

Abstract
Background
Nearly 50% of patients with heart failure (HF) have preserved LV ejection fraction, with interstitial fibrosis and cardiomyocyte hypertrophy as early manifestations of pressure overload. However, methods to assess both tissue characteristics dynamically and noninvasively with therapy are lacking. We measured the effects of mineralocorticoid receptor blockade on tissue phenotypes in LV pressure overload using cardiac magnetic resonance (CMR).
Methods and Results
Mice were randomized to l‐nitro‐ω‐methyl ester (l‐NAME, 3 mg/mL in water; n=22), or l‐NAME with spironolactone (50 mg/kg/day in subcutaneous pellets; n=21). Myocardial extracellular volume (ECV; marker of diffuse interstitial fibrosis) and the intracellular lifetime of water (τic; marker of cardiomyocyte hypertrophy) were determined by CMR T1 imaging at baseline and after 7 weeks of therapy alongside histological assessments. Administration of l‐NAME induced hypertensive heart disease in mice, with increases in mean arterial pressure, LV mass, ECV, and τic compared with placebo‐treated controls, while LV ejection fraction was preserved (>50%). In comparison, animals receiving both spironolactone and l‐NAME (“l‐NAME+S”) showed less concentric remodeling, and a lower myocardial ECV and τic, indicating decreased interstitial fibrosis and cardiomyocyte hypertrophy (ECV: 0.43±0.09 for l‐NAME versus 0.25±0.03 for l‐NAME+S, P<0.001; τic: 0.42±0.11 for l‐NAME groups versus 0.12±0.05 for l‐NAME+S group). Mice treated with a combination of l‐NAME and spironolactone were similar to placebo‐treated controls at 7 weeks.
Conclusions
Spironolactone attenuates interstitial fibrosis and cardiomyocyte hypertrophy in hypertensive heart disease. CMR can phenotype myocardial tissue remodeling in pressure‐overload, furthering our understanding of HF progression.
Keywords: cardiac magnetic resonance imaging, hypertension, hypertrophy/remodeling
Introduction
Nearly 50% of incident heart failure occurs in the presence of preserved left ventricular ejection fraction (HF‐pEF).1 In patients with HF‐pEF, both cardiomyocyte hypertrophy and elevated interstitial myocardial collagen content are prominent features of tissue remodeling.2–3 However, therapies specifically directed at interstitial fibrosis and hypertrophy (eg, phosphodiesterase‐5 inhibition and spironolactone) have had limited efficacy in treating HF‐pEF,4–5 possibly due to late application of therapies (eg, after development of irreversible tissue‐level changes). Furthermore, reliance on LV hypertrophy (LVH) and its regression to stratify risk has limitations: regression of LVH (by blood pressure control) does not fully normalize cardiac prognosis or incident HF.6–7 In this context, assessing tissue fibrosis and cardiomyocyte hypertrophy non‐invasively in pathologic LV remodeling – specifically alongside therapies that target these key pathologies – would be fundamental to pre‐clinical drug development in HF‐pEF, and for targeting patients with greatest potential benefit.
Given the importance of fibrosis as a target for prevention at the transition to HF‐pEF, increasing clinical interest has focused on the use of mineralocorticoid receptor (MR) antagonist therapy in the treatment of HF‐pEF.8 In animal models of pressure‐overload hypertrophy, spironolactone prevents interstitial fibrosis, cellular and organ‐level hypertrophy, and prevents development of HF.9 Although MR antagonism leads to regression of LV hypertrophy in hypertensive individuals,10–11 there are conflicting results on the clinical and physiological benefits of spironolactone in established HF‐pEF.5,12
In this study, we developed a noninvasive CMR‐based technique to measure the modifiability of myocardial tissue phenotypes in mice during the induction of hypertension (via l‐nitro‐ω‐methyl ester/l‐NAME‐treated mice), a well‐established model of hypertensive heart disease and HF‐pEF. This study extends our method to phenotype fibrosis and cardiomyocyte hypertrophy concurrently with a therapy – the first step in translating this imaging technique to human applications in HF‐pEF.
Methods
Animal Models
Wild‐type mice (mean body weight 37.8±6.4 g, Taconic) were randomly assigned to 2 experimental groups: (1) l‐NAME (“l‐NAME”; n=22), which included l‐NAME in the drinking water (3 mg/mL; Sigma, USA) for 7 weeks, and (2) l‐NAME and spironolactone (“l‐NAME+S”; n=21) with l‐NAME (3 mg/mL; Sigma, USA) in the drinking water, and spironolactone (50 mg/kg per day) delivered by subcutaneous pellets (Innovative Research of America) implanted in the subscapular region as previously described.13–14 (Animals in the l‐NAME group did not have subcutaneous pellets.) Animals were kept under standard conditions with normal chow and water ad libitum. Noninvasive tail blood pressures were obtained at baseline and after 7 weeks of treatment using a volume‐pressure recording tail‐cuff technique15 (CODA‐1, Kent Scientific). CMR was performed at baseline and after 7 weeks of treatment (l‐NAME and l‐NAME+S). Blood samples were collected immediately after each CMR study for blood hematocrit determination (i‐STAT). Animals were euthanized following the second CMR study, and the hearts were excised for histological analysis. The same strain of mice, undergoing the same experimental protocol as the l‐NAME group, but receiving only tap water alone for 7 weeks as placebo, had previously been studied in our laboratory as controls (n=15).16 The study protocol and animal care conformed to the Guide for the Care and Use of Laboratory Animals from the National Institutes of Health (NIH Publication No. 85‐23, Revised 1996). The Standing Committee on Animal Care and Use at Harvard University approved the study protocol.
Histopathologic Analysis
Heart tissues were fixed with buffered 10% formalin solution (Fisher Scientific). Sections stained with fluoresceinisothiocyanate‐conjugated (FITC‐) wheat germ agglutinin to delineate the cell membrane were used for cardiomyocyte size determination.17 All sections were scanned with ScanScope scanners (Aperio Technologies, Inc), and whole‐slide images were sampled to a final resolution of 1.0 μm/pixel. Measurements of (minor) cardiomyocyte diameter Dmin and major cardiomyoctye‐diameter Dmaj (equivalent to the cardiomyocyte length) were obtained by image analysis of FITC‐wheat germ agglutinin stained sections. Ten measurements of Dmaj and Dmin were made in each of the anterior, septal, lateral, and inferior wall sections of the left ventricle. Cardiomyocyte volume was calculated assuming a cell shape in the form of a prolate ellipsoid,18–21 using the median Dmin and Dmaj. Connective tissue volume fraction was quantified on sections stained with Masson's trichrome stain, using a semi‐automatic pixel color intensity algorithm in the Aperio Spectrum software to quantify pixels stained in blue.
