

Abstract
Background
Haptoglobin (Hp) is an abundant plasma protein with antioxidant properties. The Hp 2‐2 genotype has previously been linked to coronary heart disease risk in individuals with elevated glycosylated hemoglobin (HbA1c). We investigated the association of Hp and HbA1c with cardiovascular disease (CVD) in the longitudinal, population‐based Bruneck Study.
Methods and Results
Hp genotype was determined by polymerase chain reaction according to standard procedures and HbA1c concentration by a Diabetes Control and Complications Trial‐aligned assay. HbA1c was measured in 1995, 2000, and 2005. Occurrence of the combined CVD endpoint of myocardial infarction or stroke was recorded between 1995 and 2010. Outcome analyses employed the Cox proportional hazards model with HbA1c category as time‐varying covariate. At baseline in 1995, 806 subjects (male sex, 49.3%; age, mean±standard deviation, 62.70±11.08 years) were included. During follow‐up, 123 subjects experienced at least 1 CVD event (48 suffered myocardial infarction, 68 stroke, and 7 both). Among subjects with HbA1c≥6.5% (≥48 mmol/mol), those with the Hp 2‐2 genotype did not show an elevated risk of incident CVD compared with those with other genotypes (age‐ and sex‐adjusted hazard ratio [95% CI], 0.47 [0.19, 1.13], P=0.092) and a null association was also observed in subjects with HbA1c<6.5% (1.10 [0.75, 1.62], P=0.629) (P for interaction=0.082).
Conclusions
Subjects with the Hp 2‐2 genotype and elevated HbA1c compared with subjects with other Hp genotypes and elevated HbA1c did not show increased CVD risk.
Keywords: cardiovascular diseases, diabetes mellitus, genetics
Introduction
Haptoglobin (Hp) is a plasma protein that binds extracorpuscular hemoglobin (Hb) and prevents it from inflicting iron‐mediated oxidative tissue damage.1 The Hp gene has 2 major alleles, Hp1 and Hp2. Of these, the latter is a mutated version of the former and unique to humans.2 This common polymorphism (rs72294371) leads to 3 structurally and functionally distinct proteins, Hp 1‐1, 2‐1, and 2‐2, with reported genotype frequencies in subjects of Western European descent of 16%, 48%, and 36%.2–3 While Hp 1‐1 forms dimers, Hp 2‐1 and Hp 2‐2 form larger linear and cyclic polymers, respectively.2 Hp 2‐2 was reported to be less efficient than Hp 1‐1 in binding Hb and preventing oxidation by stabilizing heme iron within Hb, thereby displaying lower antioxidant activity.4–5
Based on these biological mechanisms, it has been suggested that the Hp 2‐2 genotype may confer an elevated risk of developing cardiovascular diseases (CVD). Epidemiological studies conducted in general populations, however, have shown contradictory results. On one hand, Hp 2‐2 was linked to the severity of myocardial infarction6 and to more frequent (but less severe) peripheral arterial occlusive disease.7 On the other hand, a large longitudinal population‐based survey has reported a ≈2‐fold risk for coronary artery disease (CAD) death in people with the Hp 1‐1 genotype.8 Two smaller case‐control studies found that the Hp 1 allele was more frequent in patients with CAD9 and lacunar stroke,10 compared with healthy controls.
In the past years the focus has shifted towards the role of Hp genotype in CVD of diabetic patients. Because diabetes is featured by a high level of oxidative stress, relevance of the antioxidant Hp may be considerably higher in diabetic patients.11 Actually, in a meta‐analysis of 5 studies involving 1829 patients with diabetes, the pooled odds ratio (95% CI) for CVD was 2.03 (1.46, 2.81) in a comparison of the Hp 2‐2 genotype with other genotypes.12 In line with this finding, Hp 2‐2 was associated with incident CVD in type 2 diabetes mellitus patients (odds ratio [95% CI], 4.96 [1.85, 13.33]). In conflict with these findings, the Framingham Offspring Study reported that Hp 2‐2 was associated with significantly lower CAD prevalence among diabetic patients than Hp 1‐1 (odds ratio [95% CI], 0.43 [0.21, 0.90]).13
As a potential explanation for the substantial heterogeneity in the Hp ‐ CVD association between the various studies effect modification by levels of HbA1c has been proposed.14 Indeed, the reported dysfunction of Hp 2‐2 in preventing Hb‐mediated oxidative damage was found to be accentuated with glycosylated Hb (HbA1c) in cell culture experiments.4 Testing this hypothesis in humans, Cahill and colleagues14 reported the Hp 2‐2 genotype to be associated with elevated CAD risk among subjects with elevated HbA1c in the Nurses' Health Study (odds ratio [95% CI], 10.12 [1.08, 94.97]) and validated their finding in a cohort of type 2 diabetic subjects (hazard ratio [95% CI], 7.55 [2.79, 20.47]). Based on this and earlier studies, Hp genotyping among diabetic patients and antioxidant treatment in diabetic patients with the Hp 2‐2 genotype has been propagated 2,12 but further confirmatory data are required to change clinical routine. Given its potentially large impact on diabetes management, we investigated the association of Hp genotype with CVD conditional on elevated HbA1c in the prospective, population‐based Bruneck Study.
