

Abstract
Background
Inactivation of Shox2, a member of the short‐stature homeobox gene family, leads to defective development of multiple organs and embryonic lethality as a result of cardiovascular defects, including bradycardia and severe hypoplastic sinoatrial node (SAN) and sinus valves, in mice. It has been demonstrated that Shox2 regulates a genetic network through the repression of Nkx2.5 to maintain the fate of the SAN cells. However, the functional mechanism of Shox2 protein as a transcriptional repressor on Nkx2.5 expression remains completely unknown.
Methods and Results
A specific interaction between the B56δ regulatory subunit of PP2A and Shox2a, the isoform that is expressed in the developing heart, was demonstrated by yeast 2‐hybrid screen and coimmunoprecipitation. Western blotting and immunohistochemical assays further confirmed the presence of phosphorylated Shox2a (p‐Shox2a) in cell culture as well as in the developing mouse and human SAN. Site‐directed mutagenesis and in vitro kinase assays identified Ser92 and Ser110 as true phosphorylation sites and substrates of extracellular signal‐regulated kinase 1 and 2. Despite that Shox2a and its phosphorylation mutants possessed similar transcriptional repressive activities in cell cultures when fused with Gal4 protein, the mutant forms exhibited a compromised repressive effect on the activity of the mouse Nkx2.5 promoter in cell cultures, indicating that phosphorylation is required for Shox2a to repress Nkx2.5 expression specifically. Transgenic expression of Shox2a, but not Shox2a‐S92AS110A, mutant in the developing heart resulted in down‐regulation of Nkx2.5 in wild‐type mice and rescued the SAN defects in the Shox2 mutant background. Last, we demonstrated that elimination of both phosphorylation sites on Shox2a did not alter its nuclear location and dimerization, but depleted its capability to bind to the consensus sequences within the Nkx2.5 promoter region.
Conclusions
Our studies reveal that phosphorylation is essential for Shox2a to repress Nkx2.5 expression during SAN development and differentiation.
Keywords: DNA binding, Nkx2.5 repression, pacemaking, phosphorylation, SAN development, Shox2
Introduction
Heart defects are among the most common birth defects and are the leading cause of birth defect‐related death.1 The sinoatrial node (SAN), a specialized group of cells at the junction of the superior vena cava and right atrium, functions as the primary cardiac pacemaker that controls the rhythm of the heartbeat. Sinus node dysfunctions cause cardiac defects, especially bradycardia, arrhythmias, and, in severe cases, lead to cardiac arrest, sinoatrial exit block, and sudden death.2–3 Although extensive studies have been conducted to describe the morphology, cellular components, and electrophysiological properties, structure of the normal and diseased pacemaker tissues, genetic regulation, and molecular mechanisms underlying SAN development and pathogenesis remain elusive. Elucidation of genetic mechanisms that control SAN development will shed light on the therapeutic intervention of both congenital and acquired pacemaker defects.
Several members of the homeobox and T‐box gene families have been demonstrated to be key regulators in SAN formation and function.4–5 Among them is the paired‐related homeodomain transcription factor, Shox2, 1 of the 2 members in the short‐stature homeobox gene family. During embryonic heart development, Shox2 is expressed in the sinus venosus region, including the SAN and the sinus valves.6–7 Similar expression pattern of SHOX2 was also reported in the human embryonic heart.8–9 Shox2 null mutation causes embryonic lethality between embryonic day 11.5 (E11.5) and E12.5 as a result of cardiovascular (CV) defects, including severe hypoplastic SAN and bradycardia associated with down‐regulation of the SAN‐specific marker genes, Tbx3 and Hcn4, and ectopic expression of Nkx2.5, Cx40, Cx43, and Nppa within the SAN region.6–7 Gene expression analysis indicates that Shox2 and Nkx2.5 are expressed in a mutually exclusive manner from the earliest stages of the venous pole and the SAN formation until at least E15.5.4,6–7 Nkx2.5 overexpression produced the same SAN and sinus valve phenotypes as that observed in Shox2−/− hearts, suggesting that Nkx2.5 activity is detrimental to SAN development and that Shox2 plays an essential role in SAN development through repression of Nkx2.5.4,10 However, the functional mechanism of Shox2 protein as a transcriptional repressor on Nkx2.5 expression is unknown.
Post‐translational modifications, including protein phosphorylation, regulate activities and functions of transcription factors. Protein phosphorylation can rapidly regulate the activity of many eukaryotic transcription factors during development and physiological processes at different levels, such as protein stability, cellular localization, protein‐protein interaction and oligomerization, DNA‐binding activity, and transcriptional potential.11 For example, SHOX protein, the paralogous of human SHOX2, whose mutations or deletions have been associated with syndromic short stature, such as Turner syndrome, Léri‐Weill dyschondrosteosis, and Langer mesomelic dysplasia in humans, was found to be phosphorylated in vivo on several serine residues.12 The replacement of Ser106, the major phosphorylation site, with Ala does not alter SHOX nuclear translocation and DNA‐binding capability, but completely abolishes the transcriptional activity of the protein.12
In this study, we attempted to address the functional mechanism underlying Nkx2.5 repression by Shox2a, the isoform that is expressed in the developing heart.6 We present in vitro and in vivo evidence demonstrating that phosphorylation of Shox2a is required for its specific binding to the Nkx2.5 promoter and its function to repress Nkx2.5 expression.
Methods
Animal and Human Samples
All animal experiments were approved by the Tulane University Institutional Animal Care and Use Committee (New Orleans, LA) and were conducted in strict accordance with the Guide for Care and Use of Laboratory Animals of the National Institutes of Health (NIH). Generation of transgenic (Tg) mice are described in detail in the following section. Mouse embryos (total of approximately 80) were harvested from timed pregnant females and fixed in 4% paraformaldehyde (PFA) in PBS at 4°C overnight. Surgically and medically terminated human embryos (N=4) were provided by the Hospital for Women and Children of Fujian Province (Fuzhou, China), with the permission of the Ethics Committee of Fujian Normal University (Fuzhou, China). Samples were then dehydrated through a graded ethanol series, embedded, and processed for paraffin serial sections at 7 μm, and sections were subjected to immunohistochemistry (IHC).
Generation of pMes‐Shox2a‐S92A/S110A Tg Mice
Similar to the generation of pMes‐Shox2a Tg mice reported on previously,10 Shox2a‐S92A/S110A (also named Shox2a‐DM) DNA was amplified by polymerase chain reaction (PCR) and ligated into the pMES‐STOP vector. Tg DNA fragment containing the chicken β‐actin/cytomegalovirus (CMV) enhancer promoter, the loxP flanked transcriptional STOP cassette, and Shox2a‐S92A/S110A sequences, followed by the Ires22‐Egfp sequence and a rabbit β‐globin poly(A) tail, was released by BciVI and used for pronuclear injection at the Tulane Transgenic/Knockout facility. Tg mice with tissue‐specific transgene expression (Shox2aCO or Shox2a‐DMCO) were obtained by crossing the pMes‐Shox2a or pMes‐Shox2a‐DM lines with the cTnt‐Cre Tg line, which expresses Cre recombinase in the cardiac tissue, including the SAN.10 Tg mice were identified by a PCR‐based genotyping, and binary transgenic embryos were identified by cardiac‐specific expression of enhanced green fluorescent protein (EGFP) under UV light and further confirmed by PCR genotyping.
Yeast Two‐Hybrid Screening Shox2a Interacts With the B56δ Regulatory Subunit of PP2A
MatchmarkerGAL4 Two‐Hybrid System 3 (Clontech Laboratories, Mountain View, CA) was used for screening. The bait plasmid contains the Shox2a coding sequences without its transactivation domain (C‐terminal 19 amino acids) in pGBKT7. For library screening, bail plasmid was transformed into yeast strain AH109. The transformed yeast was cultured and mated with the Matchmaker pretransformed 11‐day mEmbryo cDNA library (Catalog No.: 638868; Clontech Laboratories). Yeast colonies were selected on yeast nitrogen base medium that contains 2% glucose and lacks adenine, histidine, leucine, and trytophan. Approximately 1.15×107 cotransformants were obtained. Colonies were picked and checked for beta‐galactosidase (β‐Gal) production using a filter assay with 5‐bromo‐4‐chloro‐3‐indolyl‐β‐D‐galactopyranoside. Plasmids were isolated from positive clones, and a second round of interaction screening was performed to confirm the interactions. The inserts from positive clones were sequenced.