Cardiac Magnetic Resonance Imaging
Anesthetized mice (isofluorane≈1% to 2.5% in oxygen from a precision vaporizer) were positioned prone in a water‐heated cradle in a dedicated MRI system for small animal imaging (4.7 T Bruker BioSpin). Cine CMR images were acquired with prospective electrocardiographic and respiratory gating (model 1025L, SAII). For left ventricular (LV) size and function, short‐axis cine fast gradient echo images were acquired with full ventricular coverage (repetition time TR 5.9 ms; echo time TE 2.2 ms; temporal resolution 12 to 15 ms; in‐plane spatial resolution 100 to 120 μm×180 to 210 μm; 1 mm slice thickness, no gap). Simpson's rule was used to calculate LV volumes, mass, and LV ejection fraction (LVEF). LV volumes and mass were indexed by body weight. Gadolinium diethylenetriamine‐pentacetic acid (Magnevist, Berlex) was administered subcutaneously in multiple stages up to a cumulative dose of 0.5 mmol/kg. Myocardial and blood T1s were measured in a mid short‐axis slice once precontrast, and at least 4 times postcontrast using a Look‐Locker technique, and no earlier than 4 minutes after contrast administration as described previously22 (TR 2.5 ms; TE 1.8 ms; flip angle=10°, in‐plane resolution 190 μm, 1 mm slice thickness). Signal intensity versus time curves were used to determine T1 by non‐linear least‐squares fitting to an analytical expression for the magnitude signal measured during the inversion recovery,22 and correction for the radiofrequency pulse effects on the inversion recovery.23
The reciprocal of T1 (R1=1/T1) for each myocardial segment was analyzed as a function of R1 in the blood pool. Myocardial R1, shows a sublinear dependence on R1 in blood when the relaxation rate in blood approaches the rate of exchange of water across the transcytolemmal barrier (on the order of 1/τic)—the rate of water exchange across the cell membrane then effectively becomes a bottleneck for further increases of the myocardial R1. This allows a determination of the intracellular lifetime, using as model a 2‐space water‐exchange (2SX) model of equilibrium transcytolemmal water exchange to obtain myocardial extracellular volume (ECV) and the intracellular lifetime of water (τic). ECV and τic were both independent parameters of the 2SX model.16,22,24
The intracellular lifetime of water, τic depends on the mean time for a water molecule to diffuse to the cell membrane. It can be shown that τic is proportional to the volume‐to‐surface ratio (V/S),24–25 with V/S being on the order of the cell diameter in the case of cardiomyocytes, which have a length‐to‐diameter ratio of ≈4:1. This means that with the elongated shape of cardiomyocytes, the intracellular lifetime is primarily sensitive to changes in cell diameter.16
Statistical Analyses
Statistical analysis was performed using SAS 9.3 (SAS Institute) or R (version 3.01, R Foundation; http://www.R-project.org/). Data are presented as means±standard deviation. Continuous variables were compared between the 2 l‐NAME‐treated groups of mice using t tests. Linear mixed effects (LME) regression models (using package “lme4” in R) were built for variables with repeated measures (eg, ECV and LV parameters) to test the effects of treatment and time (ie, difference between baseline and follow‐up). Experimental group, study time‐point (baseline or follow‐up), and their interaction were included as fixed effects in each LME model. A random mouse‐specific intercept accounted for correlations of measurements within each mouse. For variables with repeated measurements P values are for comparisons of least squares means for the factors of said linear mixed effects models (using package “lmerTest” in R). Therefore, for each variable with repeated measurements, one and the same linear mixed effects model was used to obtain P values for comparisons between groups at baseline and at follow‐up, and for comparisons within a group between baseline and follow‐up. For the histological variables with only one measurement per animal, P values were obtained by Tukey's “Honest Significant Difference” Method. Linear correlations were assessed by Pearson's correlation coefficient. Multivariate linear regression models were built for myocardial ECV and intracellular lifetime of water (τic) to test whether at follow‐up the treatment effect of spironolactone was significant, with simultaneous adjustment by mean arterial pressure.
Results
LV Hypertrophy, Fibrosis and Cardiomyocyte Hypertrophy From l‐NAME
Table summarizes the hemodynamic, CMR, and histological parameters at baseline and after 7 weeks of l‐NAME therapy with and without spironolactone. An example of how the T1 measurements allowed the simultaneous determination of ECV and intracellular lifetime is shown in Figure 1. There were no significant differences in cardiac structure, function, or CMR‐derived tissue phenotype (by intracellular lifetime of water or ECV shown in Figures 2 and 3) at baseline between the 2 groups. (There were also no significant differences at baseline when the controls were included.) Administration of l‐NAME induced hypertension,26–28 as expected, with an increase in mean arterial pressure from 86.8±7.4 mm Hg at baseline to 124.4±6.1 mm Hg after 7 weeks of l‐NAME therapy (P<0.001). In response to hypertension, LV mass increased in mice treated with l‐NAME (163±19 μg for l‐NAME versus 99±14 μg for placebo‐treated controls, P<0.001), accompanied by a modest decrease in LV systolic function (LVEF 51±8% for l‐NAME versus 60±3% for controls, P<0.001). LV mass‐to‐volume increased in l‐NAME treated mice (concentric remodeling). There were no significant changes in indexed LV mass or function in the control group.
Table 1.