Methods
Study Population and Data Collection
The Bruneck Study is a prospective, population‐based survey on the epidemiology and pathogenesis of atherosclerosis and cardiovascular disease.15–18 At baseline in 1990 the study population comprised an age‐ and sex‐stratified random sample of all inhabitants of Bruneck (125 men and 125 women from each of the fifth through eighth decades of age, for an age‐ and sex‐stratified random sample of n=1000, all of western European descent). No subjects were enrolled after study initiation. In 1995, 826 subjects participated in the first quinquennial re‐examination and DNA samples for genotyping were available in 816 individuals. During follow‐up from 1995 to 2010, detailed information about fatal and nonfatal new‐onset CVD was carefully collected. Follow‐up was 100% complete for clinical endpoints, which was made possible by the extremely low population mobility of 0.2% in the Bruneck area. The study protocol was approved by the ethics committees of Bolzano and Verona and conformed to the Declaration of Helsinki. All study subjects provided written informed consent. Risk factors were assessed by means of validated standard procedures as described previously.15,19
At the baseline and follow‐up examinations in 1995, 2000, 2005, and 2010, venous blood was sampled in the morning after an overnight fast for laboratory measurements, including fasting plasma glucose and HbA1c (Diabetes Control and Complications Trial‐aligned assay; equipment and reagents from BioRad, Milan, Italy, at both baseline and follow‐up examinations). In accordance with the study of Cahill et al, HbA1c was dichotomized according to a cut‐off of 6.5% (48 mmol/mol, International Federation of Clinical Chemistry units).
Diabetes mellitus was coded present for subjects with fasting glucose levels ≥7 mmol/L (≥126 mg/dL) or a medical record confirmed prediagnosis of definite disease status. Based on the literature14 we anticipated few subjects with the Hp 1‐1 genotype and, to maximize statistical power, decided prior to analysis to pool the Hp 1‐1 and 2‐1 genotypes, forming a group of Hp1 allele carriers, which is a common approach.14
We ascertained leisure time‐related physical activity by a standardized questionnaire20 rating the intensity of activities according to the compendium of physical activities21 and calculated average metabolic equivalent hours per week to estimate long‐term physical activity. We assessed food intake by a standardized food‐frequency questionnaire (FFQ) based on the gold standard Harvard FFQ by Willett and colleagues and adapted the FFQ to the dietary peculiarities in the survey area. We validated our FFQ with a dietician‐supervised short‐term assessment of food intake. Validity was high and similar to that previously found for the same FFQ in other populations.22 The Alternative Healthy Eating Index (AHEI) was calculated from these data as a quantitative measure of healthy dietary behavior.23 Body mass index (BMI) was calculated as weight in kilograms over height in meters squared.
Haptoglobin Genotyping
The Haptoglobin genotype was determined by PCR as described previously24 with slight modifications. In brief, 20 μL reactions contained 20 ng genomic DNA, 2 units Qiagen Taq Polymerase (Qiagen), 1× Qiagen PCR buffer (Qiagen, Hilden, Germany), 1× Q solution (Qiagen), 200 μmol/L of each dNTP (Peqlab) and 0.25 μmol/L of each primer (A/B or C/D). Oligonucleotide primers A and B were used for amplification of a 1757‐bp Hp 1 allele‐specific sequence and a 3481‐bp Hp 2 allele‐specific sequence. Primers C and D were used to amplify a 349‐bp Hp 2 allele‐specific sequence. The primers A (5′‐GAGGGGAGCTTGCCTTTCCATTG‐3′), B (5′‐GAGATTTTTGAGCCCTGGCTGGT‐3′), C (5′‐CCTGCCTCGTATTAACTGCACCAT‐3′) and D (5′‐CCGAGTGCTCCACATAGCCATGT‐3′) were purchased from Microsynth. The amplification reactions were conducted on a DNA Engine Cycler (BioRad) under the following conditions: initial denaturation 3 minutes 94°C; 94°C 30 seconds, 57°C (primers A/B) and 62°C (primers C/D) 30 seconds, 72°C 2 minutes, 35 cycles; final extension 10 minutes 72°C. After amplification 8 μL PCR product A/B and 2 μL PCR product C/D were mixed and separated together on a 1% agarose gel.
Endpoints
The composite CVD endpoint included incident fatal and non‐fatal myocardial infarction and stroke. Presence of myocardial infarction was assessed by World Health Organization criteria (definite disease status),25 while stroke was classified according to the criteria of the National Survey of Stroke.26 Events were ascertained by careful review of medical records provided by general practitioners, death certificates, and Bruneck Hospital files. A major advantage of the Bruneck Study is that virtually all inhabitants of Bruneck are referred to 1 local hospital that cooperates closely with the general practitioners. This allows retrieval of complete medical information.
Subjects who had experienced CVD events before the study baseline in 1995 were included in the main analysis and observed for recurring incident CVD between 1995 and 2010, and excluded in a sensitivity analysis.
Statistical Analysis
Hardy‐Weinberg equilibrium was tested by a permutation‐based chi‐squared test. Associations with incident CVD were assessed by Cox proportional hazards regression with HbA1c or diabetes status (present/absent) as time‐varying covariates in a granularity of 5 years. This approach relates the most current measure of glycemic exposure to incident CVD and avoids potential confounding due to reliance on a single baseline measurement. Effect modification of Hp 2‐2 by HbA1c category was tested using an appropriate interaction term. To simultaneously obtain marginal effects of Hp genotype for both HbA1c groups as well as the interaction effect, a linear combination of model parameters was made. The proportional hazards assumption was tested by computing the correlation coefficient of survival time with scaled Schoenfeld residuals and was met. Base models were adjusted for age and sex (model 1) and 2 multivariable models with progressive adjustment were employed (model 2: additionally for current smoking, systolic blood pressure, low‐density lipoprotein (LDL) cholesterol, and high‐density lipoprotein (HDL) cholesterol; model 3: additionally for metabolic equivalent hours, alternative healthy eating index, statin use, and body mass index). Last observation carried forward or next observation carried backward imputation reduced the proportion of missing HbA1c values from 5.9% to 0.4%.
