

Abstract
Background
Obstructive sleep apnea (OSA) is associated with increased risk of cardiovascular and cerebrovascular disease resulting from intermittent hypoxia (IH)‐induced inflammation. Cyclooxygenase (COX)‐formed prostanoids mediate the inflammatory response, and regulate blood pressure and cerebral blood flow (CBF), but their role in blood pressure and CBF responses to IH is unknown. Therefore, this study's objective was to determine the role of prostanoids in cardiovascular and cerebrovascular responses to IH.
Methods and Results
Twelve healthy, male participants underwent three, 6‐hour IH exposures. For 4 days before each IH exposure, participants ingested a placebo, indomethacin (nonselective COX inhibitor), or Celebrex® (selective COX‐2 inhibitor) in a double‐blind, randomized, crossover study design. Pre‐ and post‐IH blood pressure, CBF, and urinary prostanoids were assessed. Additionally, blood pressure and urinary prostanoids were assessed in newly diagnosed, untreated OSA patients (n=33). Nonselective COX inhibition increased pre‐IH blood pressure (P≤0.04) and decreased pre‐IH CBF (P=0.04) while neither physiological variable was affected by COX‐2 inhibition (P≥0.90). Post‐IH, MAP was elevated (P≤0.05) and CBF was unchanged with placebo and nonselective COX inhibition. Selective COX‐2 inhibition abrogated the IH‐induced MAP increase (P=0.19), but resulted in lower post‐IH CBF (P=0.01). Prostanoids were unaffected by IH, except prostaglandin E2 was elevated with the placebo (P=0.02). Finally, OSA patients had elevated blood pressure (P≤0.4) and COX‐1 formed thromboxane A2 concentrations (P=0.02).
Conclusions
COX‐2 and COX‐1 have divergent roles in modulating vascular responses to acute and chronic IH. Moreover, COX‐1 inhibition may mitigate cardiovascular and cerebrovascular morbidity in OSA.
Clinical Trial Registration
URL: www.clinicaltrials.gov. Unique identifier: NCT01280006
Keywords: blood pressure, cerebrovascular circulation, intermittent hypoxia, obstructive sleep apnea, prostaglandins
Introduction
Obstructive sleep apnea (OSA) is a chronic medical condition affecting 3% to 7% of the population1 and is associated with an increased risk of cardiovascular and cerebrovascular disease.2 OSA is characterized by repetitive cessation of breathing during sleep due to recurrent closure of the pharynx, resulting in chronic exposure to intermittent hypoxia (IH). In healthy humans, exposure to experimental IH disrupts peripheral and cerebral vascular regulation, manifested as an increased resting blood pressure3–4 and a smaller decrease in cerebrovascular resistance during hypoxia.5 While the mechanisms implicated in vascular dysfunction with OSA are multifactorial, IH‐induced inflammation is a fundamental component leading to increased cardiovascular morbidity.6–7
Prostanoids, ubiquitous molecules formed via the catabolism of arachidonic acid by cyclooxygenase (COX) isoenzymes COX‐1 and COX‐2, are of particular interest because they are both, important mediators of the inflammatory response,8 and vital regulators of the peripheral9 and cerebral vasculatures.10–11 The increased risk of peripheral and cerebral vascular disease associated with nonselective COX inhibiting nonsteroidal anti‐inflammatory drugs (NSAIDs) and selective COX‐2 inhibiting coxibs12–13 emphasize the importance of prostanoids in vascular regulation. Additionally, although nonselective and COX‐2 selective inhibition increase the risk of cardiovascular and cerebrovascular disease,12 there is still considerable controversy regarding the importance of COX‐1 and COX‐2 formed prostanoids for vascular regulation.13–16
With IH, prostanoid concentrations are shifted towards vasoconstriction and atherogenesis.17–18 However, whether this concentration shift is involved in IH‐induced increases in blood pressure, and altered cerebral blood flow (CBF) regulation, is not known. Thus, using an experimental model of IH in healthy humans, the objective of this study was to determine the role of COX‐1 and COX‐2 formed prostanoids in modulating the vascular responses to an acute (6 hours) IH exposure. Furthermore, the impact of chronic IH exposure on prostanoid concentrations was explored through comparison with a clinical population of newly diagnosed (untreated) OSA patients.
Methods
Approvals
This study was performed according to the Declaration of Helsinki, was approved by the Conjoint Health Research Ethics Board at the University of Calgary, and is registered as a clinical trial at www.clinicaltrials.gov (NCT01280006). After initial contact, volunteers were provided a familiarization session where they were introduced to the experimental set‐up, instrumentation, and provided with an informed consent. Volunteers were then given a minimum of 24 hours to reflect on the information provided prior to signing the informed consent.
Healthy Participants
Fifteen male volunteers were assessed for eligibility. Immediate exclusion criteria included residence in Calgary, Alberta (altitude≈1101 m) for <1 year, a body mass index (BMI)≥35 kg·m−2, cigarette smoking within the past year, and any active inflammatory or musculoskeletal condition for which volunteers were currently taking any NSAIDs. Two volunteers declined to participate. The remaining 13 volunteers underwent additional screening.
Screening
Screening started with a medical history, measurement of resting blood pressure, and a 12‐lead ECG. Volunteers with a history of cardiorespiratory disease, gastrointestinal bleeding, gastritis, inflammatory bowel disease, peptic ulcers, had a sulfa allergy, had an irregular ECG, or were hypertensive (ie, systolic/diastolic blood pressure >140/90) were excluded. Next, the presence of diabetes and liver and/or kidney dysfunction were assessed via fasting venous blood and urine samples. Venous blood samples were collected from the antecubital fossa into evacuated blood collection tubes (BD Vacutainers®; 1×4.5 mL sodium citrate tube, 1×5 mL serum separator tube [SST], 1×4 mL ethylenediaminetetraacetic acid [EDTA]). Samples were analyzed for glucose with a benchtop blood gas analyzer (ABL 827 Flex; Radiometer) and alanine transaminase (ALT), aspartate transaminase (AST), alkaline phosphatase (ALP), the international normalization ratio, plasma creatinine, and complete blood counts by Calgary Laboratory Services. Urine samples were collected into sterile specimen containers (LeakBuster, Starplex Scientific) and aliquoted into 2×10 mL vials for sodium and creatinine determination by Calgary Laboratory Services and the remainder of the sample was used for urinalysis (Chemstrip 10; Roche Diagnostics). Volunteers were excluded if there was evidence of diabetes (fasting glucose >7.0 mmol·L−1), liver dysfunction (ie, ALT, AST, ALP, and INR outside normal ranges), and/or renal dysfunction (estimated glomerular filtration rate (GFR)≤60 mL·min−1∙1.73 m−2, urinary protein excretion>150 mg·24 h−1). Finally, volunteers were screened for sleep apnea and nocturnal hypoxia with a level 3 diagnostic sleep study (Remmers Sleep Recorder Model 4.2; Sagatech Electronics). The Remmers Sleep Recorder records arterial oxyhemoglobin saturation (![]() ) and heart rate via a finger pulse oximeter, nasal airflow via a nasal cannula connected to a pressure transducer, snoring via a microphone placed on the suprasternal notch, and body position (supine/non‐supine) with a body position sensor within the microphone housing. The
) and heart rate via a finger pulse oximeter, nasal airflow via a nasal cannula connected to a pressure transducer, snoring via a microphone placed on the suprasternal notch, and body position (supine/non‐supine) with a body position sensor within the microphone housing. The ![]() signal is recorded at 1 Hz and analyzed using a proprietary scoring algorithm. The respiratory disturbance index (RDI) is calculated as the number of times
signal is recorded at 1 Hz and analyzed using a proprietary scoring algorithm. The respiratory disturbance index (RDI) is calculated as the number of times ![]() decreased by ≥4%, divided by the total recording time. This system has been validated against polysomnography, the gold standard diagnostic test for OSA.19–20 Raw data from the sleep recorder (ie,
decreased by ≥4%, divided by the total recording time. This system has been validated against polysomnography, the gold standard diagnostic test for OSA.19–20 Raw data from the sleep recorder (ie, ![]() , nasal airflow, snoring, body position, and heart rate) was reviewed by a sleep medicine physician (P.J.H.) to confirm the absence of OSA and nocturnal hypoxia. Volunteers were excluded if they had a RDI>5 events·h−1 and/or a mean
, nasal airflow, snoring, body position, and heart rate) was reviewed by a sleep medicine physician (P.J.H.) to confirm the absence of OSA and nocturnal hypoxia. Volunteers were excluded if they had a RDI>5 events·h−1 and/or a mean ![]() during sleep <90%.
during sleep <90%.
Volunteers who met all inclusion criteria were randomized into the experimental protocol. Figure 1 is a CONSORT diagram showing the flow of participants through the study.
Figure 1.