Hemodynamic, MRI and Histological Measurements at Baseline and Follow‐Up in Mice Treated With l‐NAME and With or Without Spironolactone Pre‐Treatment
| Baseline | Follow‐Up (7 Weeks) | |||||||
|---|---|---|---|---|---|---|---|---|
| Placebo (n=14) | l‐NAME (n=22) | l‐NAME+S (n=21) | P Value | Placebo (n=14) | l‐NAME (n=17) | l‐NAME+S (n=18) | P Value | |
| Body weight, g | 37.6±2.5 | 37.1±2.6 | 38.8±2.16 | 0.02 | 44.28±4.5** | 40±2.0**,†† | 43±3.6** | 0.01 |
| HR, bpm | 511±112 | 502±89 | 478±88 | 0.35 | 472±60 | 447±48* | 474±51 | 0.13 |
| Mean arterial blood pressure, mm Hg | 91.0±8.0 | 86.8±7.4 | 87.0±6.8 | 0.93 | 89.2±6.4 | 124±6.1**,†† | 107±5.3** | <0.001 |
| MRI | ||||||||
| LVEF, % | 57.8±3.7 | 59.7±3.34 | 60.0±3.65 | 0.84 | 60.3±3.2 | 51.3±8.2**,†† | 52.7±7.5**,†† | 0.63 |
| LVEDV, μL | 128±30.1 | 102±9.4†† | 99.3±6.1†† | 0.46 | 111±26.2* | 118±34* | 90±24† | 0.01 |
| LVESV, μL | 54±13.1 | 41.1±5.5†† | 39.9±5.6†† | 0.55 | 44±12.0* | 57 ±19**,†† | 44±16 | 0.04 |
| LVmass, μg | 94.5±16.3 | 91.7±12.2 | 88.2±11.8 | 0.46 | 98.5±14.4 | 163±19**,†† | 96 ±13 | <0.001 |
| LV mass‐to‐volume ratio | 0.77±0.17 | 0.90±0.12 | 0.88±0.09 | 0.54 | 0.94±0.29* | 1.47±0.38**,†† | 1.14±0.33**,† | 0.01 |
| LV mass index to body weight, μg/g | 3.40±0.79 | 2.47±0.31 | 2.27±0.23 | 0.08 | 2.54±0.69* | 4.07±0.42**,†† | 2.30±0.36 | <0.001 |
| MRI extracellular volume fraction (ECV) | 0.27±0.03 | 0.27±0.04 | 0.28±0.05 | 0.59 | 0.26±0.03 | 0.43±0.09**,†† | 0.25±0.03 | <0.001 |
| MRI intracellular life time of water, s | 0.14±0.07 | 0.17±0.09 | 0.16±0.06 | 0.57 | 0.18±0.08 | 0.42±0.11**,†† | 0.12±0.05 | <0.001 |
| Histology | ||||||||
| Connective tissue fraction (histology) [%] | — | — | — | — | 2.6±0.6% | 8.5±1.6%†† | 2.7±0.8% | <0.001 |
| Cardiomyocyte area, μm2/103 | — | — | — | — | 17.0±1.4 | 23.9±2.6†† | 18.4±1.6 | <0.001 |
| Volume‐to‐surface ratio | — | — | — | — | 24.7±1.5 | 32.1±1.9†† | 25.3±1.4 | <0.001 |
| Cardiomyocyte volume by histology, 104×μm3 | — | — | — | — | 42±6 | 77±13†† | 47±6 | <0.001 |
Column denoted by “P value” shows P‐values for l‐NAME vs l‐NAME+S. All P values for variables with repeated measurements (ie, baseline and follow‐up measurements) were obtained from one and the same linear mixed effects model for the variable, which had study (baseline or follow‐up), animal group, and their interaction as fixed effects. For the histological variables with only one measurement per animal P values were obtained by Tukey's “Honest Significant Difference” Method. Data for placebo group are for reference, were part of previous publication, and were included in the analysis with mixed effects models. LVEF indicates left ventricular ejection fraction; l‐NAME, Nω–nitro‐l‐arginine‐methyl‐ester; LVEDV, left ventricular end‐diastolic volume; LVESV, left ventricular end‐systolic volume; S, spironolactone.
For comparisons of follow‐up vs baseline, *P<0.05 and **P<0.01.
For comparisons at baseline or follow‐up vs placebo group †P<0.05 and ††P<0.01 for l‐NAME and l‐NAME+S groups.
Figure 1.
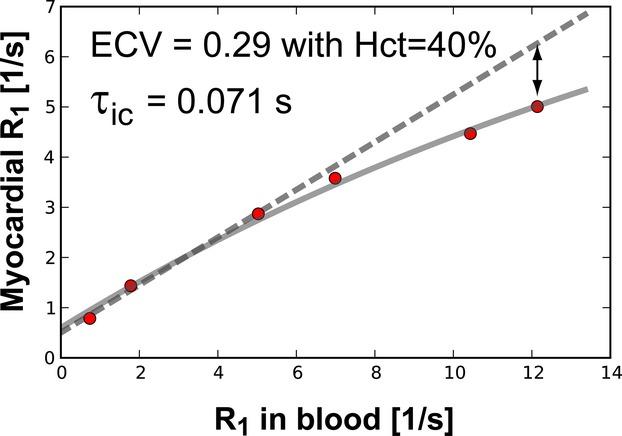
Illustration of dependence of myocardial R1 on R1 in blood. This example is from a study in a mouse at follow‐up after treatment with l‐NAME and spironolactone. The dashed line corresponds to the standard method of determining ECV from the ratio of the change of R1 in myocardial tissue divided by the change of R1 in blood (a linear model). At higher contrast concentrations the myocardial R1 increases more slowly than predicted by this linear model, because the exchange of water across the sarcolemmal membrane becomes the rate‐limiting step for myocardial relaxation, thereby resulting in a myocardial R1 that is lower than expected for fast exchange conditions (double arrow). ECV and intracellular lifetime were determined by a 2‐space model of water exchange across the (sarcolemmal) membrane between the 2 spaces. ECV indicates extracellular volume; l‐NAME, Nω–nitro‐l‐arginine‐methyl‐ester.
Figure 2.
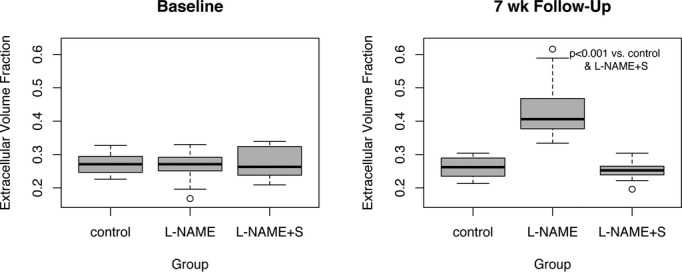
The extracellular volume fraction (ECV) increased with the build‐up of interstitial fibrosis in the mice treated with l‐NAME, compared to l‐NAME+spironolactone. At baseline there were no significant differences in ECV between controls (“placebo”), l‐NAME, and l‐NAME+spironolactone groups. The ECV data for the control group are from a previous study, using the same experimental protocol. At follow‐up ECV was not significantly different between controls and l‐NAME+spironolactone (P=0.48). l‐NAME indicates Nω–nitro‐l‐arginine‐methyl‐ester.
Figure 3.
