. PLoS One. 2010;5:e15367. doi: 10.1371/journal.pone.0015367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li W, Liu M. Distribution of 5-hydroxymethylcytosine in different human tissues. J Nucleic Acids. 2011;2011:870726. doi: 10.4061/2011/870726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ruzov A, Tsenkina Y, Serio A, Dudnakova T, Fletcher J, Bai Y, et al. Lineage-specific distribution of high levels of genomic 5-hydroxymethylcytosine in mammalian development. Cell Res. 2011;21:1332–42. doi: 10.1038/cr.2011.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ito S, D'Alessio AC, Taranova OV, Hong K, Sowers LC, Zhang Y. Role of Tet proteins in 5mC to 5hmC conversion, ES-cell self-renewal and inner cell mass specification. Nature. 2010;466:1129–33. doi: 10.1038/nature09303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Koh KP, Yabuuchi A, Rao S, Huang Y, Cunniff K, Nardone J, et al. Tet1 and Tet2 regulate 5-hydroxymethylcytosine production and cell lineage specification in mouse embryonic stem cells. Cell Stem Cell. 2011;8:200–13. doi: 10.1016/j.stem.2011.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Iqbal K, Jin SG, Pfeifer GP, Szabo PE. Reprogramming of the paternal genome upon fertilization involves genome-wide oxidation of 5-methylcytosine. Proc Natl Acad Sci U S A. 2011;108:3642–7. doi: 10.1073/pnas.1014033108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wossidlo M, Nakamura T, Lepikhov K, Marques CJ, Zakhartchenko V, Boiani M, et al. 5-Hydroxymethylcytosine in the mammalian zygote is linked with epigenetic reprogramming. Nat Commun. 2011;2:241. doi: 10.1038/ncomms1240. [DOI] [PubMed] [Google Scholar]
- 34.Mayer W, Niveleau A, Walter J, Fundele R, Haaf T. Demethylation of the zygotic paternal genome. Nature. 2000;403:501–2. doi: 10.1038/35000656. [DOI] [PubMed] [Google Scholar]
- 35.Oswald J, Engemann S, Lane N, Mayer W, Olek A, Fundele R, et al. Active demethylation of the paternal genome in the mouse zygote. Curr Biol. 2000;10:475–8. doi: 10.1016/s0960-9822(00)00448-6. [DOI] [PubMed] [Google Scholar]
- 36.Gu TP, Guo F, Yang H, Wu HP, Xu GF, Liu W, et al. The role of Tet3 DNA dioxygenase in epigenetic reprogramming by oocytes. Nature. 2011;477:606–10. doi: 10.1038/nature10443. [DOI] [PubMed] [Google Scholar]
- 37.Nakamura T, Liu YJ, Nakashima H, Umehara H, Inoue K, Matoba S, et al. PGC7 binds histone H3K9me2 to protect against conversion of 5mC to 5hmC in early embryos. Nature. 2012;486:415–9. doi: 10.1038/nature11093. [DOI] [PubMed] [Google Scholar]
- 38.Yu M, Hon GC, Szulwach KE, Song CX, Zhang L, Kim A, et al. Base-resolution analysis of 5-hydroxymethylcytosine in the mammalian genome. Cell. 2012;149:1368–80. doi: 10.1016/j.cell.2012.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Asmar F, Punj V, Christensen J, Pedersen MT, Pedersen A, Nielsen AB, et al. Genome-wide profiling identifies a DNA methylation signature that associates with TET2 mutations in diffuse large B-cell lymphoma. Haematologica. 2013;98:1912–20. doi: 10.3324/haematol.2013.088740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Stroud H, Feng S, Morey Kinney S, Pradhan S, Jacobsen SE. 5-Hydroxymethylcytosine is associated with enhancers and gene bodies in human embryonic stem cells. Genome Biol. 2011;12:R54. doi: 10.1186/gb-2011-12-6-r54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Szulwach KE, Li X, Li Y, Song CX, Han JW, Kim S, et al. Integrating 5-hydroxymethylcytosine into the epigenomic landscape of human embryonic stem cells. PLoS Genet. 2011;7:e1002154. doi: 10.1371/journal.pgen.1002154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Frauer C, Hoffmann T, Bultmann S, Casa V, Cardoso MC, Antes I, et al. Recognition of 5-hydroxymethylcytosine by the Uhrf1 SRA domain. PLoS One. 2011;6:e21306. doi: 10.1371/journal.pone.0021306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mellen M, Ayata P, Dewell S, Kriaucionis S, Heintz N. MeCP2 binds to 5hmC enriched within active genes and accessible chromatin in the nervous system. Cell. 2012;151:1417–30. doi: 10.1016/j.cell.2012.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Spruijt CG, Gnerlich F, Smits AH, Pfaffeneder T, Jansen PW, Bauer C, et al. Dynamic readers for 5-(hydroxy)methylcytosine and its oxidized derivatives. Cell. 2013;152:1146–59. doi: 10.1016/j.cell.2013.02.004. [DOI] [PubMed] [Google Scholar]
- 45.Yildirim O, Li R, Hung JH, Chen PB, Dong X, Ee LS, et al. Mbd3/NURD complex regulates expression of 5-hydroxymethylcytosine marked genes in embryonic stem cells. Cell. 2011;147:1498–510. doi: 10.1016/j.cell.2011.11.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Iurlaro M, Ficz G, Oxley D, Raiber EA, Bachman M, Booth MJ, et al. A screen for hydroxymethylcytosine and formylcytosine binding proteins suggests functions in transcription and chromatin regulation. Genome Biol. 2013;14:R119. doi: 10.1186/gb-2013-14-10-r119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hendrich B, Bird A. Identification and characterization of a family of mammalian methyl-CpG binding proteins. Mol Cell Biol. 1998;18:6538–47. doi: 10.1128/mcb.18.11.6538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Laget S, Joulie M, Le Masson F, Sasai N, Christians E, Pradhan S, et al. The human proteins MBD5 and MBD6 associate with heterochromatin but they do not bind methylated DNA. PLoS One. 2010;5:e11982. doi: 10.1371/journal.pone.0011982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bellacosa A, Cicchillitti L, Schepis F, Riccio A, Yeung AT, Matsumoto Y, et al. MED1, a novel human methyl-CpG-binding endonuclease, interacts with DNA mismatch repair protein MLH1. Proc Natl Acad Sci U S A. 1999;96:3969–74. doi: 10.1073/pnas.96.7.3969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hendrich B, Hardeland U, Ng HH, Jiricny J, Bird A. The thymine glycosylase MBD4 can bind to the product of deamination at methylated CpG sites. Nature. 1999;401:301–4. doi: 10.1038/45843. [DOI] [PubMed] [Google Scholar]
