

Abstract
Background
Right ventricular (RV) diastolic function is impaired in patients with pulmonary arterial hypertension (PAH). Our previous study showed that elevated cardiomyocyte stiffness and myofilament Ca2+ sensitivity underlie diastolic dysfunction in PAH. This study investigates protein modifications contributing to cellular diastolic dysfunction in PAH.
Methods and Results
RV samples from PAH patients undergoing heart‐lung transplantation were compared to non‐failing donors (Don). Titin stiffness contribution to RV diastolic dysfunction was determined by Western‐blot analyses using antibodies to protein‐kinase‐A (PKA), Cα (PKCα) and Ca2+/calmoduling‐dependent‐kinase (CamKIIδ) titin and phospholamban (PLN) phosphorylation sites: N2B (Ser469), PEVK (Ser170 and Ser26), and PLN (Thr17), respectively. PKA and PKCα sites were significantly less phosphorylated in PAH compared with donors (P<0.0001). To test the functional relevance of PKA‐, PKCα‐, and CamKIIδ‐mediated titin phosphorylation, we measured the stiffness of single RV cardiomyocytes before and after kinase incubation. PKA significantly decreased PAH RV cardiomyocyte diastolic stiffness, PKCα further increased stiffness while CamKIIδ had no major effect. CamKIIδ activation was determined indirectly by measuring PLN Thr17phosphorylation level. No significant changes were found between the groups. Myofilament Ca2+ sensitivity is mediated by sarcomeric troponin I (cTnI) phosphorylation. We observed increased unphosphorylated cTnI in PAH compared with donors (P<0.05) and reduced PKA‐mediated cTnI phosphorylation (Ser22/23) (P<0.001). Finally, alterations in Ca2+‐handling proteins contribute to RV diastolic dysfunction due to insufficient diastolic Ca2+ clearance. PAH SERCA2a levels and PLN phosphorylation were significantly reduced compared with donors (P<0.05).
Conclusions
Increased titin stiffness, reduced cTnI phosphorylation, and altered levels of phosphorylation of Ca2+ handling proteins contribute to RV diastolic dysfunction in PAH.
Keywords: diastole, pulmonary heart disease
Introduction
Patients with pulmonary arterial hypertension (PAH) develop severe right ventricular (RV) failure with impaired diastolic function.1 In a previous study, we showed that collagen deposition, increased cardiomyocyte stiffness, and myofilament Ca2+ sensitivity contribute to compromising RV diastolic function.1 In the present study, we investigate protein changes that underlie RV cardiomyocyte diastolic dysfunction.
Diastolic dysfunction of the left ventricle (LV) was shown to be related to increased cardiomyocyte stiffness as a consequence to functional modifications of the sarcomeric protein titin. By folding during contraction and stretching during relaxation, titin acts as a sarcomeric molecular spring and represents the major determinant of cardiomyocyte stiffness at physiological sarcomere lengths.2 Titin spans half of the sarcomere, from the Z line to the M band, and consists of a linear array of proximal and distal Ig‐like domains, together with N2B and PEVK segments.3 Its stiffness is modulated by complex mechanisms involving both fast post‐translational modification (phosphorylation) and slower changes in isoform expression.4 The effect of titin phosphorylation depends on the domain targeted. Phosphorylation of the N2B domain has been shown to lower cardiomyocyte stiffness, while PEVK domain phosphorylation exerts the opposite effect.5–7
Previously, we have shown that, unlike LV diastolic dysfunction, titin isoform composition is not changed in the RV of PAH patients. However, there is an overall decrease in titin phosphorylation in these patients.1 Therefore, the first aim of this study was to determine the particular titin phosphorylation changes specific to the RV, which could explain the increase in RV cardiomyocyte stiffness in PAH patients.
In addition to titin‐derived stiffness, RV cardiomyocytes of PAH patients are characterized by increased myofilament Ca2+‐sensitivity, which may in turn influence cardiomyocyte lusiothropy.8 Cardiac Troponin I (cTnI) and cardiac myosin binding protein C (MyBPC) are 2 important regulators of myofilament Ca2+ sensitivity.8–12 The second aim of this study was to determine whether changes in cTnI and MyBPC phosphorylation are related to the previously observed increase in RV myofilament Ca2+ sensitivity.
Furthermore, fast cytoplasmic Ca2+ clearance during diastole is crucial for a normal relaxation pattern.13 The speed of diastolic Ca2+ reuptake into the sarcoplasmic reticulum is determined by the activity of the sarcoplasmic reticulum Ca2+‐ATPase‐2a (SERCA2a).14–15 The rate at which SERCA2a transfers [Ca2+] across the sarcoplasmic reticulum membrane is enhanced by PKA‐mediated phospholamban (PLN) phosphorylation during β‐AR stimulation.16 The inhibitory coupling‐protein PLN is removed from SERCA2a secondary to PLN phosphorylation (pPLN). Ca2+ clearance is also regulated by the sodium‐calcium exchanger 1 (NCX1) that is responsible for extracellular extrusion of 1 Ca2+ ion in exchange for 3 imported Na+ ions.17 Previous animal model studies show the relation between altered Ca2+‐clearance proteins and impaired cardiac relaxation.14–15 However, the expression and function of Ca2+‐clearance proteins in the failing human RV is not known. Therefore, the third aim of this study was to determine whether Ca2+‐clearance protein expression and phosphorylation is altered in the RV of PAH patients.
To summarize, the present study investigated the protein changes involved in altering RV cardiomyocyte diastolic function in patients with PAH. Our findings reveal that increased titin stiffness, reduced cTnI phosphorylation, and altered expression levels of the Ca2+‐handling proteins contribute to RV diastolic dysfunction in PAH patients.
Methods
Tissue Samples
Explanted RV tissue samples were collected from PAH patients undergoing heart transplantation (n=11) and compared with RV tissue obtained from non‐failing donors (n=9) (Table). There were no significant differences in gender and age between the 2 groups (AgePAH=36.6±3.9, AgeDon=41.1±4.7, P=0.47, Gender(F/M)PAH=11/1, Gender(F/M)Don=6/3 P=0.28). Human cardiac tissue collection and use by collaborating universities (VU Medical Center, Amsterdam) was approved by the Human Research Ethics Committee of The University of Sydney (AU/1/961515) and the Université Paris‐Sud‐Inserm U999 (ID RBC 2008‐A00485‐50). All patients received treatment previous to cardiac transplantation corresponding to the clinical protocols present at the time of the intervention. Various treatments (acute Dobutamine administration) may be associated with the differences observed between patients. After transplantation, RV tissue samples were immediately frozen and stored in liquid nitrogen, preserving the expression and phosphorylation level of the cardiomyocytes.