Protein Coimmunoprecipitation Assay
The Z‐precipitation kit from Innovative BioSolutions LLC (Frederick, MD) was used for protein‐protein interaction assays. Shox2a or Shox2a‐S92A/S110A was cloned in frame to the Z‐tag within the MCS of the vector, following by transfection in HEK‐293T cells. Shox2a or its phosphorylation mutant proteins were expressed in fusion to the Z‐tag and interacted with potential cotransfected partners. Cells were lysed in binding buffer (50 mmol/L of Tris‐HCl [pH 8.0], 150 mmol/L of NaCl, 5 mmol/L of EDTA, 1% Nonidet P40, and protease inhibitor cocktail) and the lysates were incubated with sepharose beads for 2 hours to overnight at 4°C. After washing with binding buffer, bound proteins were released with protein loading buffer (Catalog No.: 928‐40004; LI‐COR Biosciences, Lincoln, NE) and subjected to Western blotting analysis.
Cell Culture, Transfection, and Luciferase Assay
HEK‐293T cells were cultured at 37°C in DMEM containing high glucose, supplemented with 10% FCS and antibiotics. To generate constructs expressing fusion proteins with the GAL4 DNA‐binding domain, we subcloned the following DNA fragments into the pBXG1 vector that contains the GAL4 DNA‐binding domain under the control of the SV40 enhancer/promoter: full‐length mouse Shox2a, Shox2a‐S92A, Shox2a‐S110A, and Shox2a‐S92A/S110A. To test potential transcription activities, the resultant expression vectors were cotransfected into cells with L8G5‐Luc reporter plasmid that contains 8 LexA operators with 5 GAL4‐binding sites upstream of a TATA box and the luciferase (Luc) reporter gene. The CMV‐β‐Gal plasmid was included as an internal control for transfection efficiency. Forty‐eight hours after transfection using Lipofectamine™ 2000 Reagent (Invitrogen, Carlsbad, CA), according to the manufacturer's instruction, luciferase activities were determined and normalized to β‐Gal activity. HL‐1 cells were maintained at 37°C in Claycomb medium (Catalog No.: 51800C; Sigma‐Aldrich, St. Louis, MO) supplemented with 10% FBS, 0.1 mmol/L of norepinephrine, 2 mmol/L of l‐glutamine, and antibiotics. Cells were split in two once they reached confluence on gelatin/fibronectin‐coated plates. To measure repressive activities of Shox2a or its phosphorylation mutants on the Nkx2.5 promoter, the Shox2a and its mutant expression vectors were cotransfected into cells with the Nkx2.5‐Luc reporter plasmid that harbors a 3.3‐kb mouse Nkx2.5 upstream fragment linked to the luciferase reporter gene.6 The pRL‐SV40 plasmid was included as an internal control for transfection efficiency. Transfection on HL‐1 cells was performed using PolyJet™ In Vitro DNA Transfection Reagent (SignaGen Laboratories, Gaithersburg, MD), according to the manufacturer's instruction. Forty‐eight hours after transfection, firefly luciferase activities were determined and normalized to Renilla luciferase activity. In order to test the effect of PP2A inhibitor or extracellular signal‐regulated kinase (Erk)1/2 inhibitor on Shox2a transcription activities, cotransfected cells were treated with different concentration of okadaic acid (Catalog No.: O7885‐50UG; Sigma‐Aldrich) or U0126 (Catalog No.: 9903; Cell Signaling Technology, Danvers, MA) 24 hours after transfection. Reporter assays were carried out after 24 hours in culture.
In Vitro Kinase Assay
The Shox2a, Shox2a‐92a, or Shox2a‐110A construct was transfected into HEK‐293T cells using Lipofectamine 2000 Reagent (Invitrogen). Forty‐eight hours after transfection, cells were harvested, lysed, and subjected to immunoprecipitation. Immunoprecipitated proteins were suspended in PBS and incubated with either cell division control protein kinase 2 (Cdc2) (NEB, P6020S) or Erk2 (NEB, P6080S) at 30°C for 30 minutes, following the manufacturer's instruction. Protein samples were released with protein‐loading buffer (Catalog No.: 928‐40004; LI‐COR Biosciences) and subjected to Western blotting analysis.
Immunostaining
Customized polyclonal antibodies against phosphorylated Ser92 and Ser110 of Shox2a were designed and generated by GenScript (Piscataway, NJ), respectively. IHC staining was performed by standard procedures. All samples were fixed in 4% PFA, embedded in paraffin, and sectioned at 7‐μm thickness. Sections, after rehydration, were treated with 0.3% H2O2 in PBS for 20 minutes to inhibit endogenous peroxidase, followed by incubation with 0.01 mol/L of citric buffer (pH 6.0) at 97°C for 20 minutes for antigen retrieval. Samples were then incubated at 4°C overnight with the following primary antibodies in PBS/Tween‐20 with 1% BSA: 1/100 S92 phosphorylation‐specific Shox2a antibody, 1/1000 S110 phosphorylation‐specific Shox2a antibody, 1/4000 anti‐NK2 transcription factor‐related locus 5 (Nkx2.5; Catalog No.: sc‐8697; Santa Cruz Biotechnology, Santa Cruz, CA). Samples were then rinsed with PBS (2×) and PBS‐Tween‐20 (1×) before being incubated with 1 of the following biotin‐conjugated secondary antibodies, including biotinylated anti‐rabbit immunoglobulin G (IgG; Vector BA‐1000), biotinylated anti‐rat IgG (Vector BA‐9401), and biotinylated anti‐goat IgG (Vector BA‐9500) and 1/66 serum in PBS/Tween‐20 for 40 minutes. Thereafter, all slides were rinsed with PBS 3 times and incubated with 1/400 Streptavidin‐HRP (Invitrogen 434323) in PBS for 10 minutes, then were visualized with incubated with 3,3'‐diaminobenzidine (DAB) substrate (DAB substrate Kit; Catalog No.: 00‐2014; Invitrogen).
For immunocytochemistry (ICC), transfected cells were fixed with 4% PFA in PBS for 10 minutes, washed with PBS, and treated with 1% Triton X‐100 for 15 minutes. Cells were incubated with rat monoclonal antibodies against hemagglutinin (HA; Catalog No.: 12158167001; Roche Diagnostics, Indianapolis, IN), and signals were revealed with a mouse fluorescein isothiocyanate–conjugated anti‐HA monoclonal antibody (green), together with 4',6‐diamidino‐2‐phenylindole (DAPI) counterstaining.
TaqMan Real‐Time PCR
Hearts from newborn (P0) pups were subjected to mRNA isolation with TRIzol (Invirogen). The isolated mRNAs were treated with DNase I (Invitrogen) before reverse transcription using a SuperScript kit (Invitrogen). TaqMan real‐time PCR was used to quantify expression levels of Nkx2.5 using commercially available primers (Mm01309813_S1 for Nkx2.5; Applied Biosystems, Foster City, CA). Gapdh (Mm99999915_g1; Applied Biosystems) was included as an internal control for normalization. The comparative Ct (2−ΔΔCT) method was used for calculation.13
Measurement of Heartbeat Rate
To measure embryonic heartbeat rate, we followed a previously established in vitro measurement protocol.14 Briefly, embryos at E10.5 were dissected in prewarmed ADS buffer containing glucose, and hearts were excised and cultured in DMEM containing 10% FBS and 1% antibiotics at 37°C and 10% CO2 for 24 hours. Heartbeats were counted under inverted microscope for 10 seconds 3 times for each heart. Heartbeat counts were converted to heartbeat rate per minute.