- 51.Valinluck V, Tsai HH, Rogstad DK, Burdzy A, Bird A, Sowers LC. Oxidative damage to methyl-CpG sequences inhibits the binding of the methyl-CpG binding domain (MBD) of methyl-CpG binding protein 2 (MeCP2) Nucleic Acids Res. 2004;32:4100–8. doi: 10.1093/nar/gkh739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bostick M, Kim JK, Esteve PO, Clark A, Pradhan S, Jacobsen SE. UHRF1 plays a role in maintaining DNA methylation in mammalian cells. Science. 2007;317:1760–4. doi: 10.1126/science.1147939. [DOI] [PubMed] [Google Scholar]
- 53.Sharif J, Muto M, Takebayashi S, Suetake I, Iwamatsu A, Endo TA, et al. The SRA protein Np95 mediates epigenetic inheritance by recruiting Dnmt1 to methylated DNA. Nature. 2007;450:908–12. doi: 10.1038/nature06397. [DOI] [PubMed] [Google Scholar]
- 54.Burmeister T, Meyer C, Schwartz S, Hofmann J, Molkentin M, Kowarz E, et al. The MLL recombinome of adult CD10-negative B-cell precursor acute lymphoblastic leukemia: results from the GMALL study group. Blood. 2009;113:4011–5. doi: 10.1182/blood-2008-10-183483. [DOI] [PubMed] [Google Scholar]
- 55.Ittel A, Jeandidier E, Helias C, Perrusson N, Humbrecht C, Lioure B, et al. First description of the t(10;11)(q22;q23)/MLL-TET1 translocation in a T-cell lymphoblastic lymphoma, with subsequent lineage switch to acute myelomonocytic myeloid leukemia. Haematologica. 2013;98:e166–8. doi: 10.3324/haematol.2013.096750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lorsbach RB, Moore J, Mathew S, Raimondi SC, Mukatira ST, Downing JR. TET1, a member of a novel protein family, is fused to MLL in acute myeloid leukemia containing the t(10;11)(q22;q23) Leukemia. 2003;17:637–41. doi: 10.1038/sj.leu.2402834. [DOI] [PubMed] [Google Scholar]
- 57.Ono R, Taki T, Taketani T, Taniwaki M, Kobayashi H, Hayashi Y. LCX, leukemia-associated protein with a CXXC domain, is fused to MLL in acute myeloid leukemia with trilineage dysplasia having t(10;11)(q22;q23) Cancer Res. 2002;62:4075–80. [PubMed] [Google Scholar]
- 58.Jeong M, Sun D, Luo M, Huang Y, Challen GA, Rodriguez B, et al. Large conserved domains of low DNA methylation maintained by Dnmt3a. Nat Genet. 2014;46:17–23. doi: 10.1038/ng.2836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Putiri EL, Tiedemann RL, Thompson JJ, Liu C, Ho T, Choi JH, et al. Distinct and overlapping control of 5-methylcytosine and 5-hydroxymethylcytosine by the TET proteins in human cancer cells. Genome Biol. 2014;15:R81. doi: 10.1186/gb-2014-15-6-r81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Huang Y, Chavez L, Chang X, Wang X, Pastor WA, Kang J, et al. Distinct roles of the methylcytosine oxidases Tet1 and Tet2 in mouse embryonic stem cells. Proc Natl Acad Sci U S A. 2014;111:1361–6. doi: 10.1073/pnas.1322921111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Pulakanti K, Pinello L, Stelloh C, Blinka S, Allred J, Milanovich S, et al. Enhancer transcribed RNAs arise from hypomethylated, Tet-occupied genomic regions. Epigenetics. 2013;8:1303–20. doi: 10.4161/epi.26597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hon GC, Song CX, Du T, Jin F, Selvaraj S, Lee AY, et al. 5mC oxidation by Tet2 modulates enhancer activity and timing of transcriptome reprogramming during differentiation. Mol Cell. 2014;56:286–97. doi: 10.1016/j.molcel.2014.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yin R, Mao SQ, Zhao B, Chong Z, Yang Y, Zhao C, et al. Ascorbic acid enhances Tet-mediated 5-methylcytosine oxidation and promotes DNA demethylation in mammals. J Am Chem Soc. 2013;135:10396–403. doi: 10.1021/ja4028346. [DOI] [PubMed] [Google Scholar]
- 64.Minor EA, Court BL, Young JI, Wang G. Ascorbate induces ten-eleven translocation (Tet) methylcytosine dioxygenase-mediated generation of 5-hydroxymethylcytosine. J Biol Chem. 2013;288:13669–74. doi: 10.1074/jbc.C113.464800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ko M, An J, Bandukwala HS, Chavez L, Aijo T, Pastor WA, et al. Modulation of TET2 expression and 5-methylcytosine oxidation by the CXXC domain protein IDAX. Nature. 2013;497:122–6. doi: 10.1038/nature12052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Guilhamon P, Eskandarpour M, Halai D, Wilson GA, Feber A, Teschendorff AE, et al. Meta-analysis of IDH-mutant cancers identifies EBF1 as an interaction partner for TET2. Nat Commun. 2013;4:2166. doi: 10.1038/ncomms3166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Arioka Y, Watanabe A, Saito K, Yamada Y. Activation-induced cytidine deaminase alters the subcellular localization of Tet family proteins. PLoS One. 2012;7:e45031. doi: 10.1371/journal.pone.0045031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chen Q, Chen Y, Bian C, Fujiki R, Yu X. TET2 promotes histone O-GlcNAcylation during gene transcription. Nature. 2013;493:561–4. doi: 10.1038/nature11742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Deplus R, Delatte B, Schwinn MK, Defrance M, Mendez J, Murphy N, et al. TET2 and TET3 regulate GlcNAcylation and H3K4 methylation through OGT and SET1/COMPASS. EMBO J. 2013;32:645–55. doi: 10.1038/emboj.2012.357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Vella P, Scelfo A, Jammula S, Chiacchiera F, Williams K, Cuomo A, et al. Tet proteins connect the O-linked N-acetylglucosamine transferase Ogt to chromatin in embryonic stem cells. Mol Cell. 2013;49:645–56. doi: 10.1016/j.molcel.2012.12.019. [DOI] [PubMed] [Google Scholar]
