

Abstract
Aspirin, one of the oldest and most common anti-inflammatory agents, has recently been shown to reduce cancer risks. The principal pharmacological effects of aspirin are known to arise from its covalent modification of cyclooxygenase-2 (COX-2) through acetylation of Ser530, but the detailed mechanism of its biochemical action and specificity remains to be elucidated. In this work, we have filled this gap by employing a state-of-the-art computational approach, Born–Oppenheimer molecular dynamics simulations with ab initio quantum mechanical/molecular mechanical potential and umbrella sampling. Our studies have characterized a substrate-assisted inhibition mechanism for aspirin acetylating COX: it proceeds in two successive stages with a metastable tetrahedral intermediate, in which the carboxyl group of aspirin serves as the general base. The computational results confirmed that aspirin would be 10–100 times more potent against COX-1 than against COX-2, and revealed that this inhibition specificity between the two COX isoforms can be attributed mainly to the difference in kinetics rate of the covalent inhibition reaction, not the aspirin-binding step. The structural origin of this differential inhibition of the COX enzymes by aspirin has also been elucidated.
Aspirin (acetylsalicylic acid, or ASA), an ancient anti-inflammatory agent, is a classic wonder drug.1 Besides its wide use in the treatment of inflammation, fever, and pain for over a century and its well-known benefit in the prevention/treatment of cardiovascular diseases,1f,1g regular aspirin intake has recently been convincingly shown to reduce the overall risk of certain cancers.1a−1e
Like many other nonsteroidal anti-inflammatory drugs (NSAIDs), the primary principal pharmacological molecular target for aspirin is cyclooxygenase-2 (COX-2).2 However, the biochemical mechanism of aspirin’s therapeutic action is unique: aspirin covalently modifies the COX-2 enzyme through acetylation of Ser530 near its active site, which prevents proper binding of the native substrate and thus leads to its irreversible inhibition.3 Actually, aspirin can covalently inhibit both major isoforms of COX and is 10–100 times more potent against COX-1 than against COX-2.4 In light of COX-1’s role in gastric protection5 and COX-2’s role in inflammation,2a,6 lack of COX-2 selectivity has generally been considered as a main drawback of aspirin, which accounts for aspirin’s main side effects, such as the gastric ulceration.7 In spite of significant efforts being devoted to develop aspirin-like molecules in order to improve its COX-2 selectivity8 or reduce the gastrotoxicity,9 there has been little understanding regarding how this difference in aspirin inhibition potency against the two COX isoforms is achieved. To fill this gap, here we have employed state-of-the-art computational approaches to systematically investigate aspirin covalent inhibition of both COX isoforms.
The time-dependent and irreversible inhibition of COX by aspirin is generally believed to occur in two steps, in which a rapid reversible non-covalent binding is followed by an irreversible first-order reaction,7a i.e.,
where [EI] is the non-covalent binding complex of COX and aspirin, a key intermediate in this irreversible inhibition process. Unfortunately, due to its transient nature, no structure of the COX-aspirin non-covalent complex has been determined in spite of extensive structural work on COX enzymes. Thus, our first essential task is to computationally characterize this important intermediate for both COX enzymes. Here we first docked aspirin into the active site of crystal structure of COX-1 and COX-2 (PDB codes 1Q4G and 3NT1, respectively10,11) using Autodock 4.2,12 and carried out extensive explicit water classical molecular dynamics (MD) simulations with the amber99SB force field13 and the Amber11 molecular dynamic package.14 As shown in Figure 1, our simulation results indicate that the binding mode of aspirin is very similar between two COX enzymes: aspirin is stabilized in the active site by forming hydrogen bonds with hydroxyl groups of Tyr385 and Ser530, and the carboxyl group of aspirin is open to bulk waters and forms three additional hydrogen bonds with water molecules on average. Furthermore, we have carried out classical MD simulations to determine free energy of binding between COX and aspiring during this non-covalent binding process by employing alchemical transformations.15 The calculated absolute non-covalent binding energies of aspirin are −3.5 ± 0.4 and −3.8 ± 0.5 kcal/mol for COX-1 and COX-2, respectively, in good agreement with the experimental value of −2.4 kcal/mol deduced from Ki.16 (Experimentally determined Ki = k–1/k1 = 20 mM.) Thus, our simulations are very consistent with experiment results that aspirin is a weak non-covalent binder to COX enzymes, and indicate that the observed aspirin inhibition potency difference between the two COX isoforms does not come from the reversible non-covalent binding step.
