

Abstract
Background
Dendritic cells (DC) play pivotal roles in regulating the immune system and inflammatory response. We previously reported DC infiltration in the infarcted heart and its immunoprotective roles in the post‐infarction healing process after experimental myocardial infarction (MI). However, its clinical significance has not been determined.
Methods and Results
The degree of DC infiltration and its correlation with the post‐infarction healing process in the human infarcted heart were investigated in 24 autopsy subjects after ST‐elevation MI. Patients were divided into two groups according to the presence (n=13) or absence (n=11) of cardiac rupture. The numbers of infiltrated DC and macrophages and the extent of fibrosis in the infarcted area were examined. In the rupture group, CD68+ macrophage infiltration was increased and CD209+ DC, and CD11c+ DC infiltration and the extent of reparative fibrosis were decreased compared with the non‐rupture group, under matched baseline characteristics including the time from onset to death and use of revascularization. Furthermore, there was a significant positive correlation between the number of infiltrating CD209+ DC, and CD11c+ DC and the extent of reparative fibrosis.
Conclusions
Decreased number of DC in human‐infarcted myocardial tissue was associated with increased macrophage infiltration, impaired reparative fibrosis, and the development of cardiac rupture after MI. These findings suggest a protective role of DC in post‐MI inflammation and the subsequent healing process.
Keywords: cardiac rupture, dendritic cell, inflammation, myocardial infarction, reparative fibrosis
Introduction
Early reperfusion of the infarcted tissue and reparative fibrosis are essential in preserving the structural integrity of the left ventricle (LV) after myocardial infarction (MI). Cardiac accommodation following MI occurs in different phases, which include inflammatory, early healing, and myocardial remodeling phases.1–2 An inadequate healing process after MI would result in complications such as cardiac rupture, LV aneurysm, and congestive heart failure due to exaggerated LV remodeling. However, the precise mechanisms and the potential therapeutic targets in the post‐MI healing process remain to be clarified.
We previously reported that higher concentrations of serum C‐reactive protein (CRP)3 and plasma interleukin (IL)‐64 and peripheral monocytosis5 predict a worse clinical outcome after MI, suggesting that an immune‐mediated inflammatory response may adversely affect post‐infarction healing and LV remodeling. Recently, we elucidated that dendritic cells (DC) infiltrated into myocardial tissue play immunoprotective roles in post‐infarction healing and LV remodeling in animal models.6–7
DC are professional antigen‐presenting cells (APC), which are found in all organ systems, including the myocardium. Several subtypes have been described thus far, with so‐called myeloid DC (mDC) and plasmacytoid DC (pDC) being predominant.8 We have demonstrated infiltration of mature activated mDC in the infarcted rat heart, peaking on day 7.6 To elucidate the significance of DC, we generated a mouse model with selective depletion of bone marrow‐derived DC, and demonstrated that the depletion of bone marrow‐derived DC exacerbated post‐infarction LV remodeling in association with enhanced inflammatory cytokine expression and matrix metalloproteinase (MMP)‐9 activation via marked infiltration of proinflammatory monocytes (Ly6Chigh) and classically activated M1 macrophages into the infarcted myocardium. On the contrary, decreased IL‐10, which has anti‐inflammatory activity and myocardial infiltration of anti‐inflammatory monocytes (Ly6Clow) and alternatively activated M2 macrophages were observed in a rodent model.7 These findings suggest that DC may play a protective role against post‐infarction LV remodeling by regulating the homeostasis of monocytes and macrophages during the transition from inflammation to repair. However, the presence and the clinical significance of DC in the human infarcted heart remain to be determined.
In the present study, we used immunostaining techniques to identify and quantify DC infiltration in the infarcted myocardium in human autopsy samples, to clarify the impact of DC infiltration on the post‐MI healing process and the development of cardiac rupture.
Methods
Patients
Among patients who were admitted to our institution with ST‐elevation MI (STEMI) and underwent autopsy between December 1978 and May 1998 (n=49), 24 cases with enough preserved infarcted tissue for immunohistochemical analyses were examined.
All autopsies were performed within 24 hours after death. Heart tissue samples were taken from the LV infarcted area of patients who died of cardiac rupture including free wall rupture (n=9) and ventricular septal perforation (n=4), pump failure (n=10), or fatal ventricular arrhythmias (n=1). Patients were divided into two groups according to the presence (n=13) or absence (n=11) of cardiac rupture.
Our study was approved by the ethics committee of the National Cerebral and Cardiovascular Center, and conformed to the principles of the Declaration of Helsinki.