- 71.Zhang Q, Liu X, Gao W, Li P, Hou J, Li J, et al. Differential regulation of the ten-eleven translocation (TET) family of dioxygenases by O-linked beta-N-acetylglucosamine transferase (OGT) J Biol Chem. 2014;289:5986–96. doi: 10.1074/jbc.M113.524140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Williams K, Christensen J, Helin K. DNA methylation: TET proteins-guardians of CpG islands? EMBO Rep. 2012;13:28–35. doi: 10.1038/embor.2011.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wu H, D'Alessio AC, Ito S, Xia K, Wang Z, Cui K, et al. Dual functions of Tet1 in transcriptional regulation in mouse embryonic stem cells. Nature. 2011;473:389–93. doi: 10.1038/nature09934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hanover JA, Krause MW, Love DC. Bittersweet memories: linking metabolism to epigenetics through O-GlcNAcylation. Nat Rev Mol Cell Biol. 2012;13:312–21. doi: 10.1038/nrm3334. [DOI] [PubMed] [Google Scholar]
- 75.Takayama S, Dhahbi J, Roberts A, Mao G, Heo SJ, Pachter L, et al. Genome methylation in D. melanogaster is found at specific short motifs and is independent of DNMT2 activity. Genome Res. 2014;24:821–30. doi: 10.1101/gr.162412.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Fu L, Guerrero CR, Zhong N, Amato N, Liu Y, Liu S, et al. Tet-mediated formation of 5-hydroxymethylcytosine in RNA. J Am Chem Soc. 2014;136:11582–5. doi: 10.1021/ja505305z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lu F, Liu Y, Jiang L, Yamaguchi S, Zhang Y. Role of Tet proteins in enhancer activity and telomere elongation. Genes Dev. 2014;28:2103–19. doi: 10.1101/gad.248005.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Delhommeau F, Dupont S, Della Valle V, James C, Trannoy S, Masse A, et al. Mutation in TET2 in myeloid cancers. N Engl J Med. 2009;360:2289–301. doi: 10.1056/NEJMoa0810069. [DOI] [PubMed] [Google Scholar]
- 79.Langemeijer SM, Kuiper RP, Berends M, Knops R, Aslanyan MG, Massop M, et al. Acquired mutations in TET2 are common in myelodysplastic syndromes. Nat Genet. 2009;41:838–42. doi: 10.1038/ng.391. [DOI] [PubMed] [Google Scholar]
- 80.Tefferi A, Pardanani A, Lim KH, Abdel-Wahab O, Lasho TL, Patel J, et al. TET2 mutations and their clinical correlates in polycythemia vera, essential thrombocythemia and myelofibrosis. Leukemia. 2009;23:905–11. doi: 10.1038/leu.2009.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Quivoron C, Couronne L, Della Valle V, Lopez CK, Plo I, Wagner-Ballon O, et al. TET2 inactivation results in pleiotropic hematopoietic abnormalities in mouse and is a recurrent event during human lymphomagenesis. Cancer Cell. 2011;20:25–38. doi: 10.1016/j.ccr.2011.06.003. [DOI] [PubMed] [Google Scholar]
- 82.Pronier E, Almire C, Mokrani H, Vasanthakumar A, Simon A. da Costa Reis Monte Mor B, et al. Inhibition of TET2-mediated conversion of 5-methylcytosine to 5-hydroxymethylcytosine disturbs erythroid and granulomonocytic differentiation of human hematopoietic progenitors. Blood. 2011;118:2551–5. doi: 10.1182/blood-2010-12-324707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ko M, Huang Y, Jankowska AM, Pape UJ, Tahiliani M, Bandukwala HS, et al. Impaired hydroxylation of 5-methylcytosine in myeloid cancers with mutant TET2. Nature. 2010;468:839–43. doi: 10.1038/nature09586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Figueroa ME, Abdel-Wahab O, Lu C, Ward PS, Patel J, Shih A, et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell. 2010;18:553–67. doi: 10.1016/j.ccr.2010.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Couronne L, Lippert E, Andrieux J, Kosmider O, Radford-Weiss I, Penther D, et al. Analyses of TET2 mutations in post-myeloproliferative neoplasm acute myeloid leukemias. Leukemia. 2010;24:201–3. doi: 10.1038/leu.2009.169. [DOI] [PubMed] [Google Scholar]
- 86.Couronne L, Bastard C, Bernard OA. TET2 and DNMT3A mutations in human T-cell lymphoma. N Engl J Med. 2012;366:95–6. doi: 10.1056/NEJMc1111708. [DOI] [PubMed] [Google Scholar]
- 87.Soucie E, Hanssens K, Mercher T, Georgin-Lavialle S, Damaj G, Livideanu C, et al. In aggressive forms of mastocytosis, TET2 loss cooperates with c-KITD816V to transform mast cells. Blood. 2012;120:4846–9. doi: 10.1182/blood-2011-12-397588. [DOI] [PubMed] [Google Scholar]
- 88.Mardis ER, Ding L, Dooling DJ, Larson DE, McLellan MD, Chen K, et al. Recurring mutations found by sequencing an acute myeloid leukemia genome. N Engl J Med. 2009;361:1058–66. doi: 10.1056/NEJMoa0903840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Cairns RA, Iqbal J, Lemonnier F, Kucuk C, de Leval L, Jais JP, et al. IDH2 mutations are frequent in angioimmunoblastic T-cell lymphoma. Blood. 2012;119:1901–3. doi: 10.1182/blood-2011-11-391748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Palomero T, Couronne L, Khiabanian H, Kim MY, Ambesi-Impiombato A, Perez-Garcia A, et al. Recurrent mutations in epigenetic regulators, RHOA and FYN kinase in peripheral T cell lymphomas. Nat Genet. 2014;46:166–70. doi: 10.1038/ng.2873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Sakata-Yanagimoto M, Enami T, Yoshida K, Shiraishi Y, Ishii R, Miyake Y, et al. Somatic RHOA mutation in angioimmunoblastic T cell lymphoma. Nat Genet. 2014;46:171–5. doi: 10.1038/ng.2872. [DOI] [PubMed] [Google Scholar]
