

Abstract
Background
Cerebral arteriovenous malformation (AVM) is a vascular disease exhibiting abnormal blood vessel morphology and function. miR‐18a ameliorates the abnormal characteristics of AVM‐derived brain endothelial cells (AVM‐BEC) without the use of transfection reagents. Hence, our aim was to identify the mechanisms by which miR‐18a is internalized by AVM‐BEC. Since AVM‐BEC overexpress RNA‐binding protein Argonaute‐2 (Ago‐2) we explored the clinical potential of Ago‐2 as a systemic miRNA carrier.
Methods and Results
Primary cultures of AVM‐BEC were isolated from surgical specimens and tested for endogenous miR‐18a levels using qPCR. Conditioned media (CM) was derived from AVM‐BEC cultures (AVM‐BEC‐CM). AVM‐BEC‐CM significantly enhanced miR‐18a internalization. Ago‐2 was detected using western blotting and immunostaining techniques. Ago‐2 was highly expressed in AVM‐BEC; and siAgo‐2 decreased miR‐18a entry into brain‐derived endothelial cells. Only brain‐derived endothelial cells were responsive to the Ago‐2/miR‐18a complex and not other cell types tested. Secreted products (eg, thrombospondin‐1 [TSP‐1]) were tested using ELISA. Brain endothelial cells treated with the Ago‐2/miR‐18a complex in vitro increased TSP‐1 secretion. In the in vivo angiogenesis glioma model, animals were treated with miR‐18a in combination with Ago‐2. Plasma was obtained and tested for TSP‐1 and vascular endothelial growth factor (VEGF)‐A. In this angiogenesis model, the Ago‐2/miR‐18a complex caused a significant increase in TSP‐1 and decrease in VEGF‐A secretion in the plasma.
Conclusions
Ago‐2 facilitates miR‐18a entry into brain endothelial cells in vitro and in vivo. This study highlights the clinical potential of Ago‐2 as a miRNA delivery platform for the treatment of brain vascular diseases.
Keywords: angiogenesis, Argonaute‐2, arteriovenous malformation, brain endothelial cells, miR‐18a
Introduction
Cerebral Arteriovenous Malformations (AVM) are brain vascular lesions comprising an abnormal tangle of vessels (nidus), in which arteries and veins are directly connected without an intervening capillary system.1 AVM affect approximately 300 000 people in the United States and can lead to serious neurological symptoms or death. Current medical treatments are highly invasive and can pose significant risks to nearby brain structures that regulate speech, movement, and sensory processing,2 highlighting the importance of developing more efficacious and safer therapies.
Previously, we have reported that human AVM‐derived brain endothelial cells (AVM‐BEC) have distinct and abnormal characteristics compared to normal BEC.3 Namely, AVM‐BEC proliferate more rapidly, migrate faster, and produce aberrant vessel‐like structures as compared with normal vasculature. We have also shown that AVM‐BEC express low levels of a key regulator of angiogenesis, thrombospondin‐1 (TSP‐1), and the aberrant phenotype of these cells can be normalized by restoring TSP‐1 levels.3–5 We have recently shown that these abnormal features are ameliorated with microRNA‐18a (miR‐18a) treatment. miRNAs are small non‐coding RNAs that inhibit gene expression by inducing cleavage or translational repression of messenger RNA (mRNA).6 In our study, we showed that miR‐18a inhibited TSP‐1 transcriptional repressor, Inhibitor of DNA‐binding protein‐1 (Id‐1), leading to increased TSP‐1 levels and decreased vascular endothelial growth factor (VEGF)‐A and VEGF‐D secretion. miR‐18a also regulated cell proliferation and improved tubule formation efficiency.5 Importantly, these effects were obtained with miRNA alone (naked delivery), in the absence of traditional transfection reagents, like lipofectamine, which cannot be used in vivo due to induced toxicity. Based on our previous studies, we now proceeded to explore the mechanisms of entry of naked miRNA into brain endothelial cells. Naked miRNAs have been shown to form stable complexes with circulating RNA‐binding proteins, whose function is to protect miRNA from degradation and therefore ensure efficient cell‐to‐cell communication.7 An important RNA‐binding protein is argonaute‐2 (Ago‐2),8 a member of the Argonaute protein family, which also includes Ago‐1, Ago‐3, and Ago‐4. In human cells, Ago‐2 also takes part in the RNA‐induced silencing complex (RISC) to promote endonucleolytic cleavage of mRNA.9
In the current study, we show that AVM‐BEC release Ago‐2, which can be used to enhance the entry of extracellular miR‐18a into brain endothelial cells. In vitro studies show that Ago‐2 in combination with miR‐18a is functional and able to stimulate TSP‐1 production. Furthermore, miR‐18a in combination with Ago‐2 can be delivered in vivo by intravenous administration, resulting in increased circulating serum TSP‐1 and decreased VEGF‐A. Thus Ago‐2 may be used to decrease angiogenic activity in brain endothelial cells, making Ago‐2 a biocompatible and non‐invasive miRNA‐delivery platform with great potential for treating neurovascular diseases.
Materials and Methods
Endothelial Cell Isolation and Culture
Human surgical specimens were obtained in accordance with guidelines set forth by the Institutional Review Board (HS‐04B053), at Keck School of Medicine, University of Southern California, and in accordance with Animal Research: Reporting In Vivo Experiments (ARRIVE) guidelines. AVM‐BEC were isolated from brain tissues of 6 patients who underwent microsurgical AVM resection. Control BEC were derived from the structurally normal cortex of 4 patients who underwent temporal lobectomy for intractable epilepsy.5 Tissue was gently homogenized and vessels were isolated on a 30% dextran gradient followed by enzymatic dissociation with 10% collagenase/dispase. For AVM‐BEC, no distinctions were made between venous or arterial vessels and nidus, although the surgically resected specimens principally consisted of the nidus. BEC were cultured in RPMI 1640 growth media (Mediatech Inc) supplemented with 100 ng/mL EC growth supplement (Millipore), 10 mmol/L N‐2‐hy‐droxyethylpiperazine‐N‐2‐ethanesulfonic acid (HEPES) (Invitrogen), 24 mmol/L sodium pyruvate (Invitrogen), 300 U heparin (Sigma‐Aldrich), 1x minimum essential medium (MEM) vitamin solution (Invitrogen), 1x MEM nonessential amino acids (Mediatech Inc), 1% penicillin/streptomycin (Invitrogen) and 10% fetal calf serum (FCS) (Omega Scientific Inc) in a 37°C incubator with 5% CO2. After reaching 80% confluency, cells were labeled with CD31/platelet‐EC adhesion molecule‐1‐labeled magnetic Dyna beads (Invitrogen) and purified by Mini MACS magnetic cell sorting (Miltenyl Biotec, GmbH). The purity of control BEC and AVM‐BEC was confirmed by positive immunostaining for the endothelial cell markers CD31/platelet‐EC adhesion molecule‐1 (Santa Cruz Biotechnology Inc) and von Willebrand factor (Dako North America Inc) and negative staining for glial fibrillary acidic protein (GFAP) (Chemicon International Inc) and smooth muscle actin (SMA) (Vector Laboratories) (data not shown). Only endothelial cells within passages 3 to 5 were used. Unless otherwise noted, all endothelial cells were grown on 1% gelatin‐coated surfaces. All images were captured using EVOS fl AMF‐4306 AMG microscopes. Human umbilical vein endothelial cells (HUVEC) and human dermal microvascular endothelial cell line (HMEC) were cultured in RPMI media containing fetal calf serum (FCS); however, no FCS was used in the preparation of conditioned media (CM).