Sensitivity analyses excluded subjects with missing HbA1c values, used age as the time scale and focused on individual disease endpoints. All tests were 2‐sided and P values smaller than 0.05 were considered significant. All analyses were performed using the R statistical package.
Results
Characteristics of the study population are shown in Table 1. Hp genotyping resulted in unambiguous results for 810 of 816 subjects for which DNA samples were available (Call rate, 99.3%). Of these, 4 had missing values in HbA1c concentration, which resulted in a baseline study size of 806 subjects. Duplicate measurement of 24 DNA samples yielded concordant findings in all cases. Genotypes were distributed as follows: Hp 1‐1, 10.3% (n=83), Hp 2‐1, 41.7% (n=336), Hp 2‐2, 48.0% (n=387) and were in Hardy‐Weinberg equilibrium overall (P=0.46), in subgroups of subjects with diabetes (P=0.13) and without (P=0.79), and subjects younger than 65 (P=0.52) and subjects at least 65 years old (P=0.80). There were no significant differences in prevalent diabetes (1995 baseline) or in incident diabetes (follow‐up 1995‐2010) between dichotomized Hp genotypes (P=0.71 and 0.92, respectively). For repeated measurements of HbA1c concentration we found an intra‐class correlation coefficient [95% CI] of 0.60 [0.56, 0.63].
Table 1.
Characteristics of the Study Population According to Haptoglobin Genotype
| Haptoglobin genotype | P Value | |||
|---|---|---|---|---|
| 1‐1 | 2‐1 | 2‐2 | ||
| Measurements taken in 1995 | ||||
| n (%) | 83 (10.3) | 336 (41.7) | 387 (48.0) | |
| Age, y | 63.6±12.6 | 62.1±10.3 | 63.0±11.4 | 0.417 |
| Male sex, n (%) | 40 (48.2) | 175 (52.1) | 180 (46.5) | 0.342 |
| HbA1c, % | 5.3 (5.1 to 5.7) | 5.4 (5.1 to 5.8) | 5.4 (5.1 to 5.8) | 0.496 |
| HbA1c≥6.5%, n (%) | 3 (3.6) | 15 (4.5) | 23 (5.9) | 0.575 |
| Diabetes, n (%) | 12 (14.5) | 31 (9.2) | 44 (11.4) | 0.481 |
| Hemoglobin, g/dL | 14.1±1.3 | 14.3±1.2 | 14.3±1.3 | 0.122 |
| Glucose, mg/dL | 98.0 (91.0 to 109.5) | 97.0 (90.0 to 104.0) | 98.0 (91.0 to 107.0) | 0.140 |
| Current smoking, n (%) | 13 (15.7) | 68 (20.7) | 74 (19.5) | 0.714 |
| Total cholesterol, mg/dL | 227.7±41.3 | 225.6±38.9 | 234.4±45.5 | 0.036 |
| HDL cholesterol, mg/dL | 57.4±13.9 | 58.5±15.7 | 59.3±17.0 | 0.623 |
| Systolic BP, mm Hg | 145.3±21.4 | 148.1±20.3 | 149.00±20.9 | 0.225 |
| Current smoking, n (%) | 70 (84.3) | 268 (79.8) | 311 (80.4) | 0.707 |
| Body mass index, kg/m2 | 26.0±4.4 | 25.9±3.6 | 25.4±3.9 | 0.214 |
| AHEI, score | 40.6±9.00 | 39.2±8.8 | 39.2±8.6 | 0.317 |
| MET, h/wk | 42.0 (30.4) | 42.0 (35.6) | 42.0 (38.3) | 0.782 |
| Statin use, n (%) | 1 (1.2) | 7 (2.1) | 16 (4.2) | 0.170 |
| Prior CVD, n (%) | 8 (9.6) | 21 (6.2) | 19 (4.9) | 0.304 |
| Measurements taken in 2000 | ||||
| n (%) | 68 (9.7) | 300 (42.8) | 333 (47.5) | |
| Age, y | 60.7±11.5 | 60.7±9.7 | 61.1±10.6 | 0.895 |
| Male sex, n (%) | 27 (39.7) | 154 (51.3) | 144 (43.2) | 0.067 |
| HbA1c, % | 5.7 (5.5 to 5.9) | 5.7 (5.5 to 6.0) | 5.7 (5.5 to 6.0) | 0.155 |
| HbA1c≥6.5%, n (%) | 3 (4.4) | 17 (5.7) | 34 (10.2) | 0.064 |
| Diabetes, n (%) | 8 (11.8) | 33 (11.0) | 39 (11.7) | 0.952 |
| Measurements taken in 2005 | ||||
| n (%) | 61 (10.1) | 256 (42.6) | 284 (47.3) | |
| Age, y | 59.6±10.9 | 59.7±9.5 | 59.3±9.7 | 0.826 |
| Male sex, n (%) | 25 (41.0) | 124 (48.4) | 122 (43.0) | 0.327 |
| HbA1c, % | 5.5 (5.4 to 5.7) | 5.6 (5.4 to 5.9) | 5.6 (5.4 to 5.9) | 0.539 |
| HbA1c≥6.5%, n (%) | 3 (4.9) | 20 (7.8) | 28 (9.9) | 0.328 |
| Diabetes, n (%) | 6 (9.8) | 34 (13.3) | 36 (12.7) | 0.787 |
Values are presented as mean±standard deviation, median (interquartile range), or count (percentage). P values are for tests of any difference between the 3 groups under adjustment for age and sex. AHEI indicates alternative healthy eating index; BP, blood pressure; CVD, cardiovascular disease; HbA1c, glycosylated hemoglobin; HDL, high‐density lipoprotein; MET, metabolic equivalent.