Chromatin Immunoprecipitation Assays and Electrophoretic Mobility Shift Assay
For cell culture chromatin immunoprecipitation (ChIP) assay, Flag‐Shox2a and Nkx2.5‐Luc constructs were cotransfected into HL‐1 cells. After 48 hours in culture, cells were incubated with culture media containing 1% formaldehyde (37%) at 37°C for 10 minutes, washed with Dulbecco's PBS (DPBS), and then scraped from culture dishes in DPBS containing a complete protease inhibitor cocktail. Cells were pelleted by centrifugation (2000 rpm, 4 minutes, 4°C), resuspended in SDS lysis buffer (1% SDS, 10 mmol/L of EDTA, and 50 mmol/L of Tris [pH 8.1]), and sonicated to shear DNA to lengths between 200 and 500 bps. After centrifugation (13 000 rpm, 10 minutes, 4°C), the sonicated lysate was diluted in 10‐fold ChIP dilution buffer (0.01% SDS, 1.1% Triton X‐100, 1.2 mmol/L of EDTA, 16.7 mmol/L of Tris [pH 8.1], and 167 mmol/L of NaCl), and then incubated with 60‐μL washed Dynal magnetic beads A+G (1:1) at 4°C overnight for preclearing. Meanwhile, 60‐μL Dynal magnetic beads A+G (1:1) were washed and incubated with anti‐Flag antibodies on a rotary platform at 4°C overnight. After washing with blocking solution (PBS with 0.5% BSA), the antibody/magnetic beads were mixed with the precleared cell lysate and incubated on a rotary platform at 4°C overnight. Samples were then rinsed with wash buffer (50 mmol/L HEPES [pH 7.6], 100 mmol/L of LiCl, 1 mmol/L of EDTA, 1% NP‐40, and 0.7% Na‐deoxycholate) 6 times and once with TE buffer containing 50 mmol/L of NaCl and collected by centrifugation (960g, 3 minutes, 4°C) to remove residual TE buffer. DNA‐protein complexes were eluted in elution buffer (50 mmol/L of Tris [pH 8.1], 10 mmol/L of EDTA, and 1% SDS) at 65°C for 15 minutes. The elute was collected by centrifugation (16 000g, 1 minute, room temperature) and incubated at 65°C overnight to reverse the cross‐links. Residual RNA was removed by RNase A (0.2 μg/mL) at 37°C for 2 hours and proteins removed by proteinase K (0.2 mg/mL) at 55°C for 30 minutes. Finally, the pull‐down DNA was extracted by phenol and coprecipitated with glycogen by ethanol. Regular PCR was performed to analyze the ChIP samples. The following 11 pairs of primers were used.
Forward1: 5′‐GCCAGACGAAGAGCAGAGTC‐3′
Reverse1: 5′‐ATTGGCGAGAAAGCAAACAG‐3′
Forward2: 5′‐GAGTTGCGATTCTTCCCAAA‐3′
Reverse2: 5′‐CAATCAGCCGCGAAAAGTAT‐3′
Forward3: 5′‐ATTCCGGGTGATAGTTGCAG‐3′
Reverse3: 5′‐AAGGGTTTTTGGCTCAACCT‐3′
Forward4: 5′‐AGCTACCCCGGAATTTGACT‐3′
Reverse4: 5′‐CTTTCCAACGTGGCTTCTGT‐3′
Forward5: 5′‐TTCCCAGCCATTGTAACACA‐3′
Reverse5: 5′‐CAGTGACTTTCCCACCCCTA‐3′
Forward6: 5′‐CCCAATATAGCTCCCCCAAT‐3′
Reverse6: 5′‐ACCCTCCCGAGATTGAAGAT‐3′
Forward7: 5′‐CTTTGGACTGCTTGGAGAGG‐3′
Reverse7: 5′‐TCCTTCCTGCTTTTCCTCAA‐3′
Forward8: 5′‐CCAGGAGGGACAGTTCACAC‐3′
Reverse8: 5′‐AGGCTAACCTCTGGGAGAGC‐3′
Forward9: 5′‐CTGGAATCGATGTGCCTTCT‐3′
Reverse9: 5′‐CTGCCTGGATTCTCCTGAAG‐3′
Forward10: 5′‐AGTCAATTCCCCAACAATGC‐3′
Reverse10: 5′‐TTGTTCTGGGACGAACACTG‐3′
Forward11: 5′‐TTCTGGCTTTCAATCCATCC‐3′
Reverse11: 5′‐CGCGACACATTTGGGATAGT‐3′
For small embryonic heart tissue ChIP assay, we followed a previously published protocol,15–16 with minor modifications. In short, 4 to 5 freshly dissected hearts from E12.5 mouse embryos were incubated with 300 μL of trypsin (2.5 mg/mL) in TE at 37°C for 5 minutes and further disassociated with a yellow‐tip P‐200. Cell culture media (700 μL) were added to inactive the trypsin, and the mixture was incubated with 100 μL of crossing buffer (0.1 mol/L of NaCl, 1 mmol/L of EDTA, 0.5 mmol/L of EGTA, 50 mmol/L of HEPES [pH 8.0], and 11% formaldehyde) for 30 minutes at room temperature with mild agitation on a platform shaker. The cross‐linking was stopped by adding 55 μL of 2.5 mol/L of glycine. After incubation for 5 minutes on ice, pellet was collected by centrifugation (5 minutes at 2000 rpm), rinsed with 1 mL of ice‐cold PBS, and suspended in 1 mL of lysis buffer (50 mmol/L of HEPES [pH 8.0], 0.14 mol/L of NaCl, 1 mmol/L of EDTA, 10% glycerol, 0.3% NP‐40, 0.15% Triton‐X 100, and fresh proteinase inhibitor) and placed on ice for 10 minutes before centrifugation (8 minutes at 2500 rpm). Pellet was suspended in 1 mL of buffer 2 (0.2 mol/L of NaCl, 1 mmol/L of EDTA, 0.5 mmol/L of EGTA, 10 mmol/L of Tris [pH 8.0], and proteinase inhibitor) and incubated at room temperature for 10 minutes before being spun down (8 minutes at 2500 rpm) and resuspended in 2 mL of buffer 3 (1 mmol/L of EDTA, 0.5 mmol/L of EGTA, 10 mmol/L of Tris [pH 8.0], and proteinase inhibitor). Sonication was applied to the mixture to shear DNA‐protein complex, which was then diluted to a final concentration of 0.5% with the appropriate amount of fresh 20% Sarkosyl solution, and incubated at room temperature for 10 minutes, followed by centrifugation (13 000 rpm, 10 minutes). Approximately 500 μL of supernatant was mixed with 150 μL of ChIP cocktail mix (0.43 mol/L of NaCl, 0.43% sodium deoxycholate, and 4.3% Triton‐X 100) and pretreated magnetic beads/antibody mixture were incubated on a rotary platform at 4°C for overnight. The next steps followed that for cell culture ChIP assay as described above. Regular PCR was performed to analyze the ChIP samples by using the primer pair Forward6 and Reverse6 as described above.
For gel mobility shift assays, DNA probes were generated by annealing the following 2 complementary oligonucleotides that are 5′ labeled with IRDye800CW:
Oligo6F‐800: 5′‐/5IRD800/CCAATTAAACGGTAATATTTC‐3′
Oligo6R‐800: 5′‐/5IRD800/GAAATATTACCGTTTAATTGG‐3′
Nuclear protein extracts from cultured cells 48 hours after transfection were prepared by the use of Thermo Scientific NE‐PER Nuclear and Cytoplasmic Extraction Kit. Complete Protease Inhibitor (Roche) and phosphatase inhibitor were used at a concentration of 1 tablet per 25 mL of extraction buffer.
The Odyssey® electrophoretic mobility shift assay (EMSA) buffer kit (Catalog No.: 829‐07910; LI‐COR Biosciences) was used for gel shift reactions according to the manufacturer's instruction. Reaction mixtures were preincubated with IRDye 800CW‐labeled probes at room temperature for 5 minutes before the addition of nuclear extracts. Mixtures were incubated at room temperature for 20 minutes before being loaded on a native polyacrylamide gel. In competition experiments, 10‐, 50‐, 75‐ and 100‐fold excessive unlabeled competitor DNA was included during preincubation. In supershift experiments, the nuclear extracts were preincubated with 1 μg of anti‐HA antibody at room temperature for 15 minutes before mixing with probes. Images were revealed on the Odyssey Infrared Imaging System.
Statistical Analysis
All data are expressed as mean±standard deviation, and statistical significance is established at P<0.05. In Figure 4, the luciferase activities of different Gal4‐Shox2 constructs, Shox2 and its mutants, and different treatment of the same report constructs were measured. Each assay was performed in triplicate and repeated twice. In Figure 5, the levels of Nkx2.5 mRNA expression in the hearts from wild‐type (WT) (n=5), Shox2a‐DMCO (n=4), and Shox2aCO (n=3) newborn mice were determined by real‐time reverse transcriptase (RT)‐PCR. In Figure 6, the beating rate of the isolated embryonic hearts of WT (n=6), Shox2CKO (n=4), Shox2CKO, Shox2a‐DMCO (n=4), and Shox2CKO;Shox2aCO (n=3) were measured. Comparison of the different mean values was performed using Wilcoxon's rank‐sum test.
Figure 4.
