

Abstract
Background
RV and LV have different embryologic, structural, metabolic, and electrophysiologic characteristics, but whether interventricular differences exist in β‐adrenergic (β‐AR) responsiveness is unknown. In this study, we examine whether β‐AR response and signaling differ in right (RV) versus left (LV) ventricles.
Methods and Results
Sarcomere shortening, Ca2+ transients, ICa,L and IKs currents were recorded in isolated dog LV and RV midmyocytes. Intracellular [cAMP] and PKA activity were measured by live cell imaging using FRET‐based sensors. Isoproterenol increased sarcomere shortening ≈10‐fold and Ca2+‐transient amplitude ≈2‐fold in LV midmyocytes (LVMs) versus ≈25‐fold and ≈3‐fold in RVMs. FRET imaging using targeted Epac2camps sensors revealed no change in subsarcolemmal [cAMP], but a 2‐fold higher β‐AR stimulation of cytoplasmic [cAMP] in RVMs versus LVMs. Accordingly, β‐AR regulation of ICa,L and IKs were similar between LVMs and RVMs, whereas cytoplasmic PKA activity was increased in RVMs. Both PDE3 and PDE4 contributed to the β‐AR regulation of cytoplasmic [cAMP], and the difference between LVMs and RVMs was abolished by PDE3 inhibition and attenuated by PDE4 inhibition. Finally LV and RV intracavitary pressures were recorded in anesthetized beagle dogs. A bolus injection of isoproterenol increased RV dP/dtmax≈5‐fold versus 3‐fold in LV.
Conclusion
Canine RV and LV differ in their β‐AR response due to intrinsic differences in myocyte β‐AR downstream signaling. Enhanced β‐AR responsiveness of the RV results from higher cAMP elevation in the cytoplasm, due to a decreased degradation by PDE3 and PDE4 in the RV compared to the LV.
Keywords: cAMP, dog, heart ventricles, phosphodiesterases, β‐adrenergic stimulation
Introduction
The sympathetic nervous system is responsible for adaptation of cardiac output to stress and physical exercise. This “fight‐or‐flight” response is mediated primarily by noradrenaline and adrenaline acting on β‐adrenergic receptors (β‐ARs) at the surface of cardiac myocytes. β‐ARs are coupled through Gαs to adenylyl cyclases (AC) and the generation of cAMP, which in turn activates the cAMP‐dependent protein kinase (PKA). PKA then phosphorylates key proteins involved in excitation‐contraction coupling (ECC) including sarcolemmal L‐type Ca2+ channels (ICa,L), ryanodine receptors (RyR2), phospholamban, and troponin I.1 In addition, PKA phosphorylates slowly activating delayed rectifier K+ (IKs) channels to control cardiac repolarization.2
The levels of cAMP and thus the degree of PKA activation are finely regulated by cyclic nucleotide phosphodiesterases (PDEs) that degrade the second messenger into 5′‐AMP. Cardiac PDEs degrading cAMP belong to 5 families (PDE1‐4 and PDE8), which can be distinguished by distinct enzymatic properties and pharmacology.3 In rodents, PDE3 and PDE4 are the major contributors to the total cAMP‐hydrolytic activity4–5 and PDE4 is dominant to modulate β‐AR regulation of cAMP levels.6–9 Multiple PDE4 variants associate with β‐ARs,10–12 RyR2,13 SERCA2,14–15 ICa,L,16 and IKs17 to exert local control of ECC. In larger mammals, PDE3 activity is dominant in microsomal fractions18–20 and PDE3 inhibitors exert a potent positive inotropic effect.21 Selective inhibition of PDE3 with milrinone has been shown to improve cardiac contractility in patients with congestive heart failure.22 The role of PDE4 is less well defined but evidence is emerging that PDE4 may also play an important role in these species. In the canine heart, a large PDE4 activity is found in the cytoplasm18 but PDE4 is also present in microsomal fractions, where it accounts for ≈20% of the activity.19 Recent studies have indicated that PDE4 is expressed in human ventricle where, similar to rodents, it associates with β‐ARs, RyR2 and phospholamban.5,13 Moreover, PDE4 controls ECC and arrhythmias in human atrium.23
The right (RV) and left (LV) ventricles originate from different progenitor cells24–25 and differ in several important ways. Both ventricles have different mass (under normal conditions LV mass is ≈6‐fold RV mass), volume, morphologies and pressures.26–27 Electrical heterogeneity has been well characterized between ventricular epicardial, endocardial and midmyocardial layers.28–31 Although less studied, different electrophysiological properties were also reported between the 2 ventricles. In rats and dogs, the action potential (AP) is shorter in the RV than in the LV.32–33 In dogs, the notch in phase 1 of the AP is deeper in the RV than the LV.33–34 These differences are related to larger repolarizing K+ currents, Ito and IKs in dog RV.33–34 In this species, a larger RV IKs correlates with a higher expression of KCNQ1 and KCNE1, the principal and auxiliary subunit of the IKs channel, respectively.35 Regional heterogeneity between the LV and the RV may also exist for ATP‐activated K+ current, IKATP.36–37
In contrast to these electrophysiological studies, a recent proteomic study reported no difference in the expression level of >600 proteins between the RV and the LV from pig and rabbit. Most of these were contractile/structural proteins, oxidative phosphorylation components, and enzymes from intermediary metabolism. No plasma membrane voltage‐gated ion channels were analyzed, but a few major Ca2+ handling proteins (SERCA2, RyR2) and signal transduction components (Ca2+/Calmodulin kinase IIδ, PKA type I and II) showed no variation in expression level.38
These studies raise the question of whether functional differences between the LV and the RV are limited to electrophysiological features or extend to other aspects of ventricular function. Surprisingly, only limited information is available concerning ECC and its neurohumoral regulation in RV versus LV in the healthy heart. However, a previous report in rats has shown that, despite similar interventricular expression levels, a higher proportion of the total SERCA2a pool is associated with phospholamban in RV myocytes (RVM) compared with LV myocytes (LVM), resulting in lower Ca2+ reuptake rate and prolonged Ca2+ transients.39 In mice, opposing inotropic responses to α1‐AR stimulation have been reported, and were attributed to different effects on myofilament Ca2+ sensitivity.40 In humans, recent clinical studies indicate that ventricular load increases more for the RV than the LV during exercise41 and this may reflect, at least in part, relative differences in maximal pulmonary‐arterial versus aortic pressure. It is currently unclear whether interventricular differences exist in the sympathetic responsiveness of the human heart.
In canine, while the absolute dP/dt values are far greater in LV than in RV, stimulation of the cardiac sympathetic nerves induces greater relative changes in contractile force in the RV than in the LV.42 These changes are insensitive to α1‐AR blockade by phentolamine, implicating differences in β‐AR response.43 However, several studies reported no difference in β‐AR density, AC activity and its activation by catecholamines between RV and LV under normal conditions.44–49
Intrigued by these apparent differences, we examined the β‐AR regulation of cardiac contractility in dog RV and LV in isolated myocytes and in vivo. Our data reveal enhanced sensitivity of the RV to β‐AR stimulation, and provide evidence that PDE3 and PDE4 shape distinct compartmentalized cAMP signals that underlie interventricular dispersion in β‐AR stimulation.
Methods
This investigation conformed to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85‐23, revised 1996). Animal handling was in accordance with the European Directive for the Protection of Vertebrate Animals Used for Experimental and Other Scientific Purposes (86/609/EU). The study was conducted in accordance with the Declaration of Helsinki principles, and approved by the Ethical Committees of our institutions.
In Vivo Experiments
General anesthesia was induced in 4 beagle dogs (3 F/1 mol/L; average weight 11±1 kg) by lofentanil (0.075 mg/kg body weight i.v.), scopolamine (0.015 mg/kg), succinylcholine (1.0 mg/kg), and subsequent hourly slow injections of fentanyl (0.025 mg/kg i.v.) and continuous infusion of etomidate (1.5 mg/kg per hour). Dogs were ventilated with 30% oxygen in pressurized air to normocapnia. The body temperature was kept at 37°C with a heated water mattress. ECG standard lead II was continuously recorded. Under closed‐chest conditions, LV and RV intracavitary pressures were recorded simultaneously with high‐fidelity catheter‐tip micromanometers introduced via the femoral artery and vein (Gaeltec Ltd, Dunvegan, UK and Millar Instruments Inc). In each animal a bolus injection of isoproterenol (2.5 μg/kg) was administered and repeated twice, with each next infusion given after baseline values had been stably reestablished.
Cell‐Isolation Procedure
Adult female beagle dogs were used for myocyte isolations. A total of 12 dogs were included in the study. Anesthesia was induced with 45 mg/kg of pentobarbital. Once full anesthesia was reached, the chest was opened via a left thoracotomy and the heart was excised and placed in an O2‐gassed, Ca2+‐free standard buffer solution at approximately 4°C. The cell‐isolation procedure was the same as previously described.33 Briefly, both the left‐anterior‐descending and right coronary arteries were cannulated and perfused simultaneously. After ≈20 minutes of collagenase perfusion and subsequent washout of the enzyme, the epicardial surface layer was removed from wedges of both the LV and RV until a depth of ≥3 mm was reached. Softened tissue samples were collected from the midmyocardial layer underneath, while contamination with the endocardium was avoided. Samples were gently agitated, filtered, and washed. Midmyocytes were stored at room temperature in standard buffer solution (vide infra) and only quiescent rod‐shaped cells with clear cross‐striations and without granulation were used for the experiments. Cells were used within 48 hour of isolation.