Histological and Immunohistochemical Staining
The ventricular tissue was fixed in formalin and embedded in paraffin using standard histological procedures. The tissue was cut to yield 5‐μm‐thick cross sections. The sections were subsequently stained with hematoxylin and eosin (HE) and Masson's trichrome staining to determine the extent of fibrosis.
Immunohistochemical examinations were performed on 5‐μm‐thick formalin‐fixed and paraffin‐embedded tissue sections. All steps were performed on a Leica Bond III automated system (Leica Microsystems) according to the manufacturer's instructions. In brief, specimens were deparaffinized and antigen was retrieved on the instrument. All slides were incubated with primary antibodies against CD68 (diluted 1:1000; Dako), CD209 (1:1000; BD Pharmingen), or CD11c (1:100; GeneTex) for 16 min, followed by incubation with a mouse‐rabbit‐horseradish peroxidase polymer and 3,3′‐diaminobenzidine substrate. The sections were then incubated in primer (anti‐rabbit and anti‐mouse) for 8 minutes. Antibody binding was visualized using the avidin‐biotin complex method according to the manufacturer's instructions (Vectastain ABC; Vector). The primary antibody was omitted from these protocols as a negative control. The sections were subsequently counterstained with HE.
Quantitative Analyses of Myocardial Inflammatory Cell Infiltration and Tissue Fibrosis
Stained inflammatory cells were counted in the infarcted area of each sample at a magnification of ×100 and in each of ten representative sections (0.1 mm2), which were randomly chosen from infarct tissue without hemorrhagic change, using ImageJ software (version 1.38x; National Institutes of Health). For each sample, median cell numbers were calculated. For LV tissue fibrosis, percent area fraction (%AF) was measured using ImageJ software. These quantitative analyses were performed by two trained technicians without knowledge of patients' backgrounds.
Statistical Analysis
Continuous data were expressed as mean values±SD. The two groups were compared using the Wilcoxon rank sum test for continuous variables. Categorical variables were reported as frequencies with percentages and compared between the two groups using the Fisher's exact test. The correlation among the infiltration of CD68+ macrophages, CD209+ DC, CD11c+ DC, and the extent of fibrosis were investigated by Pearson or Spearman correlation test. All statistical analyses were performed using the SPSS 13.0 for Windows (SPSS Inc). A P value of <0.05 was considered to be significant.
Results
Study Population
All patients were diagnosed with STEMI, and 50% of patients had anterior infarction. Emergent reperfusion therapy was performed in 8% of patients. Mean age was 68±9 years, and 63% of the patients were men. Baseline characteristics of the study patients including history of prior MI, time from onset to death, rate of reperfusion therapies, and traditional coronary risk factors, but not white blood cell count on admission, were similar in the rupture and non‐rupture groups (Table). The time course from onset to death was not significantly different between the two groups (Figure 1).
Table 1.
Baseline Characteristics of Patients
| Overall (n=24) | Non‐Rupture (n=11) | Rupture (n=13) | P Value | |
|---|---|---|---|---|
| Age, y | 68±9 | 68±9 | 69±10 | 0.66 |
| Male, n (%) | 15 (63) | 5 (45) | 10 (77) | 0.21 |
| Smoking, n (%) | 13 (56) | 5 (45) | 8 (62) | 0.69 |
| Hypertension, n (%) | 20 (83) | 10 (90) | 10 (77) | 0.60 |
| Diabetes, n (%) | 8 (33) | 6 (55) | 2 (15) | 0.08 |
| Dyslipidemia, n (%) | 5 (21) | 2 (18) | 3 (23) | 1.00 |
| Prior MI, n (%) | 5 (21) | 4 (36) | 1 (8) | 0.14 |
| Killip 3 or 4, n (%) | 12 (50) | 7 (64) | 5 (38) | 0.41 |
| STEMI, n (%) | 24 (100) | 11 (100) | 13 (100) | |
| Onset to death, h | 167±148 | 188±164 | 148±135 | 0.44 |
| Anterior infarction, % | 12 (50) | 6 (55) | 6 (46) | 1.00 |
| Revascularization, % | 2 (8) | 1 (9) | 1 (8) | 1.00 |
| White blood cells, ×103/μL | 13.2±5.7 | 16.5±6.1 | 10.5±3.6 | 0.01 |
| Hemoglobin, g/dL | 12.1±2.1 | 12.4±2.8 | 11.9±1.3 | 0.60 |
| Peak CPK, ×103 IU/L | 4.3±4.3 | 5.7±5.3 | 2.7±1.5 | 0.18 |
| Serum creatinine, mg/dL | 1.9±2.0 | 2.6±2.7 | 1.3±0.6 | 0.06 |
| Systolic BP, mm Hg | 97±34 | 84±38 | 108±27 | 0.07 |
| Diastolic BP, mm Hg | 61±23 | 52±24 | 67±22 | 0.19 |
| Heart rate, bpm | 78±34 | 74±45 | 82±23 | 0.56 |
Continuous variables are presented as mean±SD. Categorical variables are presented as number (percentage). BP indicates blood pressure; bpm, beats per minute; CPK, creatine phosphokinase; MI, myocardial infarction; STEMI, ST elevation MI.