Table 1.
Clinical and Demographic Characteristics
| RV Sample | Diagnosis | NYHA Class | Gender | Age (y) |
|---|---|---|---|---|
| 1 | Idiopathic PAH | IV | Female | 38 |
| 2 | Idiopathic PAH | IV | Female | 44 |
| 3 | Idiopathic PAH | IV | Female | 51 |
| 4 | PAH‐Eisenmenger | IV | Female | 46 |
| 5 | PAH‐Eisenmenger | IV | Female | 14 |
| 6 | PAH‐Eisenmenger | IV | Female | 20 |
| 7 | PAH‐Eisenmenger | IV | Female | 31 |
| 8 | PAH‐Eisenmenger | IV | Female | 21 |
| 9 | PAH‐Eisenmenger | IV | Male | 46 |
| 10 | PAH‐Eisenmenger | IV | Female | 50 |
| 11 | PAH‐Eisenmenger | IV | Female | 41 |
| 12 | Donor | Female | 41 | |
| 13 | Donor | Female | 23 | |
| 14 | Donor | Female | 19 | |
| 15 | Donor | Female | 53 | |
| 16 | Donor | Male | 65 | |
| 17 | Donor | Female | 49 | |
| 18 | Donor | Male | 45 | |
| 19 | Donor | Female | 38 | |
| 20 | Donor | Male | 37 |
PAH indicates pulmonary arterial hypertension; NYHA, New York Heart Association.
Titin Phosphorylation
To determine kinase‐specific titin phosphorylation, donor (n=7) and PAH (n=5) frozen RV tissue samples were weighed and pulverized in liquid nitrogen using a mortar and a pestle. Tissue powder was solubilized in 8 mol/L urea buffer with DTT and 50% glycerol solution with protease inhibitors (0.16 mmol/L Leupeptin, 0.04 mmol/L E‐64 and 0.2 mmol/L PMSF). Samples were loaded on 1% agarose gels stained with Coomassie Blue for protein identification. Equal titin sample dilutions were calculated derived from Myosin Heavy Chain (MHC) protein content and applied for isoform ratio determination.18
Titin phosphorylation of PKA and PKCα sites was assessed using Western blots with specific antibodies against serine 469 (Ser4185, titin N2B cardiac, UnitprotKB: Q8WZ42) on the N2B domain (PKA phosphorylation site) and serine 26 and 170 (Ser11878, UnitprotKB:Q8WZ42 and Ser12022, UnitprotKB:Q8WZ42) on the PEVK domain (PKCα phosphorylation sites). Equal sample loadings were separated on 0.8% agarose gels, transferred to PVDF membranes (Immobilon®‐FL, Cat.No.IPFL00010, 045 μm) and probed with the relevant antibodies. Membranes were scanned and analyzed using Odyssey Infrared Imaging System (Li‐COR Biosciences). N2B and N2BA protein content was determined on Ponceau‐S‐stained membranes and used to normalize for phosphorylation level.5
RV Cardiomyocyte Diastolic Stiffness
RV free wall tissue samples (between 20 and 40 mg) were defrosted in relaxing solution then sectioned in smaller pieces with a fine dissection scissors. Subsequently, single cardiomyocytes were isolated from these tissue pieces by manual mechanical homogenization. The single cells were then membrane permeabilized by adding Triton (1%) to the relaxing solution in order to wash out the lipid cellular membranes. The cell solution was repeatedly washed with relaxing solution in order to remove Triton. The cell yield is somewhat variable, depending on the initial amount of tissue, the release of cardiomyocyte from the tissue bulk during mechanical homogenization and the loss of cells during Triton washing. Cells for force measurements were chosen based on length and width (length: 50 to 100 μm, width 15 to 30 μm at rest in relaxing solutions).1 A minimum of 3 cells per sample was used to determine diastolic stiffness and the average was used for further statistic analysis. A single cell was attached with silicone adhesive between a force transducer and a piezoelectric motor. To determine cardiomyocyte stiffness a 25% shortening was performed in the relaxing solution and steady‐state stiffness measurements were recorded at increasing sarcomere lengths (1.8 to 2.4 μm).1
Cardiomyocytes were further incubated in relaxing solution with PKA (100 U/mL) (Protein Kinase A Catalytic subunit from bovine heart, P2645; Sigma‐Aldrich) (donor n=4, PAH n = 4), CamKIIδ (50 U/mL) or PKC‐subunit α (10 U/mL) (Protein Kinase C α human isozyme, P1782; Sigma‐Aldrich) (donor n=3, PAH n=3) at 20°C for 1 hour. PKC α also requires Ca2+, therefore the relaxing solution was mixed in a 1:1 concentration ratio with a Ca2+‐containing solution of pCa 5.8, obtaining therefore an end pCa of 5.9 in which the cardiomyocytes were incubated. Diastolic stiffness was recorded after PKA, PKCα, or CamKIIδ incubation.19 Individual force values were normalized by the cardiomyocyte cross‐sectional area recorded at 2.2 μm sarcomere length.
Sarcomeric Protein Phosphorylation
Sarcomeric protein phosphorylation was determined for each tissue sample (donor n=9, PAH n=11). RV tissue samples were homogenized and separated on gradient gels (NuPAGE® Bis‐Tris Gels; Life Technologies). ProQ Diamond Phosphoprotein Stain was used to determine the amount of protein phosphorylation. Gels were further fixed, washed, destained, and stained with SYPRO Ruby to determine the total amount of protein. Myofilament protein phosphorylation ratio (ProQ) was calculated relative to the corresponding SYPRO staining (ProQ/SYPRO).20
Cardiac Troponin I Phosphorylation
Proteins were separated on 1‐dimensional gel electrophoresis on NuPAGE® Bis‐Tris gels (donor n=9, PAH=11). The XCell II™ Blot Module (Life Technologies) was used for wet protein transfer from mini‐gels to ECL membranes (Hybond ECL Nitrocellulose Membrane; GE Healthcare). Blots were incubated with the following primary antibodies against specific protein or protein phosphorylation sites: cTnI dephosphorylated form (4T46, mouse monoclonal antibody, HyTest), cTnI PKA‐specific serine 22/23 site phosphorylation (4004, rabbit polyclonal antibody; Cell Signaling Technology).9–10 The amount of protein expression or phosphorylation was normalized to the concentration of Ponceau‐S stained actin.