Adenoviral Infection of Dog Ventricular Myocytes
Isolated cells were suspended in minimal essential medium (MEM: M 4780; Sigma) containing 1.2 mmol/L Ca2+, 2.5% fetal bovine serum (FBS; Invitrogen, Cergy‐Pontoise), 1% penicillin‐streptomycin, 2% HEPES (pH 7.6) and plated on 35 mm, laminin‐coated culture dishes (10 μg/mL laminin, 2 hour) at a density of 104 cells per dish. Dishes were kept in an incubator (95% O2, 5% CO2, 37°C) for 2 hours. Then the medium was replaced by 400 μL of FBS‐free MEM containing adenoviruses encoding for the cytoplasmic cAMP sensor Epac2‐camps50 at a multiplicity of infection (MOI) of 1000 pfu/cell, the plasma‐membrane targeted pmEpac2‐camps51 at an MOI of 700 pfu/cell and the cytoplasmic PKA sensor AKAR3‐NES52 at a MOI of 1000 pfu/cell.
ICa,L Recordings
The whole‐cell configuration of the patch‐clamp technique was used to record ICa,L. Patch‐electrode resistance was between 1 and 2 MΩ when filled with internal solution containing (in mmol/L): CsCl 118, EGTA 5, MgCl2 4, Na2phosphocreatine 5, Na2ATP 3.1, Na2GTP 0.42, CaCl2 0.062 (pCa 8.5), HEPES 10, adjusted to pH 7.3 with CsOH. Extracellular Cs+‐Ringer solution contained (in mmol/L): CaCl2 1.8, MgCl2 1.8, NaCl 107.1, CsCl 20, NaHCO3 4, NaH2PO4 0.8, d‐glucose 5, sodium pyruvate 5, HEPES 10, adjusted to pH 7.4 with NaOH. The cells were depolarized every 8 seconds from −50 to 0 mV during 400 ms. The use of −50 mV as holding potential allowed the inactivation of voltage‐dependent Na+ currents. K+ currents were blocked by replacing all K+ ions with external and internal Cs+. Amplifiers RK‐400 (Bio‐Logic) or Axopatch 200B (Axon Instruments, Inc.) were used for voltage clamping and acquisition. Currents were analogue filtered at 3 kHz and digitally sampled at 10 kHz using a 12‐bit analogue‐to‐digital converter (DT2827; Data Translation) connected to a compatible PC or a Digidata 1440A interface connected to a computer equipped with pClamp 10 software (Axon Instruments, Inc.). The maximal amplitude of whole‐cell ICa,L was measured as previously described.53 Currents were not compensated for capacitance and leak currents. These experiments were performed at room temperature.
IKs Recordings
The whole‐cell configuration of the patch‐clamp technique was also used to record IKs. Patch electrodes had a resistance between 0.5 and 2 MΩ when filled with internal solution containing (in mmol/L): K‐aspartate 125, KCl 20, MgCl2 1.0, MgATP 5, HEPES 5 and EGTA 10, pH 7.2 with KOH. The external solution had the following composition (mmol/L): NaCl 145, KCl 4.0, CaCl2 1.8, MgCl2 1.0, glucose 11 and HEPES 10, pH 7.4 with NaOH at 37°C. During the recordings, KCl was omitted from the external solution to increase IKs amplitude, which occurs through changes in the concentration gradient, while leaving the kinetics of IKs activation and deactivation uninfluenced.54 ICa,L was blocked with nifedipine (5 μmol/L) and IKr with dofetilide (1 μmol/L). The myocytes were depolarized from a holding potential of −50 mV to +50 mV for 3 seconds, with the tail currents on repolarization to 0 mV quantified as IKs. Ten baseline pulses were recorded followed by 3 pulses during brief (15 seconds) stimulation with Iso (±PDE inhibitor) and this was followed by a further 50 pulses during perfusion with 0 mmol/L K+ (±PDE inhibitor). Fast solution changes were achieved with the complete VC‐6 fast‐step perfusion system (Harvard Apparatus) together with a multibarrel glass pipette that was positioned ≈50 μm from the cell during drug application.
Recordings of Ca2+ Transients and Sarcomere Shortening
Myocytes were loaded with 5 μmol/L Fura‐2 AM (Invitrogen) during 15 minutes at room temperature and then washed with Ringer solution containing (in mmol/L): NaCl 121.6, KCl 5.4, MgCl2 1.8, CaCl2 1.8, NaHCO3 4, NaH2PO4 0.8, d‐glucose 5, Na‐pyruvate 5, HEPES 10, adjusted to pH 7.4 with NaOH. The loaded cells were field‐stimulated (5 V, 4 ms) at a frequency of 0.3 Hz. Then, sarcomere length and Fura‐2 ratio (measured at 512 nm upon excitation at 340 nm and 380 nm) were simultaneously recorded using an IonOptix System (IonOptix). Cell contraction was assessed as the percentage of sarcomere shortening, which is the ratio of twitch amplitude (difference of end‐diastolic and peak systolic sarcomere lengths) to end‐diastolic sarcomere length. Ca2+‐transient amplitude was assessed by the percentage of variation of the Fura‐2 ratio, by dividing the twitch amplitude (difference of end‐diastolic and peak systolic ratios) to end‐diastolic ratio. All parameters were calculated offline with dedicated software (IonWizard 6x, IonOptix). These experiments were performed at room temperature.
FRET Measurements
The same Ringer solution as described above for Ca2+ transients and cell‐shortening measurements was used in these experiments. Images were captured every 5 seconds using the 40× oil immersion objective of a Nikon TE 300 inverted microscope connected to a software‐controlled (Metafluor, Molecular Devices, Sunnyvale, CA) cooled charge coupled (CCD) camera (Sensicam PE; PCO). Cyan fluorescent protein (CFP) was excited during 150 to 300 ms by a Xenon lamp (100 W; Nikon) using a 440/20BP filter and a 455LP dichroic mirror. Dual‐emission imaging of CFP and yellow fluorescent protein (YFP) was performed using an Optosplit II emission splitter (Cairn Research) equipped with a 495LP dichroic mirror and BP filters 470/30 and 535/30, respectively. A region of interest including the entire cell was used for measurement of average CFP and YFP intensity. CFP and YFP intensities were background corrected and the YFP emission was corrected for CFP bleed through. The ratio of CFP over corrected YFP was used as an index of cAMP concentration.
Reagents
Cilostamide was from Tocris Bioscience and Ro 20‐1724 was from Calbiochem. Unless specified, all other drugs were from Sigma‐Aldrich. Isoproterenol (Iso) was first dissolved in distilled water containing 30 μmol/L ascorbic acid and then kept in the dark at 4°C until use.
Data Analysis and Statistics
All results are expressed as mean±SEM. For statistical evaluation the paired and unpaired Student's t test were used, and a difference was considered statistically significant when P<0.05. Levene and Shapiro‐Wilke tests were carried out in all the experiments in order to confirm homogeneity and normal distribution. Higher‐order ANOVA without the assumption of sphericity (Greenhouse‐Geisser correction) was used for comparison of multiple effects and to check for the absence of correlation within animals. Sidak Adjustment Multiple Comparisons was used to evaluate the significance of measurements over time.
Results
Sarcomere Shortening and Ca2+‐Transient Measurements in Response to β‐AR Stimulation in RV and LV Midmyocytes
Sarcomere shortening and Ca2+ transients were simultaneously measured in Fura‐2‐loaded RVMs and LVMs obtained from the same hearts and paced at 0.3 Hz. There was no significant difference in basal sarcomere length, basal sarcomere shortening, basal Fura‐2 ratio and basal Ca2+‐transient amplitude (Table). As shown by the individual traces of Figures 1A and 1B, a pulse application of Iso (100 nmol/L, 15 seconds) increased the amplitude of sarcomere shortening and Ca2+ transient in both LVMs and RVMs, but these effects were exacerbated in RVMs. Indeed, on average Iso increased sarcomere shortening by ≈25‐fold in RVMs versus ≈10‐fold in LVMs and increased Ca2+ transients ≈3‐fold in RVMs versus 2‐fold in LVMs (Figures 1C and 1D). The effect of Iso also lasted longer in RVMs than in LVMs, as indicated by the time to half‐maximal recovery (t1/2off) of sarcomere shortening (79±11 seconds in RVMs versus 54±3 seconds in LVMs) and Ca2+ transient (82±7 seconds in RVMs versus 49±6 seconds in LVMs) (Figures 1E and 1F). Thus, β‐AR stimulation of ECC was more efficient in dog RVMs versus LVMs.
Table 1.