Figure 1.
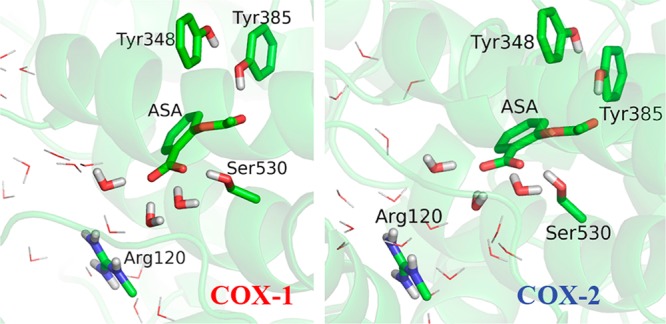
Structures of COX-aspirin noncovalent binding complexes (EI) for COX-1 and COX-2.
With modeled COX-aspirin non-covalent binding complexes, our next essential task was to characterize the irreversible acetylation step. Here we employed Born–Oppenheimer B3LYP/6-31+G* quantum mechanical/molecular mechanical (QM/MM) molecular dynamics simulations17 with umbrella sampling,18 a computational tour de force to study biochemical reactions. This state-of-the-art computational approach has been demonstrated to be powerful in characterizing the reaction mechanism for a number of complex systems.19 All our QM/MM simulations have been carried out with modified Q-Chem20 and Tinker21 programs. More computational details are presented in the Supporting Information.
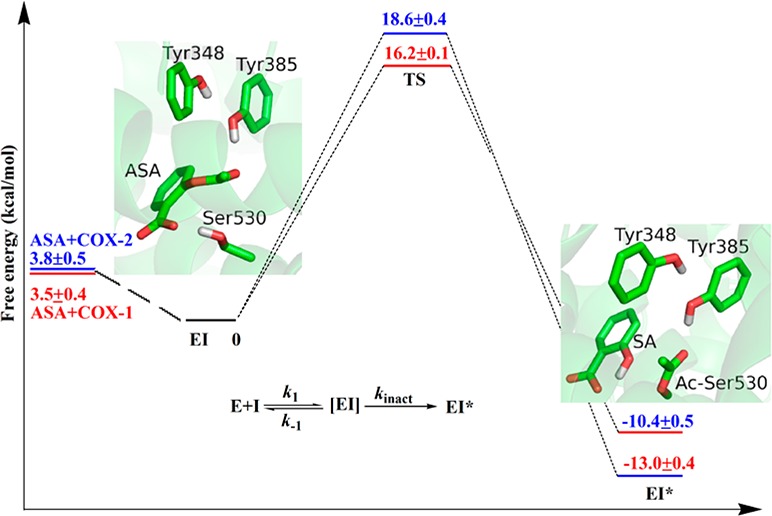
Our simulations have characterized a substrate-assisted inhibition mechanism for aspirin acetylating COX3b,16,22 as shown in Scheme 1 and Figure 2. It proceeds in two successive stages with a metastable tetrahedral intermediate (TI): in the initial step, the oxygen atom of hydroxyl group of Ser530 attacks the carbonyl carbon atom of aspirin to form TI, in which the carboxyl group of aspirin serves as the general base to abstract the proton from the hydroxyl group; in the second step, the C–O bond of aspirin carboxylic ester site breaks to yield salicylic acid (SA) and acetyl-COX (EI*) and release SA, during which the proton will transfer from the carboxylic oxygen atom to phenolic oxygen atom of SA. Comparing to the acylation mechanism employed by serine protease,19f we can see that the mechanism of aspirin acetylating COX is quite similar, in which the carboxyl group of aspirin serves as the general base and Tyr385 serves as the role of the oxyanion hole to stabilize the TI. As presented in Figure 2, and in Supporting Information, Tables S1 and S2, the overall structural characters of the active site are very similar between COX-1 and COX-2 during the covalent inhibition process. At the non-covalent binding complex state (EI), the aspirin is ideally located in a binding pocket near Ser530, with its carbonyl carbon atom positioned to be nucleophilically attacked by the serine hydroxyl oxygen OG. The two transition states during the reaction process are close to the structures of TI, and only the structures of TI are shown in Figure 2. At the TI state, the proton has already transferred to the carboxyl group of aspirin, and forms a hydrogen bond with the OG atom of Ser530 (see Scheme1 for the atom names), the scissile bond of aspirin has elongated by about 0.2 Å. For the resulted EI* structure, its active site overlaps well with that in the crystal structure of aspirin covalently bound to COX-1 (PDB code 3N8Y), as shown in Supporting Information, Figure S5. During the whole reaction, the side chain of Tyr385 forms a hydrogen bond with the carbonyl oxygen atom O2 to stabilize the negative charge on it. After a few nanoseconds of classic MD simulation for EI*, the SA will drift about 5 Å to form hydrogen bonds with the side chain of Arg120, as shown in Supporting Information, Figure S6, which is consistent with the crystal structure of the acetylated enzyme.22a One water molecule can get close to the carbonyl carbon of acetylated Ser530 (Ac-Ser530), with its oxygen atom in the position for nucleophilic attacking this carbon atom, but the hydrolysis reaction of Ac-Ser530 is hindered due to the lack of a general base.
