

Abstract
Background
DNA methylation is a major epigenetic mechanism altering gene expression in development and disease. However, its role in the regulation of gene expression during heart development is incompletely understood. The aim of this study is to reveal DNA methylation in mouse embryonic hearts and its role in regulating gene expression during heart development.
Methods and Results
We performed the genome‐wide DNA methylation profiling of mouse embryonic hearts using methyl‐sensitive, tiny fragment enrichment/massively parallel sequencing to determine methylation levels at ACGT sites. The results showed that while global methylation of 1.64 million ACGT sites in developing hearts remains stable between embryonic day (E) 11.5 and E14.5, a small fraction (2901) of them exhibit differential methylation. Gene Ontology analysis revealed that these sites are enriched at genes involved in heart development. Quantitative real‐time PCR analysis of 350 genes with differential DNA methylation showed that the expression of 181 genes is developmentally regulated, and 79 genes have correlative changes between methylation and expression, including hyaluronan synthase 2 (Has2). Required for heart valve formation, Has2 expression in the developing heart valves is downregulated at E14.5, accompanied with increased DNA methylation in its enhancer. Genetic knockout further showed that the downregulation of Has2 expression is dependent on DNA methyltransferase 3b, which is co‐expressed with Has2 in the forming heart valve region, indicating that the DNA methylation change may contribute to the Has2 enhancer's regulating function.
Conclusions
DNA methylation is developmentally regulated for genes essential to heart development, and abnormal DNA methylation may contribute to congenital heart disease.
Keywords: DNA methylation, DNA methyltransferase 3b, gene expression, heart development, hyaluronan synthase 2
Introduction
The heart is the first organ to develop during embryogenesis. In the developing mouse heart, between embryonic day (E) 11.5 and E14.5, cardiac cells undergo differentiation, migration, and proliferation driving cardiac tissue morphogenic events including chamber septation, heart valve formation, myocardial compaction, and coronary vessel formation, all essential for proper heart development.1–4 These processes are directed by cardiac transcriptional programs and endocardial‐myocardial molecular signalings.5–8 Both genetic and epigenetic mechanisms have been shown to control the expression of cardiac genes in a spatiotemporal manner during heart development.9–13
Epigenetic modifications, including DNA methylation and histone modification, regulate gene expression by changing the local chromatin structure, thus altering the interaction of chromatin and DNA‐binding proteins, such as the binding of transcription activators and repressors to gene promoters and enhancers.14–16 Different from genetic variation, epigenetic modifications regulate gene expression without altering the nucleotide sequence. In the case of DNA methylation, a methyl group is added to the carbon 5 of cytosine located at a CpG dinucleotide. It has been shown that DNA methylation is essential for gene regulation during development, especially that of tissue‐specific genes, and help to maintain cell and tissue identity.17–19 Notably, with the exception of imprinted genes, the mammalian genome is stripped of its epigenetic modifications in early embryogenesis and the epigenome is then re‐established throughout embryonic development.20–22
Globally, the patterns of DNA methylation acquired during embryogenesis remain stable throughout development and adulthood.19 However, changes in DNA methylation at individual loci do occur and can alter expression of genes with important biological functions in development and disease.23–24 DNA methylation change also occurs in response to developmental perturbations, such as hypoxia. Altered levels of 5‐methylcytosine, either genome wide or at specific gene loci, have been related to increased disease susceptibility, and dysregulation of DNA methylation has been linked to cardiovascular disease, type II diabetes, and cancer.24–26
Most studies on DNA methylation have focused on gametogenesis, development, disease, and stem cell function by demonstrating how it regulates gene expression and cell differentiation.23,27–30 Few studies, however, have been devoted to understand the roles of DNA methylation in heart development. Determining the landscape of DNA methylation in this process is an essential step for understanding how DNA methylation regulates the cardiac genes essential for heart development.
Towards this end, we have applied a genome‐wide approach in this study to profile developmental changes in DNA methylation in mouse embryonic hearts between E11.5 and E14.5. The morphogenic events occurring during this developmental window are less well studied than early morphogenic events such as the differentiation of cardiac cells. The results show that while the DNA methylome is stable during development, differential methylation occurs at a small subset of genes highly associated with cardiac tissue differentiation and heart development and reveal a regulatory relationship between differential DNA methylation and cardiac essential gene expression. Thus, these results provide new information on the regulation of cardiac gene expression and heart development by DNA methylation.