Basal Parameters Measured in RVMs and LVMs
| RVMs | n | LVMs | n | |
|---|---|---|---|---|
| Contraction | ||||
| SL, μm | 1.74±0.01 | 20 | 1.72±0.01 | 18 |
| ΔL, % | 1.30±0.27 | 20 | 1.23±0.20 | 18 |
| Calcium | ||||
| Fura‐2 ratio | 4.93±0.28 | 20 | 4.66±0.27 | 18 |
| ΔR (%) | 8.31±0.84 | 20 | 9.00±1.01 | 18 |
| Electrophysiology | ||||
| Cell capacitance, pF | 138.7±3.8 | 107 | 132.9±4.03 | 105 |
| ICa,L density, pA/pF | 2.9±0.2 | 89 | 3.3±0.2 | 92 |
| IKs density, pA/pF | 1.1±0.2 | 18 | 0.8±0.1 (P<0.05) | 13 |
LVM indicates left ventricle myocyte; RVM, right ventricle myocyte; SL, sarcomere length.
Figure 1.

β‐AR regulation of ECC in LV and RV midmyocytes. A and B, Raw traces of sarcomere shortening (A) and Ca2+ transients (B) recorded in Fura‐2 loaded LVMs and RVMs paced at 0.3 Hz at baseline (grey) and at the peak of pulse isoproterenol stimulation (Iso 100 nmol/L, 15 seconds). C and D, Average maximal effect of Iso on sarcomere shortening (C) and Ca2+ transient amplitude (D) in LVMs (n=10 cells from 4 dogs) and RVMs (n=12 cells from 4 dogs). E and F, Decay kinetics of the β‐AR response estimated by the time to 50% decrease (t1/2off) of sarcomere‐shortening amplitude (E) and Ca2+‐transient amplitude (F). The bar graphs indicate mean+SEM. Statistically significant differences between LVMs and RVMs are indicated by *, P<0.05. AR indicates adrenergic; ECC, excitation‐contraction coupling; LV, left ventricle; LVM, left ventricular myocyte; RV, right ventricle; RVM, right ventricle myocyte.
β‐AR Regulation of Cytoplasmic cAMP Signals and PKA Activity in RV and LV Midmyocytes
Because β‐AR stimulation of ECC involves cAMP mobilization, this second messenger was measured in intact LVMs and RVMs by fluorescence resonance energy transfer (FRET) using the cytoplasmic sensor Epac2‐camps.50 Figure 2A shows pseudocolor images illustrating the percent increase of the CFP/YFP ratio 1 minutes and 3 minutes after application of Iso (100 nmol/L, 15 seconds) in a LVM and a RVM expressing Epac2‐camps. As shown in the inset graph of Figure 2B, there was no difference in the basal FRET ratio between LVMs and RVMs. At 1 minute after application of Iso, the CFP/YFP ratio increased to a greater extent in RVMs than in LVMs. On average, Iso led to a transient increase in the CFP/YFP ratio that was ≈2‐fold higher in RVMs versus LVMs (P<0.001) reflecting enhanced cytoplasmic cAMP accumulation in RVMs (Figure 2B). Because PKA is the primary target of cAMP for short‐term regulation of ECC, cytoplasmic PKA activity was monitored using the PKA FRET sensor AKAR3‐NES.52 As with Epac2‐camps, there was no significant difference in the average basal FRET ratio (inset graph of Figure 2D). As shown in the pseudocolor images illustrating the relative effect of Iso (100 nmol/L, 15 seconds) in Figure 2C and on the average time course of Figure 2D, although the maximal increase in YFP/CFP ratio was identical at 1 minute, the signal decayed faster in LVMs than RVMs and became significantly higher in RVMs than LVMs after 2 minutes. This indicates that cytoplasmic PKA activity was higher in RVMs than in LVMs in response to β‐AR stimulation. The lack of difference in the YFP/CFP ratio at the beginning of the stimulation was most likely due to saturation of the AKAR3‐NES sensor at this concentration of Iso, as indicated by the lack of a sharp peak in RVMs and by previous experiments in rat ventricular myocytes showing that a ≈40% change in YFP/CFP represents the maximal FRET change that can be obtained with this sensor.55
Figure 2.
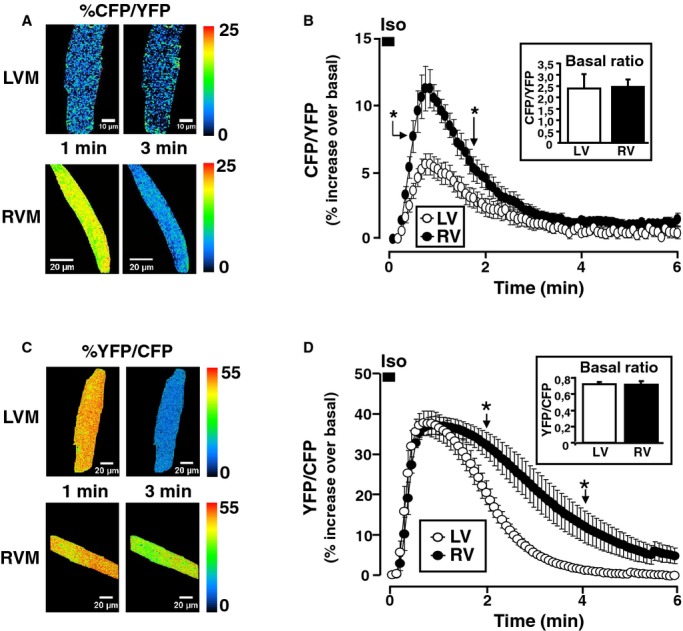
RV midmyocytes display enhanced cAMP accumulation and PKA activity in response to β‐AR stimulation. A, Pseudocolor images of a dog LVM (upper images) and a dog RVM (lower images) expressing the cytoplasmic cAMP FRET sensor Epac2‐camps. Illustrated are the percent increases of the CFP/YFP ratio over baseline at 1 minute and at 3 minutes after Iso (100 nmol/L, 15 seconds) stimulation. B, Average cytoplasmic cAMP accumulation elicited by Iso pulse stimulation in RVMs (n=36 cells from 6 dogs) and LVMs (n=37 cells from 6 dogs). Inset indicates the basal FRET ratio in LVMs and RVMs. C, Pseudocolor images of a dog LVM (upper images) and a dog RVM (lower images) expressing the cytoplasmic PKA sensor AKAR3‐NES. Illustrated is the percent increase of the YFP/CFP ratio over basal at 1 minute and at 3 minutes after the Iso pulse stimulation. D, Average PKA activation elicited by Iso pulse stimulation in RVMs (n=5 cells from 1 dog) and LVMs (n=4 cells from 1 dog). Inset indicates the basal FRET ratio in LVMs and RVMs. Statistically significant differences between LVMs and RVMs are indicated by *, P<0.05. AR indicates adrenergic; CFP, cyan fluorescent protein; FRET, fluorescence resonance energy transfer; LV, left ventricle; LVM, left ventricular myocyte; RV, right ventricle; RVM, right ventricle myocyte; PKA, protein kinase A; YFP, yellow fluorescence protein.
β‐AR Regulation of Subsarcolemmal cAMP, ICa,L and IKs in RV and LV Midmyocytes
Because subsarcolemmal cAMP plays an important role in ECC, we next used a plasma‐membrane targeted version of Epac2‐camps (pmEpac2‐camps51) to monitor cAMP specifically in this compartment. To our surprise, comparison of LVMs and RVMs revealed no difference in subsarcolemmal cAMP ([cAMP]pm) generated by β‐AR stimulation with an Iso pulse. Indeed, neither the maximal cAMP elevation nor its decay kinetics was different between LVMs and RVMs (Figure 3A). We next wondered whether the same was true for the major sarcolemmal targets of the β‐AR/cAMP/PKA pathway, ICa,L and IKs. As shown in the Table, there was no difference in basal ICa,L density between RVMs and LVMs. A pulse stimulation with Iso (100 nmol/L, 15 seconds) resulted in a similar transient increase of ICa,L between LVMs and RVMs, both in terms of amplitude and duration (Figure 3B). In contrast to ICa,L, basal IKs density was higher in RVMs than LVMs (Table), which is consistent with earlier findings.33 However, pulse β‐AR stimulation increased IKs to similar degrees in LVMs and RVMs (Figure 3C). These results indicate that (1) differences in β‐AR cAMP signals between LVMs and RVMs are compartment‐specific and (2) the increased stimulation of ECC by Iso in RVMs does not involve up‐regulation of ICa,L or down‐regulation of IKs.
Figure 3.