For cell treatments, miR‐18a (40 nmol/L), siAgo‐2 (30‐75 nmol/L), lipofectamine (2 μg/mL) (Life Technologies) were added as appropriate. A scrambled miRNA sequence (50 to 75 nmol/L) and siGFP (40 nmol/L; Life Technologies) were used as negative controls. siGFP was chosen as an additional negative control since the miR‐18a mimic is a double‐stranded RNA and these cells do not express green fluorescent protein (GFP). Cells were treated with varying concentrations of Ago‐2 (0.01 to 80 nmol/L; Abcam) for 30 minutes to determine its activity as a miRNA carrier. For inhibition of exosome‐release experiments, cells were treated with GW4869 (5 to 50 μmol/L, Sigma), an inhibitor of neutral sphingomyelinases for 24 hours.
Shear Flow Cultures
All experiments were performed under shear flow. For that purpose, endothelial cells were placed on an orbital shaker, to reproduce arterial flow (12 dyn/cm2), as reported previously.5 The apparatus was maintained in a water‐jacketed incubator with 5% CO2 at 37°C.
miRNA Extraction and Detection
After thoroughly washing with phosphate‐buffered saline (PBS), cells were lysed with lysis buffer (Ambion®, ThermoScientific) and miRNA was extracted using mirVana™ miRNA Isolation Kit (Ambion®) and transcribed using TaqMan MicroRNA Reverse Transcription Kit (Applied Biosystems), according to manufacturer's instructions. Gene expression was analyzed by qPCR using TaqMan Universal Master Mix II (Life Technologies), TaqMan® Assay for miR‐18a and TaqMan® Control miRNA Assay for RNU44 (Applied Biosystems), per manufacturer's instructions, using a Stratagene Mx3000P Bioanalyzer (Agilent Technologies). PCR products were normalized to RNU44, an endogenous small‐nucleolar RNA with stable expression within all cell types tested. For 4°C experiments, cells were maintained at 4°C for 10 minutes before treatments, and maintained at that temperature for an additional 30 minutes before cell lysis.
CM Experiments
Cells were seeded at a density of 2×104 cells/well in complete media into 24 well tray plates for 24 hours and allowed to become 70% to 80% confluent. Cells were then washed twice with PBS and incubated in serum‐free RPMI medium supplemented with penicillin and streptomycin for an additional 24‐hour period. Conditioned media was collected and centrifuged at 2000g at 4°C for 10 minutes to remove cell debris. Conditioned media was then used without dilution for subsequent experiments unless stated otherwise. In diluted conditioned media experiments, fresh media was added accordingly.
Quantitative Real‐Time Polymerase Chain Reaction
Cells were seeded at a density of 2×104 per well in 24‐well plates and treatments were added when appropriate. After thoroughly washing, cells were lysed and mRNA extracted. Gene expression was confirmed by quantitative real‐time polymerase chain reaction (qPCR) using iQ™ SYBR® Green Supermix (BioRad) according to the manufacturer's instructions using a Stratagene Mx3000PBioanalyzer (Agilent Technologies). 18S ribosomal ribonucleic acid (18S rRNA) was measured for sample normalization. Forward and reverse primers for the following genes were used: NPM‐1, 5′‐AGCACTTAGTAGCTGTGGAG‐3′; 5′‐CTGTGGAACCTTGCTACCACC‐3′; NCL, 5′‐GGTGGTTTCCCAACAAA‐3′; 5′‐GCCAGGTGTGGTAACTGCT‐3′; Ago‐2, 5′‐GTTTGACGGCAGGAAGAATCT‐3′; 5′‐AGGACACCCACTTGATGGACA‐3′; 18S rRNA, 5′‐CGGCTACCACATCCAAGGAA‐3′; 5′‐GCTGGAATTACCGCGGCT‐3′, respectively.
Western Blot
Cells were seeded at a density of 1×105 per well in 6‐well plates and treatments were added when appropriate. Total protein was extracted and quantified using the Bicinchoninic Acid Protein Assay Kit (Thermo Fisher Scientific). Equal amounts of protein (30 μg) were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis and transferred to 0.45‐μm polyvinylidene fluoride microporous membranes. Membranes were blocked with Sea Block (Thermo Fisher Scientific) for 1 hour, probed with rabbit anti‐Ago‐2 (1:1000) (Cell Signaling Technology Inc) or rabbit anti‐actin (1:1500) (Santa Cruz Biotechnology) antibodies overnight at 4°C, and incubated with the appropriate fluorescent secondary anti‐rabbit antibody (1:15 000) (Thermo Fisher Scientific) at room temperature (RT) for 2 hours. Protein bands were detected by Odyssey infrared imaging (LI‐COR Biosciences) and densitometric studies were performed using NIH free software ImageJ. Actin levels were measured for internal standardization. For the representative image depicted in Figure 2B, bands were cropped from the picture of original membrane and shown in the same order as in the graph, without image manipulation.
Figure 2.
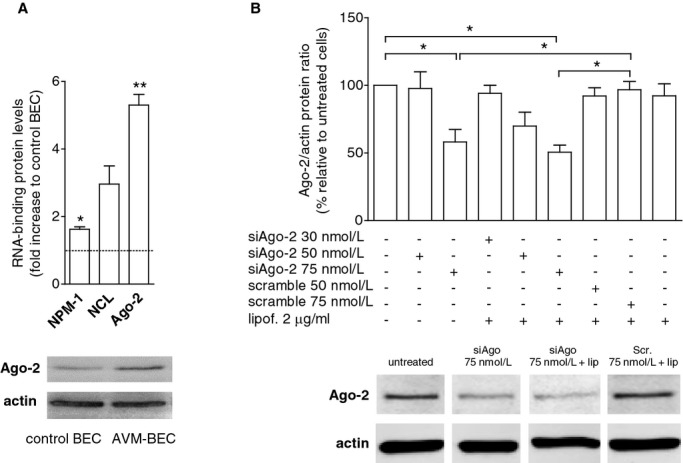
Ago‐2 is highly expressed by AVM‐BEC. A, Basal expression of RNA‐binding proteins (NPM, nucleophosmin‐1; NCL, nucleolin; Ago‐2, argonaute‐2) in AVM‐BEC and control BEC were analyzed by qPCR. Increased levels of NCL and Ago‐2 in AVM‐BEC as compared to control BEC were detected (n=3; *P<0.05; **P<0.01; paired t test). A representative western blot is depicted below showing Ago‐2 increased protein levels in AVM‐BEC compared to control BEC. B, AVM‐BEC were treated with siAgo (30 to 75 nmol/L), scrambled siAgo (50 to 75 nmol/L) and lipofectamine (2 μg/mL) and Ago‐2 protein levels were analyzed by Western blotting. siAgo‐2 (75 nmol/L) decreased ≈50% of intracellular Ago‐2 protein content (n=3; *P<0.05; paired t test). A representative image depicting the effects of siAgo‐2 (75 nmol/L) alone and in the presence of lipofectamine (2 μg/mL) is shown below. Data are presented as mean±standard error of the mean (SEM). Each human specimen‐derived cell culture represents a unit of analysis (n). AVM indicates arteriovenous malformation; Ago‐2, Argonaute‐2; BEC, brain endothelial cells.
Immunocytochemistry
Cells were fixed with 4% paraformaldehyde (PFA) and then washed with PBS. For 4°C experiments, cells were maintained at 4°C for 10 minutes before treatments, and maintained at that temperature for an additional 30 minutes before fixation. Nonspecific binding was prevented using Sea Block blocking solution, for 1 hour at RT (Thermo Fisher Scientific). Cells were kept overnight at 4°C in a primary antibody solution and incubated for 1 hour at RT with the corresponding secondary antibody. Antibodies were used as listed: rabbit anti‐Ago‐2 (1:100) (Cell Signaling Technology Inc); rabbit anti‐CD8 (1:50) (Santa Cruz Biotechnology Inc); Alexa Fluor 594 goat anti‐rabbit (1:200) (Molecular Probes, OR). For nuclear labeling, cell preparations were stained with Hoechst‐33342 (2 μg/mL) (Sigma) and mounted in Dako Fluorescence Mounting Medium (Dako North America Inc). Fluorescent images were acquired using an LSM 510 confocal microscope with a 40× objective (Carl Zeiss Inc).