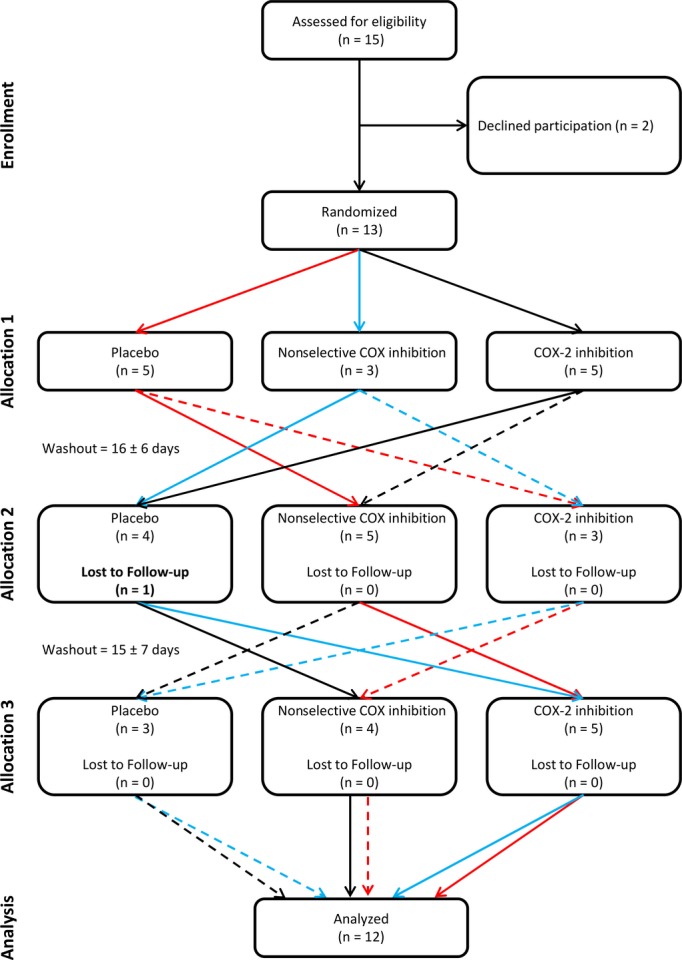
CONSORT diagram showing the flow of healthy participants exposed to intermittent hypoxia (IH).
Experimental protocol
The study used a double‐blind, placebo‐controlled, randomized, cross‐over experimental design consisting of 3 experimental IH exposures. For 4 days prior to each IH exposure, each participant ingested one of the following: a lactose placebo, the nonselective cyclooxygenase (COX) inhibitor indomethacin, or the selective COX‐2 inhibitor Celebrex®. On IH exposure days, participants were instructed to have a light breakfast prior to taking the morning dose of medication. The participant arrived in the laboratory at ≈0800 hour and provided a urine sample shortly after arrival. Next, resting brachial blood pressure and cerebral blood flow were assessed. Subsequently, the participant was exposed to 6 hours of IH. After the IH exposure, the participant provided another urine sample and resting blood pressure and cerebral blood flow measurements were repeated. A minimum of 4 days was provided between the conclusion of an IH exposure and the start of the next medication. Figure 2 shows a schematic of experimental protocol.
Figure 2.

Healthy male participants were exposed to 6 hours of isocapnic intermittent hypoxia on 3 separate occasions (IH Exposure #1 to 3). For 4 days before each IH exposure, participants ingested either 100 mg lactose placebo (3 times/day), 50 mg (3 times/day) of the nonselective cyclooxygenase (COX) inhibitor indomethacin, or 200 mg (2 times/day) of the selective COX‐2 inhibitor Celebrex® (Drug Ingestion #1 to 3), administered in a random order. For each IH Exposure, participants arrived at ≈0800 hours and immediately provided a urine sample () and were instrumented for Physiological Measurements of blood pressure and cerebral blood flow. Next, each participant was exposed to 6 hours of IH that consisted of cycling their end‐tidal partial pressure of oxygen ( ) between 88 mm Hg (normal value for the altitude (≈1101 m) at which the laboratory is located) and 45 mm Hg every 60 seconds. After each IH exposure, participants provided another urine sample () and the Physiological Measurements (ie, blood pressure and cerebral blood flow) were repeated. Each IH exposure was separated by at least 4 days to allow washout of the medication from their system (Drug Washout #1 to 3).
) between 88 mm Hg (normal value for the altitude (≈1101 m) at which the laboratory is located) and 45 mm Hg every 60 seconds. After each IH exposure, participants provided another urine sample () and the Physiological Measurements (ie, blood pressure and cerebral blood flow) were repeated. Each IH exposure was separated by at least 4 days to allow washout of the medication from their system (Drug Washout #1 to 3).
Medication dosage
The placebo (100 mg lactose placebo) was ingested 3 times a day at 0800, 1400, and 2000 hours. Similarly, the nonselective COX inhibitor indomethacin (50 mg) was also ingested 3 times a day at 0800, 1400, and 2000 hours. The selective COX‐2 inhibitor Celebrex® (200 mg) was ingested 2 times a day at 0800 and 2000 hours.21 The double‐blind study was maintained by adding a placebo as the second pill at 1400 hours when participants were taking Celebrex®. On IH exposure days (ie, day 5), the dosage regimen was maintained through the end of physiological sampling and measurement (ie, only the final dose scheduled for 2000 hour was omitted in all conditions).
Intermittent hypoxia exposure
Exposure to IH was performed in a custom‐built normobaric hypoxic room4 and consisted of cycling between 1 minute of hypoxia and 1 minute of normoxia for 6 hours, thus replicating an RDI of 30 events·h−1, which is seen in patients with moderate‐to‐severe OSA. Hypoxia was induced by maintaining the fraction of O2 within the room at a level sufficient to decrease the end‐tidal partial pressure of O2 (![]() ) to 45 mm Hg within 60 seconds and normoxia was established by administering 100% O2 to the participant at a flow rate sufficient to return
) to 45 mm Hg within 60 seconds and normoxia was established by administering 100% O2 to the participant at a flow rate sufficient to return ![]() to 88 mm Hg (normal
to 88 mm Hg (normal ![]() for the altitude at which the laboratory is located [≈1101 m]) within 60 seconds. The end‐tidal partial pressure of CO2 (
for the altitude at which the laboratory is located [≈1101 m]) within 60 seconds. The end‐tidal partial pressure of CO2 (![]() ) was maintained at normal levels by administering 100% CO2 to the participant during each hypoxic cycle.
) was maintained at normal levels by administering 100% CO2 to the participant during each hypoxic cycle.
Briefly, during all IH exposures the participants were instrumented with a nasal cannula and a non‐vented, full‐face respiratory mask (Mirage NV Full Face Mask Series 2; Resmed) attached to a 2‐way non‐rebreathing valve (2600 series; Hans Rudolph). Connected to the inspired side of the non‐rebreathing valve was a 25 cm long, wide‐bore tubing acting as a mixing chamber through which the participant could breathe the hypoxic gas within the chamber and 100% O2 and/or CO2 could be delivered to the participant as required. Respired gases were sampled continuously at a rate 150 mL·min−1 from the nasal cannula and the fractions of O2 and CO2 were measured by a dual O2 and CO2 gas analyzers (NormocapOxy; Datex‐Ohmeda); allowing for breath‐by‐breath determination of ![]() and
and ![]() . Finally, a pulse oximeter (3900p; Datex‐Ohmeda) was attached to the earlobe for monitoring of
. Finally, a pulse oximeter (3900p; Datex‐Ohmeda) was attached to the earlobe for monitoring of ![]() .
.
Blood pressure, heart rate, and cerebral blood flow measurements
Before and immediately after IH exposure participants rested for 10 minutes in a semi‐reclined position for assessment of brachial artery blood pressure, heart rate, and CBF. Brachial blood pressure was measured using an automated oscillometric blood pressure monitor (Dinamap Compact S; Critikon Inc) and the mean of at least 2 measurements was recorded. Heart rate was monitored via a 3‐lead ECG (Micromon 7142 B; Kontron Medical) and CBF was assessed by monitoring the velocity of blood travelling through the middle cerebral artery with transcranial Doppler ultrasonography.4–5 Resting heart rate and CBF were recorded as the mean rate and velocity, respectively, over the last 5 minutes of the resting period.
Urine sampling
Midstream urine samples were collected into sterile specimen containers (LeakBuster, Starplex Scientific) immediately upon arrival in the laboratory in the morning and immediately after the 6 hours of IH exposure. This permitted assessment of the urinary prostanoid production across the 6‐hour interval of IH. Each urine sample was immediately aliquoted into prefrozen, sterile centrifuge tubes as approximately eight 10 mL samples and promptly stored at −80°C for future prostanoid analyses.
OSA Patients
To extend observations from our acute model of IH to a clinical model of chronic IH, 33 newly diagnosed OSA patients were recruited from the Foothills Medical Centre Sleep Centre and a respiratory homecare company (Healthy Heart Sleep Company) between June 2011 and May 2012. Men and women, aged 18 to 70, with moderate‐to‐severe OSA and significant nocturnal hypoxia, were eligible to participate in the study. All participants underwent a medical history, physical examination, and laboratory screening. Exclusion criteria included cardiovascular disease, cerebrovascular disease, kidney disease, uncontrolled hypertension (blood pressure >140/90 despite maximal use of antihypertensive medications), diabetes, severe lung disease, current smoking, pregnancy, use of NSAIDs or exogenous sex hormones. These criteria deliberately excluded many of the co‐morbidities commonly associated with OSA.2
Determination of OSA severity
Similar to healthy participants, OSA participants performed an unattended, level 3 diagnostic sleep study (Remmers Sleep Recorder Model 4.2; Sagatech Electronics) following current guidelines and recommendations.22 Sleep apnea was defined as a RDI ≥15 as this reflects moderate‐to‐severe sleep apnea which is likely to be clinically significant.20 Significant nocturnal hypoxia was defined as ![]() ≤90% for ≥12% of the total monitoring time as used within the Sleep Heart Health Study.23 The raw data from the Remmers Sleep Recorder was reviewed by a sleep medicine physician (P.J.H.) to confirm the presence and severity of OSA.