The analyses reported in the following were adjusted for age and sex, unless specified otherwise, and utilize updates of HbA1c levels during follow‐up (time‐varying covariate). From 1995 to 2010, 123 subjects experienced at least 1 CVD event (48 suffered myocardial infarction(s), 68 stroke(s), and 7 both). Only the first CVD event was considered in the main analysis. No significant differences emerged when examining the risk of CVD by Hp genotype (hazard ratio [HR] [95% CI] for Hp 2‐2 versus Hp 2‐1/1‐1: 0.98 [0.69, 1.40]; P=0.912). However, when taking into consideration HbA1c category, Hp 2‐2 subjects with HbA1c≥6.5% showed a non‐significant trend towards lower CVD risk than Hp 1 carriers (Hp 2‐1/1‐1 subjects) with HbA1c≥6.5% (HR [95% CI], 0.47 [0.19, 1.13], P=0.092) (Figure). This result did not materially change upon progressive adjustment for HDL and LDL cholesterol, systolic blood pressure, and smoking (model 2 HR [95% CI], 0.48 [0.18, 1.26], P=0.136) and metabolic equivalent hours, alternative healthy eating index, statin use, and body mass index (model 3 HR 0.52 [0.19, 1.39], P=0.189) (Figure). No risk differences were detected among subjects with HbA1c<6.5% (HR [95% CI], 1.10 [0.75, 1.62], P=0.629; Hp 2‐2 versus other) resulting in a near significant interaction between dichotomized Hp genotype and HbA1c category (P for interaction=0.082) as illustrated in the Figure.
Figure 1.
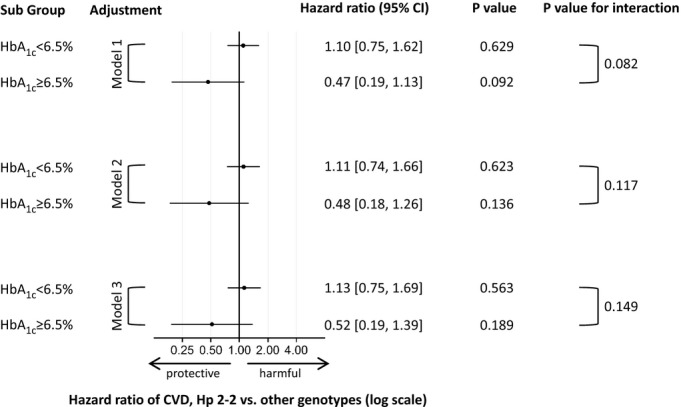
Association of Hp genotype with cardiovascular disease (CVD) risk by glycosylated hemoglobin (HbA1c) level under progressive adjustment. No. of person‐years of follow‐up/cases was 9250/102 for subjects with HbA1c<6.5% (4323/50 in the Hp 2‐2 and 4927/52 in the Hp 2‐1/1‐1 group) and 613/21 for subjects with HbA1c≥6.5% (375/8 in the Hp 2‐2 and 238/13 in the Hp 2‐1/1‐1 group). Model 1 was adjusted for age and sex, model 2 additionally for current smoking, systolic blood pressure, LDL cholesterol, and HDL cholesterol, and model 3 additionally for metabolic equivalent hours, alternative healthy eating index, statin use, and body mass index. HbA1c indicates glycosylated hemoglobin; HDL, high‐density lipoprotein cholesterol; LDL, low‐density lipoprotein cholesterol.
Finally, Hp 2‐2 was not associated with CVD risk in subjects with diabetes (P=0.944) nor in those without (P=0.950), and no statistical interaction existed between diabetes and Hp genotype (P=0.927).
Sensitivity analyses excluding subjects with missing HbA1c values, excluding subjects with prior CVD or using age as the time scale (instead of time‐on‐study) yielded similar findings (Table 2) as did analyses on individual disease endpoints (HR [95% CI] for stroke: 0.48 [0.16, 1.47], P=0.198; HR [95% CI] for myocardial infarction: 0.59 [0.16, 2.20], P=0.433; Hp 2‐2 versus other). Subgroup analyses according to sex and age (Table 2) should be interpreted cautiously given limited sample sizes in these groups.
Table 2.