Immunoprecipitation
For each sample, 200 μL of Magna Bind goat anti‐rabbit IgG Magnetic Bead slurry (Thermo Scientific) were washed with PBS solution and incubated with 10 μg of rabbit anti‐Ago‐2 (Abcam Inc) or rabbit IgG (Santa Cruz Biotechnology) antibodies for 5 hours at 4°C. The pre‐incubated beads and antibody mix was then added to 400 μL of miR‐18a (40 nmol/L), Ago‐2 (0.4 nmol/L; Abcam) or Ago‐2 and miR‐18a (0.4 and 40 nmol/L, respectively) and incubated overnight at 4°C. Beads were washed 3 times with 1% Nonidet P‐40 buffer (1% Nonidet P‐40, 50 mmol/L Tris‐HCl, pH 7.4, 150 mmol/L NaCl, 2 mmol/L EDTA) and then divided in half. One half was eluted in SDS sample buffer, followed by SDS/PAGE and immunoblotting. The other half was eluted in 100 μL of lysis buffer and processed for miRNA isolation (Ambion®) and detection (see above the protocols for miRNA detection and Western blot).
Enzyme‐Linked Immunosorbent Assay
Cells were seeded at a density of 2×104 per well in 24‐well plates and grown for 48 hours. Treatments were added as appropriate. Cell supernatants were collected, filtered through a 0.2‐μm cellulose acetate membrane (VWR International) and analyzed for TSP‐1 (R&D Systems) using commercially available enzyme‐linked immunosorbent assay (ELISA) kits per manufacturer's instructions. Remaining cells were lysed and total amount of protein was determined for normalization.
In the in vivo experiments, upon treatment conclusion, mouse blood samples (800 μL) were collected in heparin and spun down to obtain plasma. These plasma samples were analyzed for TSP‐1 and VEGF‐A using commercially available ELISA kits (R&D Systems) according to manufacturer's instructions.
In Vivo Experiments
Animal protocols were approved by Institutional Animal Care and Use Committee (IACUC) of the University of Southern California and in accordance with Animal Research: Reporting In Vivo Experiments (ARRIVE) and institutional guidelines (protocol number 11962). Renilla luciferase‐labeled U251 glioma cells (2×105 cells) were implanted intracranially into 4 to 5 week old male athymic/nude mice (Harlan Laboratories, Inc), as previously described.10 Briefly, scalp was disinfected with Betadine and an incision was made along the midline with a scalpel. With the skull exposed, scales were taken after set zero at bregma (X, AP: +1.0; Y, ML: +2.0). The skull was drilled and the syringe was lowered until the target DV (1.5). All procedures were performed under strictly sterile conditions. Animals were anesthetized with ketamine (100 mg/kg)/xylazine (10 mg/kg) administered intraperitoneally prior to implantation and treated subcutaneously with buprenorphine (0.06 mg/kg) daily for 2 days post‐implantation. Mice were imaged 3 days post‐implantation to group mice with similar imaging intensities and thus determine baseline tumor growth. The following treatment groups were established: vehicle (PBS) (n=5); miR‐18a (40 nmol/L) in combination with Ago‐2 (0.4 nmol/L) (n=5); miR‐18a alone (40 nmol/L) (n=5); Ago‐2 alone (0.4 nmol/L) (n=5). Concentrations were adjusted to the total blood volume (0.002 L) of young adult mice. Treatments (100 μL) were administered intravenously through the tail vein every 48 hours (for a total of 3 treatment sessions) and mice were imaged after treatment completion. For imaging purposes, mice were injected with 1 mg/kg ViviRen™ In Vivo Renilla Luciferase Substrate (Promega) intravenously and luminescence of selected ROI was recorded using the IVIS 200 optical imaging system (Caliper Life Sciences); data was analyzed using LIVING IMAGE software (Caliper Life Sciences).
Statistical Analysis
Statistical analysis was performed using GraphPad Prism 5.0 (GraphPad Software). Statistical significance was considered relevant for P<0.05 using paired 2‐tailed t test, repeated measures 2‐way ANOVA or 1‐way ANOVA, followed by Dunnet's Multiple Comparison Test (statistical tests are indicated in figure legends). Data are presented as mean±standard error of the mean (SEM). For cell lines, every experimental condition was tested in 3 sets of independent experiments (n) unless stated otherwise, and performed in duplicates. For primary cell cultures, every human specimen was considered the unit of analysis and accounted for an independent n value, and performed in duplicates. For every immunocytochemistry analysis, 5 independent microscopy fields were acquired per coverslip, with a 40× objective (≈40 cells per field).
Results
AVM‐BEC‐Conditioned Media Potentiates miR‐18a Entry
Previously, we have shown that miR‐18a (40 nmol/L) can be internalized, and functionally relevant, without transfection agents.5 Based on these studies, we proceeded to explore the mechanism of entry of miR‐18a into AVM‐BEC. We first analyzed the endogenous expression of miR‐18a in AVM‐BEC and control BEC using qPCR; AVM‐BEC endogenously express significantly less miR‐18a as compared to control BEC (AVM‐BEC=0.34±0.03; n=3; P<0.01) (Figure 1A). When cells were treated exogenously with miR‐18a (40 nmol/L) for increasing periods of incubation (5 minutes to 24 hours) in the presence of AVM‐BEC‐conditioned media (AVM‐BEC‐CM) (black bars) or fresh media (white bars), we observed that uptake of miR‐18a by AVM‐BEC was enhanced, particularly with AVM‐BEC‐CM after 30 minutes of incubation (AVM‐BEC‐CM30 min=17 136.0±1697.0; fresh30 min=7668.7±783.4; n=4; *P<0.05, **P<0.01) (Figure 1B). AVM‐BEC‐CM contained no FCS, to avoid contaminating nucleic acids, proteins, and transporting microvesicles. Treatment with scramble miRNA or siGFP (40 nmol/L) in the presence of AVM‐BEC‐CM, BEC‐CM or fresh media did not change intracellular miR‐18a levels (data not shown).
Figure 1.