≤90% for ≥12% of the total monitoring time as used within the Sleep Heart Health Study.23 The raw data from the Remmers Sleep Recorder was reviewed by a sleep medicine physician (P.J.H.) to confirm the presence and severity of OSA.
Experimental protocol
OSA patients were participating in a larger study assessing the impact of OSA on the renin‐angiotensin system (RAS) and instructed to consume >200 mmol of sodium per day for 3 days before each study day to ensure maximum suppression of the RAS.24 Subjects were subsequently studied while awake in the supine position in a temperature‐controlled, quiet room after an 8‐hour fast. All patients provided a second morning midstream urine sample immediately upon arrival. Premenopausal female OSA patients were studied 14 days after the first day of the last menstrual period, determined by counting days.25 Finally, patients on hypertensive medications that interfere with RAS activity were switched to a calcium‐channel blocker (amlodopine) to achieve adequate blood pressure control 2 weeks prior to the study day, as these agents are considered to have a neutral effect on the RAS.26
Blood pressure measurements
Similar to healthy participants, brachial blood pressure was measured while patients rested in a semi‐reclined position using an automated oscillometric blood pressure monitor (Critikon Dinamap Pro Care; GE Healthcare). The mean of at least 2 measurements was recorded.
Urine sampling
The second morning midstream urine sample was collected into sterile specimen containers (LeakBuster, Starplex Scientific) and immediately put on ice and subsequently stored in −80°C until analyzed for prostanoid concentrations. Thus, urine samples from the OSA patients were collected by similar methodology to the IH healthy participants.
Urinary Prostanoids
Urine samples collected from healthy participants before, and after, IH exposures and from OSA patients were thawed completely in a chilled water bath, centrifuged at 3000 rpm, and the supernatant from each sample was analyzed via enzyme immunoassays for the stable urinary metabolites of prostacyclin (PGI2; Enzo Life Sciences), prostaglandin E2 (PGE2), thromboxane A2 (TXA2), and prostaglandin F2α (PGF2α; Cayman Chemical Company). To reduce quantitation error intrinsic to the assay methodology, all samples were analyzed in triplicate, with the mean value carried forward into subsequent analyses. All assays were performed according to the manufacturer's instructions and all inter‐ and intra‐assay measures of variability, as well as quality control pools, were within expected limits as established by the manufacturer.
Statistical Analyses
The sample size of healthy participants was determined based upon previous findings from our group showing a 6.6±6.3 mm Hg (mean±SD) increase in mean arterial pressure using the same 6‐hour exposure to isocapnic IH protocol employed in the current study.4 In order to achieve a power of at least 0.85 for a 1‐tailed, paired t test with an alpha of 0.05 a sample size of 10 was predicted to be required. Considering the potential for a 20% dropout rate, 15 volunteers were assessed for participation in the study.
Dependent variables were assessed for a normal distribution using the Wilk‐Shapiro test. In healthy participants most dependent variables were found to have a normal distribution with 1 variable (PGE2) exhibiting a slight departure from normality at 2 (out of 6) time points analyzed and 2 variables (TXA2 and the PGI2:TXA2 ratio) showing a slight departure from normality at 1 time point (out of 6) analyzed. As previous research has demonstrated that analyses of variance are robust to departures from the assumption of normality even under small sample sizes,27–29 changes in blood pressure, heart rate, cerebral blood flow, and prostanoid concentrations before, and after, IH were analyzed using a 3‐by‐2 repeated measures analysis of variance (RM ANOVA) with the factors of medication (placebo, nonselective, and selective COX‐2 inhibition) and IH (pre‐ and post‐IH). Furthermore, if the assumption of Sphericity was violated, the Greenhouse‐Giesser corrected P value was reported. Finally, if there was a significant main effect, post hoc comparisons were performed incorporating a Bonferroni correction for multiple comparisons.
For OSA patients, age, BMI, diastolic blood pressure (DBP), and urinary TXA2 concentrations were normally distributed, while weight, mean arterial (MAP) and systolic blood pressure (SBP), and urinary concentrations of PGI2, PGE2, and PGF2α, and the PGI2:TXA2 ratio were not normally distributed. Therefore, comparisons between healthy participants and OSA patients for normally distributed variables were performed using independent sample Student t tests while comparisons of non‐normally distributed variables were performed using the Mann‐Whitney U test. For all comparisons, a Bonferroni correction for multiple comparisons was incorporated into the analyses. All results are provided as the mean±standard deviation and alpha was set a priori at 0.05.
Results
One healthy participant was removed from the study because of an adverse reaction to the first medication to which they were allocated (Figure 1). The remaining 12 participants completed all IH exposures and none were excluded on the basis of physiological measures. Table shows the characteristics of the healthy participants and newly diagnosed OSA patients.
Table 1.
Characteristics of Healthy Participants and Obstructive Sleep Apnea (OSA) Patients
| Healthy Participants | OSA Patients | |
|---|---|---|
| Sample size (n) | 12 | 33 |
| Gender | 12 male | 23 male |
| Age, y | 25.8±5.1 | 51.7±10.3* |
| BMI, kg m−2 | 24.9±2.5 | 34.9±6.8* |
| MAP, mm Hg | 83.0±8.3 | 96.2±10.3* |
| SBP, mm Hg | 117.2±12.4 | 132.1±18.1* |
| DBP, mm Hg | 65.9±8.7 | 78.2±8.9* |
| RDI, events h−1 | 1.8±1.1 | 47.4±21.3* |
| Mean |
94.8±1.1 | 88.8±3.9* |
| Minimum |
89.2±4.6 | 69.9±8.6* |
| Time |
0.0±0.1 | 41.1±25.5* |
BMI, body mass index; MAP, mean arterial blood pressure; SBP, systolic blood pressure; DBP, diastolic blood pressure; RDI, respiratory disturbance index; Mean SaO2, mean arterial oxyhemoglobin saturation (SaO2) for entire duration of monitoring during a level 3 sleep diagnostic test; Minimum SaO2, lowest SaO2 recorded during a level 3 diagnostic sleep test; and Time SaO2 < 90%, percentage of total monitoring time that SaO2 was less than 90% during a level 3 diagnostic sleep test. All data provided as mean±SD.
P≤0.05 versus healthy participants.
Healthy Participants
Blood pressure, heart rate, and cerebral blood flow before intermittent hypoxia
Ingestion of the lactose placebo for 4 days did not change resting MAP, SBP or DBP (P≥0.46), while ingestion of the nonselective COX inhibitor indomethacin for 4 days resulted in higher MAP, SBP, and DBP, and a lower heart rate, compared with the placebo (P≤0.05; Figure 3). Blood pressures and heart rate were not impacted by 4 days of ingestion of the selective COX‐2 inhibitor Celebrex® as blood pressures and heart rate were similar to placebo (P≥0.56; Figure 3). The blood pressure findings are consistent with the acute effects of each medication (ie, 2 to 3 hours after ingesting a single dose) on resting blood pressure (Figure 4). As salt balance may influence blood pressure,30 24‐hour sodium excretion was estimated31 from urine samples collected before each IH exposure. Sodium excretion was similar among the 3 conditions (P≥0.73; Figure 5), and thus does not explain the observed blood pressures differences.
Figure 3.

Resting mean (MAP), systolic (SBP) and diastolic (DBP) brachial blood pressures and heart rate of healthy participants before (□), and after (■) 6 hours of IH. † indicates significantly different from placebo before‐IH values with P≤0.05; * indicates significant effect of IH within each condition P≤0.05; ‡ significantly different from after‐IH placebo values with P≤0.05; and †† indicates significant difference from nonselective COX inhibition after‐IH values. IH indicates intermittent hypoxia.
Figure 4.

Using a single‐blind, placebo‐controlled, randomized, cross‐over experimental design, the acute blood pressure responses to placebo, nonselective COX inhibitor, and selective COX‐2 inhibitor medications were assessed in 10 healthy participants (9 overlap with the 12 from the IH study). Participants ingested a single oral dose of placebo (100 mg lactose), the nonselective COX inhibitor indomethacin (50 mg), and the selective COX‐2 inhibitor Celebrex® (200 mg). Mean (MAP; A), systolic (SBP; B) and diastolic blood pressures (DBP; C) brachial blood pressures were measured before (), and either 2 hours (placebo and non‐selective COX inhibition) or 3 hours (COX‐2 inhibition) after ingestion of medications (■). The mean of 3 separate blood pressure measurements taken over 10 minutes while participants were resting in a seated position was recorded. Participants ate a standardized breakfast prior to ingesting the medications. After ingestion, the participant remained in the seated, resting position watching television until post‐drug measurements were performed. The time to post‐drug measurements was based upon the pharmacokinetics of each medication and the time required to reach peak serum levels. The 3 experimental days were separated by at least 4 days. Pre‐drug blood pressures were not different between the 3 conditions (P≥0.26). MAP and SBP were significantly elevated after ingestion of the non‐selective COX inhibitor (P≤0.01). These findings support the conclusion that prostanoids formed via COX‐1 are the primary regulator of resting blood pressure in healthy individuals. Results provided as mean±SD. * significantly different from Pre‐drug values. COX indicates cyclooxygenase; IH, intermittent hypoxia.