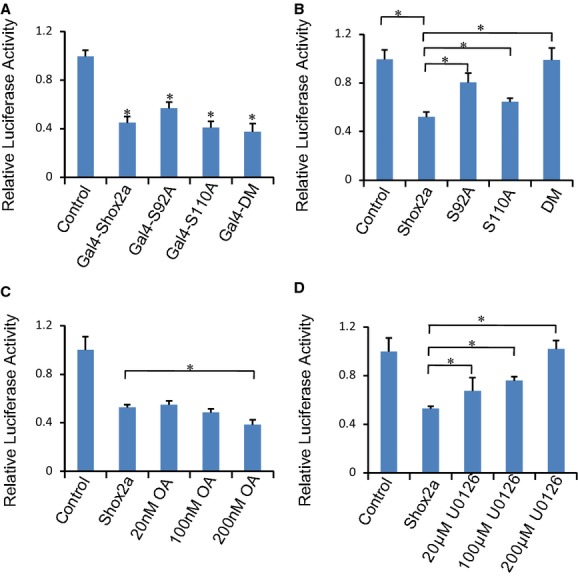
Shox2a phosphorylation is required for its specific repressive effect on the Nkx2.5 promoter. A, Luciferase assays show that Shox2a and its mutants have similar transcriptional repressive potential when fused with Gal4 DNA‐binding domain. B, Mutations on the phosphorylation sites compromise Shox2a's repressive effect on the Nkx2.5 promoter activity. C and D, Treatment of transfected cells with the PP2A inhibitor, okadaic acid, leads to a dose‐dependent increase in Shox2a's repressive function on the Nkx2.5 promoter, whereas treatment with the Erk1/2 inhibitor, U0126, results in a dose‐dependent loss of the repressive activity of Shox2a on the Nkx2.5 promoter. *P<0.05. PP2A indicates protein phosphatase 2A.
Figure 5.
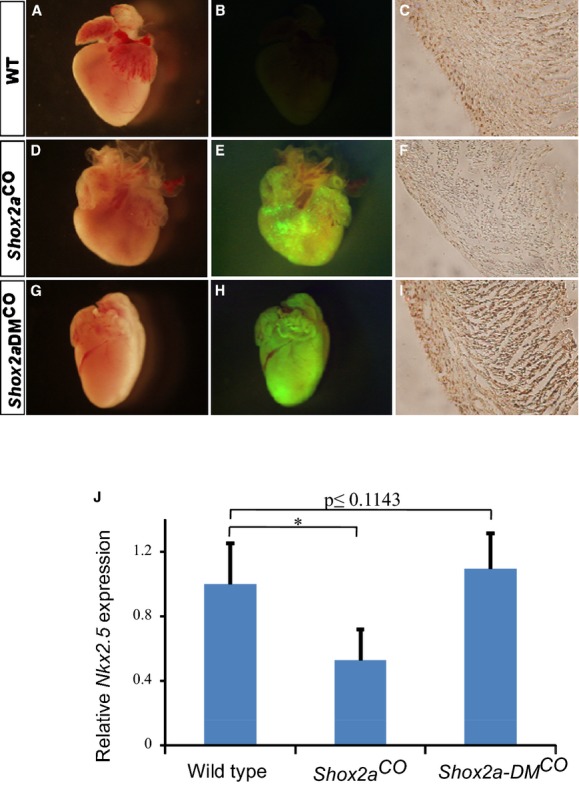
Transgenic expression of Shox2a, but not Shox2a‐DM, mutant in mouse heart results in down‐regulation of Nkx2.5 and cardiac malformation. A P0 control mouse heart shows its gross morphology (A), absent EGFP expression (B), and immunostaining of Nkx2.5 (C). A P0 cTnt‐Cre;pMes‐Shox2a (Shox2aCO) mouse heart exhibits gross blunt heart apex (D), EGFP expression (E), and reduced level of Nkx2.5 protein (F). A P0 cTnt‐Cre;pMes‐Shox2a‐DM (Shox2a‐DMCO) shows similar gross morphology (G) and unaltered Nkx2.5 expression (I), as compared to controls. Transgene expression is revealed by EGFP expression under UV light (H). J, Real‐time RT‐PCR assays show transcriptional reduction of Nkx2.5 expression in the heart from Shox2aCO transgenic mice at P0 (n=3). In contrast, the level of Nkx2.5 mRNA expression in the heart from Shox2a‐DMCO transgenic mice (n=4) remains the same as control (n=5). *P<0.05. Scale bars in red represent 1 mm and scale bars in black represent 100 μm. EGFP indicates enhanced green fluorescent protein; PCR, polymerase chain reaction; WT, wild type.
Figure 6.
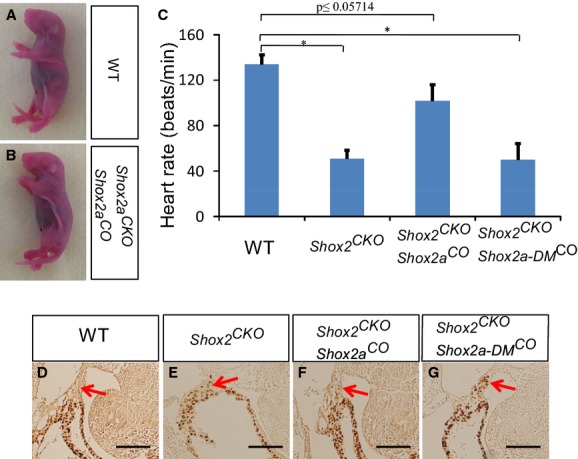
Rescue of embryonic lethality and cardiac defects of Shox2 conditional mutant by transgenic expression of Shox2a, but not Shox2a‐DM. Panoramic comparison between littermates of wild‐type (A) and cTnt‐Cre;ShoxF/F;pMes‐Shox2a (Shox2CKO;Shox2aCO) mice (B) at P0. C, Heartbeat rate measurement shows significantly reduced heartbeat rate in Shox2CKO (50±14.1 bpm) and cTnt‐Cre;Shox2F/F;pMes‐Shox2a‐DM (Shox2CKO;Shox2a‐DMCO) (51±7.4 bpm) embryos, which was less than 40% of that in wild‐type controls (134±8.3 bpm). However, Shox2CKO;Shox2aCO hearts exhibit a heartbeat rate (102±14 bpm) comparable to controls. Immunohistochemical staining shows lack of Nkx2.5 expression in an E11.5 control SAN (D), ectopic Nkx2.5 expression in an E11.5 Shox2CKO embryo (E), and rescue of ectopic Nkx2.5 expression in the SAN of Shox2CKO embryos by transgenic expression of Shox2a (F), but not Shox2a‐DM (G). Arrows point to the SAN region. *P<0.05. Scale bar=50 μm. SAN indicates sinoatrial node.
Results
Shox2a Interacts With the B56δ Regulatory Subunit of PP2A
The Shox2 gene encodes 2 alternatively spliced isoforms: Shox2a and a shorter version Shox2b.7,9,17 Our previous studies have identified the transcription activity of Shox2a and Shox2b protein within the C‐terminus, and both isoforms can act as either a transcriptional activator or a repressor in a cell‐type–specific manner.18 In order to reveal the functional mechanisms of Shox2 protein, we set out to identify Shox2 interacting proteins by a yeast 2‐hybrid screen. In order to reduce false positives from yeast 2‐hybrid screen, we used Shox2a without the OAR (opt, aristaless, rax) domain (named Shox2aT) as the bait to screen a cDNA library from E11.5 mouse embryo. Among those highest hit potential interacting proteins is the δ isoform (Ppp2r5d) of B56 regulatory subunit of protein phosphatase 2A (PP2A). Further analysis showed that the whole sequence of Shox2a, except the OAR domain, is essential for interaction with PP2A (Figure 1A). However, Shox2b failed to interact with PP2A. PP2A is a heterotrimeric serine/threonine protein phosphatase composed of 2 highly conserved subunits (the catalytic C subunit and the structural A subunit) and a highly variable regulatory B subunit. Sequence comparisons showed that the Shox2a‐interacting clones encompass all of the amino acid sequence of B56δ, which contains an A subunit‐binding domain required for interaction of B subunit with the PP2A A subunit. Coimmunoprecitpitation (Co‐IP) assays further confirmed physical interaction of Ppp2r5d with Shox2a, but not Shox2b (Figure 1B).
Figure 1.

Interaction of Shox2a with the δ isoform (Ppp2r5d) of B56 regulatory subunit of protein phosphatase 2A (PP2A). A, Shox2a interacts with Ppp2r5d determined by yeast mating. The whole sequence of Shox2a, except the 14‐amino acid motif (OAR) at the C‐terminus, is essential for interaction with PP2A. B, Z‐protein pull‐down demonstrates physical interaction of Shox2a, but not Shox2b, with Ppp2r5d. OAR indicates opt, aristaless, rax.