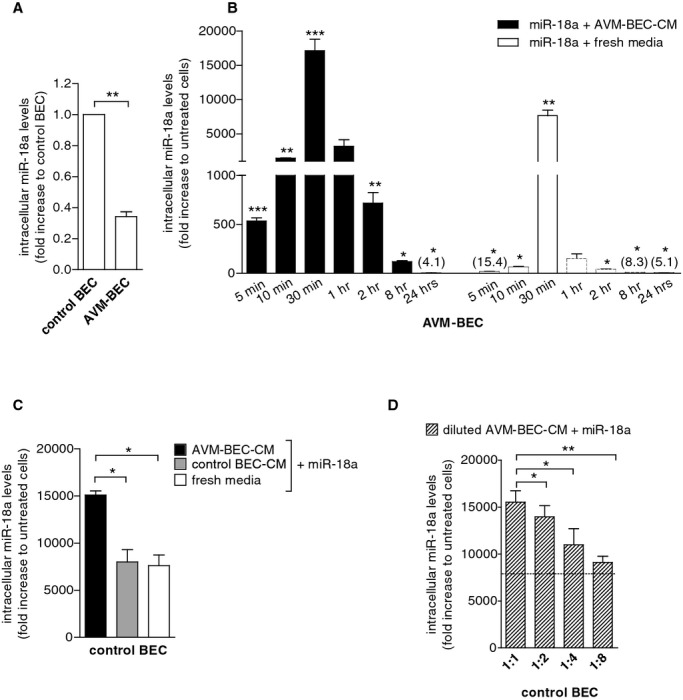
AVM‐BEC‐conditioned media (AVM‐BEC‐CM) potentiates miR‐18a internalization. A, AVM‐BEC and control BEC were analyzed for intracellular miR‐18a levels using qPCR. Control BEC were used as baseline (n=3; ***P<0.001; paired t test). B, miR‐18a (40 nmol/L) in combination with AVM‐BEC‐CM (black bars) or fresh culture media (white bars) was added to AVM‐BEC and tested for intracellular miR‐18a after 5, 10, 30 minutes (n=4), 1, 2, 8 and 24 hours (n=3). AVM‐BEC‐CM enhanced miR‐18a entry up to 30 minutes (*P<0.05, **P<0.01, ***P<0.001; paired t test). C, Control BEC were treated with miR‐18a (40 nmol/L) in combination with AVM‐BEC‐CM (black bar), control BEC‐CM (gray bar) or fresh media (white bar). Intracellular miR‐18a was analyzed (qPCR) after 30 minutes incubation; AVM‐BEC‐CM potentiated miR‐18a internalization by control BEC (n=3; *P<0.05; paired t test). D, Control BEC were treated with miR‐18a (40 nmol/L) and in the presence of serial diluted AVM‐BEC‐CM (diagonal line bars), demonstrating that progressively diluted AVM‐BEC‐CM loses its ability to enhance miR‐18a internalization (n=3, *P<0.05, **P<0.01; paired t test). Dotted line represents miR‐18a uptake by control BEC in the presence of fresh media. Data are presented as mean±standard error of the mean (SEM). Each human specimen‐derived cell culture represents a unit of analysis (n). AVM indicates arteriovenous malformation; BEC, brain endothelial cells; CM, conditioned media; qPCR, quantitative real‐time polymerase chain reaction.
These data clearly show that AVM‐BEC‐CM enhanced entry of exogenous miR‐18a. To assess whether AVM‐BEC‐CM affected miR‐18a entry in control BEC, these cells were subjected to miR‐18a treatment in the presence of AVM‐BEC‐CM (black bar), BEC‐CM (gray bar), or fresh media (white bar) for 30 minutes (Figure 1C). The results show that AVM‐BEC‐CM potentiated miR‐18a entry in control BEC, compared to BEC‐CM and fresh media (AVM‐BEC‐CM=15 106.0±419.2; BEC‐CM=8012.1±1288.2; fresh= 7606.0±1137.1; n=3; **P<0.01) (Figure 1C). Diluted AVM‐BEC‐CM (1:2, 1:4 and 1:8) was used to determine if a soluble agent, in limiting amount, was promoting miRNA entry in miR‐18a‐treated cells (Figure 1D). Increasingly diluted AVM‐BEC‐CM led to proportional decline of miR‐18 internalization (1:1=15 521.3 ±1216.0; 1:2= 13 951.4± 1197.1; 1:4=11 124.9±2112.3; 1:8=9097.8±663.5; n=3; P<0.01, P<0.001) (Figure 1D). Thus, a soluble agent secreted by AVM‐BEC in AVM‐BEC‐CM is likely responsible for enhanced uptake of miR‐18a.
AVM‐BEC Express RNA‐Binding Protein Ago‐2
Recent studies have suggested that the main mechanism for the entry of extracellular RNA into cells occurs through the formation of ribonucleoprotein complexes.11 Therefore, we screened AVM‐BEC as compared with control BEC for RNA‐binding protein expression. AVM‐BEC significantly express more nucleolin (NCL) and argonaute‐2 (Ago‐2) as compared with control BEC, while nucleophosmin 1 (NPM‐1) was not significantly different (Figure 2A). Given its high expression and its described effect as an miRNA carrier (Ago‐2=5.3±0.3; n=3; ***P<0.001), we selected Ago‐2 as a potential target for downregulation, and treated AVM‐BEC with siAgo‐2 (30 to 75 nmol/L) to determine the role of this RNA‐binding protein in miR‐18a delivery (Figure 2B). The results of this experiment showed that treatment with siAgo‐2 (75 nmol/L) alone, or siAgo‐2 (75 nmol/L) with lipofectamine had similar and significant effects in reducing Ago‐2 expression (siAgo‐2=58.2±9.2; lipof.+siAgo‐2=50.6±5.3; n=3; P<0.05). Concentrations above 75 nmol/mL did not induce a higher reduction of Ago‐2. These data showed that AVM‐BEC‐CM contains a delivery agent that works as effectively as lipofectamine (Figure 2B). Nevertheless, in subsequent experiments siAgo‐2 was used in the presence of lipofectamine to induce the maximal decrease of Ago‐2 levels. It should be noted that given the role of Ago‐2 for miRNA biogenesis a more efficient reduction in Ago‐2 levels would likely compromise cell integrity.
It was not possible to determine secreted Ago‐2 protein levels in AVM‐BEC‐CM because of the limited amount of conditioned media obtainable from these primary endothelial cell cultures (which can be used only up to passage 5); we were able to collect 15 mL of additional AVM‐BEC‐CM, which was insufficient to detect Ago‐2 protein levels by Western blot. Additionally, we tested the internalization of another miRNA mimic, miR‐128a, and found internalization, but to a lesser extent than miR‐18a (AVM‐BEC=5.9±0.1; n=3; *P<0.05; data not shown), which indicates that AVM‐BEC can efficiently internalize universal exogenous miRNA without transfection reagents.
Silencing Ago‐2 Compromises the Entry of Exogenous miR‐18a Into Brain Endothelial Cells
To determine the effects of decreased Ago‐2 secreted levels, AVM‐BEC were treated with siAgo‐2 or scrambled siRNA; siAgo‐2‐AVM‐BEC‐CM was then collected and tested in the presence of miR‐18a (Figure 3A). AVM‐BEC‐CM was more effective than siAgo2‐AVM‐BEC in raising intracellular miR‐18a levels after exogenous treatment; thus Ago‐2 is important in transporting miR‐18a. Primary endothelial cell cultures of brain origin such as AVM‐BEC, control BEC and glioma‐derived brain endothelial cells (TuBEC) showed the most significant miR‐18a internalization with AVM‐BEC‐CM and the most significant reduction of internalized miR‐18a in the presence of siAgo‐2‐AVM‐BEC‐CM (AVM‐BECAVM‐BEC‐CM=16 167.7± 600.9; AVM‐BECsiAgo2‐AVM‐BEC‐CM=11 633.2± 876.2; control BECAVM‐BEC‐CM=13 815.7±823.4; control BECsiAgo‐2‐AVM‐BEC‐CM=6856.2±545.0; TuBECAVM‐BEC‐CM=1104.1±249.1; TuBECsiAgo‐2‐AVM‐BEC=383.6±104.8; n=3; P<0.001; P<0.05). Interestingly, astrocytes did not exhibit increased intracellular miR‐18a levels with AVM‐BEC‐CM and, accordingly, Ago‐2 silencing had no effect on other brain cell types. There were also no significant differences in miRNA uptake with AVM‐BEC‐CM or siAgo‐2‐AVM‐BEC‐CM in human umbilical vein endothelial cells (HUVEC) or human dermal microvascular endothelial cell line (HMEC) (Figure 3A). These results correlated reduced Ago‐2 levels (as observed in siAgo‐2‐AVM‐BEC‐CM) with decreased miR‐18a uptake into brain endothelial cells. To further validate the role of Ago‐2 as an miRNA carrier, control BEC were treated with miR‐18a (40 nmol/L) and Ago‐2 (0.01 to 0.8 nmol/L) (Figure 3B). Increasing concentrations of exogenously applied Ago‐2 (up to 0.4 nmol/L) led to increased detection of miR‐18 intracellular levels (n=3). Additionally, miR‐18a in combination with Ago‐2 resulted in significantly lower miRNA degradation levels than miR‐18a alone, particularly after 30 and 120 minutes (mir‐18a30 min= 23.3±6.0; mir‐18a120 min= 36.7±3.3; mir‐18a+Ago‐230 min=2.7±1.2; mir‐18a+Ago‐2120 min=12.3±3.9; n=3; P<0.01) (Figure 3C). These data show that one potential mechanism by which Ago‐2 increases miR‐18a intracellular levels is that Ago‐2 protects the miRNA from degradation.