Scheme 1. Reaction Mechanism of Aspirin Acetylating Ser530 of COX.

The names for different O atoms are also illustrated.
Figure 2.
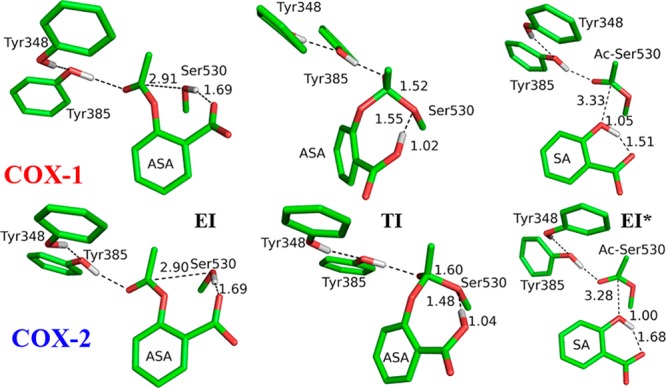
Critical structures for the acetylation reactions of COX-1 and COX-2. EI refers to the COX-aspirin noncovalent binding complex state, TI represents the tetrahedral intermediate, and EI* is the COX-aspirin covalent binding complex state.
The computed free energy profiles for aspirin acetylating COX with B3LYP(6-31+G*) QM/MM MD simulations and umbrella sampling are presented in Figure 3 for both isoforms of COX. The activation free energy barrier is 16.2 ± 0.1 kcal/mol for COX-1, which is in reasonable agreement with the experimental value of 19.2–19.7 kcal/mol derived from kinact,16,23 considering approximations in our computational methods as well as assumptions in estimating reaction barriers from experimental kinetic studies. Our calculated activation energy barrier for aspirin acetylating COX-2 is 2.4 kcal/mol higher than that for COX-1, which indicates that ratio of kincat for aspirin inhibiting COX-1 and COX-2 is around 49. This is consistent with the experimental result that aspirin would be 10–100 times more potent against COX-1 than against COX-2. From Figure 3, we can see that the free energy of EI* is more than 10 kcal/mol lower than the corresponding EI state, which indicates that the covalent inhibition reaction is exothermic and the EI* is much more stable than the EI. Thus, our simulations are consistent with experimental results that aspirin is a time-dependent and irreversible inhibitor of COX enzymes, and indicate that this inhibition specificity between the two COX isoforms mainly come from the difference in kinetics rate of the covalent inhibition reaction.
Figure 3.
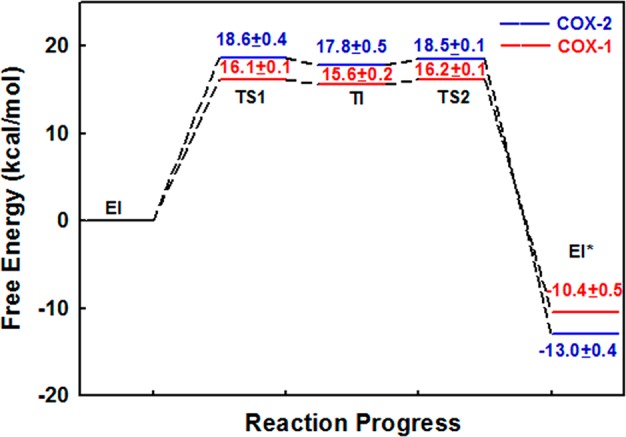
Calculated free energy changes and their statistical errors during acetylation reactions of COX-1 and COX-2 by aspirin. TS1 and TS2 refer to two transition states.
