

Abstract
Dissection of regulatory networks that control gene transcription is one of the greatest challenges of functional genomics. Using human genomic sequences, models for binding sites of known transcription factors, and gene expression data, we demonstrate that the reverse engineering approach, which infers regulatory mechanisms from gene expression patterns, can reveal transcriptional networks in human cells. To date, such methodologies were successfully demonstrated only in prokaryotes and low eukaryotes. We developed computational methods for identifying putative binding sites of transcription factors and for evaluating the statistical significance of their prevalence in a given set of promoters. Focusing on transcriptional mechanisms that control cell cycle progression, our computational analyses revealed eight transcription factors whose binding sites are significantly overrepresented in promoters of genes whose expression is cell-cycle-dependent. The enrichment of some of these factors is specific to certain phases of the cell cycle. In addition, several pairs of these transcription factors show a significant co-occurrence rate in cell-cycle-regulated promoters. Each such pair indicates functional cooperation between its members in regulating the transcriptional program associated with cell cycle progression. The methods presented here are general and can be applied to the analysis of transcriptional networks controlling any biological process.
[Supplemental material is available online at www.genome.org, including full lists of genes whose promoters were found to contain high scoring sites for any of the enriched transcription factors reported in Tables 1 and 3.]
With completion of sequencing of the human genome, focus has shifted from sequencing and mapping genes to functional genomics. The goal of functional genomics is not merely to assign genes into functional categories, but also to provide a comprehensive understanding of genetic networks—to disclose how gene products interact and regulate each other to produce coherent and coordinated physiological processes and responses to homeostatic challenges (Lockhart and Winzeler 2000). A hallmark of functional genomics is the attempt to characterize biological pathways and processes in a holistic manner (Lander and Weinberg 2000). The holistic approach has become feasible in the study of biological systems thanks to the availability of genome sequences of many organisms, the maturation of high-throughput genome-scale technologies, and the development of computational tools to analyze the rapidly accumulating volume of biological data.
Regulation of transcription is a key component of physiological networks. Indeed, it is the endpoint of many signal transduction pathways emanating from either extracellular or intracellular triggers. Transcription of genes is controlled primarily via regulatory sequence elements that are recognized and bound by transcription factors (TFs). Transcriptional regulation in eukaryotes is combinatorial in nature. The expression pattern of any particular gene is determined by an interplay among several TFs that bind its promoter. Therefore, a major task of deciphering transcriptional regulation networks is to identify combinations of TFs that cooperate in the regulation of genes and form a recurrent regulation motif, termed a “regulation module.” Recent works successfully undertook a computational approach for genome-wide mapping of transcriptional regulation modules involved in the regulation of Drosophila development (Berman et al. 2002; Halfon et al. 2002; Markstein et al. 2002). Transcriptional modules in mammalian cells were defined and identified by several pioneering works (Frech et al. 1998; Wasserman and Fickett 1998; Kel et al. 1999).
The use of DNA microarrays to study global gene expression profiles is emerging as a pivotal technology in functional genomics. Comparison of gene expression profiles under different biological conditions reveals the corresponding modifications in the cellular transcriptional programs. Microarray measurements do not, however, directly reveal the regulatory networks that underlie the observed transcriptional modulation. Combining promoter analysis with microarray results can shed light on those networks. Recent studies integrated computational promoter analysis and microarray data to identify novel transcriptional regulatory networks in Saccharoymces cerevisiae (Tavazoie et al. 1999; Jelinsky et al. 2000; Pilpel et al. 2001) These studies demonstrated that genes that are coexpressed over multiple biological conditions are often regulated via common mechanisms, and, hence, share common cis-regulatory elements in their promoters.
We developed novel computational approaches that use the human genome and data from high-throughput functional genomics technologies to dissect transcriptional regulation networks. Our methods identify TFs whose binding sites are significantly overrepresented in specific sets of promoters, as well as pairs of TFs whose binding sites exhibit a significant co-occurrence rate. Applying these methods to the analysis of cell cycle regulation in human cells disclosed key regulators in the cell cycle transcriptional program and pointed to several possible interconnections among these regulators.
RESULTS
Extraction of Putative Promoters From the Human Genome Data
As a first step in our analysis we constructed a set of putative promoter sequences of the known human genes. To this aim we downloaded the human genome data assembled into genomic contigs by the NCBI Reference Sequence project (Maglott et al. 2000; ftp://ftp.ncbi.nih.gov/genomes/H_sapiens; release of June 2001). We used the version in which human repetitive sequences are masked (mfa files). From these genomic contigs, putative promoter sequences of known human genes were extracted based on genes' start annotations provided by NCBI (gbs files provided at the same url). We determined the length of sequence around the putative TSS in which to search for transcriptional regulatory elements by examining the location distribution of 1075 empirically validated TF-binding sites in human promoters (data from TRANSFAC database; Wingender et al. 2000). Because 80% of these elements were located within 1200 bases upstream of the genes' transcription start site (TSS; data not shown), our analyses were confined to this region. Clearly, present knowledge is biased toward binding sites short distances from the TSS. Certain regulatory elements were demonstrated to act over very great distances, up to several kilobases from the TSS, but it is clear that ample information resides in sequences in close proximity to the TSS. Our promoter set contains sequences for putative promoter regions of 12,981 known human genes, each 1200 bp in length. This promoter set is referred to as the “13K set.” To estimate the accuracy of this promoter set, we compared it with experimentally validated human promoters taken from the EPD database (Praz et al. 2002). EPD contains validated promoter sequences for 247 distinct human genes. The 13K set contains promoter sequences for 180 of these genes. When the pairs of putative and validated promoters were aligned, the distance between the putative and true TSS was within 200 bp in 70% of cases (data not shown). The 13K set can be downloaded from http://www.cs.tau.ac.il/∼rshamir/prima/PRIMA.htm.