Figure 3.

Ago‐2 silencing compromises miR‐18a entry. A, Intracellular miR‐18a detection in AVM‐BEC, control BEC, tumor‐derived endothelial cells (TuBEC), human umbilical vein endothelial cells (HUVEC), human microvascular endothelial cells (HMEC) and astrocytes treated with miR‐18a (40 nmol/L) in the presence of siAgo‐2‐AVM‐BEC‐CM (dotted bars) or AVM‐BEC‐CM (black bars). Ago‐2 silencing decreased miR‐18 entry in AVM‐BEC, control BEC, and TuBEC (n=3; *P<0.05; paired t test). B, Intracellular detection of miR‐18a in control BEC (qPCR) after treating cells for 30 minutes with different concentrations of Ago‐2 (0.01 to 4 nmol/L) in combination with miR‐18a (40 nmol/L) showed that higher concentrations of Ago‐2 (up to 0.4 nmol/L) increased miR‐18a detection (n=3). Dotted line represents miR‐18a uptake by control BEC in the presence of AVM‐BEC‐CM. C, Analysis of intracellular miR‐18a (qPCR) showed that miR‐18a (40 nmol/L) in combination with Ago‐2 (0.4 nmol/L) (for 5, 30, 120 and 1440 minutes) was more resistant to degradation than miR‐18a alone; maximum effect was observed at 120 minutes (n=3; **P<0.01; repeated measures 2‐way ANOVA). D, AVM‐BEC and control BEC were exposed to miR‐18a in combination with siAgo‐2‐AVM‐BEC‐CM or AVM‐BEC‐CM. Ago‐2 staining (red) showed that cells exposed to AVM‐BEC‐CM increased Ago‐2 detection in control BEC when treated with miR‐18a (40 nmol/L) (n=3, **P<0.01). Nuclear staining is shown in blue. Data are presented as mean±standard error of the mean (SEM). Each human specimen‐derived cell culture represents a unit of analysis (n). For experiments using cell lines, (n) is represented by cell cultures obtained from different passages. ANOVA indicates analysis of variance; AVM, arteriovenous malformation; Ago‐2, Argonaute‐2; BEC, brain endothelial cells; CM, conditioned media; qPCR, quantitative real‐time polymerase chain reaction.
Immunocytochemistry analysis of AVM‐BEC and control BEC stained for Ago‐2 showed that basal Ago‐2 expression is higher in AVM‐BEC than control BEC (Figure 3D; i versus iv). However, control BEC treated with miR‐18a in the presence of AVM‐BEC‐CM showed increased intracellular Ago‐2 labeling supporting the previous concept that Ago‐2 acts as a carrier for miR‐18a (Figure 3D; ii). Conversely, control BEC treated with miR‐18a in the presence of siAgo‐2‐AVM‐BEC‐CM did not show a significant signal (Figure 3D; iii). AVM‐BEC‐CM alone, ie, without added miR‐18a, did not increase intracellular Ago‐2, emphasizing the importance of having both Ago‐2 and miR‐18a available in the media to enhance internalization (Figure 3D; vi).
To assess whether a specific transmembrane protein was involved in Ago‐2/miR‐18a crossing of the cell membrane we conducted subsequent experiments in an effort to clarify whether facilitated transport was responsible for miR‐18a entry. Hence, miR‐18a intracellular levels were quantified in recipient cells in the presence of AVM‐BEC‐CM and miR‐18a treatment (40 nmol/L) at 4°C and compared to 37°C (Figure 4A). AVM‐BEC and control BEC subjected to cold temperature showed decreased miR‐18a uptake, albeit not significantly suggesting that miRNA delivery could involve passive transport. However, at low temperatures Ago‐2 may adhere to the cell surface,12 resulting in an miR‐18a and Ago‐2 complex adsorbed onto the outer surface of the membrane; this led to an overestimate of intracellular miR‐18a levels when cell lysates were evaluated. Accordingly, confocal microscopy studies revealed that at 4°C in the presence of AVM‐BEC‐CM and miR‐18a control BEC were positive for Ago‐2 staining on the cell surface despite rigorous washing (Figure 4B). Therefore, the data showing high levels of miR‐18a at are likely the result of Ago‐2 adsorption onto the cell surface, rather than passive cellular uptake.
Figure 4.

Facilitated transport is involved in miR‐18a delivery. A, AVM‐BEC and control BEC were treated with AVM‐BEC‐CM plus miR‐18a (40 nmol/L) at 4°C and 37°C for 30 minutes. Intracellular miR‐18a was measured using qPCR as described previously, showing that at 4°C miRNA entry was only minimally compromised (n=3). B, The distribution of Ago‐2 (red) was identified using immunocytochemistry. At 4°C untreated AVM‐BEC expressed high levels of intracellular Ago‐2 (i) compared to untreated control BEC (ii). When control BEC were treated with AVM‐BEC‐CM plus miR‐18a at 4°C, Ago‐2 staining was apparent and associated with the cell membrane (iii; white arrows) (n=3). Blue staining denotes nuclear staining. C, The formation of a ribonucleoprotein complex between Ago‐2 and miR‐18a was determined by immunoprecipitation and immunoblotting (left panel) and qPCR (right panel). Ago‐2 was detected only in the 2 fractions in contact with anti‐Ago‐2, as expected. Only the fraction with both Ago‐2 and miR‐18, but not miR‐18a alone, led to the detection of miR‐18a by qPCR. Rabbit IgG served as the isotypic control. D, GW4869 (10 to 50 μmol/L), a specific inhibitor of N‐Smase‐2 (neutral sphingomyelinase‐2) did not interfere with miR‐18a uptake in AVM‐BEC. Data are presented as mean±standard error of the mean (SEM). Each human specimen‐derived cell culture represents a unit of analysis (n). AVM indicates arteriovenous malformation; Ago‐2, Argonaute‐2; BEC, brain endothelial cells; CM, conditioned media; qPCR, quantitative real‐time polymerase chain reaction.
To determine whether Ago‐2 formed a ribonucleoprotein complex with miR‐18a, we performed Ago‐2 immunoprecipitation, followed by immunoblotting for Ago‐2 and qPCR analysis for miRNA detection (Figure 4C). Ago‐2 was only detected in the 2 fractions that were in contact with anti‐Ago‐2 (Figure 4C; left panel), as expected. Accordingly, the anti‐Ago‐2‐coated beads in contact with both Ago‐2 and miR‐18a, but not with miR‐18a alone, led to the detection of miR‐18a by qPCR, because this miRNA was bound to Ago‐2 (0.13±0.03; n=3) (Figure 4C; right panel). The absence of Ago‐2 or miR‐18a in the isotypic control immunoprecipitated fraction demonstrated that miR‐18a was specifically associated with Ago‐2.
miRNA delivery reportedly may occur through other mechanisms, namely through exosome release.13 This hypothesis was excluded by using GW4869, a specific inhibitor of N‐Smase‐2 (neutral sphingomyelinase‐2), necessary for exocytosis. Experiments showed that this agent did not interfere with miR‐18a uptake (Figure 4D).