Shox2a Is Phosphorylated Exclusively on Ser92 and Ser110 In Vivo
The interaction between Shox2a and PP2A regulatory subunit B56δ suggests an involvement of protein phosphorylation on Shox2a. To verify whether Shox2a is indeed phosphorylated, we transfected HA‐tagged Shox2a expression vector (CMV‐HA‐Shox2a) into 293T cells and analyzed the expressed proteins by Western blotting analysis, which revealed 2 slightly shifted forms of Shox2a (Figure 2A). Digestion of immunoprecipitated proteins by lambda protein phosphatase (λ‐PPase) eliminated the shifted forms, confirming the presence of phosphorylated Shox2a (p‐Shox2a) (Figure 2A). Based on these observations, we reasoned that Shox2a contains 2 phosphorylation sites, with the slower migrating form bearing a single‐site phosphorylation, and the slowest migrating one carrying phosphorylation on both sites. Because no phosphorylation form of Shox2b was detected by the similar analysis (data no shown), the phosphorylation sites must reside within the N‐terminal region that is missing in Shox2b. Bioinformatic analysis revealed potential phosphorylation sites in this region (Table). We conducted a site‐directed mutagenesis assay on each of these potential sites and identified Ser92 and Ser110 as the true phosphorylation sites, as evidenced by the fact that either S92A or S110A mutation (serine to alanine) eliminated 1 phosphorylated form, and double mutation (S92A/S110A, referred as Shox2a‐DM) eliminated both shifted bands on Western blotting (Figure 2B). Based on the prediction from different kinase‐specific phosphorylation site prediction tools and analysis of the adjacent residues of Ser92 and Ser110, we reasoned that Shox2a may be the substrate of Erk1/2 and/or Cdc2. Western blotting analysis after in vitro kinase assay demonstrated that Shox2a, Shox2a‐S92A, and Shox2a‐S110A were all phosphorylated by Erk2, resulting in the disappearance of the unphosphorylated form and the increased amounts of the phosphorylated form(s) (Figure 2C through 2E). However, these proteins were not phosphorylated by Cdc2. We concluded that Shox2a is phosphorylated on serine residues 92 and 110 by Erk1/2.
Figure 2.
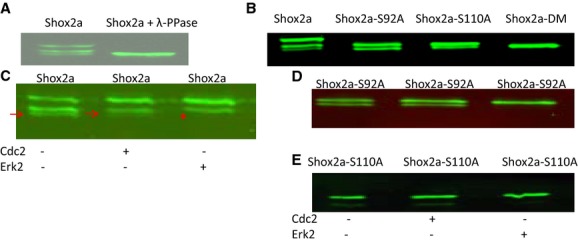
Shox2a is phosphorylated on serine residues in vivo. A, Western blotting assay shows the presence of shifted forms of Shox2a that are eliminated by incubation of Shox2a proteins with lambda protein phosphatase (λ‐PPase). B, Serine to alanine mutation on Ser92 (Shox2a‐S92A), Ser110 (Shox2a‐S110A), or both sites (Shox2a‐DM) eliminates phosphorylated Shox2a forms. Treatment of immunoprecipitated Shox2a (C), Shox2a‐S92A (D), or Shox2a‐110A (E) by Cdc2 or Erk2 demonstrates that Shox2a can be phosphorylated by Erk2, but not Cdc2. Red arrows point to the unphosphorylated band and red start indicates the disappearance of unphopshorylated band after Erk2 treatment in (C). Cdc2 indicates cell division control protein 2; Erk1/2, extracellular signal‐regulated kinase 1 and 2.
Table 1.
Shox2a Phosphorylation Site Prediction
| Pos | Context | Score | Pred |
|---|---|---|---|
| 24 | KEAITYREV | 0.963 | *T* |
| 52 | DDRSSPAVR | 0.991 | *S* |
| 92 | GGGRSPVRE | 0.900 | *S* |
| 105 | AAERSREPG | 0.939 | *S* |
| 110 | REPGSPRLT | 0.996 | *S* |
| 114 | SPRLTEVSP | 0.942 | *T* |
| 117 | LTEVSPELK | 0.984 | *S* |
| 143 | KQRRSRTNF | 0.995 | *S* |
To determine that Shox2a is indeed phosphorylated in the developing SAN, we generated antibodies that recognize phosphorylated Ser92 and Ser110 of Shox2a, respectively, whose specificity was proved by Western blotting on Shox2a, Shox2a‐S92A, Shox2a‐S110A, and Shox2a‐S92A/S110A (Shox2a‐DM) proteins (data no shown). Both antibodies detected the presence of p‐Shox2a proteins in the mouse developing SAN (Figure 3A through 3F), in an overlapping pattern with Hcn4, the molecular marker of the SAN (Figure 3C and 3G). Western blotting assay further confirmed the presence of p‐Shox2a in the mouse embryonic heart (Figure 3H). Furthermore, we also detected the presence of p‐SHOX2a in the human embryonic SAN (Figure 3D), suggesting a conserved function of Shox2a in SAN development in mice and humans.
Figure 3.
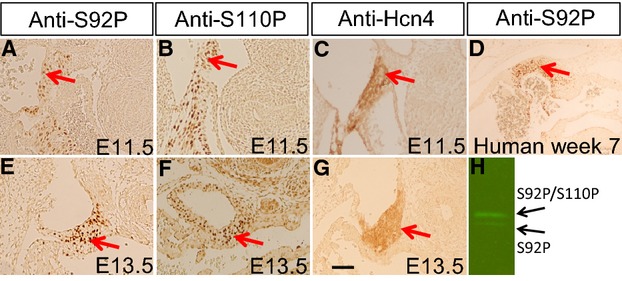
Presence of p‐Shox2a in the SAN of mouse and human embryos. Immunohistochemical staining using anti‐S92P (A, E) or anti‐S110P (B, F) specific antibodies demonstrates the presence of p‐Shox2a in the SAN of E11.5 (A, B) and E13.5 (E, F) mouse embryo. Immunohistochemical staining shows the expression of Hcn4 in the SAN of E11.5 (C) and E13.5 (G) mouse embryo. D, Immunohistochemical staining using anti‐S92P antibody reveals the presence of p‐SHOX2a in the SAN of human embryo at 7‐week gestation. F, Western blotting using anti‐S92P antibody reveals the presence of p‐Shox2a in E11.5 mouse embryonic hearts. Red arrows point to the SAN region. Scale bar=20 μm for all panels. SAN indicates sinoatrial node.
Functional Significance of Shox2a Phosphorylation
We next sought to investigate the functional significance of Shox2a phosphorylation. Because protein phosphorylation could change the conformation and hence the function of protein, we first examined whether phosphorylation would modify the transcriptional activity of Shox2a. The transcription activity assay of Gal4‐Shox2a and its mutants demonstrated that Shox2a and its phosphorylation mutants all possessed transcriptional repressive potential in HEK‐293T cells (Figure 4A), suggesting that phosphorylation does not change the transcription potential of Shox2a protein.
We reported previously that Shox2a acts as a transcriptional repressor on Nkx2.5 in vitro and in vivo.6,10 To determine whether phosphorylation confers Shox2a‐repressive capability specifically on Nkx2.5, we carried out reporter gene expression assays in the HL‐1 cardiac muscle cell line using the Nkx2.5‐Luc reporter.6 Cotransfection of the reporter construct with Shox2a or its various mutant forms demonstrated significant changes in the repressive effect of Shox2a phosphorylation mutants on Nkx2.5 promoter activity (Figure 4B). As compared to Shox2a, both Shox2a‐S92A and Shox2a‐S110A exhibited significantly compromised repressive activities on the Nkx2.5 promoter, and Shox2a‐DM even lost its repressive activity completely. Consistent with these observations, treatment of Shox2a‐transfected cells with PP2A‐specific inhibitor, okadaic acid, increased the transcriptional repressive activity of Shox2a in a dose‐dependent manner (Figure 4C), and conversely, addition of the highly selective Erk1/2 inhibitor, U0126, to cell culture led to a dose‐dependent loss of repressive activity of Shox2a (Figure 4D). All together, these observations indicate a requirement of phosphorylation on both sites for Shox2a to achieve its maximal repressive activity on the Nkx2.5 promoter in cell cultures.