In Silico Identification of TFs That Synergize With E2F
The aim of our first approach is to reveal, by in silico analysis, TFs that cooperate with any particular TF of interest. The scheme of the analysis is as follows: A set of promoters of genes that are directly regulated by the TF of interest (termed “targets” of this TF) is constructed and scanned for overrepresented binding sites corresponding to other TFs. Such overrepresentations may point to a functional link between the overrepresented TFs and the TF of interest. Here we used this scheme in an attempt to ferret out TFs that cooperate with E2F. Because robust statistics require as large a set of E2F targets as possible, we used recent results published by Ren et al. (2002), who combined ChIP (chromatin immunoprecipitation) and microarray technologies to identify 124 genes whose promoters bind either E2F1 or E2F4 in vivo. Our 13K set contains promoter sequences for 103 of these genes. This set of E2F target promoters was scanned with experimentally derived position weight matrices (PWMs) for 107 human TFs (PWMs are from the TRANSFAC database; Wingender et al. 2000). The occurrence frequency of each PWM in the E2F target set and in the 13K set, which served as a background set, was compared, and an analytical score was computed for the significance of its observed abundance in the E2F target set (see Methods for details). For those PWMs that achieved a highly significant analytical score, we applied an additional empirical test versus random promoter sets. We determined the occurrence frequency of those high-scoring PWMs on 10,000 subsets of promoters that were randomly chosen from the 13K set and with the same size as the target set (103 promoters). We report only PWMs whose abundance in the E2F target set was significantly higher than in the random sets. The screening criterion we applied corresponded to p < 0.05 after accounting for multiple testing (see Methods for details). We identified four significantly enriched PWMs in the E2F target set (Table 1). As expected, the PWM of E2F itself is highly enriched in this set. Because E2F is a true positive in this set, the identification of its PWM demonstrates the ability of our approach to detect true signals. PWMs of three TFs—NF-Y, CREB, and NRF-1—are also significantly enriched, pointing to possible functional links between these TFs and E2F.
Table 1.
Enriched TF PWMs in Promoters of E2F Target Genes
| TF | Number of promoters with hits | Number of hits | Analytical score | Rank relative to abundance in random sets |
| E2F | 28 | 35 | 1.9 × 10−10 | 1 |
| NF-Y | 44 | 64 | 1.7 × 10−14 | 1 |
| CREB | 28 | 41 | 2.5 × 10−5 | 1 |
| NRF-1 | 32 | 77 | 3.1 × 10−4 | 3 |
A set of 103 promoters corresponding to E2F target genes reported by Ren et al. (2002) was scanned for overrepresented binding sites corresponding to 107 human TF PWMs. Four significantly enriched PWMs were found. Indicated for each one are the number of promoters with hits of the PWM and the total number of hits of the PWM (some promoters have multiple hits of a PWM), the analytical score for observing such enrichment, and the rank of the PWM's abundance in the E2F target set relative to its abundance in 10,000 sets of randomly selected promoters of the same size as that of the E2F target set. Similarity score thresholds for declaring hits were stringently determined to enable identification of real enrichments in the examined set. Therefore, the number of promoters having E2F-binding sites in this E2F target set is underestimated. Nevertheless, the observed occurrence rate of E2F is highly significant. Notably, the enrichment of the NF-Y PWM in this set is even more significant than the enrichment of the E2F PWM. Full lists of genes whose promoters were found to contain high scoring sites for the enriched TFs are provided in Supplemental Tables A1–A4 (available on line at www.genome.org).
Utilization of Functional Annotation in Dissection of Regulatory Mechanisms
Hughes et al. (2000) demonstrated that groups of functionally related genes in S. cerevisiae often share common cis-regulatory elements in their promoters. Hence, analyzing promoters of genes with common function could reveal regulatory elements characteristic to specific functional categories. We examined whether this approach could be applied to human promoters, using the functional categorization of human genes provided by the LocusLink DB (Maglott et al. 2000), which uses the standard Gene Ontology vocabulary for description of biological processes (Ashburner et al. 2000). We focused on four cell-cycle-related categories: cell cycle control, mitotic cell cycle, DNA metabolism, and M phase (some genes are assigned to several functional categories, hence the groups are not mutually exclusive). The methodology described above was applied to each category, again using the 13K set as the background set and scanning with all 107 PWMs. Significantly enriched PWMs were revealed in all functional categories (Table 2). The E2F PWM is enriched in all categories, reflecting its central role in regulating these processes. Notably, it is enriched in promoters of genes known to function in the M phase of the cell cycle. This is in accordance with recent studies (Ishida et al. 2001; Polager et al. 2002) showing that E2F's role in controlling the cell cycle goes beyond its previously documented control of the entry into the S phase. NF-Y and NRF-1 PWMs are enriched in three out of the four categories, Sp1 PWM is enriched in the cell cycle control and DNA metabolism categories, and ETF and ATF PWMs are enriched in the cell cycle control and the M-phase categories, respectively.
Table 2.