- 92.Losman JA, Kaelin WG., Jr What a difference a hydroxyl makes: mutant IDH, (R)-2-hydroxyglutarate, and cancer. Genes Dev. 2013;27:836–52. doi: 10.1101/gad.217406.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.McKenney AS, Levine RL. Isocitrate dehydrogenase mutations in leukemia. J Clin Invest. 2013;123:3672–7. doi: 10.1172/JCI67266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Lu C, Thompson CB. Metabolic regulation of epigenetics. Cell Metab. 2012;16:9–17. doi: 10.1016/j.cmet.2012.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Xu W, Yang H, Liu Y, Yang Y, Wang P, Kim SH, et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of alpha-ketoglutarate-dependent dioxygenases. Cancer Cell. 2011;19:17–30. doi: 10.1016/j.ccr.2010.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Rampal R, Alkalin A, Madzo J, Vasanthakumar A, Pronier E, Patel J, et al. DNA hydroxymethylation profiling reveals that WT1 mutations result in loss of TET2 function in acute myeloid leukemia. Cell Rep. 2014;9:1841–55. doi: 10.1016/j.celrep.2014.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Fu X, Jin L, Wang X, Luo A, Hu J, Zheng X, et al. MicroRNA-26a targets ten eleven translocation enzymes and is regulated during pancreatic cell differentiation. Proc Natl Acad Sci U S A. 2013;110:17892–7. doi: 10.1073/pnas.1317397110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Zhang P, Huang B, Xu X, Sessa WC. Ten-eleven translocation (Tet) and thymine DNA glycosylase (TDG), components of the demethylation pathway, are direct targets of miRNA-29a. Biochem Biophys Res Commun. 2013;437:368–73. doi: 10.1016/j.bbrc.2013.06.082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Cheng J, Guo S, Chen S, Mastriano SJ, Liu C, D'Alessio AC, et al. An extensive network of TET2-targeting MicroRNAs regulates malignant hematopoiesis. Cell Rep. 2013;5:471–81. doi: 10.1016/j.celrep.2013.08.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Song SJ, Ito K, Ala U, Kats L, Webster K, Sun SM, et al. The oncogenic microRNA miR-22 targets the TET2 tumor suppressor to promote hematopoietic stem cell self-renewal and transformation. Cell Stem Cell. 2013;13:87–101. doi: 10.1016/j.stem.2013.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Lian CG, Xu Y, Ceol C, Wu F, Larson A, Dresser K, et al. Loss of 5-hydroxymethylcytosine is an epigenetic hallmark of melanoma. Cell. 2012;150:1135–46. doi: 10.1016/j.cell.2012.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Yan H, Parsons DW, Jin G, McLendon R, Rasheed BA, Yuan W, et al. IDH1 and IDH2 mutations in gliomas. N Engl J Med. 2009;360:765–73. doi: 10.1056/NEJMoa0808710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Lu C, Ward PS, Kapoor GS, Rohle D, Turcan S, Abdel-Wahab O, et al. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature. 2012;483:474–8. doi: 10.1038/nature10860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Letouze E, Martinelli C, Loriot C, Burnichon N, Abermil N, Ottolenghi C, et al. SDH mutations establish a hypermethylator phenotype in paraganglioma. Cancer Cell. 2013;23:739–52. doi: 10.1016/j.ccr.2013.04.018. [DOI] [PubMed] [Google Scholar]
- 105.Cancer Genome Atlas N Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487:330–7. doi: 10.1038/nature11252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Kan Z, Jaiswal BS, Stinson J, Janakiraman V, Bhatt D, Stern HM, et al. Diverse somatic mutation patterns and pathway alterations in human cancers. Nature. 2010;466:869–73. doi: 10.1038/nature09208. [DOI] [PubMed] [Google Scholar]
- 107.Dulak AM, Stojanov P, Peng S, Lawrence MS, Fox C, Stewart C, et al. Exome and whole-genome sequencing of esophageal adenocarcinoma identifies recurrent driver events and mutational complexity. Nat Genet. 2013;45:478–86. doi: 10.1038/ng.2591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Nikolaev SI, Rimoldi D, Iseli C, Valsesia A, Robyr D, Gehrig C, et al. Exome sequencing identifies recurrent somatic MAP2K1 and MAP2K2 mutations in melanoma. Nat Genet. 2012;44:133–9. doi: 10.1038/ng.1026. [DOI] [PubMed] [Google Scholar]
- 109.Krauthammer M, Kong Y, Ha BH, Evans P, Bacchiocchi A, McCusker JP, et al. Exome sequencing identifies recurrent somatic RAC1 mutations in melanoma. Nat Genet. 2012;44:1006–14. doi: 10.1038/ng.2359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Tarpey PS, Behjati S, Cooke SL, Van Loo P, Wedge DC, Pillay N, et al. Frequent mutation of the major cartilage collagen gene COL2A1 in chondrosarcoma. Nat Genet. 2013;45:923–6. doi: 10.1038/ng.2668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Pugh TJ, Morozova O, Attiyeh EF, Asgharzadeh S, Wei JS, Auclair D, et al. The genetic landscape of high-risk neuroblastoma. Nat Genet. 2013;45:279–84. doi: 10.1038/ng.2529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Cancer Genome Atlas Research N Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474:609–15. doi: 10.1038/nature10166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Sato Y, Yoshizato T, Shiraishi Y, Maekawa S, Okuno Y, Kamura T, et al. Integrated molecular analysis of clear-cell renal cell carcinoma. Nat Genet. 2013;45:860–7. doi: 10.1038/ng.2699. [DOI] [PubMed] [Google Scholar]
- 114.Peifer M, Fernandez-Cuesta L, Sos ML, George J, Seidel D, Kasper LH, et al. Integrative genome analyses identify key somatic driver mutations of small-cell lung cancer. Nat Genet. 2012;44:1104–10. doi: 10.1038/ng.2396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Stephens PJ, Tarpey PS, Davies H, Van Loo P, Greenman C, Wedge DC, et al. The landscape of cancer genes and mutational processes in breast cancer. Nature. 2012;486:400–4. doi: 10.1038/nature11017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Berger MF, Hodis E, Heffernan TP, Deribe YL, Lawrence MS, Protopopov A, et al. Melanoma genome sequencing reveals frequent PREX2 mutations. Nature. 2012;485:502–6. doi: 10.1038/nature11071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Stransky N, Egloff AM, Tward AD, Kostic AD, Cibulskis K, Sivachenko A, et al. The mutational landscape of head and neck squamous cell carcinoma. Science. 2011;333:1157–60. doi: 10.1126/science.1208130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Fenaux P, Mufti GJ, Hellstrom-Lindberg E, Santini V, Gattermann N, Germing U, et al. Azacitidine prolongs overall survival compared with conventional care regimens in elderly patients with low bone marrow blast count acute myeloid leukemia. J Clin Oncol. 2010;28:562–9. doi: 10.1200/JCO.2009.23.8329. [DOI] [PubMed] [Google Scholar]