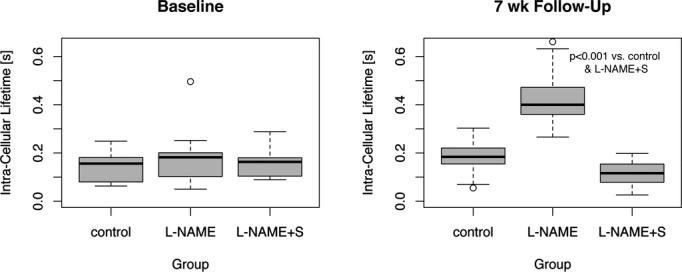
The intracellular lifetime of water (τic), a measure of cardiomyocyte diameter, increased significantly in the mice treated with l‐NAME, compared to l‐NAME+spironolactone, consistent with the histological appearance of cardiomyocyte hypertrophy. At baseline there was no significantly difference in τic between controls, l‐NAME, and l‐NAME+spironolactone groups. The τic data for the control group are from a previous study, using the same experimental protocol. At follow‐up, τic in the l‐NAME+spironolactone group was not significantly different from controls (P=0.34). l‐NAME indicates Nω–nitro‐l‐arginine‐methyl‐ester.
The myocardial ECV, a CMR marker of interstitial fibrosis, increased after 7 weeks of l‐NAME treatment (0.43±0.09 in l‐NAME at follow‐up versus 0.26±0.03 in controls at follow‐up, P<0.001). The intracellular lifetime of water by CMR (τic), a marker of cardiomyocyte hypertrophy, was increased after 7 weeks of l‐NAME as compared with controls (0.42±0.11 for l‐NAME versus 0.18±0.08 versus controls, P<0.001).
Effects of Spironolactone on Interstitial Fibrosis and Cardiomyocyte Hypertrophy
Spironolactone treatment successfully abrogated the development of hypertension and hypertensive heart disease in animals treated with l‐NAME, with a reduction in mean arterial pressure and LV mass relative to mice receiving solely l‐NAME (Table). In comparison with l‐NAME‐treated mice, animals receiving both spironolactone and l‐NAME showed a lower myocardial ECV and τic, indicating decreased interstitial fibrosis and cardiomyocyte hypertrophy (myocardial ECV: 0.43±0.09 for l‐NAME versus 0.25±0.03 for l‐NAME+S, P<0.001; τic: 0.42±0.11 for l‐NAME groups versus 0.12±0.05 for l‐NAME+S group). Indeed, at follow‐up, myocardial ECV and τic in mice receiving both l‐NAME and spironolactone were not significantly different from controls (Figures 2 and 3). The differences in ECV and τic between l‐NAME and l‐NAME+S groups remained highly significant at follow‐up when adjusted by the baseline measurements, using a LME model, and this also remained the case when the control group was included. The on‐therapy effect of spironolactone on myocardial ECV and τic remained significant after adjustment for mean arterial pressure in linear regression (myocardial ECV: multiple R2 adjusted to MAP=0.71, P<0.001; τic: multiple R2 adjusted to MAP=0.77, P<0.001). Concomitant administration of spironolactone and l‐NAME returned the myocardial tissue phenotype to that seen in controls when measured by histology or CMR.
ECV correlated strongly with the LV mass‐to‐volume ratio, a marker of concentric remodeling (r=0.62, P<0.001). The marker of cardiomyocyte hypertrophy, τic, also correlated with LV mass‐to‐volume ratio (r=0.54, P<0.001).
Histological Measurements and Relation to CMR Markers
The histological measurements of connective tissue volume fraction and the cardiomyocyte volume‐to‐surface ratio (Figure 4), showed changes between groups, which paralleled the differences for ECV and τic, respectively. Overall, there was a strong association between ECV and histological fibrosis (r=0.879, P<0.01; Figure 5A), and between τic and cardiomyocyte (minor) diameter (r=0.892, P<0.01; Figure 5B). Mice treated with l‐NAME alone averaged the highest volume‐to‐surface ratio (32.1±1.9 in l‐NAME at follow‐up versus 24.7±1.5 in controls, P<0.001, and also versus 25.3±1.4 in l‐NAME+S, P<0.001), and the highest cardiomyocyte volume (77.4±12.9 in l‐NAME versus 42.3±6.1 in controls, P<0.001 and also versus, 47±6 in l‐NAME+S, P<0.001), paralleling the increase of ECV and τic between l‐NAME+S and l‐NAME groups.
Figure 4.
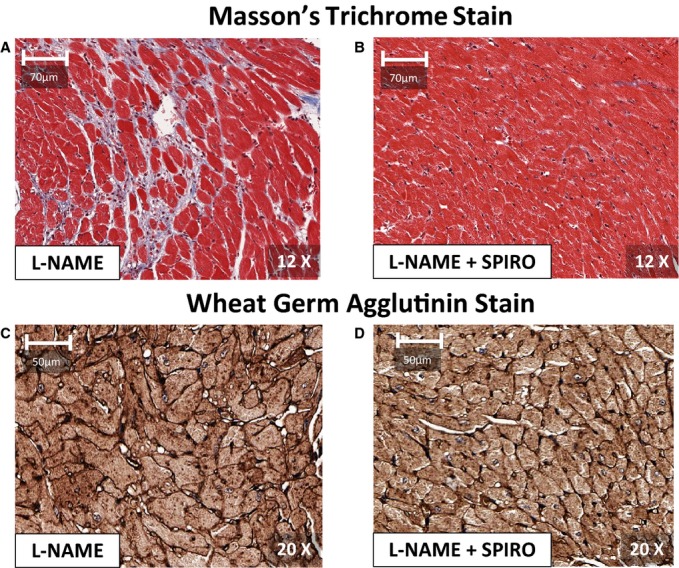
Short‐axis, mid‐level sections of cardiac tissue from the left ventricle (LV) in mice treated with l‐NAME (A and C), and spironolactone and l‐NAME (B and D), after 7 weeks of l‐NAME. Short‐axis sections stained with Masson's trichrome stains in (A) and (B) illustrate relative abundant interstitial fibrosis (appearing blue) in l‐NAME, as compared to l‐NAME+spironolactone. Adjacent slices in the same mice in (C) and (D) were stained with fluorescein isothiocyanate‐conjugated (FITC) wheat germ agglutinin to delineate cell membranes. With l‐NAME alone, mice showed cardiomyocyte hypertrophy, compared to l‐NAME+spironolactone. The cardiomyocyte diameters in the anterior, lateral, inferior and septal LV segments were significantly larger in l‐NAME compared to l‐NAME+spironolactone (P<0.001). l‐NAME indicates Nω–nitro‐l‐arginine‐methyl‐ester.
Figure 5.