The distribution of cTnI phosphorylation (unphosphorylated [P0], mono‐phosphorylated [P1], bis‐phosphorylated [P2]) was determined on acrylamide Phos‐TagTM gels.10
Ca2+‐Handling Proteins Expression and Phosphorylation
To determine SERCA2a expression, monoclonal rabbit antibody was used (courtesy of Warner S. Simonides, VU University Medical Center) (donor n=9, PAH=11). PLN binds to and inhibits SERCA2a, while phosphorylation of PLN (pPLN) removes the inhibitory binding of PLN and promotes SERCA2a activity. PLN and pPLN were determined with the following antibodies: total PLN (L15, sc21923; Santa Cruz Biotechnology, Inc), PKA‐specific PLN phosphorylation site (Ser16, sc12963; Santa Cruz Biotechnology, Inc), and CaMKIIδ‐specific PLN phosphorylation (Thr17, A010‐13AP; Badrilla). NCX1 expression was quantified using NCX1‐C2C12 antibody (ab2869; Abcam). Cardiac specific Ryanodine Receptor 2 expression was also quantified (C3‐33; ThermoFisher Scientific). The amount of protein expression or phosphorylation was normalized to the concentration of Ponceau‐S‐stained actin.
Statistical Analyses
Statistical analyses were performed using Prism 5 for Windows (GraphPad Software Inc and IBM® SPSS® Statistics 20.0; IBM Corporation). P values lower than 0.05 were considered significant. All data are presented as mean±SEM.
Age differences between patients were tested for significance by a non‐paired t test. Gender differences were tested by a Fisher's exact test. Changes in protein expression were tested for significance by a non‐paired, non‐parametric Mann Whitney U test. Phos‐Tag™ analysis was tested for significance by repeated 2‐way ANOVA followed by the Bonferroni post‐hoc test.
The effects of PKA, PKCα, and CamKIIδ incubation in PAH patients and donors at increasing sarcomere lengths were tested by using a mixed‐design ANOVA with disease as between‐group measure, sarcomere length and PKA/PKCα/CamKIIδ‐incubation as repeated measures. The greenhouse‐Geisser correction was used, because sphericity could not be assumed.
Results
PKA‐Mediated Titin Phosphorylation is Reduced in PAH RV Cardiomyocytes
Cardiomyocyte stiffness is modulated by titin isoform composition and phosphorylation. In our previous study we showed that titin isoform ratio (N2BA/N2B) is not significantly changed in PAH compared with donors (N2BA/N2BDon=0.91±0.08, N2BA/N2BPAH=0.77±0.07, P=0.20).1 Therefore, the overall increase in cardiomyocyte stiffness may be a consequence of titin N2B or PEVK domain phosphorylation.
Titin phosphorylation was determined using phospho‐specific antibodies for PKA and PKCα phosphorylation sites. We found significantly reduced levels of PKA‐dependent phosphorylation of N2B serine 469 site and PKCα‐dependent phosphorylation of PEVK serine 170 site (PKADon=1.00±0.03, PKAPAH=0.44±0.04, P=0.002; Figure 1A1) and (PKCα‐S170Don=1.00±0.06, PKCα‐S170PAH=0.46±0.06, P=0.002; Figure 1B1). No significant difference was found in PKCα‐dependent phosphorylation of PEVK serine 26 (PKCα‐S26Don=1.00±0.12, PKCα‐S26PAH=0.96±0.12, P=0.53; Figure 1B2).
Figure 1.

PKA, PKCα, and CaMKIIδ treatment effect on diastolic stiffness mediated by titin phosphorylation. A‐A1. Titin N2B domain serine 469 PKΑ‐dependent phosphorylation. Typical example of the 2 titin isoforms (upper band: N2BA; lower band N2B) immunostained with phosphospecific antibody against serine 469 site on titin N2B domain and the corresponding Ponceau S staining for total titin (nDon=7, nPAH=5). A2. Donor and PAH cardiomyocyte stiffness was measured in relaxing solution at increasing sarcomere length (1.8 to 2.4 μm)—continuous line. The same cardiomyocyte was further incubated with PKA active subunit and stiffness measurements were repeated—dotted line (nDon=4, nPAH=4). B‐B1. Titin PEVK domain serine 170 PKCα‐dependent phosphorylation. Typical example of the 2 titin isoforms (upper band: N2BA; lower band N2B) immunostained with phosphospecific antibody against serine 170 site on titin PEVK domain and the corresponding Ponceau S staining for total titin (nDon=7, nPAH=5). B2.Titin PEVK domain serine 26 PKCα‐dependent phosphorylation. Typical example of the 2 titin isoforms (upper band: N2BA; lower band N2B) immunostained with phosphospecific antibody against serine 26 site on titin PEVK domain and the corresponding Ponceau S staining for total titin (nDon=7, nPAH=5). B3. Donor and PAH cardiomyocyte stiffness was measured in relaxing solution at increasing sarcomere length (1.8 to 2.4 μm)—continuous line. The same cardiomyocyte was further incubated with PKCα active subunit and stiffness measurements were repeated—dotted line (nDon=3, nPAH=3). C‐C1. Phospholamban threonine 17 CamKIIδ‐dependent phosphorylation was used as an indirect measurement of titin CamKIIδ phosphorylation. The phosphorylation level was normalized to the total amount of Phospholamban present in the sample (nDon=7, nPAH=11). C2. Donor and PAH cardiomyocyte stiffness was measured in relaxing solution at increasing sarcomere length (1.8 to 2.4 μm)—continuous line. The same cardiomyocyte was further incubated with CamKIIδ and stiffness measurements were repeated—dotted line (nDon=3, nPAH=3). Data presented as mean±SEM. CamKIIδ indicates calmoduling‐dependent‐kinase; PAH, pulmonary arterial hypertension; PKA, protein‐kinase‐A; PLN, phospholamban.
Activation of CamKIIδ determined indirectly by assessing the level of PLN CamKIIδ‐dependent phosphorylation of the residue threonine at position 17, which is an exclusive specific site for CamKIIδ.21 We found no statistical significant difference between the 2 groups (CamKIIδDon=1.00±0.19, CamKIIδPAH=1.42±0.29, P=0.41; Figure 1C1).