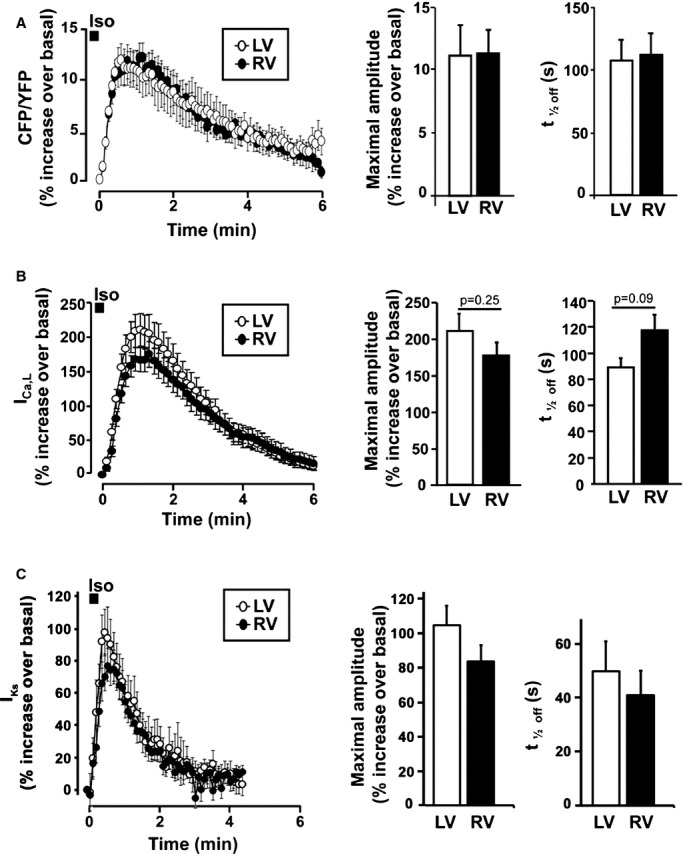
β‐AR regulation of subsarcolemmal cAMP, ICa,L and IKs in LV and RV midmyocytes. A, left, average time‐courses of Iso (100 nmol/L, 15 seconds)‐induced subsarcolemmal cAMP signals measured with pmEpac2‐camps in RVMs (n=13 from 3 dogs) and LVMs (n=7 from 3 dogs). Right, bar graphs comparing average maximal amplitude and time to 50% decrease (t1/2off) of the cAMP response. B, left, average time‐courses of ICa,L following Iso (100 nmol/L, 15 seconds) stimulation in RVMs (n=22 from 7 dogs) and LVMs (n=25 from 7 dogs). Right, bar graphs comparing average maximal amplitude and time to 50% decrease (t1/2off) of the ICa,L response. C, left, average time‐course of IKs response to Iso (100 nmol/L, 15 seconds) in RVMs (n=8 from 3 dogs) and LVMs (n=8 from 6 dogs). Right, bar graphs comparing average maximal amplitude and time to 50% decrease (t1/2off) of the IKs response. AR indicates adrenergic; cAMP, cyclic adenosine monophosphate; ICa,L, L‐type Ca2+ channel current; IKs, slow delayed rectifier K+ current; LV, left ventricle; LVM, left ventricular myocyte; RV, right ventricle; RVM, right ventricle myocyte.
PDE3 and PDE4 Shape Distinct β‐AR cAMP Signals in RVMs and LVMs
We next investigated whether phosphodiesterases (PDEs) are involved in the differences in cytoplasmic cAMP accumulation observed between RVMs and LVMs upon β‐AR stimulation. We first tested the implication of PDE3, because of its critical importance for the control of cardiac contractility in dog.21 Thus, myocytes expressing Epac2‐camps were challenged with Iso (100 nmol/L, 15 seconds) in the presence of the specific PDE3 inhibitor cilostamide (Cil, 1 μmol/L), and the inhibitor was maintained during Iso washout. Figure 4A shows that Cil doubled the maximal amplitude of the Iso‐induced cAMP transient in LVMs, while having little effect in RVMs. Cil also profoundly impaired cAMP recovery in both LVMs and RVMs, as indicated by the ≈3‐fold increase in time to half maximal decay (t1/2off) values (Figure 4A). We next tested the implication of PDE4, which represents the major soluble cAMP‐PDE activity in dog ventricle18 by using the specific PDE4 inhibitor Ro‐201724 (Ro, 10 μmol/L). Figure 4B shows that Ro induced a 3‐fold increase in the amplitude of the cAMP transient elicited by Iso in LVMs and a 2‐fold increase in RVMs. PDE4 inhibition also significantly delayed cAMP recovery, as demonstrated by a ≈2.5‐fold increase in time‐to‐half‐maximal decay (t1/2off) values. Thus, PDE3 and PDE4 regulate cytoplasmic cAMP accumulation upon β‐AR stimulation in canine ventricular midmyocytes. However, this control is less stringent in RVMs compared to LVMs, resulting in higher cytoplasmic cAMP ([cAMP]cyt) upon β‐AR stimulation.
Figure 4.

Regulation of β‐AR cytoplasmic cAMP signals by PDE3 and PDE4 in LVMs and RVMs. A, left, average time course of cAMP levels measured in the cytoplasm with Epac2‐camps following β‐AR stimulation with Iso (100 nmol/L, 15 seconds) alone (gray and black lines, data from Figure 3B) or during administration of the PDE3 inhibitor cilostamide (Cil, 1 μmol/L, white circles: LVMs, n=13 from 3 dogs; black circles: RVMs, n=17 from 7 dogs). Right, Bar graphs representing the average maximal amplitude and time to 50% recovery (t1/2off) of the cytoplasmic cAMP transients induced by Iso alone or Iso with Cil in LVMs (n=13) and RVMs (n=17). B, left, average time course of cytosolic cAMP levels following β‐AR stimulation with Iso (100 nmol/L, 15 seconds) alone or during administration of the PDE4 inhibitor Ro 20‐1724 (Ro, 10 μmol/L; white circles: LVMs, n=21 from 6 dogs; black circles: RVMs, n=24 from 5 dogs). Right, bar graphs representing the average maximal amplitude and time to 50% recovery (t1/2off) of the cAMP transients induced by Iso alone or Iso with Ro in LVMs (n=21) and LVMs (n=24). PDE inhibitors were added to the Iso solution and in the washout solution. Symbols and bar graphs indicate the mean±SEM. Statistical difference between Iso alone and Iso+PDE inhibitor in LVMs and RVMs is indicated as *, P<0.05; ***, P<0.001. AR indicates adrenergic; cAMP, cyclic adenosine monophosphate; LV, left ventricle; LVM, left ventricular myocyte; PDE, phosphodiesterase; RV, right ventricle; RVM, right ventricle myocyte.
Regulation of Membrane cAMP, ICa,L and IKs by PDE3 and PDE4 in RVMs and LVMs
Given the large contribution of PDE4 to cytoplasmic cAMP hydrolysis upon β‐AR stimulation, we compared the respective roles of PDE3 and PDE4 in the β‐AR regulation of subsarcolemmal cAMP in canine ventricular myocytes. As shown in Figure 5A, inhibition of PDE3 with 1 μmol/L Cil had no effect on the maximal cAMP elevation at the plasma membrane but delayed cAMP recovery in LVMs and RVMs. Interestingly, the difference in average t1/2off values between Iso and Iso+Cil reached statistical significance only in LVMs (t1/2off was 107±17 seconds for Iso, and 232±50 seconds for Iso+Cil, P<0.05 whereas in RVMs, t1/2off was 112±17 seconds for Iso alone and 171±39 seconds for Iso+Cil, P=0.36) suggesting a stronger contribution of membrane‐bound PDE3 in LVMs versus RVMs to cAMP hydrolysis upon β‐AR stimulation. In sharp contrast to what was observed in the cytoplasm, PDE4 inhibition with Ro at 10 μmol/L had no effect, neither on the amplitude nor on the recovery kinetics of the cAMP transient generated by Iso pulse stimulation at the plasma membrane (Figure 5B). We next investigated the respective contribution of PDE3 and PDE4 to the regulation of ICa,L and IKs. As shown in Figure 6A, PDE3 inhibition had no effect on the maximal ICa,L stimulation induced by Iso (100 nmol/L, 15 seconds), but significantly delayed ICa,L recovery in LVMs and RVMs (in LVMs, t1/2off was 91±7 seconds for Iso, and 203±22 seconds for Iso+Cil, P<0.001; in RVMs, t1/2off was 113±11 seconds for Iso alone and 176±19 seconds for Iso+Cil, P<0.05). Interestingly, PDE3 inhibition also slowed IKs recovery in RVMs and LVMs (data not shown). In contrast, inhibition of PDE4 with Ro at 10 μmol/L did not modify ICa,L (Figure 6B) nor IKs recovery. However, when PDE3 was inhibited by Cil, concomitant inhibition of PDE4 drastically prolonged the β‐AR stimulation of the current (Figure 6C), with t1/2off values reaching 381±66 seconds in RVMs (P<0.05 versus Iso+Cil) and 381±51 seconds in LVMs (P<0.01 versus Iso+Cil). These results indicate that although PDE3 is dominant for β‐AR regulation of ICa,L in canine myocytes, PDE4 becomes important when PDE3 is inhibited.
Figure 5.