We have reported previously that overexpression of Shox2a in the developing mouse heart results in a reduction of Nkx2.5 expression and abnormal cardiac formation.10 To determine whether or not Shox2a phosphorylation is also required for its activity in vivo, we generated a conditional Shox2a‐S92A/S110A Tg allele (pMes‐Shox2a‐DMCO). Tissue‐specific overexpression of transgenes in the developing mouse heart was achieved by crossing pMes‐Shox2a‐DMCO Tg mice or pMes‐Shox2aCO line to the cTnt‐Cre Tg line,19 respectively. Similar to the previous report,10 examination of the cTnt‐Cre;pMes‐Shox2aCO Tg heart (referred to as Shox2aCO) at newborn (P0) revealed a significantly round‐shaped heart and an overall reduction in Nkx2.5 expression, as determined by IHC staining, when compared to WT littermate controls (Figure 5A through 5F). In contrast, no obvious difference was observed in the cTnt‐Cre;pMes‐Shox2a‐DMCO) Tg heart (referred to as Shox2a‐DMCO), as compared to controls, in terms of heart morphology and overall Nkx2.5 protein expression (Figure 5G through 5I). Quantitative real‐time RT‐PCR assays further confirmed complete loss of repressive activity of Shox2a‐DM on Nkx2.5 mRNA expression in vivo (Figure 5J), consistent with the results from in vitro reporter assays in cell cultures (Figure 4).
It was demonstrated previously that Nkx2.5 activity is detrimental to the normal development and function of the early developing SAN and that Shox2 shields SAN cell fate by repressing Nkx2.5 expression.4,6,10 To further confirm the requirement of phosphorylation for Shox2a to repress Nkx2.5 expression in the developing SAN, we conducted rescue experiments by activating the Shox2aCO or Shox2a‐DMCO Tg allele in the heart of mice carrying cTnt‐Cre and Shox2F/Falleles.20 Whereas mice bearing cTnt‐Cre;Shox2aCO;Shox2F/F compound alleles could survive to birth and exhibited inhibition of Nkx2.5 expression in the SAN, cTnt‐Cre;Shox2a‐DMCO;Shox2F/F mice showed cardiac phenotypes resembling that noted in cTnt‐Cre;Shox2F/F(referred to as Shox2CKO) mice, including ectopic activation of Nkx2.5 in the SAN and embryonic lethality between E11.5 and E12.5 (Figure 6A, 6B, 6D through 6G). In vitro heartbeat rate measurement showed near normal beat rate of embryonic hearts from cTnt‐Cre;Shox2aCO;Shox2F/F mice and significantly reduced beat rates of embryonic hearts from both cTnt‐Cre;Shox2F/F and cTnt‐Cre;Shox2a‐DMCO;Shox2F/F mice, as compared to WT controls (Figure 6C). We recognize that this in vitro heartbeat rate measurement does not reflect real in vivo embryonic heartbeats and could present large variations. However, the direct comparison under the same condition indeed revealed a significant difference in beat rates between mice with different genotype. Nevertheless, these results indicate the in vivo functional importance of Shox2a phosphorylation during SAN development.
Phosphorylation of Shox2a Is Necessary for Its Binding to the Nkx2.5 Promoter
It has been well established that post‐translational modifications, including phosphorylation of transcription factors, regulate protein function at different levels, such as subcellular localization, dimerization, protein‐protein interactions, and DNA binding. To investigate the underlying mechanisms of how phosphorylation alters Shox2a transcriptional potential on Nkx2.5 expression, we conducted a series of experiments. ICC revealed exclusive nuclear localization of both Shox2a and Shox2a‐DM in cell cultures (Figure 7A), indicating that nuclear localization of Shox2a is not dependent on its phosphorylation. Because many homeodomain proteins form homodimers to exert their functions, and Shox2a was found to form homodimer as well,4 we tested whether phosphorylation is required for Shox2a dimerization by generating Z‐tag‐Shox2a, Myc‐Shox2a, Z‐tag‐Shox2a‐DM, and Myc‐Shox2a‐DM constructs and transfecting these constructs into HEK‐293T cells, followed by Co‐IP and Western blotting assays. The results showed that, similar to Shox2a, Shox2a‐DM is able to form homodimer, indicating that phosphorylation has no effect on dimerization of Shox2a protein. Because the replacement of S92 and Ser110 with Ala did not alter the transcriptional potential of Shox2a protein, but caused dramatic loss of its repressive activity, specifically on the Nkx2.5 promoter (Figure 4), we reasoned that Shox2a phosphorylation is likely related to its specific binding to the Nkx2.5 promoter. In order to test the hypothesis, Flag‐Shox2a and the Nkx2.5‐Luc reporter constructs were cotransfected into HL‐1 cells, and the nuclear extract was subject to ChIP PCR analysis. Eleven pairs of primers were designed to evenly cover the 3.3‐kb promoter region and 2 Shox2a‐binding fragments were identified from PCR analysis of the ChIP samples (Figure 8A and 8B). These 2 sites (site‐1: −2937 to −2727; site‐2: −2160 to −1814) both contain the SHOX AATT‐palindromic binding sequence. Bioinformatic search indicates that these 2 sequences are highly conserved across the species, including the human, rat, dog, and mouse (data not shown). Interestingly, they are the only 2 regions that are highly conserved between the human and mouse within the 3.3‐kb Nkx2.5 promoter region. Because the site‐2 contains a very typical SHOX‐binding sequence (ATTAAACGGTAAT), we picked up this site for ChIP PCR assay of E12.5 mouse embryonic heart chromatin and subsequent EMSA studies. We took advantage of a recently created Flag‐HA‐Shox2a knock‐in allele (to be reported elsewhere) in the lab and collected hearts from E12.5 embryos for ChIP assay. The results confirmed the site‐2 of the Nkx2.5 promoter as a genome‐specific element recognized by Shox2a in vivo (Figure 8C). To further confirm direct binding and the Shox2a‐binding sequence, EMSA was performed by using a 60‐bp probe that covers the AATT‐palindromic sequence in the site‐2 of the Nkx2.5 promoter. Incubation of labeled probes with the nuclear extracts of HA‐Shox2a transfected cells, but not the nuclear extracts of control cells and HA‐Shox2a‐DM transfected cells, gave rise to 2 shifted bands, most likely presenting binding of Shox2a monomers and dimers to the probes (Figure 8D). The binding specificity was confirmed by competitive EMSA with excessive cold probes (Figure 8D), as well as by supershift assay with anti‐HA antibodies (Figure 8E). These results demonstrate that Shox2a binds to the Nkx2.5 promoter directly and that phosphorylation of Shox2a is required for such binding to exert its transcriptional activity.
Figure 7.
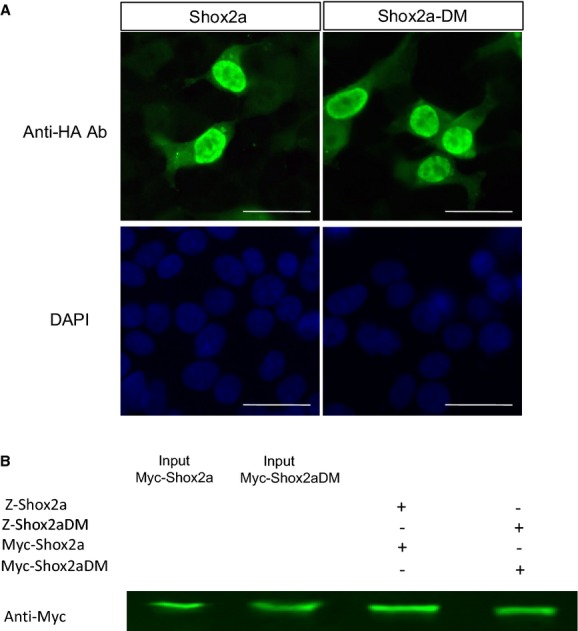
Abolishment of the phosphorylation sites in Shox2a does not alter its nuclear localization (A) and dimerization (B). Scale bar=15 μm..
Figure 8.