- 119.Fenaux P, Mufti GJ, Hellstrom-Lindberg E, Santini V, Finelli C, Giagounidis A, et al. Efficacy of azacitidine compared with that of conventional care regimens in the treatment of higher-risk myelodysplastic syndromes: a randomised, open-label, phase III study. Lancet Oncol. 2009;10:223–32. doi: 10.1016/S1470-2045(09)70003-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Janin M, Mylonas E, Saada V, Micol JB, Renneville A, Quivoron C, et al. Serum 2-hydroxyglutarate production in IDH1- and IDH2-mutated de novo acute myeloid leukemia: a study by the Acute Leukemia French Association group. J Clin Oncol. 2014;32:297–305. doi: 10.1200/JCO.2013.50.2047. [DOI] [PubMed] [Google Scholar]
- 121.Wang F, Travins J, DeLaBarre B, Penard-Lacronique V, Schalm S, Hansen E, et al. Targeted inhibition of mutant IDH2 in leukemia cells induces cellular differentiation. Science. 2013;340:622–6. doi: 10.1126/science.1234769. [DOI] [PubMed] [Google Scholar]
- 122.Shlush LI, Zandi S, Mitchell A, Chen WC, Brandwein JM, Gupta V, et al. Identification of pre-leukaemic haematopoietic stem cells in acute leukaemia. Nature. 2014;506:328–33. doi: 10.1038/nature13038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Jan M, Snyder TM, Corces-Zimmerman MR, Vyas P, Weissman IL, Quake SR, et al. Clonal evolution of preleukemic hematopoietic stem cells precedes human acute myeloid leukemia. Sci Transl Med. 2012;4:149ra18. doi: 10.1126/scitranslmed.3004315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Busque L, Patel JP, Figueroa ME, Vasanthakumar A, Provost S, Hamilou Z, et al. Recurrent somatic TET2 mutations in normal elderly individuals with clonal hematopoiesis. Nat Genet. 2012;44:1179–81. doi: 10.1038/ng.2413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Genovese G, Kahler AK, Handsaker RE, Lindberg J, Rose SA, Bakhoum SF, et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N Engl J Med. 2014;371:2477–87. doi: 10.1056/NEJMoa1409405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Jaiswal S, Fontanillas P, Flannick J, Manning A, Grauman PV, Mar BG, et al. Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med. 2014;371:2488–98. doi: 10.1056/NEJMoa1408617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Xie M, Lu C, Wang J, McLellan MD, Johnson KJ, Wendl MC, et al. Age-related mutations associated with clonal hematopoietic expansion and malignancies. Nat Med. 2014;20:1472–8. doi: 10.1038/nm.3733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Ley TJ, Ding L, Walter MJ, McLellan MD, Lamprecht T, Larson DE, et al. DNMT3A mutations in acute myeloid leukemia. N Engl J Med. 2010;363:2424–33. doi: 10.1056/NEJMoa1005143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.La Salle S, Trasler JM. Dynamic expression of DNMT3a and DNMT3b isoforms during male germ cell development in the mouse. Dev Biol. 2006;296:71–82. doi: 10.1016/j.ydbio.2006.04.436. [DOI] [PubMed] [Google Scholar]
- 130.Robertson KD, Uzvolgyi E, Liang G, Talmadge C, Sumegi J, Gonzales FA, et al. The human DNA methyltransferases (DNMTs) 1, 3a and 3b: coordinate mRNA expression in normal tissues and overexpression in tumors. Nucleic Acids Res. 1999;27:2291–8. doi: 10.1093/nar/27.11.2291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Xie S, Wang Z, Okano M, Nogami M, Li Y, He WW, et al. Cloning, expression and chromosome locations of the human DNMT3 gene family. Gene. 1999;236:87–95. doi: 10.1016/s0378-1119(99)00252-8. [DOI] [PubMed] [Google Scholar]
- 132.Mizuno S, Chijiwa T, Okamura T, Akashi K, Fukumaki Y, Niho Y, et al. Expression of DNA methyltransferases DNMT1, 3A, and 3B in normal hematopoiesis and in acute and chronic myelogenous leukemia. Blood. 2001;97:1172–9. doi: 10.1182/blood.v97.5.1172. [DOI] [PubMed] [Google Scholar]
- 133.Meyers G, Ng YS, Bannock JM, Lavoie A, Walter JE, Notarangelo LD, et al. Activation-induced cytidine deaminase (AID) is required for B-cell tolerance in humans. Proc Natl Acad Sci U S A. 2011;108:11554–9. doi: 10.1073/pnas.1102600108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Muramatsu M, Sankaranand VS, Anant S, Sugai M, Kinoshita K, Davidson NO, et al. Specific expression of activation-induced cytidine deaminase (AID), a novel member of the RNA-editing deaminase family in germinal center B cells. J Biol Chem. 1999;274:18470–6. doi: 10.1074/jbc.274.26.18470. [DOI] [PubMed] [Google Scholar]
- 135.Niederreither K, Harbers M, Chambon P, Dolle P. Expression of T:G mismatch-specific thymidine-DNA glycosylase and DNA methyl transferase genes during development and tumorigenesis. Oncogene. 1998;17:1577–85. doi: 10.1038/sj.onc.1202072. [DOI] [PubMed] [Google Scholar]
- 136.Tolley E, Craig I. Presence of two forms of fumarase (fumarate hydratase E.C. 4.2.1.2) in mammalian cells: immunological characterization and genetic analysis in somatic cell hybrids. Confirmation of the assignment of a gene necessary for the enzyme expression to human chromosome 1. Biochem Genet. 1975;13:867–83. doi: 10.1007/BF00484417. [DOI] [PubMed] [Google Scholar]