Enriched TF PWMs in Promoters of Genes That Function in the Cell Cycle
| Biological process category | Number of genes | TF | Analytical score | Rank relative to abundance in random sets |
| Cell cycle control | 223 | ETF | 1.5 × 10−7 | 1 |
| (GO 000074) | E2F | 1.5 × 10−6 | 1 | |
| NRF-1 | 2.5 × 10−5 | 1 | ||
| Sp1 | 2.5 × 10−4 | 4 (2) | ||
| Mitotic cell cycle | 175 | E2F | 1.4 × 10−9 | 1 |
| (GO 0000278) | NF-Y | 1.3 × 10−4 | 1 (2) | |
| NRF-1 | 1.6 × 10−4 | 1 | ||
| DNA metabolism | 240 | E2F | 6.7 × 10−5 | 1 |
| (GO 0006259) | NF-Y | 4.6 × 10−4 | 4 (2) | |
| Sp1 | 6.8 × 10−4 | 5 (5) | ||
| M phase | 100 | NRF-1 | 5.9 × 10−6 | 1 |
| (GO 0000279) | NF-Y | 2.5 × 10−4 | 2 (2) | |
| ATF | 3.4 × 10−4 | 4 (5) | ||
| E2F | 3.8 × 10−4 | 1 |
Promoters in the 13K set were assigned to functional categories. Functional annotations of genes were extracted from LocusLink DB, which uses the GO vocabulary (Maglott et al. 2000). Four categories related to the cell cycle, containing a total of 672 distinct genes, were analyzed (certain genes are assigned to several categories; hence the categories are not mutually exclusive). The number of promoters and the TF PWMs significantly enriched in each category are indicated. Indicated for each overrepresented PWM are the analytical score for observing such enrichment and the rank of the PWM's abundance in the functional category relative to its abundance in 10,000 sets of randomly selected promoters of the same size as that of the functional category set. Numbers in parentheses represent the number of random sets in which the PWM was equally abundant as in the functional category set.
Deciphering Regulatory Mechanisms Using Gene Expression Data
Next, we undertook the reverse engineering approach, which infers transcriptional regulatory mechanisms from gene expression data. We analyzed the human cell cycle data set published recently by Whitfield et al. (2002). Their study recorded genome-wide gene expression levels over multiple time points during the progression of the cell cycle in the HeLa human cell line; 874 genes showed periodic expression patterns over several cell cycles. Our 13K promoters set contains putative promoter sequences for 568 of these genes. Whitfield et al. (2002) partitioned the cell-cycle-regulated genes according to their expression periodicity patterns into five clusters, corresponding to cell cycle phases G1/S, S, G2, G2/M, and M/G1. We analyzed clusters of 103, 105, 122, 145, and 93 promoters, respectively.
We searched for significantly enriched PWMs in the entire set of the 568 cell cycle-regulated promoters using the 13K set as the background set. Six out of the 107 PWMs, corresponding to E2F, NF-Y, NRF-1, Sp1, ATF, and CREB TFs, were significantly overrepresented in this target set (Table 3A). We then searched for PWMs enriched only in specific phase clusters; Arnt and YY1 PWMs were specifically enriched in the G1/S and the M/G1 clusters, respectively (Table 3B). Caution must be exercised when examining whether PWMs that were enriched in the entire set favor any specific phase cluster. Given their significant overrepresentation in the entire set, random partitions of the data set are also expected to yield clusters in which these PWMs are enriched with respect to their genomic prevalence. What, therefore, should be tested is whether these PWMs favor any specific phase cluster given their prevalence in this data set rather than their genomic background prevalence. Hence, in this examination, the set of 568 cell-cycle-regulated promoters was used as the background set. The E2F PWM was found to be significantly overrepresented in the G1/S and S phases (p = 3.2 × 10−7 for the observed prevalence in these two clusters together) and underrepresented in the M/G1 cluster (p = 0.015); NF-Y PWM was overrepresented in the G2 and G2/M phases (p = 0.0096 for the observed prevalence in these two clusters together); and Sp1 PWM slightly favored the G1/S cluster (p = 0.02). NRF-1, ATF, and CREB PWMs were more uniformly distributed and showed no bias for any particular phase (Fig. 1).
Table 3.
Enriched TF PWMs in Promoters of Cell-Cycle-Regulated Genes A.
| TF | Number of promoters with hits | Number of hits | Analytical score | Rank relative to abundance in random sets |
| NF-Y | 152 | 203 | 1.2 × 10−11 | 1 |
| E2F | 78 | 92 | 1.2 × 10−8 | 1 |
| NRF-1 | 127 | 234 | 3.3 × 10−6 | 1 |
| Sp1 | 223 | 365 | 1.3 × 10−4 | 1 |
| ATF | 113 | 162 | 5.3 × 10−4 | 2 |
| CREB | 91 | 117 | 9.3 × 10−4 | 2 (1) |
| B. | |||||
| TF | Number of promoters with hits | Number of hits | Cell cycle phase | Analytical score | Rank relative to abundance in random sets |
| Arnt | 33 | 37 | G1/S | 5.1 × 10−4 | 5 (4) |
| YY1 | 20 | 25 | M/G1 | 8.1 × 10−4 | 5 (3) |
(A) A set of 568 promoters of cell cycle-regulated genes scanned for overrepresented TF PWMs, disclosing six significantly enriched PWMs. Information for each PWM is as in Table 1.
(B) Whitfield et al. (Whitfield et al. 2002) partitioned the cell cycle-regulated genes according to their expression periodicity patterns into five clusters corresponding to different phases of the cell cycle. When the promoter sequences of these clusters were scanned for enriched PWMs, two PWMs were enriched in a specific phase cluster, but not in the 568 set as a whole. Full lists of genes whose promoters were found to contain high scoring sites for the enriched TFs are provided in Supplemental Tables B1–B8 (available online at www.genome.org.)
Figure 1.

Representation of TF PWMs in the cell cycle phase clusters. The eight circles correspond to the PWMs that were highly enriched in promoters of cell-cycle-regulated genes (Table 3). Each circle is divided into five zones, corresponding to the phase clusters. The number adjacent to the zone represents the ratio of its prevalence in promoters contained in each of the cell cycle phase clusters to its prevalence in the set of 13K background promoters. Note that several TFs show a tendency toward specific cell cycle phases, for example, overrepresentation of the E2F PWM in promoters of the G1/S and S clusters and its underrepresentation in promoters of the M/G1 cluster.