Figure 1.
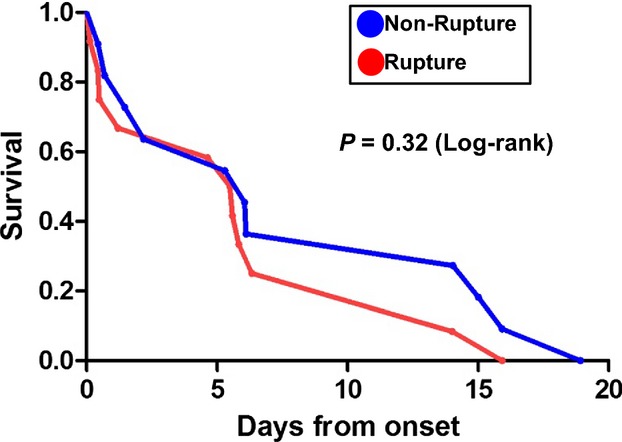
Kaplan‐Meier survival analysis in non‐rupture and rupture patients after myocardial infarction (MI).
Extent of Fibrosis and Infiltration of CD68+ Macrophages and CD209+ DC in Infarcted Myocardium
Staining with Masson's trichrome showed decreased %AF of fibrosis in patients with cardiac rupture compared to those without (Figure 2).
Figure 2.
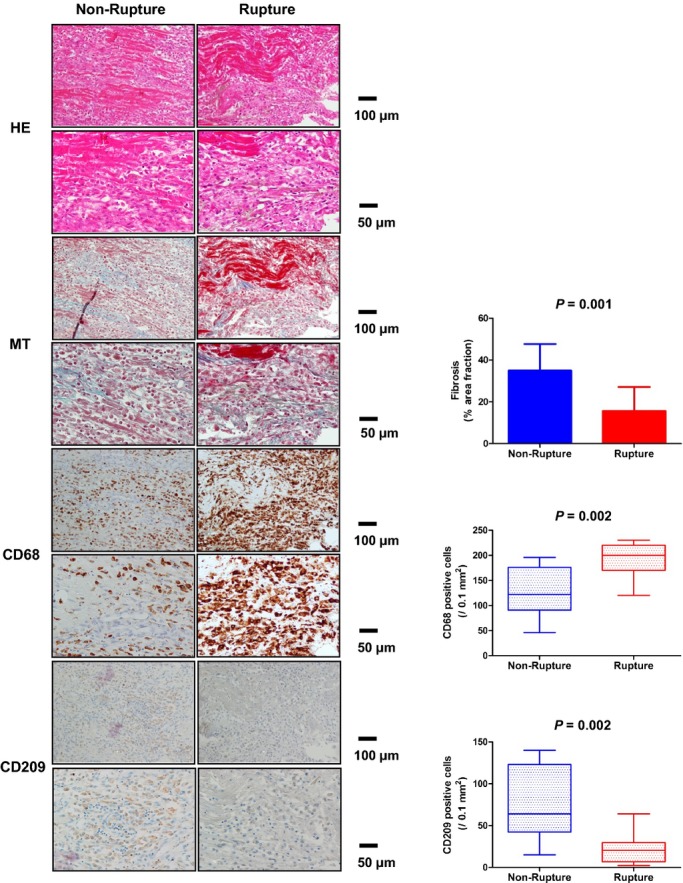
Infiltration of macrophages and DC, and extent of fibrosis in left ventricular infarcted tissue. HE staining, Masson's‐Trichrome (MT) staining, immunohistochemical staining for CD68+ macrophages and CD209+ DC in left ventricular infarcted tissue of non‐rupture and rupture patients after MI. DC indicates dendritic cells; HE, hematoxylin and eosin; MI, myocardial infarction.
Immunohistochemical staining of the infarcted myocardium showed an increase in the number of infiltrating CD68+ macrophages and a decrease in CD209+ DC in patients with cardiac rupture compared with those without (Figure 2).