- 137.Seshagiri S, Stawiski EW, Durinck S, Modrusan Z, Storm EE, Conboy CB, et al. Recurrent R-spondin fusions in colon cancer. Nature. 2012;488:660–4. doi: 10.1038/nature11282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Imielinski M, Berger AH, Hammerman PS, Hernandez B, Pugh TJ, Hodis E, et al. Mapping the hallmarks of lung adenocarcinoma with massively parallel sequencing. Cell. 2012;150:1107–20. doi: 10.1016/j.cell.2012.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Seo JS, Ju YS, Lee WC, Shin JY, Lee JK, Bleazard T, et al. The transcriptional landscape and mutational profile of lung adenocarcinoma. Genome Res. 2012;22:2109–19. doi: 10.1101/gr.145144.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Gui Y, Guo G, Huang Y, Hu X, Tang A, Gao S, et al. Frequent mutations of chromatin remodeling genes in transitional cell carcinoma of the bladder. Nat Genet. 2011;43:875–8. doi: 10.1038/ng.907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Dolnik A, Engelmann JC, Scharfenberger-Schmeer M, Mauch J, Kelkenberg-Schade S, Haldemann B, et al. Commonly altered genomic regions in acute myeloid leukemia are enriched for somatic mutations involved in chromatin remodeling and splicing. Blood. 2012;120:e83–92. doi: 10.1182/blood-2011-12-401471. [DOI] [PubMed] [Google Scholar]
- 142.Quesada V, Conde L, Villamor N, Ordonez GR, Jares P, Bassaganyas L, et al. Exome sequencing identifies recurrent mutations of the splicing factor SF3B1 gene in chronic lymphocytic leukemia. Nat Genet. 2012;44:47–52. doi: 10.1038/ng.1032. [DOI] [PubMed] [Google Scholar]
- 143.Kalender Atak Z, De Keersmaecker K, Gianfelici V, Geerdens E, Vandepoel R, Pauwels D, et al. High accuracy mutation detection in leukemia on a selected panel of cancer genes. PLoS One. 2012;7:e38463. doi: 10.1371/journal.pone.0038463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Kim MS, Kim YR, Yoo NJ, Lee SH. Mutational analysis of DNMT3A gene in acute leukemias and common solid cancers. APMIS. 2013;121:85–94. doi: 10.1111/j.1600-0463.2012.02940.x. [DOI] [PubMed] [Google Scholar]
- 145.Walter MJ, Ding L, Shen D, Shao J, Grillot M, McLellan M, et al. Recurrent DNMT3A mutations in patients with myelodysplastic syndromes. Leukemia. 2011;25:1153–8. doi: 10.1038/leu.2011.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Pugh TJ, Weeraratne SD, Archer TC, Pomeranz Krummel DA, Auclair D, Bochicchio J, et al. Medulloblastoma exome sequencing uncovers subtype-specific somatic mutations. Nature. 2012;488:106–10. doi: 10.1038/nature11329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Zhang J, Ding L, Holmfeldt L, Wu G, Heatley SL, Payne-Turner D, et al. The genetic basis of early T-cell precursor acute lymphoblastic leukaemia. Nature. 2012;481:157–63. doi: 10.1038/nature10725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Agrawal N, Jiao Y, Sausen M, Leary R, Bettegowda C, Roberts NJ, et al. Exomic sequencing of medullary thyroid cancer reveals dominant and mutually exclusive oncogenic mutations in RET and RAS. J Clin Endocrinol Metab. 2013;98:E364–9. doi: 10.1210/jc.2012-2703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Shah SP, Roth A, Goya R, Oloumi A, Ha G, Zhao Y, et al. The clonal and mutational evolution spectrum of primary triple-negative breast cancers. Nature. 2012;486:395–9. doi: 10.1038/nature10933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Morin RD, Mendez-Lago M, Mungall AJ, Goya R, Mungall KL, Corbett RD, et al. Frequent mutation of histone-modifying genes in non-Hodgkin lymphoma. Nature. 2011;476:298–303. doi: 10.1038/nature10351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Chen C, Bartenhagen C, Gombert M, Okpanyi V, Binder V, Rottgers S, et al. Next-generation-sequencing-based risk stratification and identification of new genes involved in structural and sequence variations in near haploid lymphoblastic leukemia. Genes Chromosomes Cancer. 2013;52:564–79. doi: 10.1002/gcc.22054. [DOI] [PubMed] [Google Scholar]
- 152.Roberts KG, Morin RD, Zhang J, Hirst M, Zhao Y, Su X, et al. Genetic alterations activating kinase and cytokine receptor signaling in high-risk acute lymphoblastic leukemia. Cancer Cell. 2012;22:153–66. doi: 10.1016/j.ccr.2012.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Gaal J, Burnichon N, Korpershoek E, Roncelin I, Bertherat J, Plouin PF, et al. Isocitrate dehydrogenase mutations are rare in pheochromocytomas and paragangliomas. J Clin Endocrinol Metab. 2010;95:1274–8. doi: 10.1210/jc.2009-2170. [DOI] [PubMed] [Google Scholar]
- 154.Amary MF, Bacsi K, Maggiani F, Damato S, Halai D, Berisha F, et al. IDH1 and IDH2 mutations are frequent events in central chondrosarcoma and central and periosteal chondromas but not in other mesenchymal tumours. J Pathol. 2011;224:334–43. doi: 10.1002/path.2913. [DOI] [PubMed] [Google Scholar]
- 155.Hemerly JP, Bastos AU, Cerutti JM. Identification of several novel non-p.R132 IDH1 variants in thyroid carcinomas. Eur J Endocrinol. 2010;163:747–55. doi: 10.1530/EJE-10-0473. [DOI] [PubMed] [Google Scholar]
- 156.Murugan AK, Bojdani E, Xing M. Identification and functional characterization of isocitrate dehydrogenase 1 (IDH1) mutations in thyroid cancer. Biochem Biophys Res Commun. 2010;393:555–9. doi: 10.1016/j.bbrc.2010.02.095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Ghiam AF, Cairns RA, Thoms J, Dal Pra A, Ahmed O, Meng A, et al. IDH mutation status in prostate cancer. Oncogene. 2012;31:3826. doi: 10.1038/onc.2011.546. [DOI] [PubMed] [Google Scholar]
- 158.Parsons DW, Jones S, Zhang X, Lin JC, Leary RJ, Angenendt P, et al. An integrated genomic analysis of human glioblastoma multiforme. Science. 2008;321:1807–12. doi: 10.1126/science.1164382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Balss J, Meyer J, Mueller W, Korshunov A, Hartmann C, von Deimling A. Analysis of the IDH1 codon 132 mutation in brain tumors. Acta Neuropathol. 2008;116:597–602. doi: 10.1007/s00401-008-0455-2. [DOI] [PubMed] [Google Scholar]