We examined the location distribution of the computationally identified binding sites of the enriched PWMs. The putative binding sites for E2F, NF-Y, NRF-1, Sp1, ATF, and CREB tend to concentrate in the proximity of the TSS (Fig. 2). This observation is in agreement with experimental data on the locations of in vivo binding sites of E2F (Kel et al. 2001) and NF-Y (Mantovani 1998). In addition to the fact that the positions of the computationally identified hits are not uniformly distributed, but rather concentrated near the TSSs, we also observed that their occurrence rate declines sharply downstream of the putative TSSs (data not shown). These observations provide an additional indication for the accuracy of the putative promoters we used.
Figure 2.

Distribution of locations of TF putative binding sites found in 568 cell-cycle-regulated promoters. Promoters were divided into six intervals, 200 bp each. For each of the PWMs listed in Table 3, the number of times its computationally identified binding sites appeared in each interval was counted (after accounting for the actual number of base pairs scanned in each interval; this number changes as the masked sequences are not uniformly distributed among the six intervals). Locations of NRF-1, CREB, NF-Y, Sp1, ATF, and E2F binding sites tend to concentrate in the vicinity of the TSSs (χ2 test, p < 0.01).
Identification of Co-occurring Pairs of TFs
The approach described thus far identified TF PWMs that were enriched in target sets of promoters, with the tests performed separately on each PWM. Finding several enriched PWMs on the same target set may indirectly point to functional links between the corresponding TFs. We sought a direct method to test the associations between distinct PWMs. In an effort to identify pairs of PWMs that exhibit a significant tendency to appear together in the same promoters, we examined whether the prevalence of promoters containing hits for two PWMs was significantly higher than would be expected if the PWMs occurred independently. This analysis was applied to the set of 568 promoters of cell-cycle-regulated genes. We examined all possible pairs formed by the nine PWMs found to be enriched in any of the analyses reported above. Eight pairs showed a significant tendency to co-occur in this promoter set. Each such pair constitutes a hypothetical regulatory module, or a part thereof (Fig. 3). Figure 3 suggests that NRF-1, Sp1, ETF, and E2F may constitute transcriptional modules of higher orders, that is, recurrent motifs of three or four TFs.
Figure 3.

Pairs of PWMs that co-occur significantly in promoters of genes regulated in a cell cycle manner. We examined whether the nine PWMs reported in Tables 1 can be organized into regulatory modules. For each possible pair formed by these PWMs, we tested whether the prevalence of cell-cycle-regulated promoters that contain hits for both PWMs is significantly higher than would be expected if the PWMs occurred independently. Eight significant pairs were identified, each connected by an edge. The corresponding p-value is indicated next to the edge. The edge connecting the E2F–NRF1 pair is dashed to indicate that its significance is borderline.
DISCUSSION
The computational approaches presented here use the human genome sequence and data obtained by large-scale functional genomics technologies to determine putative regulatory mechanisms that control the transcriptional program of the cell cycle in human cells. Our analyses identified eight TFs whose regulatory sequences are significantly enriched in promoters of cell-cycle-regulated genes. The enrichment of several of these TFs was shown to be specific for certain phases of the cell cycle.
The E2F family is well documented as a prime regulator of the mammalian cell cycle. Pathways that modulate the activity of E2F are frequently disrupted in human cancers, leading to misregulated cellular proliferation (Nevins 2001). The E2F PWM obtained highly significant enrichment scores in all our analyses, demonstrating the sensitivity of our methods to reveal true signals. The role of this family of TFs in the cell cycle was underscored by several recent studies showing that E2F regulates not only genes that function in the G1/S and S phases, but also many M phase genes (Ishida et al. 2001; Polager et al. 2002). Our analysis indicates that the E2F PWM is, indeed, enriched in promoters of genes that are expressed in G2, although its enrichment in promoters of genes that are expressed in G1/S and S phases is much more prominent (Fig. 1).
Published experimental data support our findings on most of the other TFs as well. NF-Y and Sp1 PWMs obtained highly significant enrichment scores. Although involved in many different aspects of cellular life, both TFs have an established role in the regulation of the cell cycle. NF-Y was demonstrated to control the expression of several key regulators of the cell cycle (Yun et al. 1999; Jung et al. 2001; Manni et al. 2001). The transcriptional activity of Sp1 is modulated in a cell-cycle-dependent manner through its phosphorylation by cyclin A–CDK complexes (Fojas de Borja et al. 2001). In addition, several cell cycle regulators were reported to be controlled by Sp1 (Eto 2000; Paskind et al. 2000; Cram et al. 2001; Martino et al. 2001).
Our analysis shows that E2F- and NF-Y-binding sites, as well as E2F- and Sp1-binding sites, significantly co-occur in promoters of cell-cycle-regulated genes, implying functional cooperation between these TFs in the regulation of cell cycle progression. Experimental evidence supports the existence of such relations. Physical interactions were demonstrated between members of the E2F and Sp1 families (Rotheneder et al. 1999), and functional cooperation between E2F and Sp1 was reported in several cell-cycle-related promoters (Rotheneder et al. 1999; Chang et al. 2001; Huang et al. 2001; Nishikawa et al. 2001; Parisi et al. 2002). As for E2F and NF-Y, co-occurrence of functional binding sites for both TFs was reported in several promoters, including Cdc2, TK, POLA, Cyclin A, and several histone genes (Matuoka and Yu Chen 1999). Functional synergism between E2F and NF-Y was demonstrated in the regulation of the E2F-1 promoter (van Ginkel et al. 1997). Our findings substantially expand the generality of these functional links, pointing to possible synergism between these TFs on dozens of cell-cycle-regulated promoters.