Silencing Ago‐2 Compromises miR‐18a‐Induced TSP‐1 Increase
Previously, we have reported that miR‐18a treatment increases TSP‐1 release by AVM‐BEC.5 Hence, we treated AVM‐BEC with siAgo‐2 (75 nmol/L) to determine if miR‐18a‐induced TSP‐1 release would be decreased in the presence of low Ago‐2 levels (Figure 5A). Thus, AVM‐BEC were treated with siAgo‐2 and then incubated with miR‐18a. The amount of TSP‐1 protein secreted by these cells was less than miR‐18a treated AVM‐BEC (Figure 5A) (untreatedAVM‐BEC=209.3±21.12; miR‐18aAVM‐BEC=425.3± 48.3; siAgo2+miR‐18aAVM‐BEC=289.4±52.34; n=4; P<0.05). In addition, when control BEC were treated with antagomir of miR‐18a (40 to 120 nmol/L), the results showed that inhibition of endogenous miR‐18a (80 nmol/L) significantly decreased TSP‐1 levels (untreatedcontrol BEC= 934.8±53.2; lipof.+antagomir80control BEC=664.5±66.7; antagomir80+ AVM‐BEC‐CMcontrol BEC=706.7±46.7; n=4; P<0.01) (Figure 5B). These data show that decreasing Ago‐2 decreases miR‐18a entry and therefore lowers TSP‐1 secretion; thus miR‐18a is internalized and is dependent on Ago‐2 availability.
Figure 5.
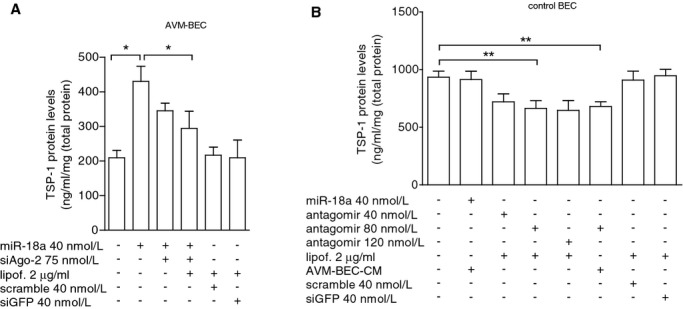
Ago‐2 silencing decreases miR‐18a‐induced TSP‐1 secretion. A, AVM‐BEC were treated with siAgo‐2 (75 nmol/L) followed by miR‐18a treatment (40 nmol/L) and cell supernatants tested for TSP‐1 (n=4; *P<0.05; paired t test). B) Control BEC were treated with varying concentrations of the miR‐18a inhibitory sequence, antagomir (40 to 120 nmol/L). Antagomir treatment (80 nmol/L) significantly decreased TSP‐1 levels (n=4; **P<0.01; paired t test). Data are presented as mean±standard error of the mean (SEM). Each human specimen‐derived cell culture represents a unit of analysis (n). AVM indicates arteriovenous malformation; Ago‐2, Argonaute‐2; BEC, brain endothelial cells; CM, conditioned media; TSP‐1, thrombospondin‐1.
Intravenous Administration of miR‐18a In Vivo Enhances Anti‐Angiogenic Properties
Currently there are no validated in vivo brain AVM models; hence, we used an alternative intracranial tumor model that also exhibited active angiogenesis to test the anti‐angiogenic potential of mir‐18a in vivo. To initiate angiogenesis, we injected renilla luciferase‐labeled human glioma cells into immune‐incompetent athymic nude mice (Figure 6). In this model, the systemic application of an anti‐angiogenic agent has greatest impact when given during the first week post‐implantation of tumor cells.14 Thus, to achieve maximal effects on angiogenesis, treatment was initiated 3 days after intracranial implantation of tumor cells. The animal groups were as follows: group 1: vehicle (PBS); group 2: miR‐18a (40 nmol/L) in combination with Ago‐2 (0.4 nmol/L); group 3: miR‐18 alone (40 nmol); group 4: Ago‐2 alone (0.4 nmol/L). Agents were administered intravenously (lateral tail vein) every 48 hours until 3 treatments were completed per group; animals were then euthanized at day 9 and blood samples were collected. Analysis of blood samples showed that the combination treatment of miR‐18a and Ago‐2 resulted in a significant change in the key angiogenic factors, TSP‐1 and VEGF‐A (Figures 6A and 6B, respectively). The combination treatment of miR‐18a and Ago‐2 led to a significant increase in TSP‐1 levels as compared to vehicle‐treated animals (vehicle=2.9±2.9; miR‐18+Ago‐2=30.0±6.6; n=5; P<0.01) (Figure 6A). Furthermore, TSP‐1 protein levels in response to miR‐18a and Ago‐2 co‐treatment was similar to levels found in healthy animals (healthy=34.6±4.6; n=3). miR‐18a alone also increased TSP‐1 levels but to a lesser extent (miR‐18a=21.0±1.3; n=5; P<0.05). The combination of miR‐18a and Ago‐2, or miR‐18a alone, led to a decrease in secretion of the pro‐angiogenic VEGF‐A (vehicle=92.1±22.6; miR‐18a+Ago‐2=43.6±8.2; miR‐18a=32.0±10.1; healthy=36.6±7.3; n=3 to 5; P<0.05) (Figure 6B). Ago‐2 treatment alone had no effect on either TSP‐1 or VEGF‐A secretion. To test whether the in vivo data resulted from activation of tumor cells rather than blood vessel cells, we analyzed the levels of miR‐18a internalized by the implanted tumor cells (U251). We observed no intracellular uptake of miR‐18a over time indicating that the effects of miR‐18 and Ago‐2 were a reflection of brain endothelial cell internalization and processing and not tumor cell internalization of miR‐18a (Figure 6C), which further supports that miR‐18a delivery occurs to the brain vasculature and not to brain cells overall. Imaging data also showed that mice treated with miR‐18a alone and in combination with Ago‐2 showed a trend towards reduced tumor growth compared to vehicle treatment, although these differences were not significant (Figures 6D and 6E). Based on a histological examination of tissue slides, we did not observe any pathology in peripheral organs in any of the groups tested. Hence, our data show that Ago‐2 in combination with miR‐18a is effective and functional in vivo.
Figure 6.