- 160.Kosmider O, Gelsi-Boyer V, Slama L, Dreyfus F, Beyne-Rauzy O, Quesnel B, et al. Mutations of IDH1 and IDH2 genes in early and accelerated phases of myelodysplastic syndromes and MDS/myeloproliferative neoplasms. Leukemia. 2010;24:1094–6. doi: 10.1038/leu.2010.52. [DOI] [PubMed] [Google Scholar]
- 161.Zhang J, Grubor V, Love CL, Banerjee A, Richards KL, Mieczkowski PA, et al. Genetic heterogeneity of diffuse large B-cell lymphoma. Proc Natl Acad Sci U S A. 2013;110:1398–403. doi: 10.1073/pnas.1205299110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Kang MR, Kim MS, Oh JE, Kim YR, Song SY, Seo SI, et al. Mutational analysis of IDH1 codon 132 in glioblastomas and other common cancers. Int J Cancer. 2009;125:353–5. doi: 10.1002/ijc.24379. [DOI] [PubMed] [Google Scholar]
- 163.Tomlinson IP, Alam NA, Rowan AJ, Barclay E, Jaeger EE, Kelsell D, et al. Germline mutations in FH predispose to dominantly inherited uterine fibroids, skin leiomyomata and papillary renal cell cancer. Nat Genet. 2002;30:406–10. doi: 10.1038/ng849. [DOI] [PubMed] [Google Scholar]
- 164.Ricketts C, Woodward ER, Killick P, Morris MR, Astuti D, Latif F, et al. Germline SDHB mutations and familial renal cell carcinoma. J Natl Cancer Inst. 2008;100:1260–2. doi: 10.1093/jnci/djn254. [DOI] [PubMed] [Google Scholar]
- 165.Bejar R, Stevenson K, Abdel-Wahab O, Galili N, Nilsson B, Garcia-Manero G, et al. Clinical effect of point mutations in myelodysplastic syndromes. N Engl J Med. 2011;364:2496–506. doi: 10.1056/NEJMoa1013343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Jankowska AM, Szpurka H, Tiu RV, Makishima H, Afable M, Huh J, et al. Loss of heterozygosity 4q24 and TET2 mutations associated with myelodysplastic/myeloproliferative neoplasms. Blood. 2009;113:6403–10. doi: 10.1182/blood-2009-02-205690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Kosmider O, Gelsi-Boyer V, Cheok M, Grabar S, Della-Valle V, Picard F, et al. TET2 mutation is an independent favorable prognostic factor in myelodysplastic syndromes (MDSs) Blood. 2009;114:3285–91. doi: 10.1182/blood-2009-04-215814. [DOI] [PubMed] [Google Scholar]
- 168.Smith AE, Mohamedali AM, Kulasekararaj A, Lim Z, Gaken J, Lea NC, et al. Next-generation sequencing of the TET2 gene in 355 MDS and CMML patients reveals low-abundance mutant clones with early origins, but indicates no definite prognostic value. Blood. 2010;116:3923–32. doi: 10.1182/blood-2010-03-274704. [DOI] [PubMed] [Google Scholar]
- 169.Tefferi A, Lim KH, Abdel-Wahab O, Lasho TL, Patel J, Patnaik MM, et al. Detection of mutant TET2 in myeloid malignancies other than myeloproliferative neoplasms: CMML, MDS, MDS/MPN and AML. Leukemia. 2009;23:1343–5. doi: 10.1038/leu.2009.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Abdel-Wahab O, Mullally A, Hedvat C, Garcia-Manero G, Patel J, Wadleigh M, et al. Genetic characterization of TET1, TET2, and TET3 alterations in myeloid malignancies. Blood. 2009;114:144–7. doi: 10.1182/blood-2009-03-210039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Grossmann V, Kohlmann A, Eder C, Haferlach C, Kern W, Cross NC, et al. Molecular profiling of chronic myelomonocytic leukemia reveals diverse mutations in >80% of patients with TET2 and EZH2 being of high prognostic relevance. Leukemia. 2011;25:877–9. doi: 10.1038/leu.2011.10. [DOI] [PubMed] [Google Scholar]
- 172.Itzykson R, Kosmider O, Renneville A, Gelsi-Boyer V, Meggendorfer M, Morabito M, et al. Prognostic score including gene mutations in chronic myelomonocytic leukemia. J Clin Oncol. 2013;31:2428–36. doi: 10.1200/JCO.2012.47.3314. [DOI] [PubMed] [Google Scholar]
- 173.Kohlmann A, Grossmann V, Klein HU, Schindela S, Weiss T, Kazak B, et al. Next-generation sequencing technology reveals a characteristic pattern of molecular mutations in 72.8% of chronic myelomonocytic leukemia by detecting frequent alterations in TET2, CBL, RAS, and RUNX1. J Clin Oncol. 2010;28:3858–65. doi: 10.1200/JCO.2009.27.1361. [DOI] [PubMed] [Google Scholar]
- 174.Kosmider O, Gelsi-Boyer V, Ciudad M, Racoeur C, Jooste V, Vey N, et al. TET2 gene mutation is a frequent and adverse event in chronic myelomonocytic leukemia. Haematologica. 2009;94:1676–81. doi: 10.3324/haematol.2009.011205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Roche-Lestienne C, Marceau A, Labis E, Nibourel O, Coiteux V, Guilhot J, et al. Mutation analysis of TET2, IDH1, IDH2 and ASXL1 in chronic myeloid leukemia. Leukemia. 2011;25:1661–4. doi: 10.1038/leu.2011.139. [DOI] [PubMed] [Google Scholar]
- 176.Makishima H, Jankowska AM, McDevitt MA, O'Keefe C, Dujardin S, Cazzolli H, et al. CBL, CBLB, TET2, ASXL1, and IDH1/2 mutations and additional chromosomal aberrations constitute molecular events in chronic myelogenous leukemia. Blood. 2011;117:e198–206. doi: 10.1182/blood-2010-06-292433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Shen Y, Zhu YM, Fan X, Shi JY, Wang QR, Yan XJ, et al. Gene mutation patterns and their prognostic impact in a cohort of 1185 patients with acute myeloid leukemia. Blood. 2011;118:5593–603. doi: 10.1182/blood-2011-03-343988. [DOI] [PubMed] [Google Scholar]
- 178.Nibourel O, Kosmider O, Cheok M, Boissel N, Renneville A, Philippe N, et al. Incidence and prognostic value of TET2 alterations in de novo acute myeloid leukemia achieving complete remission. Blood. 2010;116:1132–5. doi: 10.1182/blood-2009-07-234484. [DOI] [PubMed] [Google Scholar]
- 179.Metzeler KH, Maharry K, Radmacher MD, Mrozek K, Margeson D, Becker H, et al. TET2 mutations improve the new European LeukemiaNet risk classification of acute myeloid leukemia: a Cancer and Leukemia Group B study. J Clin Oncol. 2011;29:1373–81. doi: 10.1200/JCO.2010.32.7742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Chou WC, Chou SC, Liu CY, Chen CY, Hou HA, Kuo YY, et al. TET2 mutation is an unfavorable prognostic factor in acute myeloid leukemia patients with intermediate-risk cytogenetics. Blood. 2011;118:3803–10. doi: 10.1182/blood-2011-02-339747. [DOI] [PubMed] [Google Scholar]