Correlation Among CD68+ macrophages, CD209+ DC and Extent of Reparative Fibrosis
To reveal the possible relationship between inflammatory cell infiltration and the extent of reparative fibrosis, detailed correlation analysis was performed. No significant correlation was found between the number of CD68+ macrophages and%AF of myocardial fibrosis in the infarcted area (R2=0.01, P=0.39; Figure 3A). However, we found a significant positive correlation between the number of CD209+ DC and %AF of myocardial fibrosis (R2=0.69, P<0.0001; Figure 3B).
Figure 3.
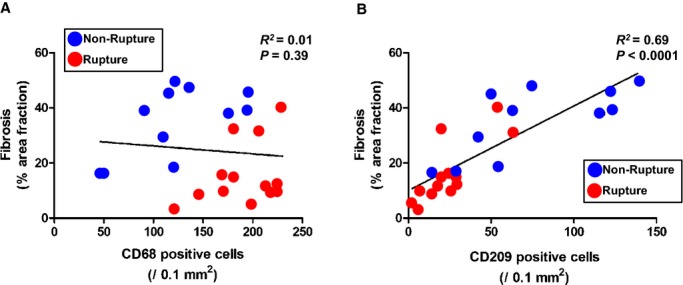
Correlation between extent of fibrosis and infiltration of macrophages and dendritic cells (DC) in left ventricular infarcted tissue. A, Correlation between extent of fibrosis and number of CD68+ macrophages. B, Number of CD209+ DC.
Extent of Fibrosis and Infiltration of CD11c+ DC in Infarcted Myocardium, and Correlation Between CD11c+ DC and Extent of Reparative Fibrosis
To confirm the relationship between DC infiltration and the extent of reparative fibrosis, the same analyses were performed using another DC marker, CD11c.
A decrease in the number of infiltrating CD11c+ DC was observed in patients with cardiac rupture compared to those without (Figure 4A). In addition, we also found a significant positive correlation between the number of CD11c+ DC and %AF of myocardial fibrosis (R2=0.37, P=0.001; Figure 4B).
Figure 4.
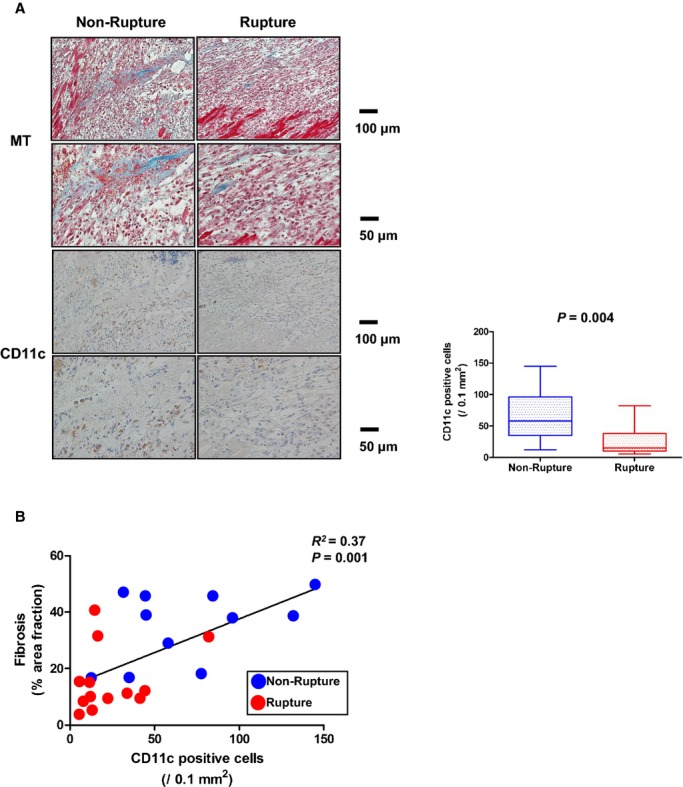
Infiltration of dendritic cells (DC) evaluated by another marker, and extent of fibrosis in left ventricular infarcted tissue. A, Masson's‐Trichrome (MT) staining, immunohistochemical staining for CD11c+ DC in left ventricular infarcted tissue. B, Correlation between extent of fibrosis and number of CD11c+ DC.
Discussion
In the present study, we demonstrated that DC infiltrated human infarcted myocardium after STEMI, and that the degree of infiltration and the extent of reparative fibrosis were significantly lower and the degree of macrophage infiltration was higher in patients with cardiac rupture compared with those without. Interestingly, there was a strong positive correlation between the number of infiltrating DC and the extent of fibrosis in the infarcted myocardium. These findings suggest that DC may play a protective role against cardiac rupture through promotion of reparative fibrosis after MI.