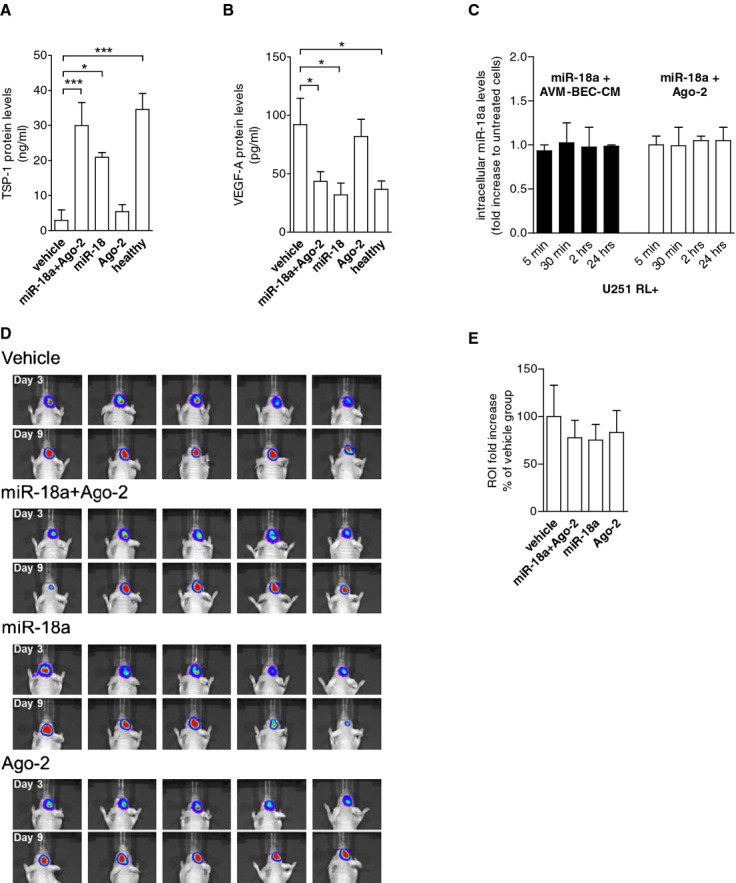
Co‐treatment of miR‐18a and Ago‐2 in vivo “normalizes” TSP‐1 and VEGF‐A plasma levels. A, Athymic nude mice were implanted with glioma cells intracranially. After 3 days, animals were treated intravenously with vehicle, miR‐18a plus Ago‐2, miR‐18a alone or Ago‐2 alone every 48 hours for 3 cycles. Subsequently, plasma was tested for TSP‐1 (A) and VEGF‐A (B). miR‐18a and Ago‐2 combination treatment caused the most significant increase of TSP‐1 levels (n=5; *P<0.05; ***P<0.001; paired 1‐way ANOVA followed by Dunnett's Multiple Comparison Test), and reduction of VEGF‐A levels (n=5; *P<0.05; paired 1‐way ANOVA followed by Dunnett's Multiple Comparison Test). Control plasma (healthy) was obtained from normal athymic mice. C, qPCR analysis of miR‐18a detection showed that tumor cells did not internalize miR‐18a in the presence of AVM‐BEC‐CM (black bars) or Ago‐2 (white bars) (n=3). D, Imaging of intracranial renilla luciferase‐positive tumor cells day 3 after implantation (beginning of treatment) and day 9 (treatment completion). E, Region of interest (ROI) analysis of detected luminescence showed that miR‐18a and Ago‐2 combination and miR‐18a treatment alone led to slower tumor growth as compared to vehicle‐treated animals, albeit not statistically significant (n=5). Data are presented as mean±standard error of the mean (SEM). ANOVA indicates analysis of variance; AVM, arteriovenous malformation; Ago‐2, Argonaute‐2; BEC, brain endothelial cells; CM, conditioned media; qPCR, quantitative real‐time polymerase chain reaction; TSP‐1, thrombospondin‐1; VEGF, vascular endothelial growth factor.
Discussion
In this study, we show that AVM‐BEC secrete RNA‐binding protein Ago‐2, which serves as a carrier for miR‐18a and mediates, in part, miR‐18a specific entry into brain endothelial cells. Furthermore, the combination of miR‐18a and Ago‐2 is active in vivo, and can modulate the expression of angiogenic factors, thereby demonstrating that intravascular delivery of miRNA to the brain vasculature is a feasible therapeutic approach.
Previously, we have shown that the exogenous application of miR‐18a can ameliorate the abnormal characteristics of AVM‐BEC without transfection reagents.5 The purpose of our study was to explore the mechanisms by which extracellular miR‐18a is efficiently internalized by brain endothelial cells. miRNA is detected extracellularly in a variety of human body fluids including blood.15–16 Although the bloodstream is enriched with nucleases, and rapid renal clearance may lead to miRNA degradation and clearance,17 systemic delivery of miRNA sequences without transfection reagents (naked delivery) has been attempted successfully, namely with inhibitors of miRNA.18 Intraperitoneal injection of anti‐miR‐182 reduced tumor metastasis in the liver of mice, showing that an antagomir without packaging system can be internalized by tumor cells.19 Additionally, intravenous injection of anti‐miR‐122 entered virally infected cells, thus inhibiting viral replication and improving virus‐induced liver disease.20 Mounting evidence has suggested that miRNA, and other nucleic acids, are spared from degradation by either being encased in apoptotic bodies, lipid vesicles, by forming complexes with high‐density lipoproteins, or with ribonucleoproteins. The latter is believed to be the main mechanism for miRNA trafficking and for that reason our study was focused on Ago‐2‐mediated delivery.11 In fact, several extracellular miRNAs, including miR‐18a, found in human blood plasma or cell culture media are associated with the RNA‐binding protein Ago‐221–22_ENREF_19. Nucleolin was also upregulated at an mRNA level, but to a smaller extent and because it has not been described to bind to miR‐18a, we focused our studies in the more biologically relevant target.
In mammalian cells, Ago‐2 proteins bind intracellularly to endogenous double‐stranded small RNAs for incorporation into RISC23 or extracellularly to exogenous miRNA, such as miR‐18a, used in our work. This activity suggests the possibility of a more general mechanism of action for Ago‐2 in miRNA delivery. Our results demonstrated that Ago‐2 was the major factor in AVM‐BEC‐CM, responsible to a great extent for delivery and uptake of miR‐18a to AVM‐BEC recipient cells, by forming a stable ribonucleoprotein complex with miR‐18a. Production of Ago‐2 was antagonized in AVM‐BEC treated with siAgo‐2 (siAgo‐2‐AVM‐BEC‐CM) resulting in compromised miR‐18a entry and activity (Figures 2 through 5).
These results were further extended by the detection of Ago‐2/miR18a complexes internalized by recipient BEC, as demonstrated by confocal fluorescence microscopy (Figure 3). This analysis clearly showed that the combination of miR‐18a with Ago‐2 was required for cellular uptake of miR‐18a. The use of a labeled miR‐18a would further validate our internalization data; however a labeled mimic may cause a structural change in the miRNA and consequently, in its properties. Structural alterations may alter miRNA binding to Ago‐2, cellular uptake by endothelial cells and the efficient crossing of the blood‐brain barrier.
The addition of either AVM‐BEC‐CM or miR‐18a alone did not result in Ago‐2 uptake or a functional response. The requirement for this binary complex also implied the specificity of a putative cell membrane receptor for the ribonucleoprotein complex prior to facilitated internalization, as shown by fluorescence microscopy (Figure 4). However, to the best of our knowledge there is no known ribonucleoprotein receptor for Ago‐2 complexes. Additional studies are needed to confirm this hypothesis.
Endothelial cells have been shown to use Ago‐2 to deliver miRNA cargoes for cell‐to‐cell communication.24 Our studies further extend this observation to show that brain endothelial cells are highly permissive for miRNA uptake compared with other endothelial cell types (eg, HMEC and HUVEC). Whether this is due to an intrinsic property of brain endothelial cells or is related to differences in ribonucleoprotein receptor density among the different endothelial cell types remains to be shown. Because normal brain endothelial cells are able to rapidly uptake miR‐18a in the presence of AVM‐BEC‐CM or exogenously applied Ago‐2, we believe that miR‐18a uptake does not rely on physical properties of these endothelial cells (eg, differences in membrane permeability); therefore a receptor is more likely involved.
We previously established that miR‐18a, through the inhibition of Id‐1 expression, derepresses TSP‐1 secretion in AVM‐BEC, thus “normalizing” the abnormal features of these cells.5 We demonstrated that miR‐18a‐induced TSP‐1 secretion is compromised in the presence of lower amounts of Ago‐2 (Figure 5), thus corroborating our previous results and extending these observations by demonstrating that Ago‐2 was needed for miRNA delivery.