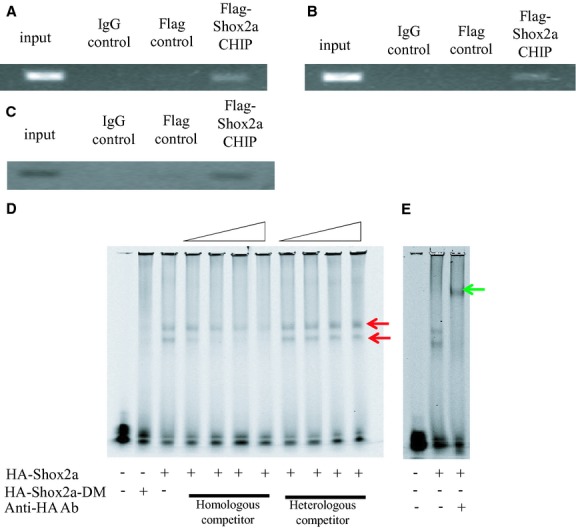
Binding of Shox2a to the Nkx2.5 regulatory elements depends on its phosphorylation. ChIP assays reveal 2 Shox2a‐binding elements, site‐1 (A) and site‐2 (B), within the 3.3‐kb Nkx2.5 promoter region. (C) ChIP assays using mouse embryonic hearts confirm binding of Shox2a to the site‐2 in vivo. D, Electrophoretic mobility shift assay (EMSA) reveals specific binding of the site‐2 sequence by nuclear extracts from cells expressing HA‐Shox2a, but not HA‐Shox2a‐DM. Red arrows point to the shift bands. (E) Supershift assay with anti‐HA antibodies demonstrates specific DNA with HA‐Shox2a. Green arrow points to the supershifted band. ChIP indicates chromatin immunoprecipitation.
Discussion
Cardiac transcription factors, such as Nkx2.5, the GATA family members, and myocyte enhancer factor 2, play essential roles in embryonic heart development and postnatal heart functions. Among them, Nkx2.5 is a central regulator.21 In mammals, Nkx2.5 is expressed in the developing heart, including the first and second heart fields, and is crucial for heart patterning and development.22 Though Nkx2.5 is expressed in cardiomyocytes throughout the whole heart, it is absent in the myocardium of the sinus horns and the SAN during early development.4,10,23 Mutation in Shox2 leads to ectopic Nkx2.5 expression in the developing SAN and disrupts the expression of the SAN genetic regulatory genes, leading to deviation of the fate of SAN cells.6–7 This phenotype was recapitulated in mice carrying Tg expression of Nkx2.5 to the SAN,10 indicating that Nkx2.5 activity is detrimental to the normal development of the SAN. Shox2 appears to play an essential role in regulating SAN development and function through the repression of Nkx2.5 expression. However, the mechanism underlying the repression of Nkx2.5 by Shox2 remained unknown. In the present study, we demonstrate the direct binding of Shox2a to the Nkx2.5 promoter. We identified 2 Shox2a‐binding sites within the 3.3‐kb mouse Nkx2.5 promoter region. Both sites contain the SHOX AATT‐palindromic consensus binding sequences and are highly conserved across the species. Two cardiac enhancers, referred to as activating region (AR) 1 and AR2, have been identified previously in the 5′‐flanking region of the mouse Nkx2.5 gene by serial deletion experiments.22,24 AR2 is located between −3 and −2.5 kb upstream of the gene and contains evolutionarily conserved binding sites for Smad, GATA, and NFAT, the key regulators of Nkx2.5 expression.25–26 This enhancer is repressed by a closely adjacent inhibitory sequence element, named inhibitory region (IR) 2. It has been shown that the cardiac activity of AR2 is inhibited when the IR2 is activated.22,24 Interestingly, the Shox2a binding site‐1 identified in this study overlaps with AR2 and the site‐2 is located within IR2. Although additional Shox2a‐binding sites very likely exist in the Nkx2.5 regulatory elements, binding of Shox2a to the site‐2 within IR2 appears to contribute to the repression of Nkx2.5.
The homeodomain‐containing transcription factors play crucial roles during embryonic development by regulating pattern formation and organogenesis.27–28 Protein phosphorylation represents a major molecular mechanism to regulate various functions of proteins in vivo.11 Phosphorylation is found to regulate the transcriptional activity of many homeodomain proteins during embryogenesis. For example, phosphorylation modulates the biological activities of human SHOX,12 another member of the SHOX family that has been shown to have a similar function as Shox2 in the regulation of SAN development and pacemaking function, in addition to several other organs.8 In the current study, we present evidence that Shox2a interacts with the δ isoform of the B56 regulatory subunit of PP2A and is phosphorylated on Ser92 and Ser110 by Erk1/2 in vitro and in vivo. The presence of p‐Shox2a in the developing SAN of mouse and human embryos suggests a conserved biological function during SAN development in both humans and mice. Indeed, the failure of the Shox2a‐DM transgene to rescue ectopic Nkx2.5 expression in the SAN as well as bradycardia in Shox2 mutant embryos further supports the functional importance of Shox2a phosphorylation. A recent study using systemic mass spectrometry also revealed the presence of p‐Shox2a (phosphorylation on Ser110) in the adult mouse brain,29 suggesting a conserved regulatory mechanism of Shox2a function by phosphorylation in other organs.
In contrast to their wide spectrum of biological functions of homeodomain transcription factors in vivo, the homeodomain itself is highly conserved and in vitro DNA‐binding studies have revealed binding of most homeodomain proteins to a similar short consensus DNA sequence containing the TAAT motif.28,30 This apparent discrepancy indicates that target gene specificity of each homeodomain protein in vivo may not depend on the homeodomain only, but may be achieved by a complex combination of different elements, such as specific expression patterns, interactions with different cofactors,31 differential DNA‐binding affinities to individual target sites,32 or post‐translational modifications.33 Our present studies demonstrate that the phosphorylation of Shox2a is not necessary for its nuclear localization and dimerization, but is essential for its specific binding to the Nkx2.5 promoter. Mutations on the phosphorylation sites of Shox2a compromise its capability to repress Nkx2.5 expression both in cell culture and in the developing heart. Nevertheless, our studies reveal a post‐translational modification mechanism of biological function of Shox2a and demonstrate that phosphorylation is essential for Shox2a to repress Nkx2.5 during SAN development and differentiation.
Clinical Perspective
Although no known human syndrome has been mapped or linked to the SHOX2 locus, studies using mouse gene‐targeting models have demonstrated an essential role for Shox2 in limb skeletogenesis, palatogenesis, temporomandibular joint formation, and CV development.4 Human SHOX2 is located on chromosome 3q25‐q26, a chromosomal region that has been implicated in Cornelia de Lange syndrome, which is characterized by growth and mental retardation, cleft palate, abnormally situated eyelids, nose and ear deformities, as well as heart and limb defects. The expression patterns of mouse Shox2 are in perfect agreement with the features noted in Cornelia de Lange syndrome. Thus, human SHOX2 is a candidate gene for Cornelia de Lange syndrome. In addition to its pivotal roles during embryogenesis, SHOX2 DNA methylation has been used as a standard biomarker for diagnosis of lung cancer in clinics.34–35 In addition, it was found recently that Shox2 plays a crucial role in determining adipose distribution in mice and humans.36 In a mouse model in which Shox2 was replaced with a human SHOXa hypomorphic allele, despite formation of a morphologically normal SAN and rescue of embryonic lethality, the mice developed bradycardia and arrhythmia, suggesting a dose requirement not only for SAN development, but also for its function.8 Given the fact that mouse Shox2 shares 99% identity with its human counterpart at the amino acid level and both exhibit similar expression patterns during embryonic development, including the SAN, SHOX2 could represent a candidate gene for mutation in human bradycardia and arrhythmia. The presence of phosphorylated SHOX2a in the SAN of human embryos would also predict an essential biological function for SHOX2a phosphorylation in SAN development and its pacemaking function.
Sources of Funding
This work was supported by an NIH grant (R01 DE17792) to Chen. Ye was supported by an American Heart Association Predoctoral Fellowship (13PRE13750003). This work was also supported by grants from the National Natural Science Foundation of China (81271102; 81100730) and by a grant from the National Health and Family Planning Commission of China (WKJ‐FJ‐24).
Disclosures
None.