Cardiac rupture is an acute life‐threatening complication that occurs in the first several days after MI. Acute myocyte loss and breakdown of extracellular matrix (ECM) promote early ventricular expansion, which is a trigger for subacute cardiac rupture and worsening cardiac function after MI.9–10 Replacement by collagen is important to provide mechanical stability to the injured tissue, and protects against increased LV dilatation.11–12 In fact, agents that inhibit collagen synthesis were shown to be associated with an increase in the risk of cardiac rupture in MI patients.13 These results suggested that appropriate reparative fibrosis is important to prevent cardiac rupture after MI. As well as collagen synthesis, inflammation is also a crucial factor in the post‐MI healing process. An excessive inflammatory response in the infarcted myocardium is related to adverse cardiac events including cardiac rupture;3–4 however, anti‐inflammatory corticosteroid therapy has been reported to increase the incidence of cardiac rupture by delaying collagen accumulation and scar formation, a far from favorable impact on the post‐MI healing process.14–15 These findings indicate that inflammation could be a prerequisite for an adequate post‐MI healing process, although excessive inflammation is harmful. Our current study, based on Masson's trichrome staining, showed that the infarcted myocardium in patients with cardiac rupture consisted of disorganized collagen fibers, suggesting the presence of impaired reparative fibrosis. Briefly, impaired reparative fibrosis can be explained by two mechanisms. The first is the degradation of ECM by augmented MMPs secreted from inflammatory cells, predominantly inflammatory monocytes and M1 macrophages, infiltrating the infarcted heart.7,16–17 Although this process is important for elimination of the necrotic tissue, excessive activation of MMPs could facilitate infarct expansion, resulting in cardiac rupture.18–21 The second is disordered collagenogenesis by myofibroblasts differentiated from cardiac resident fibroblasts, which are regulated by pro‐fibrotic cytokines, such as transforming growth factor beta (TGF‐β) secreted mainly from anti‐inflammatory monocytes and M2 macrophages during the repair process. M2 macrophages have been reported to promote differentiation of cardiac fibroblasts into myofibroblasts through production of TGF‐β.22 Therefore, adequate regulation of cellular employment is critical for the post‐infarction repair process and prevention of cardiac rupture.
DC have come to be appreciated as potent critical controllers that modulate various kinds of inflammatory cells in innate and adoptive immunity.23–25 Generally, bone‐marrow and splenic progenitors and circulating monocytes are reported to differentiate into DC and exert various influences on the immune system at the inflammatory site, such as priming of antigen‐specific immune responses, induction of tolerance, and chronic inflammation after tissue injury.26–28 In the blood, both subtypes of DC are found as circulating DC precursors that lack the expression of costimulatory molecules, so that they are unable to activate other inflammatory cells. In the infarcted myocardium, it was reported that mDC became activated in response to danger signals such as heat shock protein, which is released from necrotic tissue after MI, through the activation of toll‐like receptors (TLRs) signaling.29
Kretzchmar et al demonstrated a significant decrease in circulating mDC in patients with MI, and also showed significantly higher numbers of markers indicative of mDC and inflammatory cells such as macrophages in the infarcted compared with non‐infarcted myocardium. This was accompanied by increased serum levels of an anti‐inflammatory cytokine (IL‐10) as well as inflammatory cytokines (IL‐6, IL‐12, and TNFα) in patients with MI.30 Yilmaz et al also demonstrated that serum high sensitive CRP (hsCRP) and IL‐6 levels were decreased, and mDC were partially reconstituted 1 week after the onset in patients with MI. They also showed the number of mDC precursors was negatively correlated with serum hsCRP or IL‐6 level, while, in contrast, no significant correlation between pDC precursors and hsCRP or IL‐6 was detected.31 These results suggest that mDC could play important roles in regulating excessive inflammation in the post‐MI healing process. Recently, potential mechanisms of suppression of excessive inflammation in a hepatic ischemia‐reperfusion mouse model were reported. DC, which were activated by necrotic hepatocytes through TLR9 signaling, might restrict pro‐inflammatory monocyte function via production of IL‐10.32 We previously observed mDC infiltrated into the infarcted and border areas, peaking 1 week after MI in a rat model,6 and also found that depletion of DC resulted in enhanced inflammation and ECM degradation, through activation of pro‐inflammatory monocytes and M1 macrophages, and impaired post‐infarction healing process, through suppression of infiltration of anti‐inflammatory monocytes and M2 macrophages and expression of anti‐inflammatory cytokines such as IL‐10, in a mouse model.7 Notably, in the present study, the number of infiltrated CD68‐positive macrophages was higher, the number of CD209 and CD11c‐positive mDC was lower, and the extent of reparative fibrosis in the infarcted myocardium was less in patients with cardiac rupture compared with those without. Thus, also in human infarcted myocardium, DC may play a protective role against excessive inflammation and cardiac rupture by promoting the post‐MI healing process.