Subgroup Analyses, Endpoint‐Specific Analyses, and Sensitivity Analyses
| Model 1 | Model 2 | Model 3 | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| HbA1c Group | HR [95% CI] | P Value | P interaction | HR [95% CI] | P Value | P interaction | HR [95% CI] | P Value | P interaction | |
| Full sample | <6.5% | 1.10 [0.75, 1.62] | 0.629 | 0.082 | 1.11 [0.74, 1.66] | 0.623 | 0.117 | 1.13 [0.75, 1.69] | 0.563 | 0.149 |
| ≥6.5% | 0.47 [0.19, 1.13] | 0.092 | 0.48 [0.18, 1.26] | 0.136 | 0.52 [0.19, 1.39] | 0.189 | ||||
| Subgroups | ||||||||||
| Age<75 y | <6.5% | 0.98 [0.59, 1.63] | 0.949 | 0.715 | 0.95 [0.57, 1.58] | 0.834 | 0.767 | 0.98 [0.58, 1.64] | 0.928 | 0.564 |
| ≥6.5% | 0.79 [0.26, 2.35] | 0.666 | 0.77 [0.23, 2.65] | 0.682 | 0.66 [0.19, 2.31] | 0.511 | ||||
| Age≥75 y | <6.5% | 1.22 [0.65, 2.27] | 0.536 | 0.035 | 1.35 [0.68, 2.68] | 0.390 | 0.081 | 1.30 [0.65, 2.60] | 0.451 | 0.083 |
| ≥6.5% | 0.11 [0.01, 0.95] | 0.044 | 0.18 [0.02, 1.55] | 0.118 | 0.17 [0.02, 1.53] | 0.115 | ||||
| Men | <6.5% | 0.98 [0.59, 1.62] | 0.938 | 0.986 | 0.94 [0.55, 1.60] | 0.810 | 0.821 | 0.93 [0.54, 1.60] | 0.795 | 0.796 |
| ≥6.5% | 0.97 [0.24, 3.92] | 0.963 | 0.78 [0.17, 3.64] | 0.748 | 0.75 [0.16, 3.60] | 0.719 | ||||
| Women | <6.5% | 1.36 [0.72, 2.55] | 0.339 | 0.018 | 1.46 [0.76, 2.79] | 0.258 | 0.074 | 1.59 [0.82, 3.09] | 0.171 | 0.110 |
| ≥6.5% | 0.27 [0.08, 0.88] | 0.030 | 0.40 [0.11, 1.40] | 0.151 | 0.47 [0.13, 1.77] | 0.264 | ||||
| Individual endpoints | ||||||||||
| Stroke | <6.5% | 0.87 [0.53, 1.45] | 0.601 | 0.337 | 0.94 [0.56, 1.58] | 0.823 | 0.645 | 0.96 [0.57, 1.62] | 0.883 | 0.691 |
| ≥6.5% | 0.48 [0.16, 1.47] | 0.198 | 0.67 [0.18, 2.56] | 0.563 | 0.72 [0.19, 2.76] | 0.630 | ||||
| Myocardial infarction | <6.5% | 1.51 [0.84, 2.71] | 0.165 | 0.201 | 1.43 [0.78, 2.63] | 0.246 | 0.100 | 1.43 [0.78, 2.62] | 0.254 | 0.081 |
| ≥6.5% | 0.59 [0.16, 2.20] | 0.433 | 0.38 [0.09, 1.64] | 0.197 | 0.35 [0.08, 1.52] | 0.160 | ||||
| Sensitivity analyses | ||||||||||
| Age as time scale | <6.5% | 1.09 [0.74, 1.61] | 0.659 | 0.114 | 1.10 [0.74, 1.66] | 0.630 | 0.146 | 1.13 [0.75, 1.70] | 0.553 | 0.176 |
| ≥6.5% | 0.50 [0.21, 1.21] | 0.125 | 0.51 [0.19, 1.34] | 0.171 | 0.54 [0.20, 1.46] | 0.226 | ||||
| No HbA1c imputation | <6.5% | 1.08 [0.72, 1.61] | 0.708 | 0.255 | 1.10 [0.73, 1.67] | 0.640 | 0.252 | 1.10 [0.73, 1.67] | 0.649 | 0.346 |
| ≥6.5% | 0.60 [0.24, 1.52] | 0.281 | 0.59 [0.21, 1.60] | 0.297 | 0.64 [0.23, 1.82] | 0.408 | ||||
| Subjects with prior CVD excluded | <6.5% | 1.24 [0.80, 1.90] | 0.332 | 0.130 | 1.23 [0.79, 1.92] | 0.357 | 0.233 | 1.27 [0.82, 1.99] | 0.287 | 0.233 |
| ≥6.5% | 0.55 [0.21, 1.44] | 0.222 | 0.63 [0.23, 1.73] | 0.373 | 0.65 [0.23, 1.81] | 0.409 | ||||
HR [95% CI] and P value are for the comparison of Hp 2‐2 versus other genotypes among subjects with glycosylated hemoglobin (HbA1c) <6.5% or HbA1c≥6.5%, respectively. Pinteraction is for a difference in the effect of dichotomized Hp genotype between those with HbA1c<6.5% and those with HbA1c≥6.5%. Model 1: adjustment for age and sex; Model 2: as model 1, with additional adjustment for current smoking, systolic blood pressure, LDL cholesterol, and HDL cholesterol; Model 3: as model 2, with additional adjustment for metabolic equivalent hours, alternative healthy eating index, statin use, and body mass index. CVD indicates cardiovascular disease; HbA1c, glycosylated hemoglobin; HR, hazard ratio.
Finally, among those with HbA1c≥6.5%, risk estimates were almost identical for Hp 2‐1 compared with Hp 1‐1 subjects (HR [95% CI], 0.97 [0.25, 3.76], P=0.964) providing a post‐hoc justification for the dichotomization of Hp genotypes applied in the current study (Hp 2‐2 versus 2‐1/1‐1).
We also investigated differences in blood lipids by Hp genotype. Total cholesterol and LDL cholesterol were higher in Hp 2‐2 subjects than in those with other genotypes when adjusting for age, sex, and body mass index (P=0.008 and P=0.016, respectively). These results are shown in Table 3.
Table 3.
Blood Lipid Levels (mg/dL) by Haptoglobin Genotype
| Hp 1‐1 | Hp 2‐1 | Hp 2‐2 | P any difference | P Hp 2‐2 vs other | |
|---|---|---|---|---|---|
| n | 83 | 336 | 387 | ||
| Lipid parameter | |||||
| Total cholesterol | 227.7±41.3 | 225.6±38.8 | 234.4±45.5 | 0.028 | 0.008 |
| HDL | 57.4±13.9 | 58.5±15.7 | 59.3±17.0 | 0.788 | 0.970 |
| LDL | 144.1±35.6 | 141.6±35.7 | 148.5±40.3 | 0.053 | 0.016 |
| Triglycerides | 143.8±116.1 | 126.7±66.7 | 133.3±80.6 | 0.075 | 0.189 |
Values are presented as mean±standard deviation. P values are adjusted for age, sex, and body mass index. HDL indicates high‐density lipoprotein cholesterol; Hp, Haptoglobin; LDL, low‐density lipoprotein cholesterol.