Figure 5.
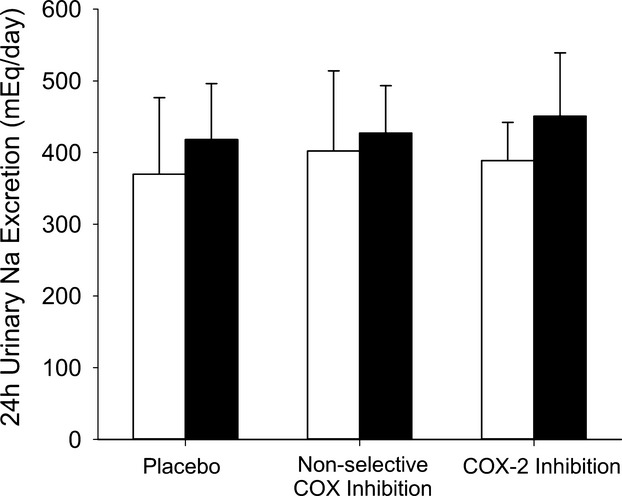
Estimated 24‐hour sodium excretion before (□), and after (■), intermittent hypoxia exposure.
Cerebral blood flow and cerebrovascular resistance (CVR=MAP/CBF) prior to IH are shown in Figure 6. CBF was lower and CVR was higher after 4 days of nonselective COX inhibition compared with the placebo, (P≤0.04). There was no difference in CBF and CVR between the selective COX‐2 inhibition and placebo conditions (P≥0.92). Again, these results are similar to the acute effects of each medication, except the acute effects of nonselective COX inhibition were much greater—≈26% decrease in CBF and ≈42% increase in CVR versus ≈9% decrease in CBF and ≈14% increase in CVR (Figure 7).
Figure 6.

Cerebral blood velocity through the middle cerebral artery (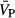 ) and cerebrovascular resistance (CVR) of healthy participants before (□), and after (■) 6 hours of IH. † indicates significantly different from placebo before‐IH values with P≤0.05; * indicates significant effect of IH within each condition P≤0.05. IH inidcates intermittent hypoxia.
) and cerebrovascular resistance (CVR) of healthy participants before (□), and after (■) 6 hours of IH. † indicates significantly different from placebo before‐IH values with P≤0.05; * indicates significant effect of IH within each condition P≤0.05. IH inidcates intermittent hypoxia.
Figure 7.

Acute cerebral blood velocity through the middle cerebral artery ( ) and cerebrovascular resistance (CVR) responses to a single dose of of placebo (100 mg lactose), nonselective COX inhibitor (50 mg indomethacin), and selective COX‐2 inhbitor (200 mg Celebrex®) medications.
) and cerebrovascular resistance (CVR) responses to a single dose of of placebo (100 mg lactose), nonselective COX inhibitor (50 mg indomethacin), and selective COX‐2 inhbitor (200 mg Celebrex®) medications. 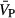 and CVR were assessed assessed before (), and either 2 hours (placebo (100 mg lactose) and nonselective COX inhibition) or 3 hours (COX‐2 inhibition) after ingesting medications (■). Pre‐drug
and CVR were assessed assessed before (), and either 2 hours (placebo (100 mg lactose) and nonselective COX inhibition) or 3 hours (COX‐2 inhibition) after ingesting medications (■). Pre‐drug  and CVR were not different between the 3 drug conditions (P≥0.24). At the post‐drug time point,
and CVR were not different between the 3 drug conditions (P≥0.24). At the post‐drug time point,  was significantly lower within all 3 drug conditions (P≤0.05) while CVR was significantly increased after ingestion of only the non‐selective COX inhibitor (P<0.01). The magnitude of the decrease in
was significantly lower within all 3 drug conditions (P≤0.05) while CVR was significantly increased after ingestion of only the non‐selective COX inhibitor (P<0.01). The magnitude of the decrease in 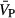 from the pre‐drug to post‐drug condition was significantly greater with nonselective COX inhibition compared to placebo (P<0.01) and selective COX‐2 inhibition (P<0.01). These findings support the conclusion that prostanoids formed via COX‐1 are the primary regulators of resting cerebral blood flow in healthy individuals. Results provided as mean±SD. * indicates significant difference from pre‐drug values with P≤0.05; ‡ significantly different from post‐drug placebo values with P≤0.05; and †† indicates significant difference from nonselective COX inhibition post‐drug values with P≤0.05. COX indicates cyclooxygenase.
from the pre‐drug to post‐drug condition was significantly greater with nonselective COX inhibition compared to placebo (P<0.01) and selective COX‐2 inhibition (P<0.01). These findings support the conclusion that prostanoids formed via COX‐1 are the primary regulators of resting cerebral blood flow in healthy individuals. Results provided as mean±SD. * indicates significant difference from pre‐drug values with P≤0.05; ‡ significantly different from post‐drug placebo values with P≤0.05; and †† indicates significant difference from nonselective COX inhibition post‐drug values with P≤0.05. COX indicates cyclooxygenase.
Blood pressure and cerebral blood flow after intermittent hypoxia
After exposure to IH, MAP was increased within the placebo (P=0.03) and nonselective COX inhibition conditions (P=0.05) to a similar degree, but was unchanged within the selective COX‐2 inhibition condition (P=0.19; Figure 3A). There was a trend for SBP to be increased in all conditions (placebo: P=0.09; nonselective COX inhibition: P=0.08; selective COX‐2 inhibition: P=0.08; Figure 3B) while DBP was increased with the placebo (P=0.03), showed a trend (P=0.07) to be elevated within the nonselective COX inhibition condition, but was unchanged from the pre‐IH level with selective COX‐2 inhibition (P=0.41; Figure 3C). As a result, MAP, SBP, and DBP with nonselective COX inhibition remained significantly higher than within the placebo condition (P≤0.01) and was higher than observed within the selective COX‐2 inhibition condition (P≤0.03) after IH. Furthermore, after the IH exposure, heart rate was similar to the pre‐IH level within the placebo (P=0.28) and nonselective inhibition conditions (P=0.39), but was increased in the selective COX‐2 inhibition condition (P=0.01; Figure 3D). Moreover, post‐IH heart rate was lower with nonselective COX inhibition compared to the placebo (P=0.01) and selective COX‐2 inhibition (P<0.01) conditions. Additionally, although IH elevated estimated 24‐hour sodium excretion32 within all conditions (P=0.05; IH main effect) the elevation was similar across all 3 conditions (P=0.54). As a result, estimated 24‐hour sodium excretion was similar between all conditions after IH (P=0.45; Figure 5).
Exposure to 6 hours of IH, did not alter CBF within the placebo and nonselective COX inhibition conditions (P≥0.54), but resulted in a significantly decreased CBF (≈10%) when COX‐2 was selectively inhibited (P=0.01; Figure 6A). Combined with the MAP changes, CVR was unchanged within the placebo and nonselective COX inhibition conditions (P≥0.29), but was elevated with selective COX‐2 inhibition (P<0.01; Figure 6B).
Urinary prostanoids
Urinary prostanoids concentrations before, and after, IH exposure are shown in Figure 8. Compared with placebo concentrations (Figure 8A), nonselective COX inhibition (Figure 8B) significantly decreased the vasodilatory prostanoids prostacyclin (PGI2) and prostaglandin E2 (PGE2; P<0.01) along with the vasoconstrictor prostanoids thromboxane A2 (TXA2) and prostaglandin F2α (PGF2α; P≤0.01). Similarly, selective COX‐2 inhibition (Figure 8C) decreased the vasodilatory prostanoids PGI2 and PGE2 (P≤0.04), but in contrast to nonselective COX inhibition, did not change concentrations of the vasoconstrictor prostanoids TXA2 and PGF2α (P=1.0 for both prostanoids). Accordingly, the PGI2:TXA2 ratio was higher after 4 days of nonselective COX inhibition and lower after 4 days of selective COX‐2 inhibition compared with the placebo (P<0.01).
Figure 8.

Urinary concentrations (normalized to creatinine) of prostacyclin (PGI2), prostaglandin E2 (PGE2), thromboxane A2 (TXA2), and prostaglandin F2α (PGF2α) and the PGI2:TXA2 ratio in healthy participants before (□), and after (■), IH exposures within the placebo (A), nonselective COX inhibition (B) and the selective COX‐2 inhibition (C) conditions. † indicates significantly different from placebo before‐IH values with P≤0.05; * indicates significant effect of IH within each condition P≤0.05; and ‡ indicates significant difference from after‐IH placebo values with P≤0.05. COX indicates cyclooxygenase; IH, intermittent hypoxia.
Within the placebo condition, IH increased PGE2 (P=0.02), but did not alter any of the other measured prostanoids (P≥0.76) or the PGI2:TXA2 ratio (P=0.25). This increase in PGE2 following IH was prevented by both, nonselective COX inhibition (P=0.45), and selective COX‐2 inhibition (P=0.13). Furthermore, similar to the placebo condition, IH did not alter any of the additional measured prostanoids within the nonselective COX and selective COX‐2 inhibition conditions (P≥0.16).