Regulation of subsarcolemmal cAMP by PDE3 and PDE4 after brief β‐AR stimulation. A, Left, average time course of cAMP levels measured at the plasma membrane with pmEpac2‐camps following β‐AR stimulation with Iso (100 nmol/L, 15 seconds) alone (gray and black lines, data from Figure 4A) and representative examples of the effect of the PDE3 inhibitor cilostamide (Cil, 1 μmol/L) in a LVM (white circles) and a RVM (black circles). Right, bar graphs representing the average maximal amplitude and time to 50% recovery (t1/2off) of the sarcolemmal cAMP transients induced by Iso alone or Iso with Cil in LVMs (n=4 from 3 dogs) and RVMs (n=5 from 2 dogs). B, Left, average time course of the plasma membrane cAMP levels following β‐AR stimulation with Iso (100 nmol/L, 15 seconds) alone (gray and black lines, data from Figure 4A) and representative examples of the effect of the PDE4 inhibitor Ro 20‐1724 (Ro, 10 μmol/L) in a LVM (white circles) and a RVM (black circles). Right, bar graphs representing the average maximal amplitude and time to 50% recovery (t1/2off) of the cAMP transients induced by Iso alone or Iso with Ro in LVMs (n=9 from 3 dogs) and RVMs (n=5 from 3 dogs). PDE inhibitors were added to the Iso solution and in the washout solution. Symbols and bar graphs indicate the mean±SEM. Statistical difference between Iso alone and Iso+PDE inhibitor in LVMs and RVMs is indicated as *, P<0.05. AR indicates adrenergic; cAMP, cyclic adenosine monophosphate; Iso, isoprenaline; LV, left ventricle; LVM, left ventricular myocyte; PDE, phosphodiesterase; RV, right ventricle; RVM, right ventricle myocyte.
Figure 6.
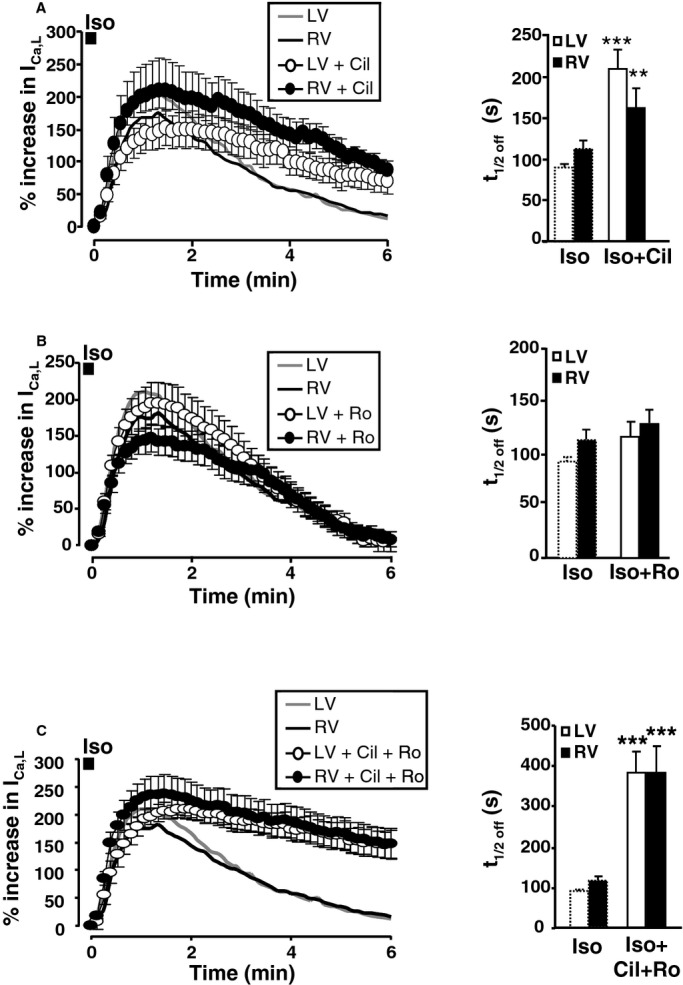
Regulation of ICa,L by PDE3 and PDE4 after brief β‐AR stimulation. Average time course of ICa,L (left) and mean values of time for 50% recovery (t1/2off) (right) in response to β‐AR stimulation with Iso (100 nmol/L, 15 seconds) alone or with A, cilostamide (Cil, 1 μmol/L; white circles: LVMs n=18 from 7 dogs; black circles: RVMs, n=11 from 6 dogs); B, Ro 20‐1724 (Ro, 10 μmol/L; white circles: RVMs, n=15 from 6 dogs; black circles: LVMs, n=11 from 6 dogs); C, the combination of Cil and Ro (Cil+Ro, white circles: LVMs, n=11 from 2 dogs; black circles: LVMs, n=11 from 2 dogs). PDE inhibitors were added to the Iso solution and in the washout solution. The symbols and bar graphs indicate the mean±SEM. Statistical difference between Iso alone and Iso+PDE inhibitor in LVMs and RVMs is indicated as **, P<0.01; ***, P<0.001. AR indicates adrenergic; cAMP, cyclic adenosine monophosphate; Iso, isoprenaline; LV, left ventricle; LVM, left ventricular myocyte; PDE, phosphodiesterase; RV, right ventricle; RVM, right ventricle myocyte.
In Vivo Inotropic Response to Isoproterenol in RV and LV
Altogether, the above results indicate that β‐AR stimulation of ECC is more efficient in RVMs versus LVMs due to a stronger contribution of compartmentalized PDE3 and PDE4 to hydrolyze cAMP in the cytoplasm. Since these results were obtained in isolated cells, it seemed important to test whether enhanced β‐AR responsiveness could be observed at a more integrated level. For this, pressure recordings were obtained simultaneously from the RV and LV at baseline and during infusion of Iso in anesthetized beagle dogs. Representative examples relative to the ECG are shown in Figure 7A. At baseline, systolic pressures and maximal dP/dt were significantly higher in the LV than in the RV (Figure 7B). For example, maximal dP/dt was 2582±194 mm Hg/s in the LV versus 534±45 mm Hg/s in the RV (P<0.001). Upon Iso, maximal dP/dt increased ≈3.4‐fold in the LV compared with almost 5‐fold in the RV (P<0.05; Figure 7B, left panel). Accordingly, the relative increase in systolic pressure by β‐AR stimulation was significantly higher in the RV than LV, reaching on average 66±19 mm Hg (+61±6%) and 172±31 mm Hg (+20±4%), respectively (P<0.01) during Iso (Figure 7B, right panel). In addition, the relative increase in maximal dP/dt and systolic pressure was more sustained in the RV than the LV after the Iso bolus infusions (Figure 7C). Diastolic pressures did not show discernible interventricular differences from baseline to Iso.
Figure 7.
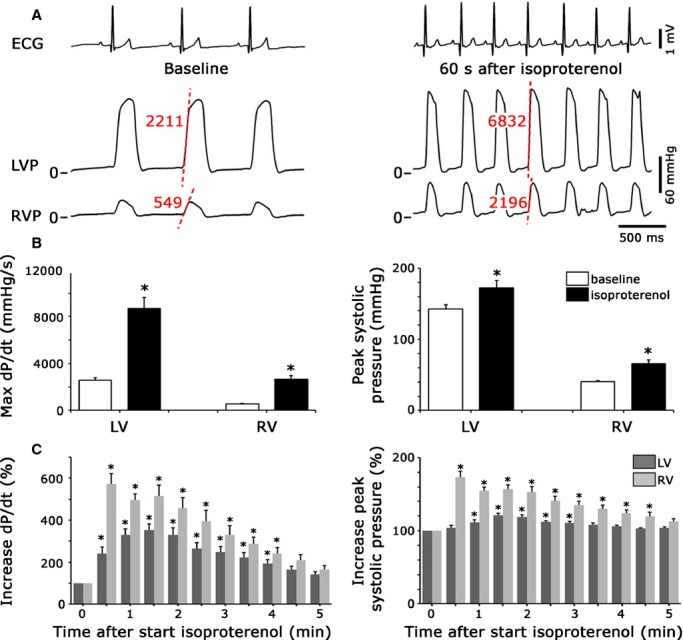
Differential response to β‐adrenergic stimulation in the LV vs RV in anesthetized dogs. A, Representative pressure recordings, together with ECG lead II, before and 60 seconds after bolus infusion of the β‐adrenergic agonist isoproterenol at 2.5 μg/kg. Representative maximal dP/dt values for LV and RV before and after Iso are indicated in red. B, Bar graphs showing average values (N=4 dogs) for maximal dP/dt and peak systolic pressures in the LV and RV before and after infusion of isoproterenol. C, Bar graphs illustrating the average time courses of alterations in maximal dP/dt and peak systolic pressures after infusion of isoproterenol, including recovery phase. Data are shown as change from baseline. *, P<0.05. ECG indicates electrocardiography; ICa,L, L‐type Ca2+ channel current; LV, left ventricle; RV, right ventricle.
Discussion
This study provides evidence that the RV is more sensitive than the LV to β‐AR stimulation, in part due to intrinsic postsynaptic differences in myocyte β‐AR signaling. The enhanced efficiency of Iso to stimulate Ca2+ transients and sarcomere shortening in RVMs compared with LVMs isolated from the same hearts is correlated with an increased positive inotropic effect of Iso observed in the RV in vivo. Enhanced β‐AR responsiveness in RVMs is not associated with modifications of β‐AR signaling at the plasma membrane, since β‐AR stimulation of subsarcolemmal [cAMP]i and of membrane currents ICa,L and IKs are similar in RVMs and LVMs, even though basal IKs density showed interventricular differences, in agreement with our previous study.33 However, RVMs display a higher cAMP accumulation and exacerbated PKA response in the cytoplasm compared to LVMs, and the difference in cAMP is abolished by inhibition of PDE3 and PDE4. Thus, PDE3 and PDE4 shape distinct compartmentalized cAMP signals that underlie regionally specific differences in the cardiac ventricles upon β‐AR stimulation.