Discussion
Our data do not confirm the recent report of Cahill and colleagues that identified the Hp 2‐2 genotype as a cardiovascular risk factor among subjects with elevated HbA1c.14 Part of this discrepancy may derive from differences in study design and population. Cahill used a nested case‐control design with 1:1 matching in female nurses with 14‐year follow‐up for their main analysis, and data from a clinical trial with 18 months of follow‐up performed in type 2 diabetic patients for confirmation, whereas we used a 15‐year prospective observational design on a random sample of the general population. Importantly, Cahill excluded participants with prior CVD, which we did not, and used a coronary heart disease (CHD) endpoint, while we used a compound of myocardial infarction and stroke in the main analysis. Another potential explanation for the discrepancy between our and Cahill's results is survival bias. However, we observed a higher frequency of Hp 2‐2 genotype (48%) than most previous studies in Caucasians did (36 to 40%),2–3,14,27 which is the opposite of what would be expected in the presence of a survival disadvantage. Furthermore, genotypes were in Hardy‐Weinberg equilibrium overall as well as in the younger and older half of our sample separately, which argues against the existence of significant survival bias.
Our results are unexpected because Hp 2‐2 as a risk genotype among diabetic subjects would be backed by a biologically plausible rationale based on its weaker antioxidant properties.2,28–30 However, not all aspects of this rationale are fully consistent. In particular, the Hp 2‐2‐Hb compared with the Hp 1‐1‐Hb complex has been reported to be taken up into macrophages via the CD163 scavenger receptor at higher31 as well as at lower4 rates. No differences in Hb binding and antioxidant potency between purified human Hp 1‐1 and Hp 2‐2 were recently found in an animal model of hemolysis.32 In addition, Hp has numerous functions apart from Hb clearance, taking part in inflammatory pathways (prostaglandin synthesis, cathepsin B activity, endothelium‐dependent vasodilation)3,33 and interfering with the functions of immune cells.34–35 Hp phenotypes differ in some of these functions.3 Notably, Hp 2‐2 is the most angiogenic phenotype,36 which was discussed as an explanation for longer walking distance in Hp 2‐2 peripheral vascular disease patients (P<0.05),7 and Hp 1‐1 was linked to decreased endothelial repair potential in lacunar stroke patients.37
In this study, subjects with the Hp 2‐2 genotype had elevated total and LDL cholesterol (Table 3), which has been reported previously.38–39 This finding may be explained by the close genetic linkage between Hp genotype and a single nucleotide polymorphism in the gene encoding haptoglobin‐related protein, which is associated with levels of total but not HDL cholesterol via apolipoprotein L.27 However, adjusting for LDL levels did not appreciably change risk estimates (Table 2), which argues against the possibility that CVD risk differences due to Hp genotype are mediated by LDL levels.
Strengths of the present study include the virtually complete, long‐term follow‐up, detailed characterization of the study population and repeated measurements of HbA1c. Another major strength of the Bruneck Study is its high representativity for the general population. It comprises predominantly low‐ and medium‐risk individuals, which are of the foremost public health interest since most CVD events happen in such individuals.40 One downside of this is a comparatively low prevalence of elevated HbA1c, which precludes more complex analyses like 3‐way interactions between HbA1c, diabetes, and Hp genotype. It merits attention that while we found a trend towards protective effects of the Hp 2‐2 genotype among subjects with elevated glycohemoglobin, this result was statistically not significant, and hazardous effects would also be compatible with our data. The largest hazardous effect that we cannot refute at the α=0.05 level is a risk elevation of ≈26% (multivariable model 2, Figure). Larger studies are required to draw definite conclusions.
In summary, this study does not confirm that the Hp 2‐2 genotype is associated with a higher CVD risk in subjects with elevated HbA1c.
Sources of Funding
Willeit, Kiechl, and Weiss are supported by the FWF (Fonds zur Förderung der wissenschaftlichen Forschung) grant TRP 188. The Bruneck Study is funded by the Pustertaler Verein zur Prävention der Herz‐ und Hirngefäßerkrankungen, Sanitätseinheit Ost, Assessorat für Gesundheit, province of Bozen, Italy.
Disclosures
None.