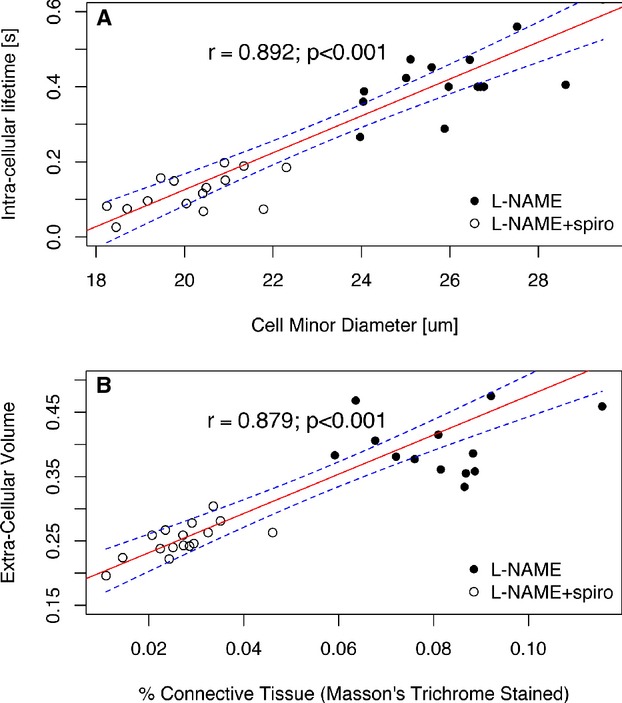
The extra‐cellular volume fraction (ECV) of myocardial tissue (A), and the intracellular lifetime of water (τic; B), both determined by cardiac magnetic resonance T1 measurements, correlated strongly with the corresponding histological measures, namely the connective tissue fraction on tissue slices stained with Masson's trichrome, and the cardiomyocyte diameters obtained from morphometric analysis of tissues slices in which the cell membranes were highlighted with wheat germ agglutinin. If the previously published data from a placebo‐treated group are included the correlations are 0.89 for (A), and 0.86 for (B). In (A) and (B) the solid line represents the least squares estimator of a linear regression model of MR derived measure (ECV or τic) as a function of corresponding histological measure, and the dashed lines represent the 95% confidence limits for the linear estimates. l‐NAME indicates Nω–nitro‐l‐arginine‐methyl‐ester; MR, mineralocorticoid receptor.
Discussion
The principal finding of our study is that CMR measurements of extracellular volume fraction and intracellular lifetime of water sensitively track changes in tissue phenotype with anti‐remodeling therapy in a mouse model of hypertensive heart disease. This work extends previous results from our laboratory validating ECV and τic in pressure overload16,22 by demonstrating the modifiability of these indices with interventions aimed at tissue remodeling in the hypertensive heart. When added to l‐NAME therapy, spironolactone blocked the increase in myocardial ECV and τic observed with l‐NAME therapy alone. Importantly, these changes were independent of on‐therapy reduction in blood pressure in mice treated with spironolactone, and occurred in the presence of a clinically normal LV ejection fraction (mean LVEF>50% in all groups). Collectively, these results establish the utility of CMR imaging to diagnose phenotypes during the transition from hypertensive heart disease to HF‐pEF.
Hypertension is the strongest modifiable risk factor for development of HF and subsequent cardiac mortality.29–30 In animal models, afterload excess from hypertension engenders a characteristic genetic and morphologic ventricular response, from cellular and organ‐level hypertrophy and interstitial fibrosis to ultimate ventricular dilatation and failure.31–33 Specifically, interstitial fibrosis may mark the transition from compensated cellular and organ‐level pathologic hypertrophy to HF in both animals and at‐risk patients.31,34–36 However, a residual risk of HF in patients after regression of LVH remains,6–7 suggesting the presence of adverse, tissue‐level phenotypes beyond LVH. In addition, the limited benefit of novel, anti‐fibrotic therapies in older patients with long‐standing hypertension and established HF‐pEF5,37–38 further suggests that demonstrating the benefits of anti‐fibrotic therapies earlier in the course of HF (eg, stage A HF) may be critical.
The imperative to identify early phenotypes in the progression to HF has partially motivated recent work utilizing T1 mapping CMR to examine interstitial fibrosis in pressure overload.39 In a study of 10 rats exposed to angiotensin II to promote LVH, Messroghli and colleagues used Look‐Locker inversion recovery T1 mapping to demonstrate elevated ECV in parallel to histologic fibrosis after induction of hypertension.40 Our group has published data demonstrating increased ECV in mice exposed to pharmacologically induced hypertension (eg, l‐NAME22) and mechanical pressure overload (eg, transverse aortic constriction16). In parallel, similar T1 mapping experiments have demonstrated increased ECV and its association with histologic fibrosis in pressure overload, including HF‐pEF22 and aortic stenosis.41 Despite this wealth of emerging data on T1 mapping in pressure overload, the extension of T1 mapping CMR into a therapeutic realm remains limited by 2 major obstacles: (1) data on the prevention or reversibility of ECV in appropriate animal models or patients is lacking and (2) the ability to look at cardiomyocyte hypertrophy (an early occurrence in pressure overload) is absent.
Accordingly, we studied the effect of mineralocorticoid antagonist therapy with spironolactone on l‐NAME‐induced hypertensive heart disease, demonstrating abrogation of interstitial fibrosis, cardiomyocyte hypertrophy, and whole‐organ LVH when spironolactone was initiated at the time of l‐NAME. Extending our previous report in this model,16,22 we demonstrate for the first time that dynamic changes in myocardial ECV and τic with spironolactone are highly associated with coordinate histological changes within the myocardium. These results establish ECV and τic as noninvasive, modifiable indices of tissue‐level changes within the heart relevant to pre‐clinical HF. Although small studies have demonstrated reduction in collagen turnover with mineralocorticoid antagonism in HF‐pEF,42 myocardial ECV provides an accurate reflection of the expansion of the extra‐cellular space due to the aggregate collagen accumulation – not turnover – within the heart, likely more specific to the underlying substrate for ventricular stiffness and clinical HF.