Recent clinical studies indicate that during intense physical exercise, the afterload of the RV increases more than that of the LV.41,56 Because the sympathetic nervous system is strongly activated during exercise, these observations suggest that the RV could be more sensitive to sympathetic stimulation than the LV in humans. Earlier studies that compared the responsiveness of the LV versus RV to sympathetic nerve stimulation in dogs are consistent with this notion: Norris and Randall showed that stimulation of the left and right ansae subclavia has a stronger inotropic effect in the RV than in the LV.42 Similarly, Abe et al43 reported stronger effects of sympathetic nerve stimulation on RV systolic pressure, which were not modified by α1‐AR blockade. Thus, previous data suggested the existence of differential β‐AR stimulation in RV versus LV, but until now its clear demonstration has been lacking and the potential mechanisms involved remain elusive. Our experiments show that Iso infusion in vivo exerts a stronger relative increase in RV contractility compared with that of the LV (Figure 7). By themselves, these in vivo data do not allow the exclusion of a differential effect of Iso on innervations or loading conditions of the 2 ventricles. However, consistent with our hypothesis, Piao et al, using RV and LV Langendorff preparations in rat, have shown recently that the inotropic effect of dobutamine is more efficient in the RV than in the LV.57 Importantly, myocytes isolated from the midmyocardial layer of the RV showed enhanced and prolonged stimulation of Ca2+ transients and contraction by Iso compared to their LV counterparts (Figure 1). These results demonstrate that the high sensitivity of the RV to β‐AR stimulation relies at least partly on an intrinsic property of RV myocytes, and raise the question of which mechanisms are involved.
β‐AR control of cardiac ECC operates mainly through activation of cAMP and PKA. Consistent with increased functional effects of β‐ARs, cytoplasmic cAMP and phosphorylation of a specific PKA sensor were found to be increased in RVMs upon Iso pulse stimulation (Figure 2). While it seems very likely that increased phosphorylation of the PKA probe was due to enhanced PKA activity in RVMs versus LVMs, the slower decay kinetics of AKAR‐NES dephosphorylation could also result from a lower phosphatase activity in RVMs. To the best of our knowledge, differences in phosphatase activity have not been documented in LV versus RV. Because we focused on the role of phosphodiesterase and compartmentalization of cAMP, we did not pursue the investigation of phosphatases. However, this point would deserve further consideration in future studies.
Interestingly, this difference was compartment‐specific since enhanced β‐AR responses were not observed at the plasma membrane. Indeed neither β‐AR regulation of subsarcolemmal cAMP signals nor that of ICa,L and IKs were modified in RV compared with LV (Figure 3). Since ICa,L was not involved in the enhanced inotropic effect of Iso in RV, other ECC proteins must be differentially regulated. Likely candidates are RyR2 and PLB, which control SR Ca2+ release and reuptake by SERCA2a, respectively. Although differential SERCA2a‐PLB association was observed in RV versus LV in rat,39 we did not observe differences in the decay kinetics of Ca2+ transients in RV versus LV dog myocytes (data not shown). To our knowledge, whether RyR2 and PLB are differentially phosphorylated in response to Iso stimulation in RV versus LV has not been investigated.
Enhanced β‐AR functional effects in RVMs could result from increased cAMP generation in RVMs. However, the lack of difference at the plasma membrane (Figure 3) does not support this idea. Moreover, previous studies in dog and humans provide compelling evidence that the density of β‐ARs, AC activity and AC stimulation by β‐ARs are similar between RV and LV.44–49 Because in rodents PDEs can generate cAMP gradients within cardiac myocytes,8–9,8–59 we tested the hypothesis that they could be involved in the interventricular differences in cytoplasmic cAMP accumulation in dogs. Our results indicate that PDE3 inhibition strongly increased maximal [cAMP]cyt in LVMs upon β‐AR stimulation, while having a weak effect on maximal [cAMP]cyt in RVMs, thus abolishing the differences between RVMs and LVMs. PDE3 inhibition also significantly prolonged the effect of Iso on [cAMP]pm in LVMs but not in RVMs (Figure 6A). PDE4 inhibition potentiated the maximal [cAMP]cyt in both RVMs and LVMs upon β‐AR stimulation, with a more pronounced effect in RVMs. As a consequence, the interventricular difference in [cAMP]cyt accumulation was attenuated upon PDE4 inhibition. We conclude that regionally specific differences in subcellular cAMP compartmentation arise from a more stringent control of β‐ARs cAMP signals by PDE3 and PDE4 in LVMs than in RVMs.
Upon β‐AR stimulation, the relatively balanced contribution of PDE3 and PDE4 to cAMP hydrolysis in the cytoplasm (Figure 4) and the dominance of PDE3 at the sarcolemma (Figure 5) is at variance with previous results in rodents, where PDE4 is dominant in both compartments.6–9 Because in dog PDE3 inhibitors have strong inotropic effects in contrast to PDE4 inhibitors,18 our results raise the question of the functional role of PDE4 in dog. When the respective role of these 2 PDEs was evaluated on the stimulation of ICa,L by Iso, inhibition of PDE3 but not PDE4 significantly prolonged ICa,L upregulation (Figure 6). This is consistent with a predominance of PDE3 in T‐tubular membranes, whereas only a fraction of PDE4 is sarcolemmal and most of the activity is soluble.18–19 However, when PDE3 was inhibited, PDE4 inhibition drastically prolonged ICa,L recovery from β‐AR stimulation (Figure 6). Thus, PDE4 can regulate ICa,L, but under normal conditions it is masked by the predominant PDE3. Therefore, the hierarchy between PDE3 and PDE4 appears to be opposite in dogs compared with rats.9
In conclusion, the current study demonstrates that RV and LV differ in their sensitivity to β‐AR stimulation both in vivo and in vitro, and reveals regionally specific differences in β‐AR coupling to PDE3 and PDE4 and subcellular cAMP compartmentation in the heart. The fact that these results were obtained in a large mammal increases the likelihood, at least to some extent, that they represent the human situation. If this is the case, such enhanced sensitivity to β‐AR stimulation may be an important factor for RV dysfunction not only in the context of sports medicine, but also in RV failure consequent to left‐sided heart failure or pulmonary hypertension (PH) where activation of the sympathetic nervous system, although beneficial on the short term, becomes detrimental for cardiac function in the long run. In this context, our results suggest that β‐blockers could be particularly effective in right heart failure. Consistent with this hypothesis, β‐adrenoceptor blockade was reported to prevent β‐AR down‐regulation and to attenuate desensitization in experimental right heart failure induced by progressive pulmonary artery banding and tricuspid avulsion in dog.48 Similarly, β‐blockers were shown to have beneficial effects on RV function and morphology in rat models of PH (reviewed in60). Interestingly, although β‐blockers are not recommended in humans with PH because of their acute negative chronotropic and inotropic effects, low doses of bisoprolol were shown to improve RV contractility and filling and to delay the progression of right heart failure in the monocrotaline rat model of PH.61 Our study also shows that the coupling of β‐AR to PDE3 and PDE4 differs in the normal LV versus RV. It will be important in future studies to determine whether this is related to a differential expression and/or localization of PDE3 and PDE4 in the 2 ventricles, and how this coupling is modified during hypertrophy and heart failure. Indeed, whereas a decrease in β1‐AR expression is known to occur in both the failing LV and RV,45 much less is known concerning PDE remodeling in right heart failure. A deeper characterization of interventricular differences in cAMP signaling compartmentation may provide new indications for therapies that preferentially target the failing RV.
Sources of Funding
This study was supported in part by a Postdoctoral Lefoulon Delalande grant from the Institut de France and a Marie Curie Intra‐European Fellowship for Career Development (PIEF‐GA‐331241) (to CEM), an ANR grant 2010 BLAN 1139‐01 (to GV) and the Fondation Leducq for the Transatlantic Network of Excellence cycAMP grant 06CVD02 (to RF). PGAV is supported by The Netherlands Heart Foundation (NHS2010B216) and a Vidi grant from the Netherlands Organization for Scientific Research (ZonMw 91710365).
Disclosures
NAG is employed by AstraZeneca and HJVDL is employed by Janssen. All other authors have reported that they have no relationships relevant to the contents of this paper to disclose.
Acknowledgments
We thank Florence Lefebvre for superb technical assistance, Dr Viacheslav Nikolaev for providing the Epac2‐camps and pmEpac2‐camps, Dr Jin Zhang for providing the AKAR3 sensor, and Dr Bertrand Crozatier for helpful discussions.