References
- 1.Hoffman JI, Kaplan S. The incidence of congenital heart disease. J Am Coll Cardiol. 2002; 39:1890-1900. [DOI] [PubMed] [Google Scholar]
- 2.Dobrzynski H, Boyett MR, Anderson RH. New insights into pacemaker activity: promoting understanding of sick sinus syndrome. Circulation. 2007; 115:1921-1932. [DOI] [PubMed] [Google Scholar]
- 3.Durham D, Worthley LI. Cardiac arrhythmias: diagnosis and management. The bradycardias. Crit Care Resusc. 2002; 4:54-60. [PubMed] [Google Scholar]
- 4.Liu H, Espinoza‐Lewis RA, Chen C, Hu X, Zhang Y, Chen Y. The role of Shox2 in SAN development and function. Pediatr Cardiol. 2012; 33:882-889. [DOI] [PubMed] [Google Scholar]
- 5.Christoffels VM, Smits GJ, Kispert A, Moorman AF. Development of the pacemaker tissues of the heart. Circ Res. 2010; 106:240-254. [DOI] [PubMed] [Google Scholar]
- 6.Espinoza‐Lewis RA, Yu L, He F, Liu H, Tang R, Shi J, Sun X, Martin JF, Wang D, Yang J, Chen Y. Shox2 is essential for the differentiation of cardiac pacemaker cells by repressing Nk2–5. Dev Biol. 2009; 327:376-385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Blaschke RJ, Hahurij ND, Kuijper S, Just S, Wisse LJ, Deissler K, Maxelon T, Anastassiadis K, Spitzer J, Hardt SE, Scholer H, Feitsma H, Rottbauer W, Blum M, Meijlink F, Rappold G. Gittenberger‐de Groot AC. Targeted mutation reveals essential functions of the homeodomain transcription factor Shox2 in sinoatrial and pacemaking development. Circulation. 2007; 115:1830-1838. [DOI] [PubMed] [Google Scholar]
- 8.Liu H, Chen CH, Espinoza‐Lewis RA, Jiao Z, Sheu I, Hu X, Lin M, Zhang Y, Chen Y. Functional redundancy between human SHOX and mouse Shox2 genes in the regulation of sinoatrial node formation and pacemaking function. J Biol Chem. 2011; 286:17029-17038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Blaschke RJ, Monaghan AP, Schiller S, Schechinger B, Rao E, Padilla‐Nash H, Ried T, Rappold GA. SHOT, a SHOX‐related homeobox gene, is implicated in craniofacial, brain, heart, and limb development. Proc Natl Acad Sci USA. 1998; 95:2406-2411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Espinoza‐Lewis RA, Liu H, Sun C, Chen C, Jiao K, Chen Y. Ectopic expression of Nkx2.5 suppresses the formation of the sinoatrial node in mice. Dev Biol. 2011; 356:359-369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ubersax JA, Ferrell JE., Jr Mechanisms of specificity in protein phosphorylation. Nat Rev Mol Cell Biol. 2007; 8:530-541. [DOI] [PubMed] [Google Scholar]
- 12.Marchini A, Daeffler L, Marttila T, Schneider KU, Blaschke RJ, Schnolzer M, Rommelaere J, Rappold G. Phosphorylation on Ser106 modulates the cellular functions of the SHOX homeodomain protein. J Mol Biol. 2006; 355:590-603. [DOI] [PubMed] [Google Scholar]
- 13.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real‐time quantitative pcr and the 2−ΔΔCT method. Methods. 2001; 25:402-408. [DOI] [PubMed] [Google Scholar]
- 14.Stieber J, Herrmann S, Feil S, Loster J, Feil R, Biel M, Hofmann F, Ludwig A. The hyperpolarization‐activated channel Hcn4 is required for the generation of pacemaker action potentials in the embryonic heart. Proc Natl Acad Sci USA. 2003; 100:15235-15240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vokes SA, Ji H, Wong WH, McMahon AP. A genome‐scale analysis of the cis‐regulatory circuitry underlying sonic hedgehog‐mediated patterning of the mammalian limb. Genes Dev. 2008; 22:2651-2663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Odom DT, Zizlsperger N, Gordon DB, Bell GW, Rinaldi NJ, Murray HL, Volkert TL, Schreiber J, Rolfe PA, Gifford DK, Fraenkel E, Bell GI, Young RA. Control of pancreas and liver gene expression by HNF transcription factors. Science. 2004; 303:1378-1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rovescalli AC, Asoh S, Nirenberg M. Cloning and characterization of four murine homeobox genes. Proc Natl Acad Sci USA. 1996; 93:10691-10696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yu L, Liu H, Yan M, Yang J, Long F, Muneoka K, Chen Y. Shox2 is required for chondrocyte proliferation and maturation in proximal limb skeleton. Dev Biol. 2007; 306:549-559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang Q, Sigmund CD, Lin JJ. Identification of cis elements in the cardiac troponin T gene conferring specific expression in cardiac muscle of transgenic mice. Circ Res. 2000; 86:478-484. [DOI] [PubMed] [Google Scholar]
- 20.Cobb J, Dierich A, Huss‐Garcia Y, Duboule D. A mouse model for human short‐stature syndromes identifies Shox2 as an upstream regulator of Runx2 during long‐bone development. Proc Natl Acad Sci USA. 2006; 103:4511-4515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Akazawa H, Komuro I. Cardiac transcription factor Csx/Nk2–5: its role in cardiac development and diseases. Pharmacol Ther. 2005; 107:252-268. [DOI] [PubMed] [Google Scholar]
- 22.Reecy JM, Li X, Yamada M, DeMayo FJ, Newman CS, Harvey RP, Schwartz RJ. Identification of upstream regulatory regions in the heart‐expressed homeobox gene Nk2–5. Development. 1999; 126:839-849. [DOI] [PubMed] [Google Scholar]
- 23.Christoffels VM, Mommersteeg MT, Trowe MO, Prall OW, de Gier‐de Vries C, Soufan AT, Bussen M, Schuster‐Gossler K, Harvey RP, Moorman AF, Kispert A. Formation of the venous pole of the heart from an Nkx2‐5‐negative precursor population requires tbx18. Circ Res. 2006; 98:1555-1563. [DOI] [PubMed] [Google Scholar]
- 24.Schwartz RJ, Olson EN. Building the heart piece by piece: modularity of cis‐elements regulating Nk2–5 transcription. Development. 1999; 126:4187-4192. [DOI] [PubMed] [Google Scholar]
- 25.Brown CO, III, Chi X, Garcia‐Gras E, Shirai M, Feng XH, Schwartz RJ. The cardiac determination factor, Nk2–5, is activated by mutual cofactors Gata‐4 and Smad1/4 via a novel upstream enhancer. J Biol Chem. 2004; 279:10659-10669. [DOI] [PubMed] [Google Scholar]
- 26.Chen Y, Cao X. NFAT directly regulates Nk2–5 transcription during cardiac cell differentiation. Biol Cell. 2009; 101:335-349. [DOI] [PubMed] [Google Scholar]
- 27.Boncinelli E. Homeobox genes and disease. Curr Opin Genet Dev. 1997; 7:331-337. [DOI] [PubMed] [Google Scholar]
- 28.Gehring WJ, Affolter M, Burglin T. Homeodomain proteins. Annu Rev Biochem. 1994; 63:487-526. [DOI] [PubMed] [Google Scholar]
- 29.Tweedie‐Cullen RY, Reck JM, Mansuy IM. Comprehensive mapping of post‐translational modifications on synaptic, nuclear, and histone proteins in the adult mouse brain. J Proteome Res. 2009; 8:4966-4982. [DOI] [PubMed] [Google Scholar]
- 30.Hayashi S, Scott MP. What determines the specificity of action of drosophila homeodomain proteins? Cell. 1990; 63:883-894. [DOI] [PubMed] [Google Scholar]
- 31.Biggin MD, McGinnis W. Regulation of segmentation and segmental identity by Drosophila homeoproteins: the role of DNA binding in functional activity and specificity. Development. 1997; 124:4425-4433. [DOI] [PubMed] [Google Scholar]
- 32.Ekker SC, Jackson DG, von Kessler DP, Sun BI, Young KE, Beachy PA. The degree of variation in DNA sequence recognition among four Drosophila homeotic proteins. EMBO J. 1994; 13:3551-3560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kasahara H, Izumo S. Identification of the in vivo casein kinase II phosphorylation site within the homeodomain of the cardiac tisue‐specifying homeobox gene product Csx/Nkx2.5. Mol Cell Biol. 1999; 19:526-536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schmidt B, Liebenberg V, Dietrich D, Schlegel T, Kneip C, Seegebarth A, Flemming N, Seemann S, Distler J, Lewin J, Tetzner R, Weickmann S, Wille U, Liloglou T, Raji O, Walshaw M, Fleischhacker M, Witt C, Field JK. Shox2 DNA methylation is a biomarker for the diagnosis of lung cancer based on bronchial aspirates. BMC Cancer. 2010; 10:600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kneip C, Schmidt B, Seegebarth A, Weickmann S, Fleischhacker M, Liebenberg V, Field JK, Dietrich D. Shox2 DNA methylation is a biomarker for the diagnosis of lung cancer in plasma. J Thorac Oncol. 2011; 6:1632-1638. [DOI] [PubMed] [Google Scholar]
- 36.Lee KY, Yamamoto Y, Boucher J, Winnay JN, Gesta S, Cobb J, Bluher M, Kahn CR. Shox2 is a molecular determinant of depot‐specific adipocyte function. Proc Natl Acad Sci USA. 2013; 110:11409-11414. [DOI] [PMC free article] [PubMed] [Google Scholar]