OSA Patients
Untreated OSA patients had significantly higher MAP, SBP, and DBP compared with healthy participants before IH exposure within the placebo condition (Table). Moreover, OSA patients had a lower urinary concentration of PGE2 (P=0.03) and a higher urinary concentration of the vasoconstrictor TXA2 (P<0.01; Figure 9) compared with healthy participants before IH exposure within the placebo condition. In contrast, OSA patients had similar concentrations of the vasodilator PGI2, and the vasoconstrictor PGF2α, as well as the PGI2:TXA2 ratio to the healthy participants before IH exposure within the placebo condition (P≥0.20).
Figure 9.

Urinary prostanoid concentrations (normalized to creatinine) in newly diagnosed OSA patients. † indicates significantly different from placebo pre‐IH values with P≤0.05 and ‡ significantly different from post‐IH placebo values with P≤0.05. IH indcates intermittent hypoxia; OSA, obstructive sleep apnea; PGE2, prostaglandin E2; PGF2α, prostaglandin F2α; PGI2, prostacyclin; TXA2, thromboxane A2.
Compared with the prostanoid concentrations of healthy participants after IH, PGI2, and PGF2α concentrations remained similar between OSA patients and healthy participants (P≥0.28), while TXA2 remained significantly higher (P<0.01) and PGE2 concentrations remained lower (P<0.01) in OSA patients. Additionally, the PGI2:TXA2 ratio for OSA patients was lower (P=0.04) than the post‐IH value of healthy participants.
Discussion
The principal findings of this double‐blind, randomized, placebo‐controlled, cross‐over study were (1) in healthy participants, 4 days of nonselective COX inhibition elevated resting blood pressure and decreased CBF, while 4 days of selective COX‐2 inhibition did not change either physiological variable; (2) in the placebo and nonselective COX inhibition conditions, IH‐mediated similar elevations in blood pressure, and selective COX‐2 inhibition prevented this increase; (3) 6 hours of IH did not change resting CBF when COX isoenzymes were uninhibited (placebo) or when both COX‐1 and COX‐2 were inhibited (nonselective inhibition), but significantly decreased CBF and increased CVR when COX‐2 was selectively inhibited; and (4) untreated OSA patients had higher blood pressures and urinary concentrations of the vasoconstrictor TXA2 compared with healthy participants.
Nonselective COX inhibition and selective COX‐2 inhibition are associated with increased risk of cardiovascular and cerebrovascular disease.12 Based upon decreases in urinary concentrations of the vasodilator, antithrombotic prostacyclin (PGI2),8 but no alteration in the vasoconstrictor, thrombotic thromboxane A2 (TXA2)8 concentrations with selective COX‐2 inhibitors and COX‐2 knockout mice,33–34 COX‐2 has been proposed as the primary COX isoenzyme responsible for endothelial production of PGI2. Accordingly, inhibition of COX‐2 with traditional NSAIDs (non‐selective COX inhibitors) is thought to be responsible for the increased cardiovascular and cerebrovascular risks associated with these medications, but this is not without controversy.14–16 A recent meta‐analysis has enhanced this debate by concluding certain nonselective NSAID medications have similar vascular risks as selective COX‐2 inhibitors.12 In our healthy participants, 4 days of nonselective COX inhibition increased blood pressure and decreased CBF. Although we also observed significantly lower urinary PGI2, but maintained TXA2 concentrations with selective COX‐2 inhibition,33,35 these did not translate into elevated blood pressure or altered CBF. The lack of change in blood pressure is similar to what has been reported previously.33 Together, these findings do not support the contention that inhibition of COX‐2 derived PGI2 is responsible for the increased cardiovascular and cerebrovascular risks with nonselective COX inhibitors. In conjunction with the observed acute effects of nonselective and COX‐2 selective inhibition on blood pressure and CBF, these findings indicate prostanoids formed via COX‐1 are the primary regulators of resting blood pressure16 and CBF in healthy humans.
Systemic inflammation induced by IH is an important mechanism for the cardiovascular and cerebrovascular sequelae of OSA by contributing to the development of endothelial dysfunction.6–7 Currently, there is mounting evidence that implicates alterations in prostanoids with IH as part of the pathway leading to vascular dysfunction.17–18 Nacher et al17 showed that rats subjected to OSA for 3 hours (60 apneas/h; 15‐second apneas) or 3 hours of IH (15‐second hypoxia and 15‐second normoxia) had decreased plasma PGI2 metabolites and elevated TXA2 metabolites compared to a control group. Regrettably, no physiological responses were reported and, consequently, the impact of these prostanoid changes on blood pressure and CBF is unknown. More recently, Gautier‐Veyret et al18 reported COX‐1 mRNA was elevated by ≈70% and COX‐2 mRNA was increased by ≈25% in ApoE knockout mice exposed to chronic IH (8 weeks; 60‐second IH cycles; 8 hours/day). In addition, IH‐induced atherosclerotic lesion size correlated with COX‐1 and thromboxane synthase (downstream enzyme from COX‐1 responsible for TXA2 formation) mRNA, and selective COX‐1 inhibition reduced atherosclerotic lesion size following IH exposure. Although the elevated COX mRNA did not translate into enhanced secretion capacity (ie, stimulated release) of PGI2 and TXA2 and basal PGI2 and TXA2 concentrations were not assessed, this study supports a greater role for COX‐1 in the etiology cardiovascular sequelae of chronic IH exposure.
Unlike previous reports, the current study specifically investigated the relationship between prostanoids derived from both COX‐1 and COX‐2, and modifications in resting blood pressure and CBF with acute IH exposure in healthy humans. In the placebo condition, both MAP and DBP were elevated after 6 hours of IH, consistent with previous findings from our group using the same model of IH.4 Although nonselective COX inhibition elevated blood pressure prior to IH exposure, the IH‐induced elevation in blood pressure was similar to the placebo condition. In contrast, selective COX‐2 inhibition prevented the increase in blood pressure with IH. This protective effect of selective COX‐2 inhibition is potentially the result of altering the interaction between COX‐2 activity and the renin angiotensin system (RAS).
Upregulation of the RAS via enhanced sympathetic activation is intimately involved in IH‐mediated elevations in blood pressure with renal denervation36 and suppression of the RAS via salt loading37 in rats preventing IH‐induced increases in blood pressure. More recently, in a pig model of OSA, renal denervation reduced post‐apneic rises in blood pressure and blunted the increase in circulating RAS components (plasma renin activity (PRA) and plasma aldosterone) associated with 4 hours of obstructive apneas (2‐minute apneas, 4/hour).38–39 In addition, we recently showed, using the same acute IH paradigm as in the present study, blockade of type 1 angiotensin‐II receptors (AT1Rs) prevents the increase in blood pressure associated with IH4 by blunting increases in oxidative stress and decreases in nitric oxide bioavailability.40 In addition to increasing superoxide generation,41 angiotensin‐II also increases COX‐2 expression in vascular smooth muscle via binding to AT1Rs, and the increased COX‐2 activity magnifies the actions of angiotensin‐II on vascular smooth muscle cells.42–43 Moreover, prostanoids mediate renin release from the kidneys in response to sympathetic activation44 and this appears to be COX‐2 dependent21 as renin release is decreased with COX‐2 inhibition.45 Therefore, our results indicate RAS‐induced increases in blood pressure with IH may be COX‐2 dependent.
Although we did not assess components of the circulating RAS in the current study, our group has previously reported, using the identical model of isocapnic‐IH during wakefulness (ie, IH exposure between ≈09:30 and 15:30 hours), that plasma renin activity (PRA) and plasma aldosterone concentration are lower after the 6 hours of IH. This decrease reflected the diurnal variation in PRA and plasma aldosterone as the decrease observed was similar to a 6‐hour sham‐IH (ie, euoxia) condition.4 Therefore, there is the potential the observed effects of IH on blood pressure may have been enhanced if the IH was administered during sleep when PRA and aldosterone concentrations are typically higher.46
Following acute IH exposure, resting CBF is typically maintained or increased4–5 despite decreases in regulators of basal CBF such as nitric oxide.4–5,40,47 Since selective COX‐2 inhibition resulted in a decreased CBF after IH exposure, the maintenance of CBF following IH may be dependent upon increased COX‐2 production of vasodilatory prostanoids (PGI2 and PGE2) involved in basal CBF regulation.47 Although speculative, an increase in PGI2 and PGE2 may be due to an augmented expression and activity of endothelial COX‐2 stimulated by greater liberation of NF‐κβ, and concentrations of IL‐1β and TNFα48–50 with IH exposure.17,51
The divergent responses to IH observed between nonselective COX and selective COX‐2 inhibition are the likely result of indomethacin having ≈50 times greater selectivity for COX‐1 compared with COX‐2.52–53 Thus, the contrasting responses of the two COX inhibitors reflect the greater role of COX‐2 in the blood pressure and CBF responses to acute IH. Figure 10 outlines putative pathways through which COX‐2 inhibition may have prevented the expected increase in blood pressure and maintained CBF with acute IH exposure.
Figure 10.