In order to elucidate structural origin of this differential inhibition of the COX enzymes by aspirin, we have calculated the individual residue contribution to the transition state stabilization during the acetylation process, and the contributions from some important residues are listed in Figure 4. The negative value indicates that the residue helps decrease the activation barrier, whereas the positive one indicates that the residue would deter the reaction. As expected, Tyr385 and Tyr348, that constitute a hydrogen-bonding network together to stabilize the accumulated negative charge on the O2 atom of aspirin during Ser530 acetylation, stabilize the TS1 for both isoforms of COX by about 4 and 2 kcal/mol, respectively. This is consistent with the site-directed mutagenesis studies of COX-2 that Tyr385Phe and Tyr348Phe mutants would decrease the inhibition of aspirin, especially Tyr385Phe, and confirmed that Tyr385 plays a vital role in COX-2 inhibition.24 However, from Figure 4, we can see that the difference in their contribution between COX-1 and COX-2 is negligible, and thus these two residues are not responsible for observed differential inhibition of the COX enzymes by aspirin.
Figure 4.
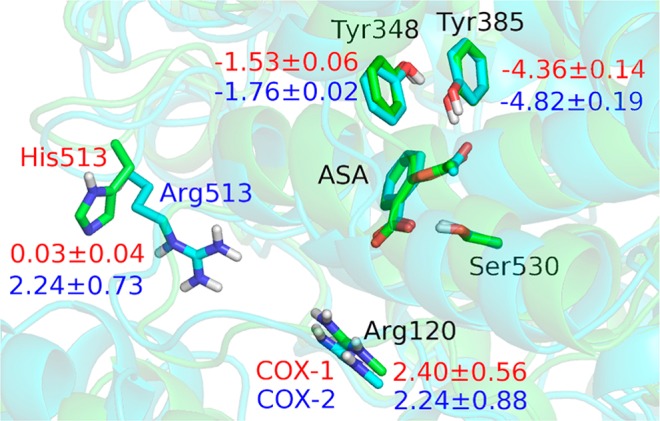
Overlap of the structures of COX-1 and COX-2 at EI state. The carbon atoms are colored green in COX-1, while cyan in COX-2. Contributions from some important residues to stabilization the transition state are also labeled in red for COX-1 (in kcal/mol), and in blue for COX-2. The negative value indicates that the residue helps decrease the activation barrier, whereas the positive one indicates that the residue would deter the reaction.
Arg120 is the only conserved positively charged residue in the COX active site, which has been shown experimentally to be important for binding of inhibitors containing a carboxylic acid moiety.25 However, the results in Figure 4 indicate that Arg120 would destabilize the transition state during the COX acetylation inhibition reaction. This brings the question how our computational studies can account for the observed role of Arg120 in aspirin inhibition of COX-125b and COX-2.24 Here we mutated Arg120 to Ala, and calculated the change in the binding free energy of mutants ΔΔG for the non-covalent binding of aspirin by thermodynamic integration.26 The calculation results indicate that the mutation of Arg120 to Ala would significantly weaken the non-covalent binding of aspiring by increasing binding free energies of 4.3 ± 0.3 and 5.6 ± 0.2 kcal/mol for COX-1 and COX-2 respectively. Thus, although Arg120 does not directly form the salt bridge with aspirin in the active site, its proximity plays an important role in facilitating the formation of the initial non-covalent COX-aspirin binding complex. Meanwhile, Arg120 is conserved between COX-1 and COX-2, and thus it is not responsible for the aspirin inhibition potency difference between COX-1 and COX-2 either.