PKA Incubation Partially Restores RV Cardiomyocyte Stiffness
Subsequently, we tested in a subgroup of samples the functional relevance titin PKA PKCα and CamKIIδ‐mediated phosphorylation. For this purpose, membrane‐permeabilized cardiomyocytes in relaxing solution were used to minimize the influence of additional determinants of cardiomyocyte stiffness such as membrane and sarcoplasmic reticulum Ca2+‐handling. Therefore, cardiomyocyte stiffness is attributed solely to the sarcomeric protein titin.
RV cardiomyocyte stiffness was measured at increasing sarcomere lengths, starting at 1.8 μm and stretched to 2.0, 2.2, and 2.4 μm. After PKA incubation we recorded a significant decrease in PAH cardiomyocyte stiffness. Donor cardiomyocyte stiffness was minimally affected by PKA incubation (Pinteraction PKA×disease=0.01; Figure 1A2). PKCα incubation significantly increased cardiomyocyte stiffness in PAH samples and had little effect on donor cardiomyocyte stiffness (Pinteraction PKCα*disease=0.09; Figure 1B3). CamKIIδ incubation decreased cardiomyocyte stiffness of both Don and PAH samples (Pinteraction CamKIIδ*disease=0.08; Figure 1C2). Similar stretch or incubation in relaxing solutions without kinases would not modify baseline cardiomyocyte stiffness.
Reduced Phosphorylation of Sarcomeric cTnI
Measurements of myofilament Ca2+ sensitivity revealed a higher sensitivity in PAH compared with donor samples. To investigate whether reduced phosphorylation of the sarcomeric protein cTnI could play a role in determining high myofilament Ca2+ sensitivity and contribute to RV diastolic dysfunction in PAH, overall sarcomeric protein phosphorylation was determined. A significant decrease in cTnI and MyBPC phosphorylation was found in PAH compared with donors (cTnIDon=1.00±0.15, cTnIPAH=0.58±0.11, P=0.03; MyBPCDon=1.00±0.15, MyBPCPAH=0.63±0 .09, P=0.06; Figure 2). Cardiac troponin T (cTnT) phosphorylation appeared to be higher in PAH compared with donors; however, this was not statistically significant (cTnTDon=1.00±0.07, cTnTPAH=1.29±0.13, P=0.07). No difference was found in desmin phosphorylation (DesminDon=1.00±0.07, DesminPAH=0.93±0.06, P=0.4).
Figure 2.
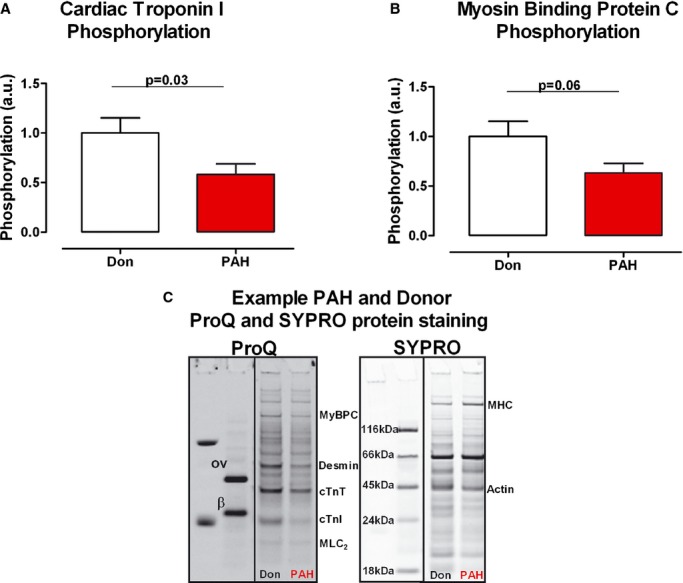
Sarcomeric protein phosphorylation. A, cTnI total phosphorylation was determined by ProQ‐Diamond staining, while total protein content was determined by SYPRO‐Ruby staining (nDon=10, nPAH=10). B, MyBPC total phosphorylation was determined by ProQ staining, while total protein content was determined by SYPRO staining (nDon=10, nPAH=10). C, Typical example of donor and PAH samples ProQ and SYPRO staining and the relative location of specific sarcomeric protein. ProQ staining annotations—ov, ovalbumin; β, β‐casein; MyBPC, myosin binding protein C; cTnT, cardiac troponin T; cTnI, cardiac troponin I; MLC2, myosin light chain 2. SYPRO staining annotations—MHC, myosin heavy chain. Data presented as mean±SEM. PAH inidcates pulmonary arterial hypertension.
Reduced cTnI phosphorylation was further confirmed by Western blot and Phos‐Tag™ analyses. The amount of dephosphorylated cTnI was significantly higher in PAH compared with donors (dephos‐cTnIDon=1.00±0.17, dephos‐cTnIPAH=1.67±0.17, P=0.02; Figure 3A). The cTnI PKA specific phosphorylation site (serine 22/23) showed significantly lower phosphorylation in PAH samples compared with donors (cTnI‐S22/23Don=1.00±0.08, cTnI‐S22/23PAH=0.41±0.11, P=0.002; Figure 3B).
Figure 3.
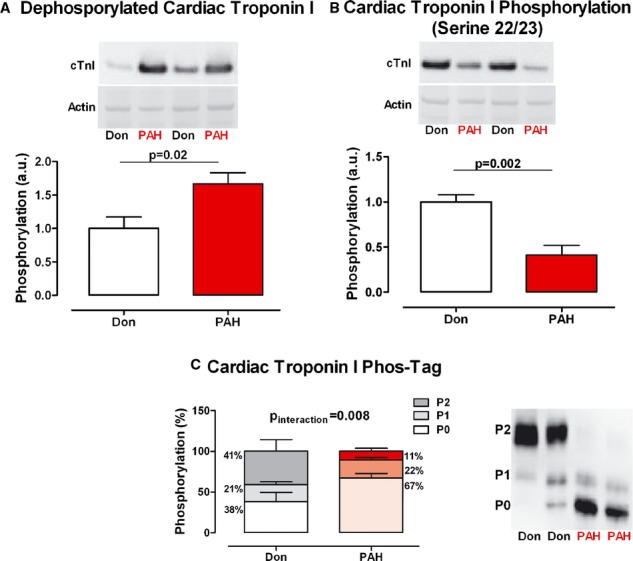
cTnI phosphorylation. A, Typical example of donor and PAH samples immunostained against the unphosphorylated form of cTnI and normalized for Ponceau‐S stained actin. cTnI total phosphorylation was determined by Western blot analysis in donor and PAH samples (nDon=8, nPAH=9). B, Typical example of donor and PAH samples immunostained against cTnI serine 22/23 residue and normalized for Ponceau‐S stained actin. cTnI serine 22/23 phosphorylation was determined by Western blot analysis in donor and PAH samples (nDon=9, nPAH=11). C, Typical example of donor and PAH samples immunostained against unphosphorylated (P0) cTnI, mono‐phosphorylated (P1) cTnI and bis‐phosphorylated (P2) cTnI. cTnI relative phosphorylation distribution was determined by Phos‐Tag analysis in donor and PAH samples (nDon=5, nPAH=9). Data presented as mean±SEM. cTnI indicates cardiac troponin I; cTnT, cardiac troponin T; PAH, pulmonary arterial hypertension.