Putative pathways through which selective COX‐2 inhibition may have prevented the IH‐induced blood pressure elevation and resulted in a decreased cerebral blood flow. With IH exposure, there is an up regulation of the renin‐angiotensin system (RAS) via increased sympathetic nervous system activation resulting in increased renin activity and angiotensin‐II formation36–37 (Right; A). Subsequently, angiotensin‐II binds to angiotensin type 1 receptors (AT1r) on vascular smooth muscle cells causing vasoconstriction and an increase in blood pressure (Right; B). Via binding to the AT1r, angiotensin‐II increases COX‐2 expression, which enhances the vascular effects of angiotensin‐II on the vascular smooth muscle cells42–43 (Right; C). Additionally, since renin release is COX‐2 dependent,21,45 IH induced increases in COX‐2 may also enhances renin activity and formation of angiotensin‐II which, in turn, will further enhance COX‐2 expression (Right; D). Thus, it is proposed that the required magnitude of RAS up regulation to produce an increase in blood pressure with IH is dependent upon the augmenting effects of COX‐2. In addition, COX‐2 expression is enhanced via IH induced inflammation (eg, IL‐1β, TNFα, and NFκβ—Right; E). Therefore, selectively inhibiting COX‐2 may have prevented the augmentation of the vascular effects of angiotensin‐II as well as minimizing renin activity. As a result, the RAS system was not sufficiently up regulated by IH resulting in maintenance of blood pressure. In contrast, within the cerebral vasculature, an elevation of NF‐κβ, IL‐1β, and TNFα may lead to augmented expression and activity of endothelial COX‐248–50 and enhanced release of vasodilatory prostanoids involved in regulating resting CBF47 leading to the maintenance of CBF after IH4–5 (Left; F). Selective inhibition of COX‐2 may have blocked this increase in vasodilatory prostanoids and caused CBF to decrease with IH exposure. CBF inidcates cerebral blood flow; COX, cyclooxygenase; IH, intermittent hypoxia; RAS, renin‐angiotensin system.
To our knowledge, this is the first study to assess the impact of acute IH on prostanoid formation in healthy humans. The lack of change in urinary prostanoid formation with IH, except for an elevated PGE2 within the placebo condition, contrasts to an animal study reporting decreased PGI2 and elevated TXA2 metabolites after only 3 hours of IH.17 These conflicting findings may be due to differences in the body fluid analyzed as well as species differences. In our study, we measured urinary prostanoids as a measure of systemic prostanoid production,54 whereas the previous study17 assessed plasma prostanoid concentrations in rats.
In contrast to acute IH, chronic IH exposure (eg, untreated OSA) was associated with increased urinary TXA2 and decreased PGE2 concentrations. Three prior studies have compared urinary prostanoid concentrations between healthy controls and OSA patients,18,55–56 and all focused on PGI2 and TXA2. All studies report an elevated TXA2 in untreated OSA patients with one study reporting an elevated TXA2 in only OSA patients with cardiovascular risk factors such as obesity, hypertension, dyslipidemia, smoking, and metabolic syndrome.18 Thromboxane A2 is preferentially formed via platelet COX‐1,8 indicating a greater influence of chronic IH on COX‐1 activity and derived prostanoids. The direct mechanism underlying the increased TXA2 and decreased PGE2 concentrations with chronic IH is unknown, but in addition to IH‐induced inflammation, it may also be related to the chronic sympatho‐excitation associated with untreated OSA.57 Acute sympathetic activation increases systemic vascular TXA2 concentrations58 and with repetitive sympathetic stimulation, PGE2 (along with PGI2) concentrations decline.59 Thus, in untreated OSA patients, chronic IH‐induced sympathetic activation may contribute to the observed increased TXA2 and decreased PGE2 concentrations.
Finally, limitations of the study must be acknowledged. First, although the IH model replicates the profile and severity of hypoxia experienced by patients with moderate‐to‐severe OSA it lacks the ancillary features associated with obstructive apneas such as increased negative intrathoracic pressure (which leads to greater sympathetic activation and post‐apneic blood pressure rises than IH alone60), hypercapnia, and sleep fragmentation.2 Although this may temper extrapolation of our findings to OSA, our IH model provides the opportunity to evaluate the impact of IH on prostanoid formation without the confounding effects of these ancillary features. Furthermore, animal18,61 and human5,62 studies have shown experimental IH produces similar physiological responses to those observed in OSA. Second, only healthy male participants were exposed to IH because the female sex may be protective against cardiovascular consequences of IH.63 Consequently, results may not be generalized to the female population. Third, our healthy controls were younger and lighter than the OSA patients. Although these differences may confound some of our results, neither age nor BMI were related to MAP or TXA2 concentration within the healthy participants (P≥0.412) and OSA patients (P≥0.349). Fourth, prostanoid‐stable metabolites were assessed in urine as validated markers of systemic prostanoid production,54 but this provides an integrated measure over the time of urine production, rather than an instantaneous measure of vascular prostanoid concentrations.64 Fifth, a potential limitation of the sample size of 12 healthy participants is a lack of statistical power. An a priori power calculation based upon previous findings4 indicated this sample size would provide a power of ≈0.85 to observe a ≈6.6 mm Hg increase in MAP following IH within the placebo condition. Although, the 2.9±4.9 mm Hg increase in MAP after IH exposure within placebo condition is smaller than expected and a post hoc power calculation revealed the current study had a power of 0.61, this increase in MAP was significant (P=0.03), indicating the study still had sufficient power to observe the enhancing effect of IH on MAP.
In conclusion, this study showed that, in healthy, male individuals, COX‐1 formed prostanoids are the principal regulators of resting blood pressure and cerebral blood flow. Conversely, as outlined in Figure 10, COX‐2 appears to be the primary COX isoenzyme contributing to the increase in blood pressure and maintenance of CBF following acute IH exposure. Furthermore, OSA is associated with elevated levels of the predominantly COX‐1 derived vasoconstrictor TXA2. Hence, COX‐2 and COX‐1 appear to have divergent roles in modulating vascular responses to acute versus chronic IH. These findings indicate COX‐2 inhibition may be beneficial for individuals exposed to acute IH (eg, altitude training), while traditional nonselective COX inhibiting NSAIDs may help prevent cardiovascular and cerebrovascular morbidity and mortality in OSA via inhibition of COX‐1,18 although enhanced patient monitoring may be required as a result of the cardiovascular risks associated with nonselective COX inhibitors.12
Author Contributions
The work outlined in this manuscript was led by a multi‐group collaboration of senior team members including Hanly (obstructive sleep apnea; phanly@ucalgary.ca), Wynne‐Edwards (prostanoids; k.wynne-edwards@ucalgary.ca), Ahmed (renal function and the renin angiotensin system; sofia.ahmed@albertahealthservices.ca), and Poulin (human integrative physiology and intermittent hypoxia; poulin@ucalgary.ca). Beaudin, Pun, Slater, Ahmed, Hanly, and Poulin conceived the experimental design of studies conducted in healthy humans. Nicholl, Ahmed, Hanly, and Poulin conceived the experimental design of studies conducted in patients with obstructive sleep apnea (OSA). Wynne‐Edwards led, guided and oversaw the analytical (development, validation, final analyses) work on prostanoid quantification, which was conducted by Beaudin, Pun, and Yang. Primary supervision for Beaudin was provided by Hanly and Poulin; primary supervision for Pun by Ahmed and Poulin; primary supervision for Yang by Poulin; primary supervision for Nicholl by Ahmed and Hanly. The experiments in healthy humans were performed by Beaudin, Pun, Yang, and Steinback while the experiments in patients with OSA were performed by Nicholl. All co‐authors contributed to the interpretation of the data. Beaudin wrote the first draft of the manuscript and Pun, Steinback, Slater, Hanly, Wynne‐Edwards, Ahmed, and Poulin edited the manuscript.
Sources of Funding
Beaudin is supported by an Alberta Innovates‐Health Solutions (AI‐HS) doctoral fellowship and the Canadian Institutes of Health Research (CIHR)‐Heart and Stroke Foundation of Canada Focus on Stroke doctoral fellowship. Pun is supported by a William H. Davies Medical Research Scholarship (University of Calgary); Yang is supported by a Markin Undergraduate Student Research Program in Health and Wellness and an AI‐HS undergraduate summer studentship. Nicholl is supported by a Cosmopolitan International Club of Calgary Graduate Scholarship, an American Society of Nephrology Student Scholar Grant and a Foothills Medical Centre Sleep Centre Development Fund. Steinback is supported by Natural Sciences and Engineering Research Council of Canada (NSERC) and AI‐HS post‐doctoral fellowships. Slater is supported by AI‐HS and CIHR. Wynne‐Edwards is the Jack Manns Professor of Comparative Endocrinology (Faculty of Veterinary Medicine). Ahmed is supported by AI‐HS, CIHR, and a joint initiative between Alberta Health and Wellness and the Universities of Alberta and Calgary. The funding for the studies in healthy humans and all biochemical analyses was provided by CIHR (PI=Poulin, Co‐Applicant=Hanly) and NSERC (PI=Poulin). The funding for the studies in OSA patients was provided by AI‐HS (PI=Ahmed).
Disclosures
None.
Acknowledgments
The authors thank Lea Bond, MSc for her technical guidance with performing enzyme immunoassay analyses, Darlene Y. Sola, BScN, RN for assisting in OSA patient recruitment and data collection, and Drs Todd J. Anderson and Jaideep Bains for their feedback on the manuscript. Finally, the authors would like to thank Dr. Tolulope Sajobi for the very helpful statistical discussions.