Methods
Animals (Mice)
ICR wild‐type mice were bred in‐house for timed pregnancies. Noontime on the day of first observing vaginal plugs was designated as embryonic day (E) 0.5. For Dnmt3b knockout studies, endocardial specific Cre mice (Nfatc1Cre)9 were crossed with floxed Dnmt3b (DNA methyltransferase 3b) mice (obtained from The Jackson Laboratory) to delete Dnmt3b in heart valves. Conditional knockout (CKO) and control embryos were identified via PCR genotyping. All mouse experiments were performed according to the protocol approved by the Institutional Animal Care and Use Committee of Albert Einstein College of Medicine.
Methyl Sensitive Tiny Fragment Enrichment/Massively Parallel Sequencing (MSFE/MPS)
Embryonic hearts from E11.5 or E14.5 were isolated from pregnant mice and non‐cardiac tissues were removed. Genomic DNA was extracted from 4 groups of pooled hearts as described previously.31 A total of 5 μg of extracted DNA from each group was used for a modified HELP‐tagging assay.32 We modified the original assay by replacing HpaII with HpyCH4IV, the restriction enzyme recognizing 5′‐ACGT‐3′ sites and sensitive to methylation at the CG. After HpyCH4IV digestion, the sequencing libraries were generated using the Ligation Mediated PCR Assay (LMPA).33 The generated libraries were submitted to the Epigenomics Shared Facility at the Albert Einstein College of Medicine for massively parallel sequencing. Sequencing was performed on individual libraries prepared from 2 biological replicates for each group. The quality of the sequencing results was determined by the parameters of length and peak value of sequence reads. The raw and processed data have been submitted to GEO (accession number: GSE55141).
Luminometric Methylation Assay to Validate the Global DNA Methylation
Global DNA methylation for each biological replicate, at both E11.5 and E14.5, was confirmed using the Luminometric Methylation Assay (LUMA) as described previously.34–35 Genomic DNA was digested for 4 hours with HpyCH4IV and EcoRI, purified and pyrosequenced at the Einstein Genomics Core.
MassArray
Loci with differential methylation, ranging from 0% to 100% determined by the massively parallel sequencing, were randomly selected and validated using Sequenom's MassArray.36 Primers were designed using MethPrimer and T7 tags were added as per the Sequenom MassArray protocol (Table 1). Genomic DNA (0.6 μg) from 3 replicates (for technical validation) and 2 replicates (for experimental validation) was bisulfite converted using the Zymo Research EZ DNA Methylation Kit prior to amplification using the MassArray Primers. Amplified bisulfite‐converted genomic DNA was then subjected to MassArray on a Sequenom machine.
Table 1.
Loci and Primers (With T7 Tags, Lower Cases) for Initial Technical Validation by MassArray
| ACGT Location | Forward Primer With T7 Balance (Top) Reverse Primer With T7 Tag (Bottom) |
|---|---|
| Chr4 145913471 | aggaagagagAGTTTTTATGTTTTTTGTAAGGTTATTTGA cagtaatacgactcactatagggagaaggctCCAAACTCCTAATCCCAATCTAAC |
| Chr7 147043670 | aggaagagagTTGGGGGTTTTTAATTAAGATAGTTT cagtaatacgactcactatagggagaaggctTTCCCTCTAATATATCCCATTTTACC |
| Chr12 62103733 | aggaagagagAGTATTAGGGTTAAGTATTGAATAAATTTA cagtaatacgactcactatagggagaaggctAATCAAAATAAAAAATCAAAAAAAA |
| Chr11 4347732 | aggaagagagTTTAGGTATATATTATTATATTTGATTTTT cagtaatacgactcactatagggagaaggctAATTACCACAAAACCTAACAC |
| Chr2 38354047 | aggaagagagAGAGTGGTATTTGTGTTAGAGAGGA cagtaatacgactcactatagggagaaggctTTCCAAAAAAAACCAAAAAAAA |
| Chr6 64724205 | aggaagagagTGTTTTTGTAATTTAGATAAGATTATTTTA cagtaatacgactcactatagggagaaggctAATCACATATCACTAACCAAACAATATC |
| Chr2 165155478 | aggaagagagGGGTGATAGGAAGTTGTAGAGATTAGA cagtaatacgactcactatagggagaaggctAAAAAAACACTACCCAAACTTAAATAACA |
| Chr11 3036940 | aggaagagagGGGTATTTGTTTAAGATATTTTTGATTTAT cagtaatacgactcactatagggagaaggctACAATAACCAAATAAAAAACACACCA |
| Chr12 101338922 | aggaagagagTGGTGTTTTAGTTGTTAAAATGTTATAGG cagtaatacgactcactatagggagaaggctCAAAAATATTCCCCAAATATCAAAA |
| Chr18 55862932 | aggaagagagTAGAAAAATAGGGAGAATGTGATATT cagtaatacgactcactatagggagaaggctATATCTAACTTCCCTACACCCACTAAAA |
| Chr1 25839243 | aggaagagagTTTTAGGATTGAATAAAATTTTAAGA cagtaatacgactcactatagggagaaggctATTTAATTTACTCATTCTCTCTATATAC |
Individual sites representing 0%, 25%, 50%, 75% or 100% methylation, that were consistent between the 2 assayed samples, were chosen for validation by Sequenom's MassArray to generate a standard by which to make calls on methylation levels. Primers were designed using MethPrimer and T7 tags were added as per the Sequenom guide.