Phos‐Tag™ analysis demonstrated that the distribution of cTnI phosphorylation (unphosphorylated [P0], mono‐phosphorylated [P1], and bis‐phosphorylated [P2]) in PAH‐cardiomyocytes was shifted to more unphosphorylated cTnI in comparison to donors, evident from higher levels of unphosphorylated cTnI and lower levels of bis‐phosphorylated cTnI (PDon 0/1/2=38.38±11.55%/20.78±3.57%/40.84±14.21%, PPAH 0/1/2=67.28±5.86%,/21.55±3.12%/11.17±3.69%, Pinteraction=0.008; Figure 3C).
Altered Expression of Ca2+‐Handling Proteins
SERCA2a expression was significantly lower in PAH cardiomyocytes (SERCA2aDon=1.00±0.14, SERCA2aPAH=0.56±0.09, P=0.02; Figure 4A). Total PLN protein levels were higher in PAH, however, the difference failed to reach statistical significance (PLNDon=1.00±0.13, PLNPAH=2.44±0.65, P=0.23; Figure 4B). PLN phosphorylation was determined as a ratio calculated from total PLN protein level in relation to the amount of phosphorylated PLN. pPLN was significantly lower in PAH compared with donors (pPLN/PLNDon=1.00±0.12, pPLN/PLNPAH=0.55±0.10, P=0.02; Figure 4C). The inhibitory effect of PLN on SERCA2a was determined by the PLN/SERCA2a ratio, which was higher in PAH compared with donors (PLN/SERCA2aDon=1.5±0.47, PLN/SERCA2aPAH=5.71±1.67, P=0.19; Figure 4D) In addition, the lower level of NCX1 protein level in PAH was not statistically significant (NCX1Don=1.00±0.28, NCX1PAH=0.55±0.12, P=0.36; Figure 4E). The expression level of RyR2, responsible for systolic Ca2+ release from the sarcoplasmic reticulum was not significantly different in the 2 groups (RyR2Don=1.00±0.05, RyR2PAH=1.26±0.13, P=0.11; Figure 4F).
Figure 4.
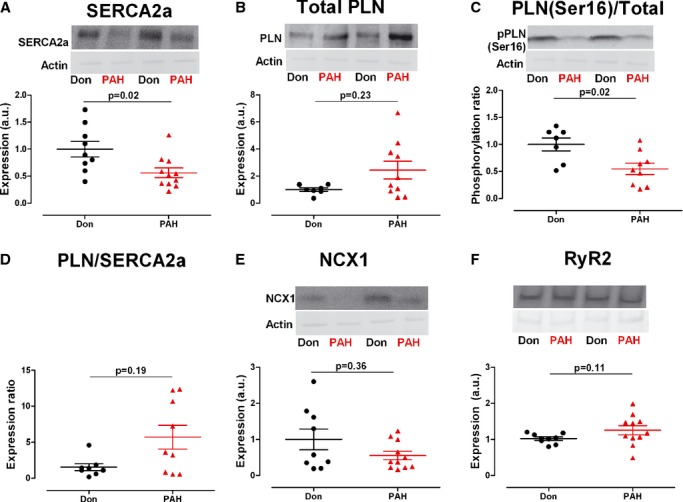
Ca2+‐handling proteins expression and phosphorylation. Typical example of donor and PAH samples immunostained against the corresponding Ca2+‐handeling protein and normalized for Ponceau‐S stained actin. A, SERCA2a: Sarco/Endoplasmic Reticulum Ca2+‐ATPase 2a (cardiac isoform) expression level (nDon=9, nPAH=11). B, Total PLN: phospholamban expression level (nDon=7, nPAH=10). C, PLN (Ser16)/Total PKA‐dependent phosphorylation level of serine 16 residue on PLN normalized to total PLN protein content (nDon=7, nPAH=9). D, PLN/SERCA2a inhibitory effect of PLN on SERCA2a quantified by the ratio between 2 (nDon=8, nPAH=9). E, NCX1, Na+/Ca2+ exchanger 1 (cardiac isoform) expression level (nDon=9, nPAH=11). F, RyR2, ryanodine receptor 2 (cardiac isoform) expression level (nDon=9, nPAH=11). Data presented as mean±SEM. PAH indicates pulmonary arterial hypertension; PLA, protein‐kinase‐A; PLN, phospholamban.
Discussion
This study demonstrates that cellular RV diastolic function in PAH is altered as a consequence of:
Reduced PKA‐mediated titin phosphorylation resulting in increased RV cardiomyocyte stiffness.
Decreased cTnI phosphorylation, increasing Ca2+ sensitivity.
Decreased levels of SERCA2a and PLN phosphorylation, suggesting reduced diastolic Ca2+ clearance.
Titin Determined Cardiomyocyte Stiffness
This is the first human study to show altered titin phosphorylation and its functional consequence in the failing right ventricle. We demonstrate that in RV tissue from PAH patients, titin serine 469 (PKA site) and serine 170 (PKCα site) phosphorylation are significantly reduced. No change was observed in titin serine 26 (PKCα site) phosphorylation between PAH and donor. Interestingly, these findings differ from those previously observed in LV pressure overload animal models. Hudson et al investigated titin phosphorylation in a mouse model of hypertensive left heart failure induced by transverse aortic constriction.22 They concluded that increased PKCα‐mediated phosphorylation of serine 26 in the PEVK domain was the main contributor to left ventricular cardiomyocyte stiffness. No significant change in PKA‐mediated phosphorylation of the N2B domain was found in this animal model of left heart failure. Kötter et al used end‐stage human LV tissue obtained during heart transplantation from hypertrophic cardiomyopathy (HCM) and idiopathic dilated cardiomyopathy (iDCM) patients and concluded that the significant increase in PEVK domain serine 26 (PKCα site) phosphorylation, together with altered titin N2B domain phosphorylation, determine the increase cardiomyocyte stiffness in LV failure.23 This suggests that while in LV failure PKCα‐dependent hyperphosphorylation of titin PEVK domain serine 26 plays a key role in increasing stiffness, in RV failure secondary to pressure overload, PKA‐dependent hypophosphorylation of N2B domain serine 469 in central in increasing cardiomyocyte stiffness.