References
- 1.Punjabi NM. The epidemiology of adult obstructive sleep apnea. Proc Am Thorac Soc. 2008; 5:136-143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Somers VK, White DP, Amin R, Abraham WT, Costa F, Culebras A, Daniels S, Floras JS, Hunt CE, Olson LJ, Pickering TG, Russell R, Woo M, Young T. Sleep apnea and cardiovascular disease: an american Heart Association/American College of Cardiology Foundation Scientific Statement from the American Heart Association Council for High Blood Pressure Research Professional Education Committee, Council on Clinical Cardiology, Stroke Council, and Council on Cardiovascular Nursing. In Collaboration with the National Heart, Lung, and Blood Institute National Center on Sleep Disorders Research (National Institutes of Health). Circulation. 2008; 118:1080-1111. [DOI] [PubMed] [Google Scholar]
- 3.Tamisier R, Pepin JL, Remy J, Baguet JP, Taylor JA, Weiss JW, Levy P. 14 nights of intermittent hypoxia elevate daytime blood pressure and sympathetic activity in healthy humans. Eur Respir J. 2011; 37:119-128. [DOI] [PubMed] [Google Scholar]
- 4.Foster GE, Hanly PJ, Ahmed SB, Beaudin AE, Pialoux V, Poulin MJ. Intermittent hypoxia increases arterial blood pressure in humans through a renin‐angiotensin‐system dependent mechanism. Hypertension. 2010; 56:369-377. [DOI] [PubMed] [Google Scholar]
- 5.Foster GE, Brugniaux JV, Pialoux V, Duggan CT, Hanly PJ, Ahmed SB, Poulin MJ. Cardiovascular and cerebrovascular responses to acute hypoxia following exposure to intermittent hypoxia in healthy humans. J Physiol. 2009; 587:3287-3299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ryan S, Taylor CT, McNicholas WT. Systemic inflammation: a key factor in the pathogenesis of cardiovascular complications in obstructive sleep apnoea syndrome? Thorax. 2009; 64:631-636. [DOI] [PubMed] [Google Scholar]
- 7.Lavie L. Oxidative stress and inflammation in OSA. Sleep Apnoea. 2010:360-380. [Google Scholar]
- 8.Ricciotti E, FitzGerald GA. Prostaglandins and inflammation. Arterioscler Thromb Vasc Biol. 2011; 31:986-1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Smyth EM, Grosser T, Wang M, Yu Y, FitzGerald GA. Prostanoids in health and disease. J Lipid Res. 2009; 50suppl:S423-S428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pickard JD, Mackenzie ET. Inhibition of prostaglandin synthesis and the response of baboon cerebral circulation to carbon dioxide. Nat New Biol. 1973; 245:187-188. [DOI] [PubMed] [Google Scholar]
- 11.Wennmalm Å, Eriksson S, Wahren J. Effect of indomethacin on basal and carbon dioxide stimulated cerebral blood flow in man. Clin Physiol. 1981; 1:227-234. [Google Scholar]
- 12.Coxib and traditional NSAID Trialists' (CNT) Collaboration. Bhala N, Emberson J, Merhi A, Abramson S, Arber N, Baron JA, Bombardier C, Cannon C, Farkouh ME, Fitzgerald GA, Goss P, Halls H, Hawk E, Hawkey C, Hennekens C, Hochberg M, Holland LE, Kearney PM, Laine L, Lanas A, Lance P, Laupacis A, Oates J, Patrono C, Schnitzer TJ, Solomon S, Tugwell P, Wilson K, Wittes J, Baigent C. Vascular and upper gastrointestinal effects of non‐steroidal anti‐inflammatory drugs: meta‐analyses of individual participant data from randomised trials. Lancet. 2013; 382:769-779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Grosser T, Fries S, FitzGerald GA. Biological basis for the cardiovascular consequences of cox‐2 inhibition: therapeutic challenges and opportunities. J Clin Invest. 2006; 116:4-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mitchell JA, Warner TD. Reply to ricciotti et al.: evidence for vascular cox isoforms. Proc Natl Acad Sci USA. 2013; 110:E184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ricciotti E, Yu Y, Grosser T, Fitzgerald GA. Cox‐2, the dominant source of prostacyclin. Proc Natl Acad Sci USA. 2013; 110:E183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kirkby NS, Lundberg MH, Harrington LS, Leadbeater PD, Milne GL, Potter CM, Al‐Yamani M, Adeyemi O, Warner TD, Mitchell JA. Cyclooxygenase‐1, not cyclooxygenase‐2, is responsible for physiological production of prostacyclin in the cardiovascular system. Proc Natl Acad Sci USA. 2012; 109:17597-17602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nacher M, Farre R, Montserrat JM, Torres M, Navajas D, Bulbena O, Serrano‐Mollar A. Biological consequences of oxygen desaturation and respiratory effort in an acute animal model of obstructive sleep apnea (OSA). Sleep Med. 2009; 10:892-897. [DOI] [PubMed] [Google Scholar]
- 18.Gautier‐Veyret E, Arnaud C, Bäck M, Pépin J‐L, Petri MH, Baguet J‐P, Tamisier R, Lévy P, Stanke‐Labesque F. Intermittent hypoxia‐activated cyclooxygenase pathway: role in atherosclerosis. Eur Respir J. 2013; 42:404-413. [DOI] [PubMed] [Google Scholar]
- 19.Issa FG, Morrison D, Hadjuk E, Iyer A, Feroah T, Remmers JE. Digital monitoring of sleep‐disordered breathing using snoring sound and arterial oxygen saturation. Am Rev Respir Dis. 1993; 148:1023-1029. [DOI] [PubMed] [Google Scholar]
- 20.Vazquez JC, Tsai WH, Flemons WW, Masuda A, Brant R, Hajduk E, Whitelaw WA, Remmers JE. Automated analysis of digital oximetry in the diagnosis of obstructive sleep apnoea. Thorax. 2000; 55:302-307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stichtenoth DO, Marhauer V, Tsikas D, Gutzki FM, Frolich JC. Effects of specific COX‐2‐inhibition on renin release and renal and systemic prostanoid synthesis in healthy volunteers. Kidney Int. 2005; 68:2197-2207. [DOI] [PubMed] [Google Scholar]
- 22.Collop NA, Anderson WM, Boehlecke B, Claman D, Goldberg R, Gottlieb DJ, Hudgel D, Sateia M, Schwab R. Portable Monitoring Task Force of the American Academy of Sleep M. Clinical guidelines for the use of unattended portable monitors in the diagnosis of obstructive sleep apnea in adult patients. Portable Monitoring Task Force of the American Academy of Sleep Medicine. J Clin Sleep Med. 2007; 3:737-747. [PMC free article] [PubMed] [Google Scholar]
- 23.Nieto FJ, Young TB, Lind BK, Shahar E, Samet JM, Redline S, D'Agostino RB, Newman AB, Lebowitz MD, Pickering TG. Association of sleep‐disordered breathing, sleep apnea, and hypertension in a large community‐based study. Sleep heart health study. JAMA. 2000; 283:1829-1836. [DOI] [PubMed] [Google Scholar]
- 24.Shoback DM, Williams GH, Swartz SL, Davies RO, Hollenberg NK. Time course and effect of sodium intake on vascular and hormonal responses to enalapril (MK 421) in normal subjects. J Cardiovasc Pharmacol. 1983; 5:1010-1018. [DOI] [PubMed] [Google Scholar]
- 25.Chidambaram M, Duncan JA, Lai VS, Cattran DC, Floras JS, Scholey JW, Miller JA. Variation in the renin angiotensin system throughout the normal menstrual cycle. J Am Soc Nephrol. 2002; 13:446-452. [DOI] [PubMed] [Google Scholar]
- 26.Seifarth C, Trenkel S, Schobel H, Hahn EG, Hensen J. Influence of antihypertensive medication on aldosterone and renin concentration in the differential diagnosis of essential hypertension and primary aldosteronism. Clin Endocrinol (Oxf). 2002; 57:457-465. [DOI] [PubMed] [Google Scholar]
- 27.Lix LM, Keselman JC, Keselman HJ. Consequences of assumption violations revisited: a quantitative review of alternatives to the one‐way analysis of variance “f” test. Rev Educ Res. 1996; 66:579-619. [Google Scholar]
- 28.Glass GV, Peckham PD, Sanders JR. Consequences of failure to meet assumptions underlying the fixed effects analyses of variance and covariance. Rev Educ Res. 1972; 42:237-288. [Google Scholar]
- 29.Harwell MR, Rubinstein EN, Hayes WS, Olds CC. Summarizing monte carlo results in methodological research: the one‐ and two‐factor fixed effects anova cases. J Educ Behav Stat. 1992; 17:315-339. [Google Scholar]
- 30.Alderman MH. Salt, blood pressure, and human health. Hypertension. 2000; 36:890-893. [DOI] [PubMed] [Google Scholar]