References
- 1.Gutteridge JMC. The antioxidant activity of haptoglobin towards haemoglobin‐stimulated lipid peroxidation. Biochim Biophys Acta. 1987; 917:219-223. [DOI] [PubMed] [Google Scholar]
- 2.Levy AP, Asleh R, Blum S, Levy NS, Miller‐Lotan R, Kalet‐Litman S, Anbinder Y, Lache O, Nakhoul FM, Asaf R, Farbstein D, Pollak M, Soloveichik YZ, Strauss M, Alshiek J, Livshits A, Schwartz A, Awad H, Jad K, Goldenstein H. Haptoglobin: basic and clinical aspects. Antioxidants Redox Signal. 2010; 12:293-304. [DOI] [PubMed] [Google Scholar]
- 3.Langlois MR, Delanghe JR. Biological and clinical significance of haptoglobin polymorphism in humans. Clin Chem. 1996; 42:1589-1600. [PubMed] [Google Scholar]
- 4.Asleh R, Marsh S, Shilkrut M, Binah O, Guetta J, Lejbkowicz F, Enav B, Shehadeh N, Kanter Y, Lache O, Cohen O, Levy NS, Levy AP. Genetically determined heterogeneity in hemoglobin scavenging and susceptibility to diabetic cardiovascular disease. Circ Res. 2003; 92:1193-1200. [DOI] [PubMed] [Google Scholar]
- 5.Schaer CA, Schoedon G, Imhof A, Kurrer MO, Schaer DJ. Constitutive endocytosis of CD163 mediates hemoglobin‐heme uptake and determines the noninflammatory and protective transcriptional response of macrophages to hemoglobin. Circ Res. 2006; 99:943-950. [DOI] [PubMed] [Google Scholar]
- 6.Chapelle J‐P, Albert A, Smeets J‐P, Heusghem C, Kulbertus HE. Effect of the haptoglobin phenotype on the size of a myocardial infarct. N Engl J Med. 1982; 307:457-463. [DOI] [PubMed] [Google Scholar]
- 7.Delanghe J, Langlois M, Duprez D, De Buyzere M, Clement D. Haptoglobin polymorphism and peripheral arterial occlusive disease. Atherosclerosis. 1999; 145:287-292. [DOI] [PubMed] [Google Scholar]
- 8.De Bacquer D, De Backer G, Langlois M, Delanghe J, Kesteloot H, Kornitzer M. Haptoglobin polymorphism as a risk factor for coronary heart disease mortality. Atherosclerosis. 2001; 157:161-166. [DOI] [PubMed] [Google Scholar]
- 9.Golabi P, Kshatriya GK, Kapoor AK. Association of genetic markers with coronary heart disease (myocardial infarction)—a case‐control study. J Indian Med Assoc. 1999; 97:6-7. [PubMed] [Google Scholar]
- 10.Staals J, Pieters B, Knottnerus I, Rouhl R, van Oostenbrugge R, Delanghe J, Lodder J. Haptoglobin polymorphism and lacunar stroke. Curr Neurovasc Res. 2008; 5:153-158. [DOI] [PubMed] [Google Scholar]
- 11.Steinberg D, Witztum JL. Is the oxidative modification hypothesis relevant to human atherosclerosis? Do the antioxidant trials conducted to date refute the hypothesis? Circulation. 2002; 105:2107-2111. [DOI] [PubMed] [Google Scholar]
- 12.Vardi M, Blum S, Levy AP. Haptoglobin genotype and cardiovascular outcomes in diabetes mellitus—natural history of the disease and the effect of vitamin E treatment. Meta‐analysis of the medical literature. Eur J Intern Med. 2012; 23:628-632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Levy AP, Larson MG, Corey D, Lotan R, Vita JA, Benjamin EJ. Haptoglobin phenotype and prevalent coronary heart disease in the Framingham offspring cohort. Atherosclerosis. 2004; 172:361-365. [DOI] [PubMed] [Google Scholar]
- 14.Cahill LE, Levy AP, Chiuve SE, Jensen MK, Wang H, Shara NM, Blum S, Howard BV, Pai JK, Mukamal KJ, Rexrode KM, Rimm EB. Haptoglobin genotype is a consistent marker of coronary heart disease risk among individuals with elevated glycosylated hemoglobin. J Am Coll Cardiol. 2013; 61:728-737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kiechl S, Lorenz E, Reindl M, Wiedermann CJ, Oberhollenzer F, Bonora E, Willeit J, Schwartz DA. Toll‐like receptor 4 polymorphisms and atherogenesis. N Engl J Med. 2002; 347:185-192. [DOI] [PubMed] [Google Scholar]
- 16.Kiechl S, Wittmann J, Giaccari A, Knoflach M, Willeit P, Bozec A, Moschen AR, Muscogiuri G, Sorice GP, Kireva T, Summerer M, Wirtz S, Luther J, Mielenz D, Billmeier U, Egger G, Mayr A, Oberhollenzer F, Kronenberg F, Orthofer M, Penninger JM, Meigs JB, Bonora E, Tilg H, Willeit J, Schett G. Blockade of receptor activator of nuclear factor‐κB (RANKL) signaling improves hepatic insulin resistance and prevents development of diabetes mellitus. Nat Med. 2013; 19:358-363. [DOI] [PubMed] [Google Scholar]
- 17.Bonora E, Kiechl S, Oberhollenzer F, Egger G, Bonadonna RC, Muggeo M, Willeit J. Impaired glucose tolerance, Type II diabetes mellitus and carotid atherosclerosis: prospective results from the Bruneck Study. Diabetologia. 2000; 43:156-164. [DOI] [PubMed] [Google Scholar]
- 18.Willeit P, Willeit J, Mayr A, Weger S, Oberhollenzer F, Brandstätter A, Kronenberg F, Kiechl S. Telomere length and risk of incident cancer and cancer mortality. J Am Med Assoc. 2010; 304:69-75. [DOI] [PubMed] [Google Scholar]
- 19.Kiechl S, Schett G, Schwaiger J, Seppi K, Eder P, Egger G, Santer P, Mayr A, Xu Q, Willeit J. Soluble receptor activator of nuclear factor‐κb ligand and risk for cardiovascular disease. Circulation. 2007; 116:385-391. [DOI] [PubMed] [Google Scholar]
- 20.Baecke JA, Burema J, Frijters JE. A short questionnaire for the measurement of habitual physical activity in epidemiological studies. Am J Clin Nutr. 1982; 36:936-942. [DOI] [PubMed] [Google Scholar]