To demonstrate the functional relevance of altered serine 469 and serine 170 phosphorylation, we incubated RV cardiomyocytes with the catalytic subunits of PKA and PKCα and measured the effects on RV cardiomyocyte stiffness. We observed that PKA incubation lowered stiffness only in PAH and largely restored RV cardiomyocyte stiffness to values observed in donors. In contrast, PKCα incubation increased cardiomyocyte stiffness in both PAH but far less in donor. At sarcomere lengths longer than physiological (>2.2 μm), PKCα incubation resulted in a large increase in RV cardiomyocyte stiffness in PAH patients. These findings suggest that titin serine 469 (PKA site) hypophosphorylation is the main contributor to RV diastolic stiffness in PAH, rather than titin serine 170 (PKCα site) hypophosphorylation. The latter may be a compensatory mechanism to prevent further increase in titin stiffness in RV cardiomyocytes in PAH patients.
In addition to PKA‐, PKCα‐, and CaMKIIδ‐ dependent phosphorylation, titin stiffness is subject to the fine‐tuning of a number of different kinases with opposite or complementary effects. In humans, protein kinase G (PKG) was shown to phosphorylate N2B serine 469 with the same functional effect as PKA.24 In addition to PKA and PKG, recent data show that extracellular‐signal‐regulated kinase 2 (ERK2) can decrease titin stiffness. However, the phosphorylation site is still to be resolved.25 Furthermore CaMKIIδ was shown to phosphorylate N2B (other sites than PKA) and PEVK domains and overlap with PKCα phosphorylation sites on the PEVK domain.21,26 However, the functional role of these novel phosphorylation pathways was not yet shown in humans.27
cTnI Modulated Ca2+‐Sensitivity
An increase in sarcomere Ca2+ sensitivity can lead to incomplete actin‐myosin detachment, despite low [Ca2+] levels, impairing the relaxation phase.28 Previously, it was demonstrated that sarcomeric protein phosphorylation plays an important role in determining Ca2+ sensitivity.10–12 In our study, we observed reduced cTnI and MyBPC phosphorylation. Site‐specific analysis further revealed that PKA‐mediated cTnI phosphorylation of serine 22/23 was less phosphorylated in PAH. This was further confirmed by Phos‐Tag™ analyses showing a higher level of unphosphorylated (P0) cTnI and reduced level of bisphosphorylated (P2) cTnI. Although in our previous study only a small increase in Ca2+ sensitivity was observed, this could further contribute to the RV diastolic impairment in PAH patients.
Diastolic Ca2+‐Clearance
For proper cardiomyocyte relaxation, cytosolic [Ca2+] must promptly drop, followed by myofilament detachment and sarcomere elongation to diastolic length.13 Therefore, perturbations in diastolic Ca2+ clearance are central to the development of diastolic dysfunction.28–29 Substantial decrease in SERCA2a protein levels and PKA‐mediated PLN phosphorylation would imply that cellular relaxation pattern is altered in PAH patients, due to increased residual diastolic [Ca2+]. However, the actual correlation between altered protein expression or phosphorylation and their functional relevance could not be determined in the present study.
PAH and Excessive Neurohormonal Activation
Although the 3 mechanisms discussed here clearly affect diastolic function in a distinct way and therefore may have different relevance in vivo, we observe a common factor to all 3 mechanisms: reduced PKA‐mediated phosphorylation. Decreased PKA phosphorylation determined increased titin‐derived RV cardiomyocyte stiffness, increased myofilament cTnI dependent Ca2+ sensitivity, and altered Ca2+ clearance due to reduced PLN phosphorylation. In the setting of heart failure, disturbed PKA phosphorylation is attributed to impaired β‐AR signaling as a consequence of increased neurohormonal stimulation and compensatory receptor β‐AR downregulation.28 In PAH, however, the consequences of increased neurohormonal activation are less well understood.30 Bristow et al observed decreased β‐AR density in failing RV myocardium.31 In addition, reducing neurohormonal activity in an experimental model of PAH resulted in improved RV diastolic function and partially restored sarcomeric protein phosphorylation (cTnI and cMyBPC).32 Therefore, we propose that in PAH patients the reduced PKA‐mediated phosphorylation of titin, cTnI, and PLN are at least partially caused by increased neurohormonal activation.
Clinical Implications
Beta‐blocker therapy is known to counteract the loss of function of β‐AR signaling by restoring β‐AR density, followed by the restoration of PKA‐mediated phosphorylation.32–33 In a previous experimental PAH model, rats receiving beta‐blocker therapy showed a significant reduction in RV diastolic stiffness and increase in cTnI and cMyBPC phsophorylation compared with PAH rats receiving placebo. These effects are likely due to reduced neurohormonal activation in PAH rats receiving beta‐blockers, normalization of β‐adrenergic stimulation, and increased PKA‐mediated titin, cTnI, and MyBPC phosphorylation. Although well tolerated in PAH rats, beta‐blockers are currently not recommended in PAH due to possible negative inotropic effects. Nevertheless, based on the beneficial effects of beta‐blockers in PAH experimental models, we have initiated a phase II clinical study to investigate the safety and efficacy of beta‐blocker (Bisoprolol) in patients with PAH (Clinicaltrials.gov identifier: NCT01246037).32 The future results of this study will reveal whether indeed, β‐AR/PKA signaling pathway is a novel therapeutic target for RV diastolic impairment in PAH patients.