- 31.Kahraman L, Thach BT. Inhibitory effects of hyperthermia on mechanisms involved in autoresuscitation from hypoxic apnea in mice: a model for thermal stress causing sids. J Appl Physiol. 2004; 97:669-674. [DOI] [PubMed] [Google Scholar]
- 32.Tanaka T, Okamura T, Miura K, Kadowaki T, Ueshima H, Nakagawa H, Hashimoto T. A simple method to estimate populational 24‐h urinary sodium and potassium excretion using a casual urine specimen. J Hum Hypertens. 2002; 16:97-103. [DOI] [PubMed] [Google Scholar]
- 33.Catella‐Lawson F, McAdam B, Morrison BW, Kapoor S, Kujubu D, Antes L, Lasseter KC, Quan H, Gertz BJ, FitzGerald GA. Effects of specific inhibition of cyclooxygenase‐2 on sodium balance, hemodynamics, and vasoactive eicosanoids. J Pharmacol Exp Ther. 1999; 289:735-741. [PubMed] [Google Scholar]
- 34.Yu Y, Ricciotti E, Scalia R, Tang S, Grant G, Yu Z, Landesberg G, Crichton I, Wu W, Puré E, Funk C, FitzGerald G. Vascular COX‐2 modulates blood pressure and thrombosis in mice. Sci Transl Med. 2012; 4:132ra154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McAdam BF, Catella‐Lawson F, Mardini IA, Kapoor S, Lawson JA, FitzGerald GA. Systemic biosynthesis of prostacyclin by cyclooxygenase (COX)‐2: the human pharmacology of a selective inhibitor of COX‐2. Proc Natl Acad Sci USA. 1999; 96:272-277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bao G, Metreveli N, Li R, Taylor A, Fletcher EC. Blood pressure response to chronic episodic hypoxia: role of the sympathetic nervous system. J Appl Physiol. 1997; 83:95-101. [DOI] [PubMed] [Google Scholar]
- 37.Fletcher EC, Orolinova N, Bader M. Blood pressure response to chronic episodic hypoxia: the renin‐angiotensin system. J Appl Physiol. 2002; 92:627-633. [DOI] [PubMed] [Google Scholar]
- 38.Linz D, Hohl M, Nickel A, Mahfoud F, Wagner M, Ewen S, Schotten U, Maack C, Wirth K, Böhm M. Effect of renal denervation on neurohumoral activation triggering atrial fibrillation in obstructive sleep apnea. Hypertension. 2013; 62:767-774. [DOI] [PubMed] [Google Scholar]
- 39.Linz D, Mahfoud F, Schotten U, Ukena C, Neuberger HR, Wirth K, Bohm M. Renal sympathetic denervation suppresses postapneic blood pressure rises and atrial fibrillation in a model for sleep apnea. Hypertension. 2012; 60:172-178. [DOI] [PubMed] [Google Scholar]
- 40.Pialoux V, Foster GE, Ahmed SB, Beaudin AE, Hanly PJ, Poulin MJ. Losartan abolishes oxidative stress induced by intermittent hypoxia in humans. J Physiol. 2011; 589:5529-5537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rajagopalan S, Kurz S, Munzel T, Tarpey M, Freeman BA, Griendling KK, Harrison DG. Angiotensin II‐mediated hypertension in the rat increases vascular superoxide production via membrane nadh/nadph oxidase activation. Contribution to alterations of vasomotor tone. J Clin Invest. 1996; 97:1916-1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Young W, Mahboubi K, Haider A, Li I, Ferreri NR. Cyclooxygenase‐2 is required for tumor necrosis factor‐α– and angiotensin II–mediated proliferation of vascular smooth muscle cells. Circ Res. 2000; 86:906-914. [DOI] [PubMed] [Google Scholar]
- 43.Hu ZW, Kerb R, Shi XY, Wei‐Lavery T, Hoffman BB. Angiotensin II increases expression of cyclooxygenase‐2: implications for the function of vascular smooth muscle cells. J Pharmacol Exp Ther. 2002; 303:563-573. [DOI] [PubMed] [Google Scholar]
- 44.Campbell WB, Graham RM, Jackson EK. Role of renal prostaglandins in sympathetically mediated renin relase in the rat. J Clin Invest. 1979; 64:448-456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Harris RC. Interactions between COX‐2 and the renin–angiotensin system in the kidney. Acta Physiol Scand. 2003; 177:423-427. [DOI] [PubMed] [Google Scholar]
- 46.Hurwitz S, Cohen RJ, Williams GH. Diurnal variation of aldosterone and plasma renin activity: timing relation to melatonin and cortisol and consistency after prolonged bed rest. J Appl Physiol. 1985; 2004:1406-1414. [DOI] [PubMed] [Google Scholar]
- 47.Andresen J, Shafi NI, Bryan RM., Jr Endothelial influences on cerebrovascular tone. J Appl Physiol. 2006; 100:318-327. [DOI] [PubMed] [Google Scholar]
- 48.Osuka K, Suzuki Y, Watanabe Y, Dogan A, Takayasu M, Shibuya M, Yoshida J. Vasodilator effects on canine basilar artery induced by intracisternal interleukin‐1 beta. J Cereb Blood Flow Metab. 1997; 17:1337-1345. [DOI] [PubMed] [Google Scholar]
- 49.Nadjar A, Tridon V, May MJ, Ghosh S, Dantzer R, Amedee T, Parnet P. NFkappaB activates in vivo the synthesis of inducible cox‐2 in the brain. J Cereb Blood Flow Metab. 2005; 25:1047-1059. [DOI] [PubMed] [Google Scholar]
- 50.Rivest S. What is the cellular source of prostaglandins in the brain in response to systemic inflammation? Facts and controversies. Mol Psychiatry. 1999; 4:500-507. [PubMed] [Google Scholar]
- 51.Ryan S, Taylor CT, McNicholas WT. Selective activation of inflammatory pathways by intermittent hypoxia in obstructive sleep apnea syndrome. Circulation. 2005; 112:2660-2667. [DOI] [PubMed] [Google Scholar]
- 52.FitzGerald GA, Patrono C. The coxibs, selective inhibitors of cyclooxygenase‐2. N Engl J Med. 2001; 345:433-442. [DOI] [PubMed] [Google Scholar]
- 53.Mitchell JA, Akarasereenont P, Thiemermann C, Flower RJ, Vane JR. Selectivity of nonsteroidal antiinflammatory drugs as inhibitors of constitutive and inducible cyclooxygenase. Proc Natl Acad Sci USA. 1993; 90:11693-11697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Capra V, Back M, Barbieri SS, Camera M, Tremoli E, Rovati GE. Eicosanoids and their drugs in cardiovascular diseases: focus on atherosclerosis and stroke. Med Res Rev. 2013; 33:364-438. [DOI] [PubMed] [Google Scholar]
- 55.Krieger J, Benzoni D, Sforza E, Sassard J. Urinary excretion of prostanoids during sleep in obstructive sleep apnoea patients. Clin Exp Pharmacol Physiol. 1991; 18:551-555. [DOI] [PubMed] [Google Scholar]
- 56.Kimura H, Niijima M, Abe Y, Edo H, Sakabe H, Kojima A, Hasako K, Masuyama S, Tatsumi K, Kuriyama T. Compensatory excretion of prostacyclin and thromboxane metabolites in obstructive sleep apnea syndrome. Intern Med. 1998; 37:127-133. [DOI] [PubMed] [Google Scholar]
- 57.Somers VK, Dyken ME, Clary MP, Abboud FM. Sympathetic neural mechanisms in obstructive sleep apnea. J Clin Invest. 1995; 96:1897-1904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Neri Serneri GG, Gensini GF, Abbate R, Prisco D, Rogasi PG, Castellani S, Casolo GC, Matucci M, Fantini F, Donato MD, Dabizzi RP. Spontaneous and cold pressor test‐induced prostaglandin biosynthesis by human heart. Am Heart J. 1985; 110:50-55. [DOI] [PubMed] [Google Scholar]
- 59.Neri Serneri GG, Castellani S, Scarti L, Trotta F, Chen JL, Carnovali M, Poggesi L, Masotti G. Repeated sympathetic stimuli elicit the decline and disappearance of prostaglandin modulation and an increase of vascular resistance in humans. Circ Res. 1990; 67:580-588. [DOI] [PubMed] [Google Scholar]
- 60.Morgan BJ, Denahan T, Ebert TJ. Neurocirculatory consequences of negative intrathoracic pressure vs. asphyxia during voluntary apnea. J Appl Physiol. 1993; 74:2969-2975. [DOI] [PubMed] [Google Scholar]
- 61.Fletcher EC, Lesske J, Qian W, Miller CC, III, Unger T. Repetitive, episodic hypoxia causes diurnal elevation of blood pressure in rats. Hypertension. 1992; 19:555-561. [DOI] [PubMed] [Google Scholar]
- 62.Tamisier R, Gilmartin GS, Launois SH, Pepin JL, Nespoulet H, Thomas R, Levy P, Weiss JW. A new model of chronic intermittent hypoxia in humans: effect on ventilation, sleep, and blood pressure. J Appl Physiol. 2009; 107:17-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hinojosa‐Laborde C, Mifflin SW. Sex differences in blood pressure response to intermittent hypoxia in rats. Hypertension. 2005; 46:1016-1021. [DOI] [PubMed] [Google Scholar]
- 64.Warner TD, Mitchell JA. Cox‐2 selectivity alone does not define the cardiovascular risks associated with non‐steroidal anti‐inflammatory drugs. Lancet. 2008; 371:270-273. [DOI] [PubMed] [Google Scholar]