- 181.Weissmann S, Alpermann T, Grossmann V, Kowarsch A, Nadarajah N, Eder C, et al. Landscape of TET2 mutations in acute myeloid leukemia. Leukemia. 2012;26:934–42. doi: 10.1038/leu.2011.326. [DOI] [PubMed] [Google Scholar]
- 182.Liang DC, Liu HC, Yang CP, Jaing TH, Hung IJ, Yeh TC, et al. Cooperating gene mutations in childhood acute myeloid leukemia with special reference on mutations of ASXL1, TET2, IDH1, IDH2, and DNMT3A. Blood. 2013;121:2988–95. doi: 10.1182/blood-2012-06-436782. [DOI] [PubMed] [Google Scholar]
- 183.Langemeijer SM, Jansen JH, Hooijer J, van Hoogen P, Stevens-Linders E, Massop M, et al. TET2 mutations in childhood leukemia. Leukemia. 2011;25:189–92. doi: 10.1038/leu.2010.243. [DOI] [PubMed] [Google Scholar]
- 184.Kosmider O, Delabesse E, de Mas VM, Cornillet-Lefebvre P, Blanchet O, Delmer A, et al. TET2 mutations in secondary acute myeloid leukemias: a French retrospective study. Haematologica. 2011;96:1059–63. doi: 10.3324/haematol.2011.040840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Konstandin N, Bultmann S, Szwagierczak A, Dufour A, Ksienzyk B, Schneider F, et al. Genomic 5-hydroxymethylcytosine levels correlate with TET2 mutations and a distinct global gene expression pattern in secondary acute myeloid leukemia. Leukemia. 2011;25:1649–52. doi: 10.1038/leu.2011.134. [DOI] [PubMed] [Google Scholar]
- 186.Tefferi A, Levine RL, Lim KH, Abdel-Wahab O, Lasho TL, Patel J, et al. Frequent TET2 mutations in systemic mastocytosis: clinical, KITD816V and FIP1L1-PDGFRA correlates. Leukemia. 2009;23:900–4. doi: 10.1038/leu.2009.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Alayed K, Patel KP, Konoplev S, Singh RR, Routbort MJ, Reddy N, et al. TET2 mutations, myelodysplastic features, and a distinct immunoprofile characterize blastic plasmacytoid dendritic cell neoplasm in the bone marrow. Am J Hematol. 2013;88:1055–61. doi: 10.1002/ajh.23567. [DOI] [PubMed] [Google Scholar]
- 188.Jardin F, Ruminy P, Parmentier F, Troussard X, Vaida I, Stamatoullas A, et al. TET2 and TP53 mutations are frequently observed in blastic plasmacytoid dendritic cell neoplasm. Br J Haematol. 2011;153:413–6. doi: 10.1111/j.1365-2141.2010.08556.x. [DOI] [PubMed] [Google Scholar]
- 189.Menezes J, Acquadro F, Wiseman M, Gomez-Lopez G, Salgado RN, Talavera-Casanas JG, et al. Exome sequencing reveals novel and recurrent mutations with clinical impact in blastic plasmacytoid dendritic cell neoplasm. Leukemia. 2014;28:823–9. doi: 10.1038/leu.2013.283. [DOI] [PubMed] [Google Scholar]
- 190.Meissner B, Kridel R, Lim RS, Rogic S, Tse K, Scott DW, et al. The E3 ubiquitin ligase UBR5 is recurrently mutated in mantle cell lymphoma. Blood. 2013;121:3161–4. doi: 10.1182/blood-2013-01-478834. [DOI] [PubMed] [Google Scholar]
- 191.Cools J. RHOA mutations in peripheral T cell lymphoma. Nat Genet. 2014;46:320–1. doi: 10.1038/ng.2937. [DOI] [PubMed] [Google Scholar]
- 192.Lemonnier F, Couronne L, Parrens M, Jais JP, Travert M, Lamant L, et al. Recurrent TET2 mutations in peripheral T-cell lymphomas correlate with TFH-like features and adverse clinical parameters. Blood. 2012;120:1466–9. doi: 10.1182/blood-2012-02-408542. [DOI] [PubMed] [Google Scholar]
- 193.Jones DT, Jager N, Kool M, Zichner T, Hutter B, Sultan M, et al. Dissecting the genomic complexity underlying medulloblastoma. Nature. 2012;488:100–5. doi: 10.1038/nature11284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Guichard C, Amaddeo G, Imbeaud S, Ladeiro Y, Pelletier L, Maad IB, et al. Integrated analysis of somatic mutations and focal copy-number changes identifies key genes and pathways in hepatocellular carcinoma. Nat Genet. 2012;44:694–8. doi: 10.1038/ng.2256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Kan Z, Zheng H, Liu X, Li S, Barber TD, Gong Z, et al. Whole-genome sequencing identifies recurrent mutations in hepatocellular carcinoma. Genome Res. 2013;23:1422–33. doi: 10.1101/gr.154492.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Barber LJ, Rosa Rosa JM, Kozarewa I, Fenwick K, Assiotis I, Mitsopoulos C, et al. Comprehensive genomic analysis of a BRCA2 deficient human pancreatic cancer. PLoS One. 2011;6:e21639. doi: 10.1371/journal.pone.0021639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Biankin AV, Waddell N, Kassahn KS, Gingras MC, Muthuswamy LB, Johns AL, et al. Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature. 2012;491:399–405. doi: 10.1038/nature11547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Lindberg J, Mills IG, Klevebring D, Liu W, Neiman M, Xu J, et al. The mitochondrial and autosomal mutation landscapes of prostate cancer. Eur Urol. 2013;63:702–8. doi: 10.1016/j.eururo.2012.11.053. [DOI] [PubMed] [Google Scholar]
- 199.Grasso CS, Wu YM, Robinson DR, Cao X, Dhanasekaran SM, Khan AP, et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature. 2012;487:239–43. doi: 10.1038/nature11125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Wang K, Kan J, Yuen ST, Shi ST, Chu KM, Law S, et al. Exome sequencing identifies frequent mutation of ARID1A in molecular subtypes of gastric cancer. Nat Genet. 2011;43:1219–23. doi: 10.1038/ng.982. [DOI] [PubMed] [Google Scholar]
- 201.Zang ZJ, Cutcutache I, Poon SL, Zhang SL, McPherson JR, Tao J, et al. Exome sequencing of gastric adenocarcinoma identifies recurrent somatic mutations in cell adhesion and chromatin remodeling genes. Nat Genet. 2012;44:570–4. doi: 10.1038/ng.2246. [DOI] [PubMed] [Google Scholar]