Limitations
Efficient diastolic Ca2+ clearance is vital for ensuring proper relaxation for the sarcomere; therefore in this study we quantified the expression level of the most important proteins involved in Ca2+ handling. However, the functional relevance of these changes was not assessed. Functional relevance can only be determined in freshly harvested myocardial tissue/whole hearts, usually from animal models where changes in protein levels are consequently related to the functional data. In our study we used human myocardial tissue preserved by freezing, which did not alter the protein level and phosphorylation status, however made functional assessments impossible. Nevertheless, previous animal model studies show a clear relation between expression levels and function of Ca2+‐handling proteins; therefore we speculate that this is also the case in our study.13
There are several other mechanisms, which regulate diastolic dysfunction such as: increased radical oxygen species production, T‐tubules loss, or disorganization of cardiomyocyte cytoarchitecture. Although important for diastolic function, this study only focused on the cardiomyocyte stiffness, myofilament Ca2+ sensitivity, and altered Ca2+‐clearance protein levels.34
Conclusions
The present study provides novel insight into the molecular mechanism underlying RV diastolic impairment in patients with PAH. We observed that reduced PKA‐mediated phosphorylation of the giant sarcomeric protein titin contributed significantly to RV cardiomyocyte stiffness. In addition, phosphorylation of the sarcomeric protein cTnI was significantly reduced in PAH. Finally, reduced PLN phosphorylation and SERCA2a protein levels may indicate altered diastolic Ca2+ clearance.
Sources of Funding
Frances S. de Man, Coen Ottenheijm, Jolanda van der Velden, and Anton Vonk‐Noordegraaf were supported by a VENI (916.14.099) and VIDI (917.12.319; 917.11.344; 917.96.306) grant from the Dutch Foundation for Scientific Research. Silvia Rain and Anton Vonk‐Noordegraaf were supported by The Netherlands organization for Health Research and Development (ZonMw 95110079). Silvia Rain was supported by the European Society of Cardiology “First initiative travel grant”. Harm‐Jan Bogaard was supported by the National Institute of Health (NIH HL062881). Denielli Bos and M. Louis Handoko were supported by CAPES – Science Without Borders grant, CNPq Brasil.
Disclosures
None.
References
- 1.Rain S, Handoko ML, Trip P, Gan CT‐J, Westerhof N, Stienen GJ, Paulus WJ, Ottenheijm CAC, Marcus JT, Dorfmüller P, Guignabert C, Humbert M, Macdonald P, Dos Remedios C, Postmus PE, Saripalli C, Hidalgo CG, Granzier HL, Vonk‐Noordegraaf A, van der Velden J, de Man FS. Right ventricular diastolic impairment in patients with pulmonary arterial hypertension. Circulation. 2013; 128:2016-2025. [DOI] [PubMed] [Google Scholar]
- 2.De Tombe PP, Granzier HL. The cytoskeleton and the cellular transduction of mechanical strain in the heart: a special issue. Pflugers Arch. 2011; 462:1-2. [DOI] [PubMed] [Google Scholar]
- 3.Fukuda N, Granzier HL, Ishiwata S, Kurihara S. Physiological functions of the giant elastic protein titin in mammalian striated muscle. J Physiol Sci. 2008; 58:151-159. [DOI] [PubMed] [Google Scholar]
- 4.Li S, Guo W, Schmitt BM, Greaser ML. Comprehensive analysis of titin protein isoform and alternative splicing in normal and mutant rats. J Cell Biochem. 2012; 113:1265-1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hidalgo C, Hudson B, Bogomolovas J, Zhu Y, Anderson B, Greaser M, Labeit S, Granzier H. PKCΑ phosphorylation of titin's PEVK element: a novel and conserved pathway for modulating myocardial stiffness. Circ Res. 2009; 105:631-638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yamasaki R, Wu Y, McNabb M, Greaser M, Labeit S, Granzier H. Protein kinase A phosphorylates titin's cardiac‐specific N2B domain and reduces passive tension in rat cardiac myocytes. Circ Res. 2002; 90:1181-1188. [DOI] [PubMed] [Google Scholar]
- 7.Borbély A, Falcao‐Pires I, van Heerebeek L, Hamdani N, Edes I, Gavina C, Leite‐Moreira AF, Bronzwaer JGF, Papp Z, van der Velden J, Stienen GJM, Paulus WJ. Hypophosphorylation of the Stiff N2B titin isoform raises cardiomyocyte resting tension in failing human myocardium. Circ Res. 2009; 104:780-786. [DOI] [PubMed] [Google Scholar]
- 8.Boontje NM, Merkus D, Zaremba R, Versteilen A, de Waard MC, Mearini G, de Beer VJ, Carrier L, Walker LA, Niessen HWM, Dobrev D, Stienen GJM, Duncker DJ, van der Velden J. Enhanced myofilament responsiveness upon β‐adrenergic stimulation in post‐infarct remodeled myocardium. J Mol Cell Cardiol. 2011; 50:487-499. [DOI] [PubMed] [Google Scholar]
- 9.Wijnker PJM, Boknik P, Gergs U, Müller FU, Neumann J, dos Remedios C, Schmitz W, Sindermann JR, Stienen GJM, van der Velden J, Kirchhefer U. Protein phosphatase 2A affects myofilament contractility in non‐failing but not in failing human myocardium. J Muscle Res Cell Motil. 2011; 32:221-233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kooij V, Saes M, Jaquet K, Zaremba R, Foster DB, Murphy AM, Dos Remedios C, van der Velden J, Stienen GJM. Effect of troponin I Ser23/24 phosphorylation on Ca2+‐sensitivity in human myocardium depends on the phosphorylation background. J Mol Cell Cardiol. 2010; 48:954-963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Solaro RJ, Moir AJ, Perry SV. Phosphorylation of troponin I and the inotropic effect of adrenaline in the perfused rabbit heart. Nature. 1976; 262:615-617. [DOI] [PubMed] [Google Scholar]