Several limitations of this study warrant mention. First, the number of study patients was relatively small. The statistical power might thus not be adequate for any negative results. Second, there were insufficient clinical data regarding inflammatory biomarkers such as monocytes, CRP, and inflammatory cytokines. Third, although we used CD209 (DC‐SIGN) in addition to CD11c for identifying DC infiltrated into infarcted myocardium, several potential markers other than DC‐SIGN have been reported for identifying DC. DC‐SIGN is a type II transmembrane protein that belongs to a family of calcium‐dependent lectins diversely used by human APC, such as tissue‐residing mDC, alveolar and lymph node macrophages, and endothelial cells from liver sinusoids,33–36 and was identified as a novel DC‐specific adhesion receptor on human DC that is essential in several key functions throughout the life cycle of DC.33 Therefore, it was assumed that CD209 was appropriate for identifying mDC infiltrated in human infarcted myocardium. Finally, there is potential for reverse causation and/or confounding factors such as white blood cell count that could also affect inflammatory response including DC recruitment. In addition, patients with cardiac rupture may have had histologic changes due to greater wall tension, such as less collateralization. Since we could not conclude that there was a causal relationship among DC, reparative fibrosis, and cardiac rupture, further study is warranted.
In conclusion, we identified DC infiltration in human infarcted myocardium, and observed a strong association between the number of DC and impaired reparative fibrosis and the development of cardiac rupture, suggesting a protective role of DC during the post‐MI healing process.
Sources of Funding
This work was supported by a Grant from the Japan Cardiovascular Research Foundation (Anzai).
Disclosures
None.
Acknowledgments
We thank Nobuyoshi Imai, Hiroshi Sagane, and Hiroyuki Hatsuyama (National Cerebral and Cardiovascular Center) for excellent technical assistance.
References
- 1.Ertl G, Frantz S. Healing after myocardial infarction. Cardiovasc Res. 2005; 66:22-32. [DOI] [PubMed] [Google Scholar]
- 2.Frantz S, Bauersachs J, Ertl G. Post‐infarct remodelling: contribution of wound healing and inflammation. Cardiovasc Res. 2009; 81:474-481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Anzai T, Yoshikawa T, Shiraki H, Asakura Y, Akaishi M, Mitamura H, Ogawa S. C‐reactive protein as a predictor of infarct expansion and cardiac rupture after a first Q‐wave acute myocardial infarction. Circulation. 1997; 96:778-784. [DOI] [PubMed] [Google Scholar]
- 4.Takahashi T, Anzai T, Yoshikawa T, Maekawa Y, Asakura Y, Satoh T, Mitamura H, Ogawa S. Serum C‐reactive protein elevation in left ventricular remodeling after acute myocardial infarction—role of neurohormones and cytokines. Int J Cardiol. 2003; 88:257-265. [DOI] [PubMed] [Google Scholar]
- 5.Maekawa Y, Anzai T, Yoshikawa T, Asakura Y, Takahashi T, Ishikawa S, Mitamura H, Ogawa S. Prognostic significance of peripheral monocytosis after reperfused acute myocardial infarction: a possible role for left ventricular remodeling. J Am Coll Cardiol. 2002; 39:241-246. [DOI] [PubMed] [Google Scholar]
- 6.Naito K, Anzai T, Sugano Y, Maekawa Y, Kohno T, Yoshikawa T, Matsuno K, Ogawa S. Differential effects of GM‐CSF and G‐CSF on infiltration of dendritic cells during early left ventricular remodeling after myocardial infarction. J Immunol. 2008; 181:5691-5701. [DOI] [PubMed] [Google Scholar]
- 7.Anzai A, Anzai T, Nagai S, Maekawa Y, Naito K, Kaneko H, Sugano Y, Takahashi T, Abe H, Mochizuki S, Sano M, Yoshikawa T, Okada Y, Koyasu S, Ogawa S, Fukuda K. Regulatory role of dendritic cells in postinfarction healing and left ventricular remodeling. Circulation. 2012; 125:1234-1245. [DOI] [PubMed] [Google Scholar]