Other TFs that were significantly overrepresented in cell-cycle-related promoters in our analyses have not been established as prominent regulators of the cell cycle, but data indicate they are involved in regulation of cellular proliferation. ATF/CREB is a family of more than a dozen TFs that bind a common regulatory element, the ATF/CRE (cAMP response element) motif. One member of the family, CREB, undergoes cell-cycle-regulated phosphorylation (Saeki et al. 1999), and was recently reported to control the expression of multiple cell cycle regulatory genes (Klemm et al. 2001). Overexpression of another family member, ATF2, inhibits the G1/S phase transition in a human cancer cell line (Crowe and Shemirani 2000), and is directly involved in the regulation of Cyclin A (Djaborkhel et al. 2000) and Cyclin D1 (Recio and Merlino 2002).
YY1 was reported to control several S-phase-induced genes (Johansson et al. 1998; Wu and Lee 2001). Overexpression of YY1 was reported to induce DNA synthesis (Petkova et al. 2001). Furthermore, a cell-cycle-regulated physical interaction between YY1 and pRb was reported in the same study. These findings link YY1 to induction of the S phase. In contrast, we found the YY1 PWM to be underrepresented in the S phase, but significantly enriched in the M/G1 cluster.
Arnt forms a dimeric TF with the aryl hydrocarbon receptor (AhR). It is implicated in developmental processes and tissue homeostasis. Several studies linked the AhR–Arnt dimer to cell cycle regulation. Activation of AhR was reported to induce G1 arrest (Weiss et al. 1996; Puga et al. 2000). Recently, this negative regulation was shown to depend on physical interaction between AhR and pRb (Elferink et al. 2001). In agreement, we find the enrichment of the Arnt PWM in the G1/S cluster.
Transition of cells from quiescence to proliferation increases the cell demand for energy. One way of responding to the increased demand for ATP is to modulate the activity of the respiratory chain components. NRF-1 regulates the expression of many genes required for mitochondrial respiratory function (Evans and Scarpulla 1990). A recent study demonstrated that NRF-1 activity is enhanced by phosphorylation upon serum-induced proliferation, leading to transcriptional induction of cytochrome c, a major component of the respiratory apparatus (Herzig et al. 2000). The induction of cytochrome c was associated with enhanced energy production by the mitochondria in preparation for entry to the cell cycle. The induction of cytochrome c in response to serum was shown to be mediated by both NRF-1 and CREB (Herzig et al. 2000). Interestingly, this is one of the pairs we identified, and is possibly involved in the cellular metabolic transition to the proliferative phase. In addition, our analysis suggests that NRF-1, together with Sp1, ETF, and E2F, form a recurrent motif of three or four TFs (Fig. 3).
Using genome-wide in silico computational analyses of promoters, we identified key regulators of the transcriptional program of the cell cycle in human cells. Several pairs of these TFs showed a significant co-occurrence rate on promoters of cell-cycle-regulated genes. We expect that our findings will provide guidelines for experimental dissection of the regulatory mechanisms controlling the cell cycle in mammalian cells. Moreover, the methods demonstrated here are general and can be applied to the analysis of transcriptional networks controlling any biological process. We anticipate that this type of transcriptional regulation network dissection will become an integral part of the analysis of data obtained from gene expression microarrays and large-scale chromatin immunoprecipitation studies, not only in low eukaryotes but also in mammals.
METHODS
A Set of Known Human TF Position Weight Matrices
Binding sites that are recognized and bound by TFs are commonly modeled by consensus sequences or position weight matrices (PWMs). As the latter are more informative, we used this type of model in our promoter analysis. PWMs for known human TF-binding sites were obtained from the TRANSFAC database (Wingender et al. 2000; release 5.4, April 2002). A total of 107 PWMs that correspond to distinct TFs (according to the TF name's field in the PWM entry) were used in our analyses. Because some TFs recognize similar binding sites, this PWM set might contain correlated matrices. All PWMs we used are based on at least five binding sites.
Scanning a Set of Promoters for Overrepresented PWMs
We developed a program, called PRIMA (PRomoter Integration in Microarray Analysis), written in Perl and C, for scanning a given set of promoters for TF-binding sites and identifying PWMs that are significantly overrepresented in the examined set in comparison with a background set of promoters. Given a PWM P of length l, both strands of each promoter are scanned by sliding a window of length l along the promoter. At each position of the window, a similarity score is computed between P and the corresponding subsequence of the promoter. We denote by p(i, j) the frequency of base i at position j in the PWM P. Given a promoter subsequence s1s2 … sl, we define its similarity to P as follows:
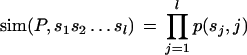 |
To identify putative binding sites, or “hits,” of a TF, a threshold T(P) for the similarity score of the TF's PWM Pis determined. Subsequences with a similarity score above T(P) are regarded as hits of P. The threshold T(P) is controlled by two parameters, α and β. The first parameter controls the rate of hits of P in random sequences as follows: A set of 400 random promoters of the same length as the real promoters is generated by an order-2 Markov model learned from the background promoters. A threshold T1 is computed, such that α percent of the random promoters contain one or more sites whose similarity score to P is above T1. The second parameter, β, controls the rate of hits of P in a background set of promoters. A threshold T2 is computed, such that β background promoters contain one or more sites whose similarity score to P is above T2. The threshold T(P) is set as the minimum of T1 and T2. Unless otherwise stated in the text, in the reported experiments, the 13K set was used as the background set of promoters, α = 10%, and β = 1000. Although the choice of these particular parameter values is somewhat arbitrary, the choice of other values gave similar results.