Bioinformatic Analysis to Profile Genome‐Wide DNA Methylation
The sequencing reads were aligned to the mouse genome (mm9) and the number of mapped reads with their 5′ ends starting at each ACGT site was recorded using the automated data analysis pipeline created by the Epigenomics Center and the Computational and Statistical Epigenomics Group at Albert Einstein College of Medicine.32,37 The read counts at individual ACGT sites from E11.5 and E14.5 were compared and sites with significantly different counts were determined by EdgeR, a Bioconductor package designed for analysis of count based genome‐wide sequencing data.38 The resultant sites were associated with genes if they were located to promoters, gene bodies, or within 50 kb of genes.
Gene Expression Analysis
Custom TaqMan Array 96‐Well Fast Plates (Applied Biosystems) were designed for candidate genes prioritized based on degree of differential methylation, function, and presence of multiple‐associated differentially methylated ACGT sites. RNA was extracted from pooled embryonic hearts from E11.5 or E14.5 (n=3 for each stage) and atrioventricular junctions isolated from 3 wild‐type or 3 CKO embryos at E11.5 and E14.5 using Trizol Reagent (Invitrogen) and reverse transcribed using the SuperScript II reverse transcription kit (Invitrogen). ΔCt values were calculated, normalizing to an endogenous control, and fold change was calculated using the 2−ΔΔCt method.39
RNA In Situ Hybridization
RNA in situ hybridization for Has2 expression in E11.5 or E14.5 hearts was carried out as described previously.5
Immunohistochemistry
Immunohistochemistry (IHC) was carried out to determine expression of Dnmt3b in the developing heart using mouse monoclonal Dnmt3b antibody (Abcam 52A1018) (1:250), according to the Vector Labs mouse‐on‐mouse (M.O.M.) basic kit.
Statistical Analysis
Pearson correlation was used to evaluate the overall similarity of MSFE/MPS tag counts between E11.5 and E14.5 samples. A linear regression was used to fit the relationship between tag counts and DNA methylation levels at 14 selected ACGT sites. Differentially methylated (DM) sites were identified by the program EdgeR,38 with <5% FDR. A two‐sided t test was used to evaluate the difference of tag counts at ACGT sites located to different genomic contents, while hypergeometric test was used to evaluate the enrichment of DM sites in promoters and gene bodies. The statistical analysis of differential gene expression was performed using Microsoft Excel and the data were presented as mean±standard error (SE). Student t test was used for comparison between groups and P values <0.05 were considered as significant, Bonferonni's correction was applied to account for multiple testing in gene expression analysis. For the expression analysis of 350 genes, Mann–Whitney test was also performed (data not shown). While the significance observed for the top 15 up‐ and down‐regulated genes in Figure 5B remained, a few other differentially expressed genes included in Figure 5A would lose statistical support.
Figure 5.
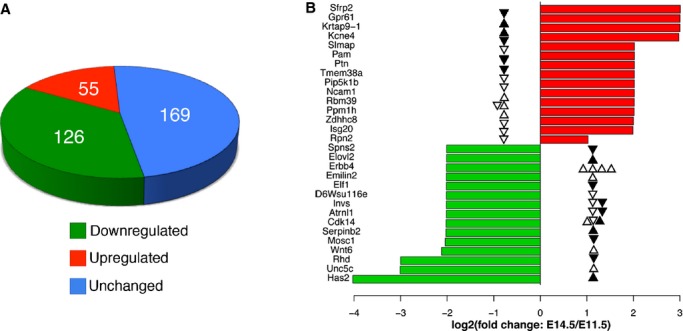
Expression changes for selected genes with differentially methylated sites. A, Gene expression analysis (n=3) of 350 differentially methylated genes between E11.5 and E14.5 showing that 181 genes are differentially expressed and 79 of those genes show consistent changes in DNA methylation. B, The top 15 upregulated genes (red) and downregulated genes (green) in E14.5. The solid and open triangles mark differentially methylated sites in regulatory regions (promoter proximal or distal) and gene bodies, respectively. The up and down directions of triangles indicate increased and decreased methylation, respectively.