As Figure 4 shows, close to the binding pocket of aspirin in COX-2, there is another positive residue Arg513, while the corresponding residue in COX-1 is His513. Our analysis indicates that the presence of Arg513 in COX-2 would increase the activation barrier of acetylation reaction, while the corresponding His513 in COX-1 plays almost no role in transition state destabilization. By examining structures, we can see that, in comparison with His513 of COX-1, the interaction between the positively charged guanidinium group of Arg513 of COX-2 and the negatively charged carboxylic group of aspirin would disfavor the protonation of aspirin’s carboxylic group during the reaction process and thus could slow down the covalent inhibition reaction of COX-2. This is consistent with experimental results that the derivatization of the carboxylate moiety of the inhibitor can always increase the COX-2 selectivity.25a Thus, it would be interesting to experimentally study R513H mutant of COX-2, including its enzyme activity and its inhibition by aspirin.
In summary, we have provided detailed insights into the biochemical inner workings of aspirin by employing state-of-the-art computational approaches. The covalent inhibition of COX by aspirin proceeds in two successive stages with a metastable tetrahedral intermediate. The difference in aspirin inhibition potency against the two COX isoforms is found to mainly come from the difference in kinetics rate of the covalent inhibition reaction, not from the non-covalent aspirin-binding step. Our results suggest that the presence of Arg513 in COX-2 (the corresponding residue in COX-1 is His513) would increase the activation barrier for the aspirin acetylation reaction, which is likely to be an important factor that makes aspirin a weaker covalent inhibitor against COX-2 than against COX-1.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (Grant No. 21203090 to Y. Zhou) and by the U.S. National Institutes of Health (R01-GM079223 to Y. Zhang). We thank the High Performance Computing Center of Nanjing University and NYU-ITS for providing computational resources.
Supporting Information Available
Computational details, Figures S1–S6, and Tables S1 and S2. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- a Lissa D.; Senovilla L.; Rello-Varona S.; Vitale I.; Michaud M.; Pietrocola F.; Boileve A.; Obrist F.; Bordenave C.; Garcia P.; Michels J.; Jemaa M.; Kepp O.; Castedo M.; Kroemer G. Proc. Natl. Acad. Sci. U.S.A. 2014, 111, 3020. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Bateman L. A.; Zaro B. W.; Miller S. M.; Pratt M. R. J. Am. Chem. Soc. 2013, 135, 14568. [DOI] [PubMed] [Google Scholar]; c Kaiser J. Science 2012, 337, 1471. [DOI] [PubMed] [Google Scholar]; d Pathak R. K.; Marrache S.; Choi J. H.; Berding T. B.; Dhar S. Angew. Chem., Int. Ed. 2014, 53, 1963. [DOI] [PubMed] [Google Scholar]; e Thun M. J.; Jacobs E. J.; Patrono C. Nat. Rev. Clin. Oncol. 2012, 9, 259. [DOI] [PubMed] [Google Scholar]; f Ann. Int. Med. 2009, 150, 396.19293072 [Google Scholar]; g Fowkes F. G. R.; Price J. F.; Stewart M. C. W.; Butcher I.; Leng G. C.; Pell A. C. H.; Sandercock P. A. G.; Fox K. A. A.; Lowe G. D. O.; Murray G. D. JAMA—J. Am. Med. Assoc. 2010, 303, 841. [DOI] [PubMed] [Google Scholar]