- 12.Solaro RJ, Rosevear P, Kobayashi T. The unique functions of cardiac troponin I in the donor of cardiac muscle contraction and relaxation. Biochem Biophys Res Commun. 2008; 369:82-87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Moon MR, Aziz A, Lee AM, Moon CJ, Okada S, Kanter EM, Yamada KA. Differential calcium handling in two canine models of right ventricular pressure overload. J Surg Res. 2012; 178:554-562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hadri L, Kratlian RG, Benard L, Maron BA, Dorfmüller P, Ladage D, Guignabert C, Ishikawa K, Aguero J, Ibanez B, Turnbull IC, Kohlbrenner E, Liang L, Zsebo K, Humbert M, Hulot J‐S, Kawase Y, Hajjar RJ, Leopold JA. Therapeutic Efficacy of AAV1.SERCA2a in Monocrotaline‐Induced Pulmonary Arterial Hypertension. Circulation. 2013; 128:512-523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Quaile MP, Rossman EI, Berretta RM, Bratinov G, Kubo H, Houser SR, Margulies KB. Reduced sarcoplasmic reticulum Ca(2+) load mediates impaired contractile reserve in right ventricular pressure overload. J Mol Cell Cardiol. 2007; 43:552-563. [DOI] [PubMed] [Google Scholar]
- 16.Sande JB, Sjaastad I, Hoen IB, Bøkenes J, Tønnessen T, Holt E, Lunde PK, Christensen G. Reduced level of serine(16) phosphorylated phospholamban in the failing rat myocardium: a major contributor to reduced SERCA2 activity. Cardiovasc Res. 2002; 53:382-391. [DOI] [PubMed] [Google Scholar]
- 17.Wang Z, Nolan B, Kutschke W, Hill JA. Na+‐Ca2+ exchanger remodeling in pressure overload cardiac hypertrophy. J Biol Chem. 2001; 276:17706-17711. [DOI] [PubMed] [Google Scholar]
- 18.Lahmers S, Wu Y, Call DR, Labeit S, Granzier H. Developmental donor of titin isoform expression and passive stiffness in fetal and neonatal myocardium. Circ Res. 2004; 94:505-513. [DOI] [PubMed] [Google Scholar]
- 19.Van der Velden J, de Jong JW, Owen VJ, Burton PB, Stienen GJ. Effect of protein kinase A on calcium sensitivity of force and its sarcomere length dependence in human cardiomyocytes. Cardiovasc Res. 2000; 46:487-495. [DOI] [PubMed] [Google Scholar]
- 20.Hamdani N, Paulus WJ, van Heerebeek L, Borbély A, Boontje NM, Zuidwijk MJ, Bronzwaer JGF, Simonides WS, Niessen HWM, Stienen GJM, van der Velden J. Distinct myocardial effects of beta‐blocker therapy in heart failure with normal and reduced left ventricular ejection fraction. Eur Heart J. 2009; 30:1863-1872. [DOI] [PubMed] [Google Scholar]
- 21.Hidalgo CG, Chung CS, Saripalli C, Methawasin M, Hutchinson KR, Tsaprailis G, Labeit S, Mattiazzi A, Granzier HL. The multifunctional Ca(2+)/calmodulin‐dependent protein kinase II delta (CaMKIIδ) phosphorylates cardiac titin's spring elements. J Mol Cell Cardiol. 2013; 54:90-97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hudson B, Hidalgo C, Saripalli C, Granzier H. Hyperphosphorylation of mouse cardiac titin contributes to transverse aortic constriction‐induced diastolic dysfunction. Circ Res. 2011; 109:858-866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kötter S, Gout L, Von Frieling‐Salewsky M, Müller AE, Helling S, Marcus K, Dos Remedios C, Linke WA, Krüger M. Differential changes in titin domain phosphorylation increase myofilament stiffness in failing human hearts. Cardiovasc Res. 2013; 99:648-656. [DOI] [PubMed] [Google Scholar]
- 24.Krüger M, Kötter S, Grützner A, Lang P, Andresen C, Redfield MM, Butt E, dos Remedios CG, Linke WA. Protein kinase G modulates human myocardial passive stiffness by phosphorylation of the titin springs. Circ Res. 2009; 104:87-94. [DOI] [PubMed] [Google Scholar]
- 25.Raskin A, Lange S, Banares K, Lyon RC, Zieseniss A, Lee LK, Yamazaki KG, Granzier HL, Gregorio CC, McCulloch AD, Omens JH, Sheikh F. A novel mechanism involving four‐and‐a‐half LIM domain protein‐1 and extracellular signal‐regulated kinase‐2 regulates titin phosphorylation and mechanics. J Biol Chem. 2012; 287:29273-29284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hamdani N, Krysiak J, Kreusser MM, Neef S, Dos Remedios CG, Maier LS, Krüger M, Backs J, Linke WA. Crucial role for Ca2(+)/calmodulin‐dependent protein kinase‐II in regulating diastolic stress of normal and failing hearts via titin phosphorylation. Circ Res. 2013; 112:664-674. [DOI] [PubMed] [Google Scholar]
- 27.Hidalgo C, Granzier H. Tuning the molecular giant titin through phosphorylation: role in health and disease. Trends Cardiovasc Med. 2013; 23:165-171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Van der Velden J. Diastolic myofilament dysfunction in the failing human heart. Pflugers Arch. 2011; 462:155-163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bers DM. Cardiac excitation‐contraction coupling. Nature. 2002; 415:198-205. [DOI] [PubMed] [Google Scholar]
- 30.De Man FS, Handoko ML, Guignabert C, Bogaard HJ, Vonk‐Noordegraaf A. Neurohormonal axis in patients with pulmonary arterial hypertension: friend or foe? Am J Respir Crit Care Med. 2013; 187:14-19. [DOI] [PubMed] [Google Scholar]
- 31.Bristow MR, Ginsburg R, Umans V, Fowler M, Minobe W, Rasmussen R, Zera P, Menlove R, Shah P, Jamieson S. Beta 1‐ and beta 2‐adrenergic‐receptor subpopulations in nonfailing and failing human ventricular myocardium: coupling of both receptor subtypes to muscle contraction and selective beta 1‐receptor down‐regulation in heart failure. Circ Res. 1986; 59:297-309. [DOI] [PubMed] [Google Scholar]
- 32.De Man FS, Handoko ML, van Ballegoij JJM, Schalij I, Bogaards SJP, Postmus PE, van der Velden J, Westerhof N, Paulus WJ, Vonk‐Noordegraaf A. Bisoprolol delays progression towards right heart failure in experimental pulmonary hypertension. Circ Heart Fail. 2012; 5:97-105. [DOI] [PubMed] [Google Scholar]
- 33.Bogaard HJ, Natarajan R, Mizuno S, Abbate A, Chang PJ, Chau VQ, Hoke NN, Kraskauskas D, Kasper M, Salloum FN, Voelkel NF. Adrenergic receptor blockade reverses right heart remodeling and dysfunction in pulmonary hypertensive rats. Am J Respir Crit Care Med. 2010; 182:652-660. [DOI] [PubMed] [Google Scholar]
- 34.McCain ML, Parker KK. Mechanotransduction: the role of mechanical stress, myocyte shape, and cytoskeletal architecture on cardiac function. Pflugers Arch. 2011; 462:89-104. [DOI] [PubMed] [Google Scholar]