Once a similarity score threshold is set, the PWM P is used to scan the promoters. Given a set B of n background promoters, and a subset T of m target promoters, we compute an analytical score for the observed enrichment of PWM P in T with respect to its abundance in B. Suppose there are h hits of P in T, where at most three hits are counted per promoter. Let n1, n2, and n3 denote the number of background promoters containing one, two, or at least three hits, respectively. Assuming that T is randomly chosen out of B, the analytical score for the probability of observing at least h hits in T is:
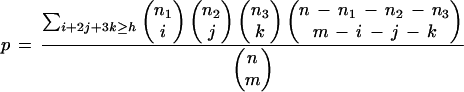 |
We used the computed analytical score as a first filter. PWMs that achieved p ≤ 0.001 were subjected to an empirical statistical test. We tested how often each of these PWMs received at least h hits on 10,000 random sets of promoters. Each set was generated by randomly choosing a subset of m background promoters from B. We report the PWMs whose observed abundance in T ranked among the top five within the 10,000 random sets. The implied significance level of this cutoff is 0.05, when applying Bonferroni correction for multiple testing of 107 distinct PWMs.
PRIMA software can be downloaded from http://www.cs.tau.ac.il/∼rshamir/prima/PRIMA.htm.
Identification of Co-occurring Pairs of PWMs
Given a set of m promoters, and a pair of PWMs, Pa and Pb, we denote by fa and fb the number of promoters that contain a hit for Pa and Pb, respectively. Let fab be the number of promoters with a hit for both Pa and Pb. The p-value for observing fab or more promoters containing hits for both PWMs is:
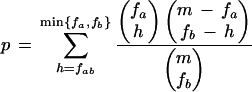 |
In this analysis we used α = 20%, β = 2000. Overlapping hits of Pa and Pb were omitted from counting. We only report pairs that remain significant (p < 0.05) after accounting for the multiple testing performed (36 pairs were tested).
Accession Numbers of Reported PWMs
The accession nos. in the TRANSFAC database of the reported transcription factor position weight matrices (PWMs) are E2F, M00516; Sp1, M00196; NF-Y, M00185; NRF-1, M00652; ETF, M00695; ATF, M00338; CREB, M00113; Arnt, M00236; YY1, M00069.
WEB SITE REFERENCES
ftp://ftp.ncbi.nih.gov/ genomes/H_sapiens; Human Genome data at NCBI.
http://genome-www.stanford.edu/Human-CellCycle/Hela/data.shtml; human cell cycle microarray data set.
http://www.cs.tau.ac.il/∼rshamir/prima/PRIMA.htm; PRIMA Web site.
http://www.gene-regulation.de; TRANSFAC database.
http://www.ncbi.nlm.nih.gov/LocusLink; LocusLink database.
http://www.epd.isb-sib.ch; EPD database.
Acknowledgments
R. Elkon is a Joseph Sassoon Fellow. R. Sharan was supported by a Fullbright grant. This study was supported by a research grant from the Ministry of Science and Technology, Israel. This work was carried out in partial fulfillment of the requirements for the Ph.D. degree of R. Elkon.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
E-MAIL yossih@post.tau.ac.il; FAX 972-3-6407471.
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.947203.
REFERENCES
- 1.Ashburner M., Ball, C.A., Blake, J.A., Botstein, D., Butler, H., Cherry, J.M., Davis, A.P., Dolinski, K., Dwight, S.S., Eppig, J.T., et al. 2000. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 25: 25-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Berman B.P., Nibu, Y., Pfeiffer, B.D., Tomancak, P., Celniker, S.E., Levine, M., Rubin, G.M., and Eisen, M.B. 2002. Exploiting transcription factor binding site clustering to identify cis-regulatory modules involved in pattern formation in the Drosophila genome. Proc. Natl. Acad. Sci. 99: 757-762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chang Y.C., Illenye, S., and Heintz, N.H. 2001. Cooperation of E2F–p130 and Sp1–pRb complexes in repression of the Chinese hamster dhfr gene. Mol. Cell. Biol. 21: 1121-1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cram E.J., Liu, B.D., Bjeldanes, L.F., and Firestone, G.L. 2001. Indole-3-carbinol inhibits CDK6 expression in human MCF-7 breast cancer cells by disrupting Sp1 transcription factor interactions with a composite element in the CDK6 gene promoter. J. Biol. Chem. 276: 22332-22340. [DOI] [PubMed] [Google Scholar]
- 5.Crowe D.L. and Shemirani, B. 2000. The transcription factor ATF-2 inhibits extracellular signal regulated kinase expression and proliferation of human cancer cells. Anticancer Res. 20: 2945-2949. [PubMed] [Google Scholar]
- 6.Djaborkhel R., Tvrdik, D., Eckschlager, T., Raska, I., and Muller, J. 2000. Cyclin A down-regulation in TGFβ1-arrested follicular lymphoma cells. Exp. Cell Res. 261: 250-259. [DOI] [PubMed] [Google Scholar]
- 7.Elferink C.J., Ge, N.L., and Levine, A. 2001. Maximal aryl hydrocarbon receptor activity depends on an interaction with the retinoblastoma protein. Mol. Pharmacol. 59: 664-673. [DOI] [PubMed] [Google Scholar]
- 8.Eto I. 2000. Molecular cloning and sequence analysis of the promoter region of mouse cyclin D1 gene: Implication in phorbol ester-induced tumour promotion. Cell Prolif. 33: 167-187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Evans M.J. and Scarpulla, R.C. 1990. NRF-1: A trans-activator of nuclear-encoded respiratory genes in animal cells. Genes & Dev. 4: 1023-1034. [DOI] [PubMed] [Google Scholar]