Results
Global DNA Methylation is Stable in the Developing Heart
To study the importance of DNA methylation in heart development, we carried out a genome‐wide cytosine methylation analysis of E11.5 and E14.5 mouse embryonic hearts using MSFE/MPS with HpyCH4IV, a methylation‐sensitive restriction enzyme recognizing ACGT. A total of 75 278 236 and 28 496 681 sequencing reads were obtained from the 2 E11.5 replicates that were mapped to 1 522 872 and 1 447 993 ACGT sites, respectively, while 89 562 687 and 65 198 805 reads were generated from the 2 E14.5 replicates that were mapped to 1 442 766 and 1 490 463 sites, respectively. At both stages, the reads from the 2 replicates covered >83% of the total 1 756 340 ACGT sites in the mouse genome. The Pearson's correlation coefficients were 0.894 and 0.896 between the 2 replicates for E11.5 and E14.5, respectively (data not shown); indicating that the quality of the data was high. The majority of ACGT sites had highly similar and correlated tag counts between E11.5 and E14.5 (Figure 1A), suggesting at the global level no significant methylation changes occurred between the 2 developmental stages. This finding of genome‐wide stable DNA methylation in the developing hearts was supported by the LUMA data (Figure 1B).
Figure 1.
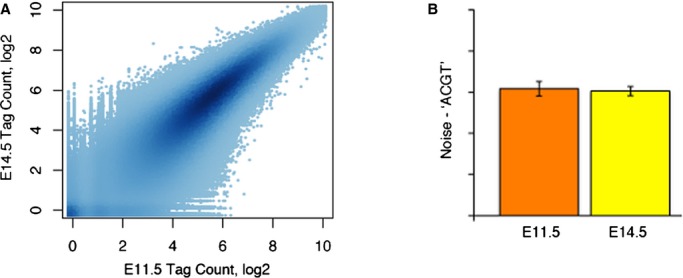
Limited DNA methylation changes between E11.5 and E14.5 hearts. A, The tag counts for all ACGT sites in E11.5 (x‐axis) and E14.5 (y‐axis) are highly correlated. The depth of the color represents the density of points in a plotting area (n=2). B, Confirmation of stable global methylation by Luminometric Methylation Assay (LUMA) (n=2). Error bars represent standard error.
Differential DNA Methylation Occurs Locally in the Developing Heart
We then catalogued the ACGT sites into genic sites (−50 kb of transcription start sites [TSS] to +0.5 kb of transcription end sites [TES] and intergenic sites) (Figure 2A), with the former further separated into 3 types: promoter proximal sites (−5 kb to +0.5 kb of TSS), gene body sites (+0.5 kb of TSS to TES), and promoter distal or enhancer sites (Figure 2B, top panel), and also determined the distribution of ACGT tag counts across the genome by intersecting the genome‐wide ACGT methylation profiles with several genomic features, including CpG islands, CTCF‐binding sites, RefSeq genes, repetitive elements, and regulatory elements (Figure 2B, bottom panel). The gene annotation, CpG islands, and repeats were downloaded from the UCSC browser. Additionally, lists of regulatory elements for embryonic hearts were obtained from previous studies, including 3596 P300 binding sites identified for E11.5 hearts, 69 073 P300‐marked enhancers, 14 874 CTCF sites, and 45 981 regions with the H3K27ac modification (a histone mark for active enhancer) for E14.5.40–42
Figure 2.

Distribution of DNA methylation across the mouse genome and various types of genomic elements in the developing heart. A, Cartoon depicting the genic regions (enhancer, promoter and genebody). B, Violin plot (a combination of a box plot and a kernel density plot) showing the distributions of tag counts for all ACGT (top) or differential methylated (bottom) sites in different genomic regions. As number of tag counts is inversely correlated to level of CG methylation, these plots indicate that gene promoters and regulatory regions exhibit significantly lower levels of DNA methylation than genomic background, and repetitive sequences are highly methylated. Plots in orange and yellow are for data from E11.5 and E14.5, respectively. TES indicates transcription end sites; TSS, transcription start sites.