- a Masferrer J. L.; Zweifel B. S.; Manning P. T.; Hauser S. D.; Leahy K. M.; Smith W. G.; Isakson P. C.; Seibert K. Proc. Natl. Acad. Sci. U.S.A. 1994, 91, 3228. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Pennisi E. Science 1998, 280, 1191. [DOI] [PubMed] [Google Scholar]
- a Flower R. J. Nat. Rev. Drug Discovery 2003, 2, 179. [DOI] [PubMed] [Google Scholar]; b Blobaum A. L.; Marnett L. J. J. Med. Chem. 2007, 50, 1425. [DOI] [PubMed] [Google Scholar]
- a Vane J. R.; Bakhle Y. S.; Botting R. M. Annu. Rev. Pharmacol. Toxicol. 1998, 38, 97. [DOI] [PubMed] [Google Scholar]; b Mitchell J. A.; Akarasereenont P.; Thiemermann C.; Flower R. J.; Vane J. R. Proc. Natl. Acad. Sci. U.S.A. 1993, 90, 11693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Kulmacz R. J.; van der Donk W. A.; Tsai A. L. Prog. Lipid Res. 2003, 42, 377. [DOI] [PubMed] [Google Scholar]; b Rouzer C. A.; Marnett L. J. Biochem. Biophys. Res. Commun. 2005, 338, 34. [DOI] [PubMed] [Google Scholar]
- Vane J. R.; Mitchell J. A.; Appleton I.; Tomlinson A.; Bishopbailey D.; Croxtall J.; Willoughby D. A. Proc. Natl. Acad. Sci. U.S.A. 1994, 91, 2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Gierse J. K.; Koboldt C. M.; Walker M. C.; Seibert K.; Isakson P. C. Biochem. J. 1999, 339, 607. [PMC free article] [PubMed] [Google Scholar]; b Meade E. A.; Smith W. L.; Dewitt D. L. J. Biol. Chem. 1993, 268, 6610. [PubMed] [Google Scholar]
- a Kalgutkar A. S.; Crews B. C.; Rowlinson S. W.; Garner C.; Seibert K.; Marnett L. J. Science 1998, 280, 1268. [DOI] [PubMed] [Google Scholar]; b Kalgutkar A. S.; Kozak K. R.; Crews B. C.; Hochgesang G. P.; Marnett L. J. J. Med. Chem. 1998, 41, 4800. [DOI] [PubMed] [Google Scholar]
- a Tosco P.; Lazzarato L. ChemMedChem 2009, 4, 939. [DOI] [PubMed] [Google Scholar]; b Lazzarato L.; Donnola M.; Rolando B.; Marini E.; Cena C.; Coruzzi G.; Guaita E.; Morini G.; Fruttero R.; Gasco A.; Biondi S.; Ongini E. J. Med. Chem. 2008, 51, 1894. [DOI] [PubMed] [Google Scholar]; c Deeb R. S.; Cheung C.; Nuriel T.; Lamon B. D.; Upmacis R. K.; Gross S. S.; Hajjar D. P. J. Am. Chem. Soc. 2010, 132, 3914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta K.; Selinsky B. S.; Kaub C. J.; Katz A. K.; Loll P. J. J. Mol. Biol. 2004, 335, 503. [DOI] [PubMed] [Google Scholar]
- Duggan K. C.; Walters M. J.; Musee J.; Harp J. M.; Kiefer J. R.; Oates J. A.; Marnett L. J. J. Biol. Chem. 2010, 285, 34950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris G. M.; Huey R.; Lindstrom W.; Sanner M. F.; Belew R. K.; Goodsell D. S.; Olson A. J. J. Comput. Chem. 2009, 30, 2785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Cornell W. D.; Cieplak P.; Bayly C. I.; Gould I. R.; Merz K. M.; Ferguson D. M.; Spellmeyer D. C.; Fox T.; Caldwell J. W.; Kollman P. A. J. Am. Chem. Soc. 1995, 117, 5179. [Google Scholar]; b Hornak V.; Abel R.; Okur A.; Strockbine B.; Roitberg A.; Simmerling C. Proteins 2006, 65, 712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Case D. A.; Darden T. A.; Cheatham T. E. III; Simmerling C. L.; Wang J.; Duke R. E.; Luo R.; ; Walker R. C.; Zhang W.; Merz K. M.; Roberts B.; Wang B.; Hayik S.; Roitberg A.; Seabra G.; Kolossváry I.; Wong K. F.; Paesani F.; Vanicek J.; Liu J. Wu X.; Brozell S. R.; Steinbrecher T.; Gohlke H.; Cai Q.; Ye X.; Wang J.; Hsieh M.-J.; Cui G.; Roe D. R.; Mathews D. H.; Seetin M. G.; Sagui C.; Babin V.; Luchko T.; Gusarov S.; Kovalenko A.; Kollman P. A.. Amber 11; University of California: San Francisco, 2010.