References
- 1.Bers DM. Cardiac excitation‐contraction coupling. Nature. 2002; 415:198-205. [DOI] [PubMed] [Google Scholar]
- 2.Marx SO, Kurokawa J, Reiken S, Motoike H, D'Armiento J, Marks AR, Kass RS. Requirement of a macromolecular signaling complex for â adrenergic receptor modulation of the KCNQ1‐KCNE1 potassium channel. Science. 2002; 295:496-499. [DOI] [PubMed] [Google Scholar]
- 3.Mika D, Leroy J, Vandecasteele G, Fischmeister R. PDEs create local domains of cAMP signaling. J Mol Cell Cardiol. 2012; 52:323-329. [DOI] [PubMed] [Google Scholar]
- 4.Rochais F, Abi‐Gerges A, Horner K, Lefebvre F, Cooper DMF, Conti M, Fischmeister R, Vandecasteele G. A specific pattern of phosphodiesterases controls the cAMP signals generated by different Gs‐coupled receptors in adult rat ventricular myocytes. Circ Res. 2006; 98:1081-1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Richter W, Xie M, Scheitrum C, Krall J, Movsesian MA, Conti M. Conserved expression and functions of PDE4 in rodent and human heart. Basic Res Cardiol. 2011; 106:249-262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mongillo M, McSorley T, Evellin S, Sood A, Lissandron V, Terrin A, Huston E, Hannawacker A, Lohse MJ, Pozzan T, Houslay MD, Zaccolo M. Fluorescence resonance energy transfer‐based analysis of cAMP dynamics in live neonatal rat cardiac myocytes reveals distinct functions of compartmentalized phosphodiesterases. Circ Res. 2004; 95:65-75. [DOI] [PubMed] [Google Scholar]
- 7.Rochais F, Vandecasteele G, Lefebvre F, Lugnier C, Lum H, Mazet J‐L, Cooper DMF, Fischmeister R. Negative feedback exerted by PKA and cAMP phosphodiesterase on subsarcolemmal cAMP signals in intact cardiac myocytes. An in vivo study using adenovirus‐mediated expression of CNG channels. J Biol Chem. 2004; 279:52095-52105. [DOI] [PubMed] [Google Scholar]
- 8.Nikolaev VO, Bunemann M, Schmitteckert E, Lohse MJ, Engelhardt S. Cyclic AMP imaging in adult cardiac myocytes reveals far‐reaching ß1‐adrenergic but locally confined ß2‐adrenergic receptor‐mediated signaling. Circ Res. 2006; 99:1084-1091. [DOI] [PubMed] [Google Scholar]
- 9.Leroy J, Abi‐Gerges A, Nikolaev VO, Richter W, Lechêne P, Mazet J‐L, Conti M, Fischmeister R, Vandecasteele G. Spatiotemporal dynamics of ß‐adrenergic cAMP signals and L‐type Ca2+ channel regulation in adult rat ventricular myocytes: role of phosphodiesterases. Circ Res. 2008; 102:1091-1100. [DOI] [PubMed] [Google Scholar]
- 10.Baillie GS, Sood A, McPhee I, Gall I, Perry SJ, Lefkowitz RJ, Houslay MD. beta‐Arrestin‐mediated PDE4 cAMP phosphodiesterase recruitment regulates beta‐adrenoceptor switching from Gs to Gi. Proc Natl Acad Sci USA. 2003; 100:941-945. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 11.Richter W, Day P, Agraval R, Bruss MD, Granier S, Wang YL, Rasmussen SGF, Horner K, Wang P, Lei T, Patterson AJ, Kobilka BK, Conti M. Signaling from ß1‐and ß2‐adrenergic receptors is defined by differential interactions with PDE4. EMBO J. 2008; 27:384-393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.De Arcangelis V, Liu R, Soto D, Xiang Y. Differential association of phosphodiesterase 4D isoforms with ß2‐adrenoceptor in cardiac myocytes. J Biol Chem. 2009; 284:33824-33832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lehnart SE, Wehrens XHT, Reiken S, Warrier S, Belevych AE, Harvey RD, Richter W, Jin SLC, Conti M, Marks A. Phosphodiesterase 4D deficiency in the ryanodine receptor complex promotes heart failure and arrhythmias. Cell. 2005; 123:23-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kerfant BG, Zhao D, Lorenzen‐Schmidt I, Wilson LS, Cai S, Chen SR, Maurice DH, Backx PH. PI3KY is required for PDE4, not PDE3, activity in subcellular microdomains containing the sarcoplasmic reticular calcium ATPase in cardiomyocytes. Circ Res. 2007; 101:400-408. [DOI] [PubMed] [Google Scholar]
- 15.Beca S, Helli PB, Simpson JA, Zhao D, Farman GP, Jones P, Tian X, Wilson LS, Ahmad F, Chen SR, Movsesian MA, Manganiello V, Maurice DH, Conti M, Backx PH. Phosphodiesterase 4D regulates baseline sarcoplasmic reticulum Ca2+ release and cardiac contractility, independently of L‐type Ca2+ current. Circ Res. 2011; 109:1024-1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Leroy J, Richter W, Mika D, Castro LRV, Abi‐Gerges A, Xie M, Scheitrum C, Lefebvre F, Schittl J, Westenbroek R, Catterall WA, Charpentier F, Conti M, Fischmeister R, Vandecasteele G. Phosphodiesterase 4B in the cardiac L‐type Ca2+ channel complex regulates Ca2+ current and protects against ventricular arrhythmias. J Clin Invest. 2011; 121:2651-2661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Terrenoire C, Houslay MD, Baillie GS, Kass RS. The cardiac IKs potassium channel macromolecular complex includes the phosphodiesterase PDE4D3. J Biol Chem. 2009; 284:9140-9146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Weishaar RE, Kobylarz‐Singer DC, Steffen RP, Kaplan HR. Subclasses of cyclic AMP‐specific phosphodiesterase in left ventricular muscle and their involvement in regulating myocardial contractility. Circ Res. 1987; 61:539-547. [DOI] [PubMed] [Google Scholar]
- 19.Lugnier C, Muller B, Lebec A, Beaudry C, Rousseau E. Characterization of indolidan‐sensitive and rolipram‐sensitive cyclic nucleotide phosphodiesterases in canine and human cardiac microsomal fractions. J Pharmacol Exp Ther. 1993; 265:1142-1151. [PubMed] [Google Scholar]
- 20.Smith CJ, Huang R, Sun D, Ricketts S, Hoegler C, Ding JZ, Moggio RA, Hintze TH. Development of decompensated dilated cardiomyopathy is associated with decreased gene expression and activity of the milrinone‐sensitive cAMP phosphodiesterase PDE3A. Circulation. 1997; 96:3116-3123. [DOI] [PubMed] [Google Scholar]
- 21.Osadchii OE. Myocardial phosphodiesterases and regulation of cardiac contractility in health and cardiac disease. Cardiovasc Drugs Ther. 2007; 21:171-194. [DOI] [PubMed] [Google Scholar]
- 22.Monrad ES, Baim DS, Smith HS, Lanoue AS, Silverman KJ, Gervino EV, Grossman W. Assessment of long‐term therapy with milrinone and the effects of milrinone withdrawal. Circulation. 1986; 73:III205-III212. [PubMed] [Google Scholar]
- 23.Molina CE, Leroy J, Xie M, Richter W, Lee I‐O, Maack C, Rucker‐Martin C, Donzeau‐Gouge P, Verde I, Hove‐Madsen L, Barriga M, Conti M, Vandecasteele G, Fischmeister R. Cyclic AMP phosphodiesterase type 4 protects against atrial arrhythmias. J Am Coll Cardiol. 2012; 59:2182-2190. [DOI] [PubMed] [Google Scholar]
- 24.Zaffran S, Kelly RG, Meilhac SM, Buckingham ME, Brown NA. Right ventricular myocardium derives from the anterior heart field. Circ Res. 2004; 95:261-268. [DOI] [PubMed] [Google Scholar]
- 25.Verzi MP, McCulley DJ, De Val S, Dodou E, Black BL. The right ventricle, outflow tract, and ventricular septum comprise a restricted expression domain within the secondary/anterior heart field. Dev Biol. 2005; 287:134-145. [DOI] [PubMed] [Google Scholar]
- 26.Ho SY, Nihoyannopoulos P. Anatomy, echocardiography, and normal right ventricular dimensions. Heart. 2006; 92suppl 1:i2-i13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Haddad F, Hunt SA, Rosenthal DN, Murphy DJ. Right ventricular function in cardiovascular disease, part I: anatomy, physiology, aging, and functional assessment of the right ventricle. Circulation. 2008; 117:1436-1448. [DOI] [PubMed] [Google Scholar]
- 28.Sicouri S, Antzelevitch C. A subpopulation of cells with unique electrophysiological properties in the deep subepicardium of the canine ventricle. The M cell. Circ Res. 1991; 68:1729-1741. [DOI] [PubMed] [Google Scholar]
- 29.Antzelevitch C, Sicouri S, Litovsky SH, Lukas A, Krishnan SC, Didiego JM, Gintant GA, Liu DW. Heterogeneity within the ventricular wall ‐Electrophysiology and pharmacology of epicardial, endocardial, and M‐Cells. Circ Res. 1991; 69:1427-1449. [DOI] [PubMed] [Google Scholar]
- 30.Zygmunt AC, Goodrow RJ, Antzelevitch C. INaCa contributes to electrical heterogeneity within the canine ventricle. Am J Physiol Heart Circ Physiol. 2000; 278:H1671-H1678. [DOI] [PubMed] [Google Scholar]