The results showed that gene promoters and regulatory regions, represented by either CpG islands or enhancers (defined by P300 occupancy in E11.5 or H3K27ac enrichment in E14.5) had significantly lower levels of DNA methylation than genomic background, as ACGT sites within these regions had increased numbers of tag counts (P<2.2e‐16, t test). Similarly, CTCF‐binding sites generally have low levels of methylation. This is consistent with previous reports that CTCF is associated with hypomethylated regions.43 In contrast, repetitive regions showed significantly higher levels of DNA methylation, as ACGT sites in these regions had decreased numbers of tag counts (P<2.2e‐16, t test). Unexpectedly, the E14.5 cardiac enhancers exhibited higher DNA methylation than the elements marked by either P300 or H3K27ac at E14.5 (Figure 2B). This is probably due to the fact that those enhancers were identified based largely on H3K4me1 modifications, which is enriched in both active and poised enhancers.44–45
Next, we chose up to 14 ACGT sites with a range of different tag counts, representing 0%, 25%, 50%, 75%, or 100% methylation by massively parallel sequencing, and determined their levels of methylation by MassArray. The results indicated that tag count was inversely correlated with the percentage of cytosine methylation (Figure 3A), thereby confirming the precision of the MSFE/MPS in quantifying methylation level, ie, tag counts measured accurately both the global and regional DNA methylation. We then set out to investigate how much methylation changed in the developing hearts between E11.5 and E14.5. After normalization by sequencing depths, ACGT sites with at least 1 sequencing tag in any of the 4 samples were evaluated for differential methylation using 2 complementary approaches. We used EdgeR, which modeled the tag counts by a negative binomial distribution, to determine ACGT sites that showed differential methylation. The result indicated that the majority of the ACGT sites were not differentially methylated in the developing hearts between the 2 stages, as <1% of sites were found to have different tags (nominal P value <0.05) (Figure 3B). Among the small fraction (2901) of the ≈1.64 million analyzed ACGT sites that were differentially methylated, 1946 (67.1%) and 955 (32.9%) sites exhibited increased and decreased methylation in the late stage hearts, respectively (FDR<0.05) (Figure 3C).38 Of note, for the majority of these sites, the degree of difference was <50%, with no sites switching from a fully methylated to an unmethylated state.
Figure 3.
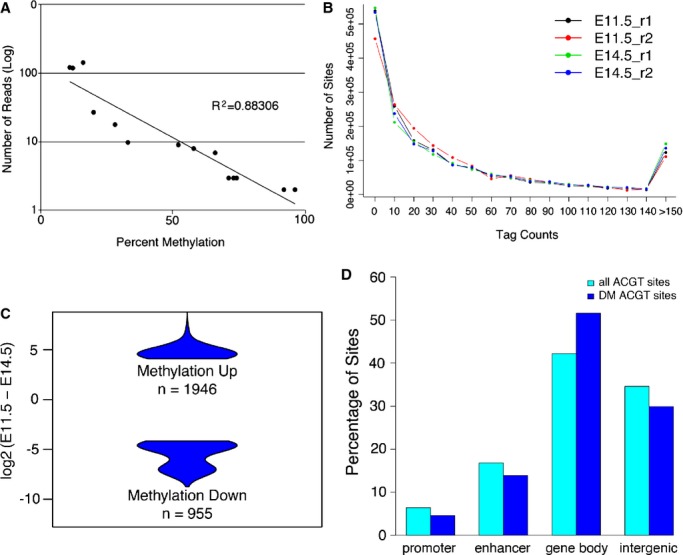
Analysis of the level of DNA methylation and differential methylation in the developing mouse heart. A, Methyl sensitive tiny fragment enrichment/massively parallel sequencing (MSFE/MPS) accurately detects levels of methylation as confirmed by Sequenom's MassArray of analyzed sites representing 0%, 25%, 50%, 75%, and 100% methylation determined by MSFE/MPS. B, Distribution of all ACGT sites with different numbers of tag counts from MSFE/MPS analysis. C, Violin plots showing the difference in tag counts for the 2901 ACGT sites that were significantly differentially methylated between E11.5 and E14.5. D, The distribution of all and differentially methylated (DM) ACGT sites in relation to gene annotation.
We also compared the percentage of differentially methylated ACGT sites at various genic and intergenic regions with the percentage of the total analyzed ACGT sites located within the same defined regions. We found that the differentially methylated sites were significantly enriched in gene bodies (P<2.2e‐16, hypergeometric test), as 51.6% of the differentially methylated sites versus 42.2% of all assayed sites were located to gene bodies (Figure 3D). On the contrary, differentially methylated ACGT sites were under‐represented in promoter (4.6%) and enhancer sites (13.9%), while 6.4% and 16.7% of all ACGT sites were in promoter‐proximal and enhancer regions, respectively. In total, 2032 (70%) of the 2901 sites were associated with genes, with 65.1% of them showing increased methylation at E14.5.