- 10.Fojas de Borja P., Collins, N.K., Du, P., Azizkhan-Clifford, J., and Mudryj, M. 2001. Cyclin A–CDK phosphorylates Sp1 and enhances Sp1-mediated transcription. EMBO J. 20: 5737-5747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Frech K., Quandt, K., and Werner, T. 1998. Muscle actin genes: A first step towards computational classification of tissue specific promoters. In Silico Biol. 1: 29-38. [PubMed] [Google Scholar]
- 12.Halfon M.S., Grad, Y., Church, G.M., and Michelson, A.M. 2002. Computation-based discovery of related transcriptional regulatory modules and motifs using an experimentally validated combinatorial model. Genome Res. 12: 1019-1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Herzig R.P., Scacco, S., and Scarpulla, R.C. 2000. Sequential serum-dependent activation of CREB and NRF-1 leads to enhanced mitochondrial respiration through the induction of cytochrome c. J. Biol. Chem. 275: 13134-13141. [DOI] [PubMed] [Google Scholar]
- 14.Huang D., Jokela, M., Tuusa, J., Skog, S., Poikonen, K., and Syvaoja, J.E. 2001. E2F mediates induction of the Sp1-controlled promoter of the human DNA polymerase epsilon B-subunit gene POLE2. Nucleic Acids Res. 29: 2810-2821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hughes J.D., Estep, P.W., Tavazoie, S., and Church, G.M. 2000. Computational identification of cis-regulatory elements associated with groups of functionally related genes in Saccharomyces cerevisiae. J. Mol. Biol. 296: 1205-1214. [DOI] [PubMed] [Google Scholar]
- 16.Ishida S., Huang, E., Zuzan, H., Spang, R., Leone, G., West, M., and Nevins, J.R. 2001. Role for E2F in control of both DNA replication and mitotic functions as revealed from DNA microarray analysis. Mol. Cell. Biol. 21: 4684-4699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jelinsky S.A., Estep, P., Church, G.M., and Samson, L.D. 2000. Regulatory networks revealed by transcriptional profiling of damaged Saccharomyces cerevisiae cells: Rpn4 links base excision repair with proteasomes. Mol. Cell. Biol. 20: 8157-8167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Johansson E., Hjortsberg, K., and Thelander, L. 1998. Two YY-1-binding proximal elements regulate the promoter strength of the TATA-less mouse ribonucleotide reductase R1 gene. J. Biol. Chem. 273: 29816-29821. [DOI] [PubMed] [Google Scholar]
- 19.Jung M.S., Yun, J., Chae, H.D., Kim, J.M., Kim, S.C., Choi, T.S., and Shin, D.Y. 2001. p53 and its homologues, p63 and p73, induce a replicative senescence through inactivation of NF-Y transcription factor. Oncogene 20: 5818-5825. [DOI] [PubMed] [Google Scholar]
- 20.Kel A., Kel-Margoulis, O., Babenko, V., and Wingender, E. 1999. Recognition of NFATp/AP-1 composite elements within genes induced upon the activation of immune cells. J. Mol. Biol. 288: 353-376. [DOI] [PubMed] [Google Scholar]
- 21.Kel A.E., Kel-Margoulis, O.V., Farnham, P.J., Bartley, S.M., Wingender, E., and Zhang, M.Q. 2001. Computer-assisted identification of cell cycle-related genes: New targets for E2F transcription factors. J. Mol. Biol. 309: 99-120. [DOI] [PubMed] [Google Scholar]
- 22.Klemm D.J., Watson, P.A., Frid, M.G., Dempsey, E.C., Schaack, J., Colton, L.A., Nesterova, A., Stenmark, K.R., and Reusch, J.E. 2001. cAMP response element-binding protein content is a molecular determinant of smooth muscle cell proliferation and migration. J. Biol. Chem. 276: 46132-46141. [DOI] [PubMed] [Google Scholar]
- 23.Lander E.S. and Weinberg, R.A. 2000. Genomics: Journey to the center of biology. Science 287: 1777-1782. [DOI] [PubMed] [Google Scholar]
- 24.Lockhart D.J. and Winzeler, E.A. 2000. Genomics, gene expression and DNA arrays. Nature 405: 827-836. [DOI] [PubMed] [Google Scholar]
- 25.Maglott D.R., Katz, K.S., Sicotte, H., and Pruitt, K.D. 2000. NCBI's LocusLink and RefSeq. Nucleic Acids Res. 28: 126-128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Manni I., Mazzaro, G., Gurtner, A., Mantovani, R., Haugwitz, U., Krause, K., Engeland, K., Sacchi, A., Soddu, S., and Piaggio, G. 2001. NF-Y mediates the transcriptional inhibition of the cyclin B1, cyclin B2, and cdc25C promoters upon induced G2 arrest. J. Biol. Chem. 276: 5570-5576. [DOI] [PubMed] [Google Scholar]
- 27.Mantovani R. 1998. A survey of 178 NF-Y binding CCAAT boxes. Nucleic Acids Res. 26: 1135-1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Markstein M., Markstein, P., Markstein, V., and Levine, M.S. 2002. Genome-wide analysis of clustered Dorsal binding sites identifies putative target genes in the Drosophila embryo. Proc. Natl. Acad. Sci. 99: 763-768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Martino A., Holmes, J.H.T., Lord, J.D., Moon, J.J., and Nelson, B.H. 2001. Stat5 and Sp1 regulate transcription of the cyclin D2 gene in response to IL-2. J. Immunol. 166: 1723-1729. [DOI] [PubMed] [Google Scholar]