- 21.Ainsworth BE, Haskell WL, Whitt MC, Irwin ML, Swartz AM, Strath SJ, O'Brien WL, Bassett DR, Jr, Schmitz KH, Emplaincourt PO, Jacobs DR, Jr, Leon AS. Compendium of physical activities: an update of activity codes and MET intensities. Med Sci Sports Exerc. 2000; 32:S498-S504. [DOI] [PubMed] [Google Scholar]
- 22.Willett WC, Sampson L, Stampfer MJ, Rosner B, Bain C, Witschi J, Hennekens CH, Speizer FE. Reproducibility and validity of a semiquantitative food frequency questionnaire. Am J Epidemiol. 1985; 122:51-65. [DOI] [PubMed] [Google Scholar]
- 23.McCullough ML, Feskanich D, Stampfer MJ, Giovannucci EL, Rimm EB, Hu FB, Spiegelman D, Hunter DJ, Colditz GA, Willett WC. Diet quality and major chronic disease risk in men and women: moving toward improved dietary guidance. Am J Clin Nutr. 2002; 76:1261-1271. [DOI] [PubMed] [Google Scholar]
- 24.Koch W, Latz W, Eichinger M, Roguin A, Levy AP, Schömig A, Kastrati A. Genotyping of the common haptoglobin Hp 1/2 polymorphism based on PCR. Clin Chem. 2002; 48:1377-1382. [PubMed] [Google Scholar]
- 25. Nomenclature and criteria for diagnosis of ischemic heart disease. Report of the Joint International Society and Federation of Cardiology/World Health Organization task force on standardization of clinical nomenclature. Circulation. 1979; 59:607-609. [DOI] [PubMed] [Google Scholar]
- 26.Walker AE, Robins M, Weinfeld FD. The National Survey of Stroke. Clinical findings. Stroke. 1981; 12:I13-I44. [PubMed] [Google Scholar]
- 27.Guthrie PAI, Rodriguez S, Gaunt TR, Lawlor DA, Smith GD, Day INM. Complexity of a complex trait locus: HP, HPR, haemoglobin and cholesterol. Gene. 2012; 499:8-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Asleh R, Guetta J, Kalet‐Litman S, Miller‐Lotan R, Levy AP. Haptoglobin genotype– and diabetes‐dependent differences in iron‐mediated oxidative stress in vitro and in vivo. Circ Res. 2005; 96:435-441. [DOI] [PubMed] [Google Scholar]
- 29.Moreno PR, Purushothaman KR, Purushothaman M, Muntner P, Levy NS, Fuster V, Fallon JT, Lento PA, Winterstern A, Levy AP. Haptoglobin genotype is a major determinant of the amount of iron in the human atherosclerotic plaque. J Am Coll Cardiol. 2008; 52:1049-1051. [DOI] [PubMed] [Google Scholar]
- 30.Levy AP, Levy JE, Kalet‐Litman S, Miller‐Lotan R, Levy NS, Asaf R, Guetta J, Yang C, Purushothaman KR, Fuster V, Moreno PR. Haptoglobin genotype is a determinant of iron, lipid peroxidation, and macrophage accumulation in the atherosclerotic plaque. Arterioscler Thromb Vasc Biol. 2007; 27:134-140. [DOI] [PubMed] [Google Scholar]
- 31.Kristiansen M, Graversen JH, Jacobsen C, Sonne O, Hoffman H‐J, Law SKA, Moestrup SK. Identification of the haemoglobin scavenger receptor. Nature. 2001; 409:198-201. [DOI] [PubMed] [Google Scholar]
- 32.Lipiski M, Deuel JW, Baek JH, Engelsberger WR, Buehler PW, Schaer DJ. Human Hp1‐1 and Hp2‐2 phenotype‐specific haptoglobin therapeutics are both effective in vitro and in guinea pigs to attenuate hemoglobin toxicity. Antioxidants Redox Signal. 2013; 19:1619-1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Edwards DH, Griffith TM, Ryley HC, Henderson AH. Haptoglobin‐haemoglobin complex in human plasma inhibits endothelium dependent relaxation: evidence that endothelium derived relaxing factor acts as a local autocoid. Cardiovasc Res. 1986; 20:549-556. [DOI] [PubMed] [Google Scholar]
- 34.Wagner L, Gessl A, Parzer SB, Base W, Waldhäusl W, Pasternack MS. Haptoglobin phenotyping by newly developed monoclonal antibodies. Demonstration of haptoglobin uptake into peripheral blood neutrophils and monocytes. J Immunol. 1996; 156:1989-1996. [PubMed] [Google Scholar]
- 35.Berkova N, Gilbert C, Goupil S, Yan J, Korobko V, Naccache PH. TNF‐induced haptoglobin release from human neutrophils: pivotal role of the TNF p55 receptor. J Immunol. 1999; 162:6226-6232. [PubMed] [Google Scholar]
- 36.Cid MC, Grant DS, Hoffman GS, Auerbach R, Fauci AS, Kleinman HK. Identification of haptoglobin as an angiogenic factor in sera from patients with systemic vasculitis. J Clin Invest. 1993; 91:977-985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rouhl RPW, van Oostenbrugge RJ, Damoiseaux JGMC, Debrus‐Palmans LL, Theunissen ROMFIH, Knottnerus ILH, Staals JEA, Delanghe JR, Tervaert JWC, Lodder J. Haptoglobin phenotype may alter endothelial progenitor cell cluster formation in cerebral small vessel disease. Curr Neurovasc Res. 2009; 6:32-41. [DOI] [PubMed] [Google Scholar]
- 38.Saha N, Liu Y, Tay JS, Basair J, Ho CH. Association of haptoglobin types with serum lipids and apolipoproteins in a Chinese population. Clin Genet. 1992; 42:57-61. [DOI] [PubMed] [Google Scholar]
- 39.Braeckman L, De Bacquer D, Delanghe J, Claeys L, De Backer G. Associations between haptoglobin polymorphism, lipids, lipoproteins and inflammatory variables. Atherosclerosis. 1999; 143:383-388. [DOI] [PubMed] [Google Scholar]
- 40.Cooney M‐T, Dudina A, Whincup P, Capewell S, Menotti A, Jousilahti P, Njølstad I, Oganov R, Thomsen T, Tverdal A, Wedel H, Wilhelmsen L, Graham I. Re‐evaluating the Rose approach: comparative benefits of the population and high‐risk preventive strategies. Eur J Cardiovasc Prev Rehabil. 2009; 16:541-549. [DOI] [PubMed] [Google Scholar]