Differential DNA Methylation in the Developing Heart Links to Heart Development
To investigate the functional importance of the small set of genes exhibiting differential methylation, we used the software GREAT to characterize the 2901 differentially methylated sites for their potential regulatory roles. Of the only 7 significantly associated gene ontology (GO) terms for biological processes returned by GREAT, 4 of them were related to heart development and cardiac tissue growth (Figure 4), indicating a significant enrichment of cardiac essential genes that have differential DNA methylation during heart development. These genes include Erbb4, Gata6, Foxp1, Fgf2, Fgf9, Has2, Invs, Mef2c, Robo2, and Wnt2. For example, Foxp1 is important in cardiomyocyte proliferation,42,46 while signaling from Gata6 to Wnt2 plays an important role in early cardiogenesis and inflow tract development,47–48 Mef2c plays an essential role in heart development as a regulator of cardiac myogenesis within the right ventricle,49–50 and Has2 plays a role in heart valve development.6,51 GREAT also reported that the affected genes were highly expressed in the cardiovascular system (P=5.3e‐5) and they were implicated in vascular disease (P=1.4e‐4) based on an analysis of Disease Ontology.52
Figure 4.
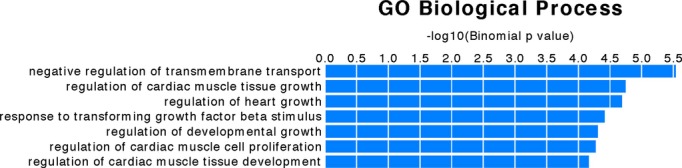
Gene ontology (GO) terms for the genes with differential DNA methylation during heart development. The software GREAT was used to characterize the 2901 differentially methylated sites for function. Four of the only 7 GO terms returned are involved in heart development and cardiac tissue growth.
Differential DNA Methylation Corresponds to Changes in Gene Expression in the Developing Heart
To directly test how the observed differential DNA methylation is related to gene expression changes in the developing heart, we picked 350 genes from the 1697 genes linked to the 2901 differentially methylated sites and performed qPCR to determine their expression levels in E11.5 and E14.5 hearts. These genes were chosen because they contained ACGT sites with a ≥50% change in methylation between the 2 stages, their known function (such as roles in embryonic development preferentially heart development), and/or presence of multiple differentially methylated sites. Change in mRNA level was calculated using the 2−ΔΔCt method and genes were ranked based on fold change.39 Of the 350 genes assayed in the gene expression analysis, 181 (51.7%) genes, including Erbb4, Has2, Invs, Robo2, and Vegfc, were differentially expressed between E11.5 and E14.5 (>1.2‐fold change and P<0.05; adjusted for multiple testing), and among these, expression of 55 genes was upregulated whereas expression of 126 genes was downregulated (Figure 5A, Table 2).
Table 2.
List of Genes With Differential Expression and Methylation
| Gene Name | ACGT Chr Location | Region | % DM | Fold Change | Function |
|---|---|---|---|---|---|
| AI661453 | Chr17 47582589 | Genebody | 74%** | −2.02 | Cellular component |
| Acaca | Chr11 84088161 | Genebody | 73%** | −2.01 | Long chain fatty acid biogenesis |
| Abi1 | Chr2 22839713 | Genebody | 73%** | −2.03 | Negative regulation of cell growth and transformation, Ras signaling, Cardiovascular and placental development |
| Aff2 | ChrX 66755393 | Genebody | 70%* | −2.02 | RNA‐binding protein |
| Atf6 | Chr1 172773438 | Genebody | 79%* | −2.00 | Unfolded protein response during ER stress |
| Arhgef12 | Chr9 42820787 | Genebody | 74%* | −2.02 | Acts as a guanine nucleotide exchange factor |
| Ank | Chr15 27482616 | Genebody | 59%** | −2.03 | Osteoblast/osteoclast differentiation, hypoxia responsive/regulated by Hif1α |
| Ank | Chr15 27513341 | Genebody | 78%** | −2.03 | |
| Ank | Chr15 27427229 | Genebody | 21%* | −2.03 | |