- 30.Matuoka K. and Yu Chen, K. 1999. Nuclear factor Y (NF-Y) and cellular senescence. Exp. Cell Res. 253: 365-371. [DOI] [PubMed] [Google Scholar]
- 31.Nevins J.R. 2001. The Rb/E2F pathway and cancer. Hum. Mol. Genet. 10: 699-703. [DOI] [PubMed] [Google Scholar]
- 32.Nishikawa N., Izumi, M., Yokoi, M., Miyazawa, H., and Hanaoka, F. 2001. E2F regulates growth-dependent transcription of genes encoding both catalytic and regulatory subunits of mouse primase. Genes Cells 6: 57-70. [DOI] [PubMed] [Google Scholar]
- 33.Parisi T., Pollice, A., Di Cristofano, A., Calabro, V., and La Mantia, G. 2002. Transcriptional regulation of the human tumor suppressor p14(ARF) by E2F1, E2F2, E2F3, and Sp1-like factors. Biochem. Biophys. Res. Commun. 291: 1138-1145. [DOI] [PubMed] [Google Scholar]
- 34.Paskind M., Johnston, C., Epstein, P.M., Timm, J., Wickramasinghe, D., Belanger, E., Rodman, L., Magada, D., and Voss, J. 2000. Structure and promoter activity of the mouse CDC25A gene. Mamm. Genome 11: 1063-1069. [DOI] [PubMed] [Google Scholar]
- 35.Petkova V., Romanowski, M.J., Sulijoadikusumo, I., Rohne, D., Kang, P., Shenk, T., and Usheva, A. 2001. Interaction between YY1 and the retinoblastoma protein. Regulation of cell cycle progression in differentiated cells. J. Biol. Chem. 276: 7932-7936. [DOI] [PubMed] [Google Scholar]
- 36.Pilpel Y., Sudarsanam, P., and Church, G.M. 2001. Identifying regulatory networks by combinatorial analysis of promoter elements. Nat. Genet. 29: 153-159. [DOI] [PubMed] [Google Scholar]
- 37.Polager S., Kalma, Y., Berkovich, E., and Ginsberg, D. 2002. E2Fs up-regulate expression of genes involved in DNA replication, DNA repair and mitosis. Oncogene 21: 437-446. [DOI] [PubMed] [Google Scholar]
- 38.Praz V., Perier, R., Bonnard, C., and Bucher, P. 2002. The Eukaryotic Promoter Database, EPD: New entry types and links to gene expression data. Nucleic Acids Res. 30: 322-324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Puga A., Barnes, S.J., Dalton, T.P., Chang, C., Knudsen, E.S., and Maier, M.A. 2000. Aromatic hydrocarbon receptor interaction with the retinoblastoma protein potentiates repression of E2F-dependent transcription and cell cycle arrest. J. Biol. Chem. 275: 2943-2950. [DOI] [PubMed] [Google Scholar]
- 40.Recio J.A. and Merlino, G. 2002. Hepatocyte growth factor/scatter factor activates proliferation in melanoma cells through p38 MAPK, ATF-2 and cyclin D1. Oncogene 21: 1000-1008. [DOI] [PubMed] [Google Scholar]
- 41.Ren B., Cam, H., Takahashi, Y., Volkert, T., Terragni, J., Young, R.A., and Dynlacht, B.D. 2002. E2F integrates cell cycle progression with DNA repair, replication, and G2/M checkpoints. Genes & Dev. 16: 245-256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rotheneder H., Geymayer, S., and Haidweger, E. 1999. Transcription factors of the Sp1 family: Interaction with E2F and regulation of the murine thymidine kinase promoter. J. Mol. Biol. 293: 1005-1015. [DOI] [PubMed] [Google Scholar]
- 43.Saeki K., Yuo, A., and Takaku, F. 1999. Cell-cycle-regulated phosphorylation of cAMP response element-binding protein: Identification of novel phosphorylation sites. Biochem. J. 338 (Pt 1): 49-54. [PMC free article] [PubMed] [Google Scholar]
- 44.Tavazoie S., Hughes, J.D., Campbell, M.J., Cho, R.J., and Church, G.M. 1999. Systematic determination of genetic network architecture. Nat. Genet. 22: 281-285. [DOI] [PubMed] [Google Scholar]
- 45.van Ginkel P.R., Hsiao, K.M., Schjerven, H., and Farnham, P.J. 1997. E2F-mediated growth regulation requires transcription factor cooperation. J. Biol. Chem. 272: 18367-18374. [DOI] [PubMed] [Google Scholar]
- 46.Wasserman W.W. and Fickett, J.W. 1998. Identification of regulatory regions which confer muscle-specific gene expression. J. Mol. Biol. 278: 167-181. [DOI] [PubMed] [Google Scholar]
- 47.Weiss C., Kolluri, S.K., Kiefer, F., and Gottlicher, M. 1996. Complementation of Ah receptor deficiency in hepatoma cells: Negative feedback regulation and cell cycle control by the Ah receptor. Exp. Cell Res. 226: 154-163. [DOI] [PubMed] [Google Scholar]
- 48.Whitfield M.L., Sherlock, G., Saldanha, A.J., Murray, J.I., Ball, C.A., Alexander, K.E., Matese, J.C., Perou, C.M., Hurt, M.M., Brown, P.O., et al. 2002. Identification of genes periodically expressed in the human cell cycle and their expression in tumors. Mol. Biol. Cell 13: 1977-2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wingender E., Chen, X., Hehl, R., Karas, H., Liebich, I., Matys, V., Meinhardt, T., Pruss, M., Reuter, I., and Schacherer, F. 2000. TRANSFAC: An integrated system for gene expression regulation. Nucleic Acids Res. 28: 316-319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wu F. and Lee, A.S. 2001. YY1 as a regulator of replication-dependent hamster histone H3.2 promoter and an interactive partner of AP-2. J. Biol. Chem. 276: 28-34. [DOI] [PubMed] [Google Scholar]
- 51.Yun J., Chae, H.D., Choy, H.E., Chung, J., Yoo, H.S., Han, M.H., and Shin, D.Y. 1999. p53 negatively regulates cdc2 transcription via the CCAAT-binding NF-Y transcription factor. J. Biol. Chem. 274: 29677-29682. [DOI] [PubMed] [Google Scholar]