- 31.Gaborit N, Le Bouter S, Szuts V, Varro A, Escande D, Nattel S, Demolombe S. Regional and tissue specific transcript signatures of ion channel genes in the non‐diseased human heart. J Physiol. 2007; 582:675-693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Watanabe T, Delbridge LM, Bustamante JO, McDonald TF. Heterogeneity of the action potential in isolated rat ventricular myocytes and tissue. Circ Res. 1983; 52:280-290. [DOI] [PubMed] [Google Scholar]
- 33.Volders PG, Sipido KR, Carmeliet E, Spatjens RL, Wellens HJ, Vos MA. Repolarizing K+ currents ITO1 and IKs are larger in right than left canine ventricular midmyocardium. Circulation. 1999; 99:206-210. [DOI] [PubMed] [Google Scholar]
- 34.Di Diego JM, Sun ZQ, Antzelevitch C. Ito and action potential notch are smaller in left vs. right canine ventricular epicardium. Am J Physiol. 1996; 271:H548-H561. [DOI] [PubMed] [Google Scholar]
- 35.Ramakers C, Vos MA, Doevendans PA, Schoenmakers M, Wu YS, Scicchitano S, Iodice A, Thomas GP, Antzelevitch C, Dumaine R. Coordinated down‐regulation of KCNQ1 and KCNE1 expression contributes to reduction of IKs in canine hypertrophied hearts. Cardiovasc Res. 2003; 57:486-496. [DOI] [PubMed] [Google Scholar]
- 36.Pandit SV, Kaur K, Zlochiver S, Noujaim SF, Furspan P, Mironov S, Shibayama J, Anumonwo J, Jalife J. Left‐to‐right ventricular differences in IKATP underlie epicardial repolarization gradient during global ischemia. Heart Rhythm. 2011; 8:1732-1739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Choi SW, Ahn JS, Kim HK, Kim N, Choi TH, Park SW, Ko EA, Park WS, Song DK, Han J. Increased expression of ATP‐sensitive K+ channels improves the right ventricular tolerance to hypoxia in rabbit hearts. Korean J Physiol Pharmacol. 2011; 15:189-194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Phillips D, Aponte AM, Covian R, Neufeld E, Yu ZX, Balaban RS. Homogenous protein programming in the mammalian left and right ventricle free walls. Physiol Genomics. 2011; 43:1198-1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sathish V, Xu A, Karmazyn M, Sims SM, Narayanan N. Mechanistic basis of differences in Ca2+ ‐handling properties of sarcoplasmic reticulum in right and left ventricles of normal rat myocardium. Am J Physiol Heart Circ Physiol. 2006; 291:H88-H96. [DOI] [PubMed] [Google Scholar]
- 40.Wang GY, McCloskey DT, Turcato S, Swigart PM, Simpson PC, Baker AJ. Contrasting inotropic responses to alpha1‐adrenergic receptor stimulation in left versus right ventricular myocardium. Am J Physiol Heart Circ Physiol. 2006; 291:H2013-H2017. [DOI] [PubMed] [Google Scholar]
- 41.La Gerche A, Heidbuchel H, Burns AT, Mooney DJ, Taylor AJ, Pfluger HB, Inder WJ, Macisaac AI, Prior DL. Disproportionate exercise load and remodeling of the athlete's right ventricle. Med Sci Sports Exerc. 2011; 43:974-981. [DOI] [PubMed] [Google Scholar]
- 42.Norris JE, Randall WC. Responses of the canine myocardium to stimulation of thoracic cardiac nerves. Am J Physiol. 1977; 232:H485-H494. [DOI] [PubMed] [Google Scholar]
- 43.Abe Y, Saito D, Tani H, Nakatsu T, Kusachi S, Haraoka S, Nagashima H. The effect of cardiac sympathetic nerve stimulation on the right ventricle in canine heart. Jpn Circ J. 1987; 51:535-542. [DOI] [PubMed] [Google Scholar]
- 44.Vatner DE, Homcy CJ, Sit SP, Manders WT, Vatner SF. Effects of pressure overload, left ventricular hypertrophy on ß‐adrenergic receptors, and responsiveness to catecholamines. J Clin Invest. 1984; 73:1473-1482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bristow MR, Ginsburg R, Umans V, Fowler M, Minobe W, Rasmussen R, Zera P, Menlove R, Shah P, Jamieson S, Stinson EB. Beta 1‐and beta 2‐adrenergic‐receptor subpopulations in nonfailing and failing human ventricular myocardium: coupling of both receptor subtypes to muscle contraction and selective beta 1‐receptor down‐regulation in heart failure. Circ Res. 1986; 59:297-309. [DOI] [PubMed] [Google Scholar]
- 46.Fan TH, Liang CS, Kawashima S, Banerjee SP. Alterations in cardiac beta‐adrenoceptor responsiveness and adenylate cyclase system by congestive heart failure in dogs. Eur J Pharmacol. 1987; 140:123-132. [DOI] [PubMed] [Google Scholar]
- 47.Calderone A, Bouvier M, Li K, Juneau C, de Champlain J, Rouleau JL. Dysfunction of the ß‐and alpha‐adrenergic systems in a model of congestive heart failure. The pacing‐overdrive dog. Circ Res. 1991; 69:332-343. [DOI] [PubMed] [Google Scholar]
- 48.Liang CS, Frantz RP, Suematsu M, Sakamoto S, Sullebarger JT, Fan TM, Guthinger L. Chronic beta‐adrenoceptor blockade prevents the development of beta‐adrenergic subsensitivity in experimental right‐sided congestive heart failure in dogs. Circulation. 1991; 84:254-266. [DOI] [PubMed] [Google Scholar]
- 49.White M, Roden R, Minobe W, Khan MF, Larrabee P, Wollmering M, Port JD, Anderson F, Campbell D, Feldman AM, Bristow MR. Age‐related changes in ß‐adrenergic neuroeffector systems in the human heart. Circulation. 1994; 90:1225-1238. [DOI] [PubMed] [Google Scholar]
- 50.Nikolaev VO, Bunemann M, Hein L, Hannawacker A, Lohse MJ. Novel single chain cAMP sensors for receptor‐induced signal propagation. J Biol Chem. 2004; 279:37215-37218. [DOI] [PubMed] [Google Scholar]
- 51.Wachten S, Masada N, Ayling LJ, Ciruela A, Nikolaev VO, Lohse MJ, Cooper DM. Distinct pools of cAMP centre on different isoforms of adenylyl cyclase in pituitary‐derived GH3B6 cells. J Cell Sci. 2010; 123:95-106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Allen MD, Zhang J. Subcellular dynamics of protein kinase A activity visualized by FRET‐based reporters. Biochem Biophys Res Commun. 2006; 348:716-721. [DOI] [PubMed] [Google Scholar]
- 53.Verde I, Vandecasteele G, Lezoualc'h F, Fischmeister R. Characterization of the cyclic nucleotide phosphodiesterase subtypes involved in the regulation of the L‐type Ca2+ current in rat ventricular myocytes. Br J Pharmacol. 1999; 127:65-74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sanguinetti MC, Jurkiewicz NK. Role of external Ca2+ and K+ in gating of cardiac delayed rectifier K+ currents. Pflügers Arch. 1992; 420:180-186. [DOI] [PubMed] [Google Scholar]
- 55.Haj Slimane Z, Bedioune I, Lechêne P, Varin A, Lefebvre F, Mateo P, Domergue‐Dupont V, Dewenter M, Richter W, Conti M, El‐Armouche A, Zhang J, Fischmeister R, Vandecasteele G. Control of cytoplasmic and nuclear protein kinase A activity by phosphodiesterases and phosphatases in cardiac myocytes. Cardiovasc Res. 2014; 102:97-106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.La Gerche A, Burns AT, Mooney DJ, Inder WJ, Taylor AJ, Bogaert J, Macisaac AI, Heidbuchel H, Prior DL. Exercise‐induced right ventricular dysfunction and structural remodelling in endurance athletes. Eur Heart J. 2012; 33:998-1006. [DOI] [PubMed] [Google Scholar]
- 57.Piao L, Fang YH, Parikh KS, Ryan JJ, D'Souza KM, Theccanat T, Toth PT, Pogoriler J, Paul J, Blaxall BC, Akhter SA, Archer SL. GRK2‐mediated inhibition of adrenergic and dopaminergic signaling in right ventricular hypertrophy: therapeutic implications in pulmonary hypertension. Circulation. 2012; 126:2859-2869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jurevicius J, Fischmeister R. cAMP compartmentation is responsible for a local activation of cardiac Ca2+ channels by ß‐adrenergic agonists. Proc Natl Acad Sci USA. 1996; 93:295-299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zaccolo M, Pozzan T. Discrete microdomains with high concentration of cAMP in stimulated rat neonatal cardiac myocytes. Science. 2002; 295:1711-1715. [DOI] [PubMed] [Google Scholar]
- 60.de Man FS, Handoko ML, Guignabert C, Bogaard HJ, Vonk‐Noordegraaf A. Neurohormonal axis in patients with pulmonary arterial hypertension: friend or foe? Am J Respir Crit Care Med. 2013; 187:14-19. [DOI] [PubMed] [Google Scholar]
- 61.de Man FS, Handoko ML, van Ballegoij JJ, Schalij I, Bogaards SJ, Postmus PE, van der Velden J, Westerhof N, Paulus WJ, Vonk‐Noordegraaf A. Bisoprolol delays progression towards right heart failure in experimental pulmonary hypertension. Circ Heart Fail. 2012; 5:97-105. [DOI] [PubMed] [Google Scholar]