| Ank | Chr15 27430132 | Genebody | 52% | −2.03 | |
| Atrnl1 | Chr19 58000444 | Genebody | 84%** | −4.06 | G‐protein coupled receptor signaling, may regulate energy homeostasis |
| Atrnl1 | Chr19 57683177 | Promoter | 29% | −4.06 | |
| Atrnl1 | Chr19 58054451 | Genebody | 35% | −4.06 | |
| Atxn1 | Chr13 45770455 | Genebody | 67%** | −2.02 | Chromatin binding factor that represses Notch signaling. |
| Brdt | Chr5 107807029 | Genebody | 75%** | −2.00 | Chromocenter organization, Spermatogenesis |
| Cacna2d3 | Chr14 30485237 | Genebody | 74%** | −2.03 | Voltage‐gated calcium channel activity, Regulated by promoter methylation, contractility of ventricular myocytes |
| Cacna2d3 | Chr14 30303225 | Genebody | 22% | −2.03 | |
| Cd180 | Chr13 103491174 | Genebody | 66%* | −2.00 | Innate immune response, life/death decision of B‐cells |
| Cdk14 | Chr5 4822015 | Genebody | 76%** | −4.07 | Cell cycle regulation |
| Cdk14 | Chr5 5428041 | Enhancer | 23% | −4.07 | |
| Casz1 | Chr4 148329448 | Enhancer | 80%** | −2.00 | Blood vessel development and lumen morphogenesis, differentially methylated in a tissue specific manner |
| Cdh4 | Chr2 179440423 | Genebody | 75%** | −2.01 | Calcium dependent cell adhesion, may play a role in retinal development, Regulated by methylation |
| Cdh4 | Chr2 179292446 | Genebody | 22% | −2.01 | |
| Cdh4 | Chr2 179316146 | Genebody | 21% | −2.01 | |
| Cdh4 | Chr2 179385054 | Genebody | 19%* | −2.01 | |
| Clasp1 | Chr1 120296717 | Genebody | 75% | −2.03 | Regulation of microtubule dynamics, mitosis |
| Chrna9 | Chr5 66367625 | Genebody | 71%** | −1.94 | Ion transport, cochlea hair development |
| Chrna9 | Chr5 66348791 | Enhancer | 20%** | −1.94 | |
| Col9a1 | Chr1 24203890 | Genebody | 63%** | −2.02 | Expressed in developing heart, differentially methylated in cancer |
| Clasp2 | Chr9 113641090 | Enhancer | 69%** | −2.02 | Microtubule stabilization, mitosis |
| Creb1 | Chr1 64569233 | Enhancer | 81%** | −2.02 | Gene transcription, HIF‐1‐alpha transcription factor network |
| D6Wsu116e | Chr6 116164345 | Genebody | 76%** | −4.05 | N/A |
| Csde1 | Chr3 102840451 | Genebody | 59%** | −2.00 | RNA binding and transcriptionally coupled mRNA turnover |
| Disc1 | Chr8 127751886 | Genebody | 78%** | −2.02 | Multiple roles in embryonic and adult neurogenesis. Associated with schizophrenia |
| Dach2 | ChrX 110423200 | Genebody | 65%** | −2.04 | Eye and limb development and sex determination |
| Dgkb | Chr12 38744479 | Genebody | 73%** | −1.88 | Brain development |
| Dnajc11 | Chr4 151311759 | Genebody | 78%** | −2.03 | Heat shock binding protein |
| Dlx5 | Chr6 6859827 | Enhancer | 77%* | −2.03 | Transcriptional activator during bone development, Promotes cell proliferation, osteoblast differentiation. Cell type specific expression regulated by methylation |
| Dnajc2 | Chr5 21291210 | Promoter | 69%** | 2.00 | DNA replication |
| Dlg2 | Chr7 99358525 | Genebody | 60%** | −2.00 | Regulation of synaptic stability, Chronic pain perception |
| Eif2ak4 | Chr2 118271831 | Genebody | 86%* | −2.01 | Hypoxia response |
| Emilin2 | Chr17 71603514 | Genebody | 78%** | −4.05 | Extracellular matrix component, regulates methylation in breast cancer |
| Emilin2 | Chr17 71606328 | Genebody | 74%** | −4.05 | |
| Fam73a | Chr3 151956508 | Genebody | 81%** | −2.01 | Integral to membrane |
| Fam111a | Chr19 12661528 | Genebody | 78%** | −2.02 | Simian virus 40 (SV40) host range restriction factor |
| Fbxw7 | Chr3 84630582 | Genebody | 77** | −2.00 | Mediates ubiquitination and subsequent proteasomal degradation of target proteins including NOTCH1, inactivated by promoter hypermethylation |
| Fancl | Chr11 26311200 | Genebody | 73%* | −2.04 | Mediates monoubiquitination, may play a role in primordial germ cell proliferation |