The potential therapeutic use of Ago‐2 as a miRNA carrier bypasses the many challenges faced by in vivo delivery of miRNA, primarily protection from serum nuclease degradation.25 Because endothelial cells are in direct and exclusive contact with the bloodstream, intravascular miRNA delivery can achieve high target specificity with negligible side effects. An interesting result is that brain endothelial cells are highly permissive for uptake of miR‐18a, raising the possibility of intravascular delivery of therapeutic miRNA to AVM‐BEC and possibly other brain vascular dysfunctions. As a preliminary demonstration of an in vivo response to intravenous Ago‐2/miR‐18a delivery we used an intracranial tumor model, which gives rise to highly vascularized tumors (Figure 6). This approach was used because there are no in vivo brain AVM models. To demonstrate the anti‐angiogenic potential of miR‐18a and Ago‐2 co‐treatment blood samples were collected from Ago‐2/miR‐18a‐treated animals to evaluate key angiogenic markers, namely TSP‐1 and VEGF‐A.5 miR‐18a alone and miR‐18a and Ago‐2 co‐treatment significantly increased TSP‐1, and decreased VEGF‐A secretion to levels comparable to healthy controls. Administration of miR‐18a alone also showed an anti‐angiogenic effect; however, this could be attributed to the presence of circulating Ago‐2 in the bloodstream. However, it should be noted that miR‐18a degradation may raise some concerns (80% in less than 24 hours) for successful naked miRNA delivery to the brain. Nevertheless, in mice a complete blood circulation circuit is accomplished in <15 seconds and in humans, in 1 to 2 minutes. Because miR‐18a internalization peaks at 30 minutes, the use of naked miRNA, particularly in combination with Ago‐2, is a feasible approach to target the brain vasculature.
In conclusion, AVM‐BEC secrete RNA‐binding protein Ago‐2, which is involved in part in the mediation of miR‐18a delivery to brain endothelial cells. Moreover, miR‐18a delivered by Ago‐2 is functionally active both in vitro and in vivo. Thus, Ago‐2 can be used as an efficient vehicle for miRNA, supporting the development of potentially safer and more efficient brain endothelial cell‐targeted therapies.
Sources of Funding
Work was supported by the Neithart Foundation (Ferreira, Giannotta), the Haile Foundation (Chen) and NIH grant HL111372 (Tahara).
Disclosures
None.
References
- 1.Yashar P, Amar AP, Giannotta SL, Yu C, Pagnini PG, Liu CY, Apuzzo ML. Cerebral arteriovenous malformations: issues of the interplay between stereotactic radiosurgery and endovascular surgical therapy. World Neurosurg. 2011; 75:638-647. [DOI] [PubMed] [Google Scholar]
- 2.Haw CS, terBrugge K, Willinsky R, Tomlinson G. Complications of embolization of arteriovenous malformations of the brain. J Neurosurg. 2006; 104:226-232. [DOI] [PubMed] [Google Scholar]
- 3.Jabbour MN, Elder JB, Samuelson CG, Khashabi S, Hofman FM, Giannotta SL, Liu CY. Aberrant angiogenic characteristics of human brain arteriovenous malformation endothelial cells. Neurosurgery. 2009; 64:139-146. [DOI] [PubMed] [Google Scholar]
- 4.Stapleton CJ, Armstrong DL, Zidovetzki R, Liu CY, Giannotta SL, Hofman FM. Thrombospondin‐1 modulates the angiogenic phenotype of human cerebral arteriovenous malformation endothelial cells. Neurosurgery. 2011; 68:1342-1353. [DOI] [PubMed] [Google Scholar]
- 5.Ferreira R, Santos T, Amar A, Tahara SM, Chen TC, Giannotta SL, Hofman FM. Microrna‐18a improves human cerebral arteriovenous malformation endothelial cell function. Stroke. 2014; 45:293-297. [DOI] [PubMed] [Google Scholar]
- 6.Bartel DP. Micrornas: genomics, biogenesis, mechanism, and function. Cell. 2004; 116:281-297. [DOI] [PubMed] [Google Scholar]
- 7.Etheridge A, Lee I, Hood L, Galas D, Wang K. Extracellular microrna: a new source of biomarkers. Mutat Res. 2011; 717:85-90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Boon RA, Vickers KC. Intercellular transport of micrornas. Arterioscler Thromb Vasc Biol. 2013; 33:186-192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu J, Carmell MA, Rivas FV, Marsden CG, Thomson JM, Song JJ, Hammond SM, Joshua‐Tor L, Hannon GJ. Argonaute2 is the catalytic engine of mammalian rnai. Science. 2004; 305:1437-1441. [DOI] [PubMed] [Google Scholar]
- 10.Virrey JJ, Golden EB, Sivakumar W, Wang W, Pen L, Schonthal AH, Hofman FM, Chen TC. Glioma‐associated endothelial cells are chemoresistant to temozolomide. J Neurooncol. 2009; 95:13-22. [DOI] [PubMed] [Google Scholar]
- 11.Creemers EE, Tijsen AJ, Pinto YM. Circulating micrornas: novel biomarkers and extracellular communicators in cardiovascular disease? Circ Res. 2012; 110:483-495. [DOI] [PubMed] [Google Scholar]
- 12.Lundberg M, Johansson M. Positively charged DNA‐binding proteins cause apparent cell membrane translocation. Biochem Biophys Res Commun. 2002; 291:367-371. [DOI] [PubMed] [Google Scholar]
- 13.Kosaka N, Iguchi H, Yoshioka Y, Takeshita F, Matsuki Y, Ochiya T. Secretory mechanisms and intercellular transfer of micrornas in living cells. J Biol Chem. 2010; 285:17442-17452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rubenstein JL, Kim J, Ozawa T, Zhang M, Westphal M, Deen DF, Shuman MA. Anti‐vegf antibody treatment of glioblastoma prolongs survival but results in increased vascular cooption. Neoplasia. 2000; 2:306-314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fabbri M. Tlrs as mirna receptors. Cancer Res. 2012; 72:6333-6337. [DOI] [PubMed] [Google Scholar]
- 16.Weber JA, Baxter DH, Zhang S, Huang DY, Huang KH, Lee MJ, Galas DJ, Wang K. The microrna spectrum in 12 body fluids. Clin Chem. 2010; 56:1733-1741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pecot CV, Calin GA, Coleman RL, Lopez‐Berestein G, Sood AK. RNA interference in the clinic: challenges and future directions. Nat Rev Cancer. 2011; 11:59-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nana‐Sinkam SP, Croce CM. Micrornas as therapeutic targets in cancer. Transl Res. 2011; 157:216-225. [DOI] [PubMed] [Google Scholar]
- 19.Huynh C, Segura MF, Gaziel‐Sovran A, Menendez S, Darvishian F, Chiriboga L, Levin B, Meruelo D, Osman I, Zavadil J, Marcusson EG, Hernando E. Efficient in vivo microrna targeting of liver metastasis. Oncogene. 2011; 30:1481-1488. [DOI] [PubMed] [Google Scholar]
- 20.Lanford RE, Hildebrandt‐Eriksen ES, Petri A, Persson R, Lindow M, Munk ME, Kauppinen S, Orum H. Therapeutic silencing of microrna‐122 in primates with chronic hepatitis c virus infection. Science. 2010; 327:198-201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Turchinovich A, Weiz L, Langheinz A, Burwinkel B. Characterization of extracellular circulating microrna. Nucleic Acids Res. 2011; 39:7223-7233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Arroyo JD, Chevillet JR, Kroh EM, Ruf IK, Pritchard CC, Gibson DF, Mitchell PS, Bennett CF, Pogosova‐Agadjanyan EL, Stirewalt DL, Tait JF, Tewari M. Argonaute2 complexes carry a population of circulating micrornas independent of vesicles in human plasma. Proc Natl Acad Sci USA. 2011; 108:5003-5008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Czech B, Hannon GJ. Small rna sorting: matchmaking for argonautes. Nat Rev Genet. 2011; 12:19-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhou J, Li YS, Nguyen P, Wang KC, Weiss A, Kuo YC, Chiu JJ, Shyy JY, Chien S. Regulation of vascular smooth muscle cell turnover by endothelial cell‐secreted microrna‐126: role of shear stress. Circ Res. 2013; 113:40-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dominska M, Dykxhoorn DM. Breaking down the barrierssirna delivery and endosome escape. J Cell Sci. 2010; 123:1183-1189. [DOI] [PubMed] [Google Scholar]