- 8.Steinman RM. Lasker Basic Medical Research Award. Dendritic cells: versatile controllers of the immune system. Nat Med. 2007; 13:1155-1159. [DOI] [PubMed] [Google Scholar]
- 9.Pfeffer MA, Braunwald E. Ventricular remodeling after myocardial infarction. Experimental observations and clinical implications. Circulation. 1990; 81:1161-1172. [DOI] [PubMed] [Google Scholar]
- 10.Sutton MG, Sharpe N. Left ventricular remodeling after myocardial infarction: pathophysiology and therapy. Circulation. 2000; 101:2981-2988. [DOI] [PubMed] [Google Scholar]
- 11.Jugdutt BI, Amy RW. Healing after myocardial infarction in the dog: changes in infarct hydroxyproline and topography. J Am Coll Cardiol. 1986; 7:91-102. [DOI] [PubMed] [Google Scholar]
- 12.Whittaker P, Boughner DR, Kloner RA. Role of collagen in acute myocardial infarct expansion. Circulation. 1991; 84:2123-2134. [DOI] [PubMed] [Google Scholar]
- 13.Peuhkurinen K, Risteli L, Jounela A, Risteli J. Changes in interstitial collagen metabolism during acute myocardial infarction treated with streptokinase or tissue plasminogen activator. Am Heart J. 1996; 131:7-13. [DOI] [PubMed] [Google Scholar]
- 14.Hammerman H, Kloner RA, Hale S, Schoen FJ, Braunwald E. Dose‐dependent effects of short‐term methylprednisolone on myocardial infarct extent, scar formation, and ventricular function. Circulation. 1983; 68:446-452. [DOI] [PubMed] [Google Scholar]
- 15.Silverman HS, Pfeifer MP. Relation between use of anti‐inflammatory agents and left ventricular free wall rupture during acute myocardial infarction. Am J Cardiol. 1987; 59:363-364. [DOI] [PubMed] [Google Scholar]
- 16.Frangogiannis NG, Smith CW, Entman ML. The inflammatory response in myocardial infarction. Cardiovasc Res. 2002; 53:31-47. [DOI] [PubMed] [Google Scholar]
- 17.Cleutjens JP, Blankesteijn WM, Daemen MJ, Smits JF. The infarcted myocardium: simply dead tissue, or a lively target for therapeutic interventions. Cardiovasc Res. 1999; 44:232-241. [DOI] [PubMed] [Google Scholar]
- 18.Heymans S, Luttun A, Nuyens D, Theilmeier G, Creemers E, Moons L, Dyspersin GD, Cleutjens JP, Shipley M, Angellilo A, Levi M, Nube O, Baker A, Keshet E, Lupu F, Herbert JM, Smits JF, Shapiro SD, Baes M, Borgers M, Collen D, Daemen MJ, Carmeliet P. Inhibition of plasminogen activators or matrix metalloproteinases prevents cardiac rupture but impairs therapeutic angiogenesis and causes cardiac failure. Nat Med. 1999; 5:1135-1142. [DOI] [PubMed] [Google Scholar]
- 19.Rohde LE, Ducharme A, Arroyo LH, Aikawa M, Sukhova GH, Lopez‐Anaya A, McClure KF, Mitchell PG, Libby P, Lee RT. Matrix metalloproteinase inhibition attenuates early left ventricular enlargement after experimental myocardial infarction in mice. Circulation. 1999; 99:3063-3070. [DOI] [PubMed] [Google Scholar]
- 20.Ducharme A, Frantz S, Aikawa M, Rabkin E, Lindsey M, Rohde LE, Schoen FJ, Kelly RA, Werb Z, Libby P, Lee RT. Targeted deletion of matrix metalloproteinase‐9 attenuates left ventricular enlargement and collagen accumulation after experimental myocardial infarction. J Clin Invest. 2000; 106:55-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Creemers EE, Davis JN, Parkhurst AM, Leenders P, Dowdy KB, Hapke E, Hauet AM, Escobar PG, Cleutjens JP, Smits JF, Daemen MJ, Zile MR, Spinale FG. Deficiency of TIMP‐1 exacerbates LV remodeling after myocardial infarction in mice. Am J Physiol Heart Circ Physiol. 2003; 284:H364-H371. [DOI] [PubMed] [Google Scholar]
- 22.Li Y, Zhang C, Wu Y, Han Y, Cui W, Jia L, Cai L, Cheng J, Li H, Du J. Interleukin‐12p35 deletion promotes CD4 T‐cell‐dependent macrophage differentiation and enhances angiotensin II‐Induced cardiac fibrosis. Arterioscler Thromb Vasc Biol. 2012; 32:1662-1674. [DOI] [PubMed] [Google Scholar]