- a Gumbart J. C.; Roux B.; Chipot C. J. Chem. Theory Comput. 2013, 9, 794. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Gumbart J. C.; Roux B.; Chipot C. J. Chem. Theory Comput. 2013, 9, 3789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dewitt D. L.; Elharith E. A.; Kraemer S. A.; Andrews M. J.; Yao E. F.; Armstrong R. L.; Smith W. L. J. Biol. Chem. 1990, 265, 5192. [PubMed] [Google Scholar]
- a Ganguly A.; Thaplyal P.; Rosta E.; Bevilacqua P. C.; Hammes-Schiffer S. J. Am. Chem. Soc. 2014, 136, 1483. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Lans I.; Medina M.; Rosta E.; Hummer G.; Garcia-Viloca M.; Lluch J. M.; Gonzalez-Lafont A. J. Am. Chem. Soc. 2012, 134, 20544. [DOI] [PubMed] [Google Scholar]; c Meier K.; Choutko A.; Dolenc J.; Eichenberger A. P.; Riniker S.; van Gunsteren W. F. Angew. Chem., Int. Ed. 2013, 52, 2820. [DOI] [PubMed] [Google Scholar]; d Zhang R.; Lev B.; Cuervo J. E.; Noskov S. Y.; Salahub D. R. In Combining Quantum Mechanics and Molecular Mechanics. Some Recent Progresses in QM/MM Methods; Advances in Quantum Chemistry59; Sabin J. R., Brändas E., Eds.; Elsevier: Amsterdam, 2010; p 353. [Google Scholar]
- a Patey G. N.; Valleau J. P. J. Chem. Phys. 1975, 63, 2334. [Google Scholar]; b Roux B. Comput. Phys. Commun. 1995, 91, 275. [Google Scholar]; c Boczko E. M.; Brooks C. L. J. Phys. Chem. 1993, 97, 4509. [Google Scholar]
- a Wu R. B.; Lu Z. Y.; Cao Z. X.; Zhang Y. K. J. Am. Chem. Soc. 2011, 133, 6110. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Rooklin D. W.; Lu M.; Zhang Y. K. J. Am. Chem. Soc. 2012, 134, 15595. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Ke Z. H.; Smith G. K.; Zhang Y. K.; Guo H. J. Am. Chem. Soc. 2011, 133, 11103. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Wu R.; Wang S.; Zhou N.; Cao Z.; Zhang Y. J. Am. Chem. Soc. 2010, 132, 9471. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Zhou Y. Z.; Wang S. L.; Zhang Y. K. J. Phys. Chem. B 2010, 114, 8817. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Zhou Y.; Zhang Y. Chem. Commun. 2011, 47, 1577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao Y.; Molnar L. F.; Jung Y.; et al. Phys. Chem. Chem. Phys. 2006, 8, 3172. [DOI] [PubMed] [Google Scholar]
- Ponder J. W.TINKER 4.2, Software Tools for Molecular Design; June 2004. [DOI] [PMC free article] [PubMed]
- a Loll P. J.; Picot D.; Garavito R. M. Nat. Struct. Biol. 1995, 2, 637. [DOI] [PubMed] [Google Scholar]; b Tosco P.; Lazzarato L. ChemMedChem 2009, 4, 939. [DOI] [PubMed] [Google Scholar]; c Toth L.; Muszbek L.; Komaromi I. J. Mol. Graphics Model. 2013, 40, 99. [DOI] [PubMed] [Google Scholar]
- a Rome L. H.; Lands W. E. M. Proc. Natl. Acad. Sci. U.S.A. 1975, 72, 4863. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Ouellet M.; Riendeau D.; Percival M. D. Proc. Natl. Acad. Sci. U.S.A. 2001, 98, 14583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hochgesang G. P.; Rowlinson S. W.; Marnett L. J. J. Am. Chem. Soc. 2000, 122, 6514. [Google Scholar]
- a Kalgutkar A. S.; Crews B. C.; Rowlinson S. W.; Marnett A. B.; Kozak K. R.; Remmel R. P.; Marnett L. J. Proc. Natl. Acad. Sci. U.S.A. 2000, 97, 925. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Mancini J. A.; Riendeau D.; Falgueyret J. P.; Vickers P. J.; Oneill G. P. J. Biol. Chem. 1995, 270, 29372. [DOI] [PubMed] [Google Scholar]; c Bhattacharyya D. K.; Lecomte M.; Rieke C. J.; Garavito R. M.; Smith W. L. J. Biol. Chem. 1996, 271, 2179. [DOI] [PubMed] [Google Scholar]
- a Kollman P. Chem. Rev. 1993, 93, 2395. [Google Scholar]; b de Ruiter A.; Boresch S.; Oostenbrink C. J. Comput. Chem. 2013, 34, 1024. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.