Our results are timely in the context of increasing attention on the role of mineralcorticoid (MR) antagonist therapy in HF‐pEF.8 MR antagonist therapy reduces interstitial fibrosis and cellular hypertrophy in animal models, improving ventricular performance and survival independent of blood pressure lowering or serum aldosterone concentration.9,43 In the largest published clinical study of spironolactone in HF‐pEF, Edelmann and colleagues reported a reduction of LV mass with slight improvement in echocardiographic diastolic dysfunction with spironolactone; unfortunately, this did not translate to improvement in functional capacity.5,37 In small studies, circulating markers of myocardial fibrosis are reduced with MR antagonism.42 Given the complex, life‐long exposure to profibrotic comorbidities that track closely with HF‐pEF (eg, hypertension, diabetes, obesity), these results suggest that true benefits of MR antagonism may be realized with preventative application earlier in the course of disease, before irreversible interstitial fibrosis has taken hold. In this light, the prevalence and reversibility of an aberrant myocardial tissue phenotype by CMR demonstrated here provides a backdrop for future investigations in antifibrotic therapies at an earlier stage of disease, possibly allowing the direct observation of therapeutic efficacy before frank LV dysfunction, hypertrophy, or HF. In this context, we eagerly await the results of TOPCAT (Treatment of Preserved Cardiac Function after Heart Failure with an Aldosterone Antagonist study; clinical trials identifier NCT00094302).8
Our results should be viewed in the context of study design. Although we do not have spironolactone drug levels to confirm efficacy, we observe a lower average blood pressure with spironolactone. While we do not measure reversibility of established fibrosis, our demonstration of the modifiability of myocardial ECV and τic is the first, essential step in determining a “threshold” beyond which MR antagonism fails to affect the heart. In addition, the placebo‐treated controls used in this study were previously published and not imaged concurrently with l‐NAME‐treated mice; however, given the large experience with normal mice,44 there is no systematic reason why concurrent imaging would yield different results. Finally, more detailed assessments of diastolic function (eg, CMR tagging), HF (via lung‐heart weight), or changes in myocardial stiffness (eg, by invasive pressure‐volume loop analysis) were not performed in this study. However, prevention of fibrosis with spironolactone (versus l‐NAME alone) with a preserved LVEF suggests that spironolactone normalized the structural myocardial phenotype.
The measurements of ECV and τic reported in this study relied on multiple (4 to 5) measurements of T1 after contrast injection. This is more time‐consuming than protocols utilized in previous studies for determination of ECV alone, which generally rely on two T1 measurements (1 pre‐ and 1 post‐contrast).45 The shorter protocol based on 2 T1 measurements assumes that the exchange of water across the sarcolemmal membrane (described by the intracellular lifetime of water τic) is negligible.22 For determination of τic, it is necessary to measure T1 when fast water exchange conditions no longer apply in order to observe an effect from the finite intracellular lifetime. The model for fitting the change of the myocardial R1 as a function of R1 in blood has both ECV and τic as adjustable parameters and requires multiple postcontrast measurements for a reliable estimation of τic.16 A further limitation is the fact that in myocardial tissue, cardiomyocytes account for approximately 90% of the intracellular volume, meaning the measured intracellular lifetime of water will have some slight bias from the contribution of other cell types.
To our knowledge, our results are the first noninvasive demonstration that a comprehensive myocardial tissue phenotype quantifying cardiomyocyte hypertrophy and interstitial fibrosis are variable with therapy in a model of pressure overload. These results extend the use of CMR to detect early disease myocardial changes into a therapeutic realm: the ability to noninvasively phenotype tissue‐based LV remodeling with therapy – otherwise inaccessible markers early in the development of HF – is fundamental to furthering our understanding of pathobiology of HF and its reversal.
Sources of Funding
The Research reported in this publication was supported by the National Heart, Lung, and Blood Institute of the National Institutes of Health under Award Number R01HL090634. Drs Coelho‐Filho and Shah are supported by Post‐Doctoral Fellowships from the American Heart Association (AHA 11POST5550053 to OCF and AHA 11POST110033 to RVS). Dr Neilan is supported by an American Heart Association Fellow‐to‐Faculty grant (12FTF12060588).
Disclosures
None.
Acknowledgments
We would like to thank Deborah Burstein, PhD, director of the Beth Israel Deaconess Small Animal Imaging Facility, and Reza Akhavan, MS, for their support.
References
- 1.Borlaug BA, Paulus WJ. Heart failure with preserved ejection fraction: pathophysiology, diagnosis, and treatment. Eur Heart J. 2011; 32:670-679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.van Heerebeek L, Borbely A, Niessen HW, Bronzwaer JG, van der Velden J, Stienen GJ, Linke WA, Laarman GJ, Paulus WJ. Myocardial structure and function differ in systolic and diastolic heart failure. Circulation. 2006; 113:1966-1973. [DOI] [PubMed] [Google Scholar]
- 3.Shah AM. Ventricular remodeling in heart failure with preserved ejection fraction. Curr Heart fail Rep. 2013; 10:341-349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Redfield MM, Borlaug BA, Lewis GD, Mohammed SF, Semigran MJ, Lewinter MM, Deswal A, Hernandez AF, Lee KL, Braunwald E. PhosphdiesteRasE‐5 Inhibition to Improve CLinical Status and EXercise Capacity in Diastolic Heart Failure (RELAX) trial: rationale and design. Circ Heart Fail. 2012; 5:653-659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Edelmann F, Wachter R, Schmidt AG, Kraigher‐Krainer E, Colantonio C, Kamke W, Duvinage A, Stahrenberg R, Durstewitz K, Loffler M, Dungen HD, Tschope C, Herrmann‐Lingen C, Halle M, Hasenfuss G, Gelbrich G, Pieske B. Effect of spironolactone on diastolic function and exercise capacity in patients with heart failure with preserved ejection fraction: the Aldo‐DHF randomized controlled trial. JAMA. 2013; 309:781-791. [DOI] [PubMed] [Google Scholar]