- 23.Banchereau J, Steinman RM. Dendritic cells and the control of immunity. Nature. 1998; 392:245-252. [DOI] [PubMed] [Google Scholar]
- 24.Pulendran B, Tang H, Manicassamy S. Programming dendritic cells to induce T(H)2 and tolerogenic responses. Nat Immunol. 2010; 11:647-655. [DOI] [PubMed] [Google Scholar]
- 25.Banchereau J, Briere F, Caux C, Davoust J, Lebecque S, Liu YJ, Pulendran B, Palucka K. Immunobiology of dendritic cells. Annu Rev Immunol. 2000; 18:767-811. [DOI] [PubMed] [Google Scholar]
- 26.Sallusto F, Lanzavecchia A. Mobilizing dendritic cells for tolerance, priming, and chronic inflammation. J Exp Med. 1999; 189:611-614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Swirski FK, Nahrendorf M, Etzrodt M, Wildgruber M, Cortez‐Retamozo V, Panizzi P, Figueiredo JL, Kohler RH, Chudnovskiy A, Waterman P, Aikawa E, Mempel TR, Libby P, Weissleder R, Pittet MJ. Identification of splenic reservoir monocytes and their deployment to inflammatory sites. Science. 2009; 325:612-616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Peng Y, Latchman Y, Elkon KB. Ly6C(low) monocytes differentiate into dendritic cells and cross‐tolerize T cells through PDL‐1. J Immunol. 2009; 182:2777-2785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Maekawa Y, Mizue N, Chan A, Shi Y, Liu Y, Dawood S, Chen M, Dawood F, de Couto G, Li GH, Suzuki N, Yeh WC, Gramolini A, Medin JA, Liu PP. Survival and cardiac remodeling after myocardial infarction are critically dependent on the host innate immune interleukin‐1 receptor‐associated kinase‐4 signaling: a regulator of bone marrow‐derived dendritic cells. Circulation. 2009; 120:1401-1414. [DOI] [PubMed] [Google Scholar]
- 30.Kretzschmar D, Betge S, Windisch A, Pistulli R, Rohm I, Fritzenwanger M, Jung C, Schubert K, Theis B, Petersen I, Drobnik S, Mall G, Figulla HR, Yilmaz A. Recruitment of circulating dendritic cell precursors into the infarcted myocardium and pro‐inflammatory response in acute myocardial infarction. Clin Sci (Lond). 2012; 123:387-398. [DOI] [PubMed] [Google Scholar]
- 31.Yilmaz A, Weber J, Cicha I, Stumpf C, Klein M, Raithel D, Daniel WG, Garlichs CD. Decrease in circulating myeloid dendritic cell precursors in coronary artery disease. J Am Coll Cardiol. 2006; 48:70-80. [DOI] [PubMed] [Google Scholar]
- 32.Bamboat ZM, Ocuin LM, Balachandran VP, Obaid H, Plitas G, DeMatteo RP. Conventional DCs reduce liver ischemia/reperfusion injury in mice via IL‐10 secretion. J Clin Invest. 2010; 120:559-569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Geijtenbeek TB, Kwon DS, Torensma R, van Vliet SJ, van Duijnhoven GC, Middel J, Cornelissen IL, Nottet HS, KewalRamani VN, Littman DR, Figdor CG, van Kooyk Y. DC‐SIGN, a dendritic cell‐specific HIV‐1‐binding protein that enhances trans‐infection of T cells. Cell. 2000; 100:587-597. [DOI] [PubMed] [Google Scholar]
- 34.Lai WK, Sun PJ, Zhang J, Jennings A, Lalor PF, Hubscher S, McKeating JA, Adams DH. Expression of DC‐SIGN and DC‐SIGNR on human sinusoidal endothelium: a role for capturing hepatitis C virus particles. Am J Pathol. 2006; 169:200-208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tailleux L, Pham‐Thi N, Bergeron‐Lafaurie A, Herrmann JL, Charles P, Schwartz O, Scheinmann P, Lagrange PH, de Blic J, Tazi A, Gicquel B, Neyrolles O. DC‐SIGN induction in alveolar macrophages defines privileged target host cells for mycobacteria in patients with tuberculosis. PLoS Med. 2005; 2:e381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.van Lent PL, Figdor CG, Barrera P, van Ginkel K, Sloetjes A, van den Berg WB, Torensma R. Expression of the dendritic cell‐associated C‐type lectin DC‐SIGN by inflammatory matrix metalloproteinase‐producing macrophages in rheumatoid arthritis synovium and interaction with intercellular adhesion molecule 3‐positive T cells. Arthritis Rheum. 2003; 48:360-369. [DOI] [PubMed] [Google Scholar]