- 6.Mathew J, Sleight P, Lonn E, Johnstone D, Pogue J, Yi Q, Bosch J, Sussex B, Probstfield J, Yusuf S. Reduction of cardiovascular risk by regression of electrocardiographic markers of left ventricular hypertrophy by the angiotensin‐converting enzyme inhibitor ramipril. Circulation. 2001; 104:1615-1621. [DOI] [PubMed] [Google Scholar]
- 7.Devereux RB, Wachtell K, Gerdts E, Boman K, Nieminen MS, Papademetriou V, Rokkedal J, Harris K, Aurup P, Dahlof B. Prognostic significance of left ventricular mass change during treatment of hypertension. JAMA. 2004; 292:2350-2356. [DOI] [PubMed] [Google Scholar]
- 8.Shah SJ, Heitner JF, Sweitzer NK, Anand IS, Kim HY, Harty B, Boineau R, Clausell N, Desai AS, Diaz R, Fleg JL, Gordeev I, Lewis EF, Markov V, O'Meara E, Kobulia B, Shaburishvili T, Solomon SD, Pitt B, Pfeffer MA, Li R. Baseline characteristics of patients in the treatment of preserved cardiac function heart failure with an aldosterone antagonist trial. Circ Heart Fail. 2013; 6:184-192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kuster GM, Kotlyar E, Rude MK, Siwik DA, Liao R, Colucci WS, Sam F. Mineralocorticoid receptor inhibition ameliorates the transition to myocardial failure and decreases oxidative stress and inflammation in mice with chronic pressure overload. Circulation. 2005; 111:420-427. [DOI] [PubMed] [Google Scholar]
- 10.Pitt B, Reichek N, Willenbrock R, Zannad F, Phillips RA, Roniker B, Kleiman J, Krause S, Burns D, Williams GH. Effects of eplerenone, enalapril, and eplerenone/enalapril in patients with essential hypertension and left ventricular hypertrophy: the 4E‐left ventricular hypertrophy study. Circulation. 2003; 108:1831-1838. [DOI] [PubMed] [Google Scholar]
- 11.Sato A, Hayashi M, Saruta T. Relative long‐term effects of spironolactone in conjunction with an angiotensin‐converting enzyme inhibitor on left ventricular mass and diastolic function in patients with essential hypertension. Hypertens Res. 2002; 25:837-842. [DOI] [PubMed] [Google Scholar]
- 12.Mottram PM, Haluska B, Leano R, Cowley D, Stowasser M, Marwick TH. Effect of aldosterone antagonism on myocardial dysfunction in hypertensive patients with diastolic heart failure. Circulation. 2004; 110:558-565. [DOI] [PubMed] [Google Scholar]
- 13.Cassis LA, Helton MJ, Howatt DA, King VL, Daugherty A. Aldosterone does not mediate angiotensin II‐induced atherosclerosis and abdominal aortic aneurysms. Br J Pharmacol. 2005; 144:443-448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Crowley SD, Zhang J, Herrera M, Griffiths R, Ruiz P, Coffman TM. Role of AT(1) receptor‐mediated salt retention in angiotensin II‐dependent hypertension. Am J Physiol Renal Physiol. 2011; 301:F1124-F1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Feng M, Whitesall S, Zhang Y, Beibel M, D'Alecy L, DiPetrillo K. Validation of volume‐pressure recording tail‐cuff blood pressure measurements. Am J Hypertens. 2008; 21:1288-1291. [DOI] [PubMed] [Google Scholar]
- 16.Coelho‐Filho OR, Shah RV, Mitchell R, Neilan TG, Moreno H, Jr, Simonson B, Kwong R, Rosenzweig A, Das S, Jerosch‐Herold M. Quantification of cardiomyocyte hypertrophy by cardiac magnetic resonance: implications for early cardiac remodeling. Circulation. 2013; 128:1225-1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xu J, Kimball TR, Lorenz JN, Brown DA, Bauskin AR, Klevitsky R, Hewett TE, Breit SN, Molkentin JD. GDF15/MIC‐1 functions as a protective and antihypertrophic factor released from the myocardium in association with SMAD protein activation. Circ Res. 2006; 98:342-350. [DOI] [PubMed] [Google Scholar]
- 18.Yaniv Y, Juhaszova M, Wang S, Fishbein KW, Zorov DB, Sollott SJ. Analysis of mitochondrial 3D‐deformation in cardiomyocytes during active contraction reveals passive structural anisotropy of orthogonal short axes. PLoS One. 2011; 6:e21985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Thomas TA, Kuzman JA, Anderson BE, Andersen SM, Schlenker EH, Holder MS, Gerdes AM. Thyroid hormones induce unique and potentially beneficial changes in cardiac myocyte shape in hypertensive rats near heart failure. Am J Physiol Heart Circ Physiol. 2005; 288:H2118-H2122. [DOI] [PubMed] [Google Scholar]
- 20.Boyett MR, Frampton JE, Kirby MS. The length, width and volume of isolated rat and ferret ventricular myocytes during twitch contractions and changes in osmotic strength. Exp Physiol. 1991; 76:259-270. [DOI] [PubMed] [Google Scholar]
- 21.Sorenson AL, Tepper D, Sonnenblick EH, Robinson TF, Capasso JM. Size and shape of enzymatically isolated ventricular myocytes from rats and cardiomyopathic hamsters. Cardiovasc Res. 1985; 19:793-799. [DOI] [PubMed] [Google Scholar]
- 22.Coelho‐Filho OR, Mongeon FP, Mitchell R, Moreno H, Jr, Nadruz W, Jr, Kwong R, Jerosch‐Herold M. Role of transcytolemmal water‐exchange in magnetic resonance measurements of diffuse myocardial fibrosis in hypertensive heart disease. Circ Cardiovasc Imaging. 2013; 6:134-141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Deichmann R, Haase A. Quantification of T1 values by SNAPSHOT‐FLASH NMR imaging. J Magn Reson. 1992; 96:608-612. [Google Scholar]
- 24.Landis CS, Li X, Telang FW, Molina PE, Palyka I, Vetek G, Springer CS., Jr Equilibrium transcytolemmal water‐exchange kinetics in skeletal muscle in vivo. Magn Reson Med. 1999; 42:467-478. [DOI] [PubMed] [Google Scholar]
- 25.Yankeelov TE, Rooney WD, Huang W, Dyke JP, Li X, Tudorica A, Lee JH, Koutcher JA, Springer CS., Jr Evidence for shutter‐speed variation in CR bolus‐tracking studies of human pathology. NMR Biomed. 2005; 18:173-185. [DOI] [PubMed] [Google Scholar]
- 26.Arnal JF, el Amrani AI, Chatellier G, Menard J, Michel JB. Cardiac weight in hypertension induced by nitric oxide synthase blockade. Hypertension. 1993; 22:380-387. [DOI] [PubMed] [Google Scholar]
- 27.Moreno H, Jr, Metze K, Bento AC, Antunes E, Zatz R, de Nucci G. Chronic nitric oxide inhibition as a model of hypertensive heart muscle disease. Basic Res Cardiol. 1996; 91:248-255. [DOI] [PubMed] [Google Scholar]
- 28.Pechanova O, Bernatova I, Pelouch V, Babal P. l‐NAME‐induced protein remodeling and fibrosis in the rat heart. Physiol Res. 1999; 48:353-362. [PubMed] [Google Scholar]
- 29.Haider AW, Larson MG, Benjamin EJ, Levy D. Increased left ventricular mass and hypertrophy are associated with increased risk for sudden death. J Am Coll Cardiol. 1998; 32:1454-1459. [DOI] [PubMed] [Google Scholar]
- 30.Levy D, Anderson KM, Savage DD, Kannel WB, Christiansen JC, Castelli WP. Echocardiographically detected left ventricular hypertrophy: prevalence and risk factors. The Framingham Heart Study. Ann Intern Med. 1988; 108:7-13. [DOI] [PubMed] [Google Scholar]
- 31.Boluyt MO, O'Neill L, Meredith AL, Bing OH, Brooks WW, Conrad CH, Crow MT, Lakatta EG. Alterations in cardiac gene expression during the transition from stable hypertrophy to heart failure. Marked upregulation of genes encoding extracellular matrix components. Circ Res. 1994; 75:23-32. [DOI] [PubMed] [Google Scholar]
- 32.Hein S, Arnon E, Kostin S, Schonburg M, Elsasser A, Polyakova V, Bauer EP, Klovekorn WP, Schaper J. Progression from compensated hypertrophy to failure in the pressure‐overloaded human heart: structural deterioration and compensatory mechanisms. Circulation. 2003; 107:984-991. [DOI] [PubMed] [Google Scholar]
- 33.Lorell BH, Carabello BA. Left ventricular hypertrophy: pathogenesis, detection, and prognosis. Circulation. 2000; 102:470-479. [DOI] [PubMed] [Google Scholar]
- 34.Hall AP, Elcombe CR, Foster JR, Harada T, Kaufmann W, Knippel A, Kuttler K, Malarkey DE, Maronpot RR, Nishikawa A, Nolte T, Schulte A, Strauss V, York MJ. Liver hypertrophy: a review of adaptive (adverse and non‐adverse) changes–conclusions from the 3rd International ESTP Expert Workshop. Toxicol Pathol. 2012; 40:971-994. [DOI] [PubMed] [Google Scholar]
- 35.Maron BJ, Pelliccia A, Spirito P. Cardiac disease in young trained athletes. Insights into methods for distinguishing athlete's heart from structural heart disease, with particular emphasis on hypertrophic cardiomyopathy. Circulation. 1995; 91:1596-1601. [DOI] [PubMed] [Google Scholar]
- 36.Mujumdar VS, Tyagi SC. Temporal regulation of extracellular matrix components in transition from compensatory hypertrophy to decompensatory heart failure. J Hypertens. 1999; 17:261-270. [DOI] [PubMed] [Google Scholar]
- 37.Redfield MM, Chen HH, Borlaug BA, Semigran MJ, Lee KL, Lewis G, LeWinter MM, Rouleau JL, Bull DA, Mann DL, Deswal A, Stevenson LW, Givertz MM, Ofili EO, O'Connor CM, Felker GM, Goldsmith SR, Bart BA, McNulty SE, Ibarra JC, Lin G, Oh JK, Patel MR, Kim RJ, Tracy RP, Velazquez EJ, Anstrom KJ, Hernandez AF, Mascette AM, Braunwald E. Effect of phosphodiesterase‐5 inhibition on exercise capacity and clinical status in heart failure with preserved ejection fraction: a randomized clinical trial. JAMA. 2013; 309:1268-1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shah RV, Desai AS, Givertz MM. The effect of renin‐angiotensin system inhibitors on mortality and heart failure hospitalization in patients with heart failure and preserved ejection fraction: a systematic review and meta‐analysis. J Cardiac Fail. 2010; 16:260-267. [DOI] [PubMed] [Google Scholar]
- 39.Maceira AM, Mohiaddin RH. Cardiovascular magnetic resonance in systemic hypertension. J Cardiovasc Magn Reson. 2012; 14:28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Messroghli DR, Nordmeyer S, Dietrich T, Dirsch O, Kaschina E, Savvatis K, D Oh‐I, Klein C, Berger F, Kuehne T. Assessment of diffuse myocardial fibrosis in rats using small‐animal Look‐Locker inversion recovery T1 mapping. Circ Cardiovasc Imaging. 2011; 4:636-640. [DOI] [PubMed] [Google Scholar]
- 41.Mascherbauer J, Marzluf BA, Tufaro C, Pfaffenberger S, Graf A, Wexberg P, Panzenbock A, Jakowitsch J, Bangert C, Laimer D, Schreiber C, Karakus G, Hulsmann M, Pacher R, Lang IM, Maurer G, Bonderman D. Cardiac magnetic resonance post‐contrast T1 time is associated with outcome in patients with heart failure and preserved ejection fraction. Circ Cardiovasc Imaging. 2013; 6:1056-1065. [DOI] [PubMed] [Google Scholar]
- 42.Sado DM, Flett AS, Banypersad SM, White SK, Maestrini V, Quarta G, Lachmann RH, Murphy E, Mehta A, Hughes DA, McKenna WJ, Taylor AM, Hausenloy DJ, Hawkins PN, Elliott PM, Moon JC. Cardiovascular magnetic resonance measurement of myocardial extracellular volume in health and disease. Heart. 2012; 98:1436-1441. [DOI] [PubMed] [Google Scholar]
- 43.Deswal A, Richardson P, Bozkurt B, Mann DL. Results of the Randomized Aldosterone Antagonism in Heart Failure with Preserved Ejection Fraction trial (RAAM‐PEF). J Card Fail. 2011; 17:634-642. [DOI] [PubMed] [Google Scholar]
- 44.Nagata K, Obata K, Xu J, Ichihara S, Noda A, Kimata H, Kato T, Izawa H, Murohara T, Yokota M. Mineralocorticoid receptor antagonism attenuates cardiac hypertrophy and failure in low‐aldosterone hypertensive rats. Hypertension. 2006; 47:656-664. [DOI] [PubMed] [Google Scholar]
- 45.Neilan TG, Coelho‐Filho OR, Shah RV, Abbasi SA, Heydari B, Watanabe E, Chen Y, Mandry D, Pierre‐Mongeon F, Blankstein R, Kwong RY, Jerosch‐Herold M. Myocardial extracellular volume fraction from T1 measurements in healthy volunteers and mice: relationship to aging and cardiac dimensions. JACC Cardiovasc Imaging. 2013; 6:672-683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Flett AS, Hayward MP, Ashworth MT, Hansen MS, Taylor AM, Elliott PM, McGregor C, Moon JC. Equilibrium contrast cardiovascular magnetic resonance for the measurement of diffuse myocardial fibrosis: preliminary validation in humans. Circulation. 2010; 122:138-144. [DOI] [PubMed] [Google Scholar]