| Fancl | Chr11 26368061 | Genebody | 96%* | −2.04 | |
| Fbxw8 | Chr5 118558780 | Genebody | 92%* | −2.02 | Mediates ubiquitination and subsequent proteasomal degradation of target proteins, placental development |
| Ftsjd2 | Chr17 29820177 | Genebody | 67%** | −2.01 | Methyltransferase that mediates mRNA cap1 |
| Fscn1 | Chr5 143721993 | Promoter | 67%* | −2.03 | Cell migration, motility, adhesion and cellular interactions |
| Has2 | Chr15 56549727 | Enhancer | 65%* | −16.31 | Heart valve development |
| Grik4 | Chr9 42754379 | Promoter | 79%* | −2.04 | Glutamate receptor in CNS |
| Invs | Chr4 48429137 | Genebody | 67%** | −4.05 | Embryonic heart tube left development and right pattern formation |
| Invs | Chr4 48288832 | Promoter | 24%* | −4.05 | |
| Il6st | Chr13 113256622 | Genebody | 77%* | −2.01 | Signal transduction. Plays a role in embryonic development, vascular endothelial growth |
| Hipk2 | Chr6 38796532 | Genebody | 61%* | −2.00 | Angiogenesis, marked for degradation by hif1‐a in cancer |
| Itgb1 | Chr8 131241575 | Genebody | 78%** | −2.03 | Promotes endothelial cell motility and angiogenesis, Hif1 regulated in wound healing |
| Itsn1 | Chr16 91786820 | Genebody | 79%* | −2.02 | Adaptor protein linking endocytic membrane traffic and actin assembly machinery |
| Isg20 | Chr7 86061520 | Genebody | 62%** | 3.97 | Viral response |
| Itga1 | Chr13 115765151 | Genebody | 66%** | −2.04 | Integrin and Collagen Binding, rapid methylation leading to initiation of megakaryocyte differentiation |
| Itga1 | Chr13 115800600 | Genebody | 20%** | −2.04 | |
| Kcne4 | Chr1 78770239 | Enhancer | 77%** | 7.87 | Potassium voltage channel, cardiac function (cardiomyopathy) |
| Kif26b | Chr1 180479937 | Genebody | 81%** | −2.00 | Embryonic kidney development, plays a role in compact adhesion between mesenchymal cells |
| Klhl2 | Chr8 67366115 | Genebody | 76%* | −2.02 | Mediate ubiquitination of target proteins, Plays a role in the reorganization of actin cytoskeleton |
| Ksr1 | Chr11 79001767 | Enhancer | 82%** | −2.04 | Promotes MEK and RAF phosphorylation and activity |
| Ksr1 | Chr11 78904857 | Genebody | 20% | −2.04 | |
| Limk1 | Chr5 135156130 | Genebody | 80%* | −2.04 | Regulation of actin filament dynamics, cell motility, cell cycle progression and differentiation |
| Lmf1 | Chr17 25771254 | Genebody | 82%** | −2.04 | Maturation and transport of lipoprotein lipase through the secretory pathway |
| Mkrn2 | Chr6 115567892 | Genebody | 62%* | 2.01 | Neurogenesis |
| Ncoa7 | Chr10 30373619 | Genebody | 75%** | 2.02 | Co‐activation of several nuclear receptors |
| Ncam1 | Chr9 49570879 | Genebody | 61%* | 4.06 | Neural adhesion, pathological angiogenesis in oxygen induced retinopathy, ventricular wall thickening in hypertension, cardiac protection |
| Odz3 | Chr8 49397492 | Genebody | 73%** | −2.00 | Signal transduction, neuronal growth and tumorigenesis |
| Odz3 | Chr8 49336206 | Genebody | 47%** | −2.00 | |
| Odz3 | Chr8 49341811 | Genebody | 21% | −2.00 | |
| Pam | Chr1 99716878 | Enhancer | 79%** | 4.07 | Heart development and hypoxia response |
| Pde5a | Chr3 122538715 | Genebody | 79** | 2.02 | Signal transduction, cardiac muscle contraction and hypertrophy, hypoxia response |
| Pde11a | Chr2 75840713 | Genebody | 70%* | 1.98 | Signal transduction, may play a role in vascular smooth muscle proliferation and contraction, cardiac contractility and immune cell activation |
| Pcgf5 | Chr19 36450106 | Promoter | 71%* | 2.02 | Maintenance of transcriptional repressive state in development, including that of Hox genes |
| Pfkfb3 | Chr2 11406131 | Genebody | 78%** | 2.03 | Induced by Hif1α |
| Pet112 l | Chr3 85403998 | Genebody | 70%* | 2.03 | Glutamyl‐tRNA amidotransferase complex, Functions in mitochondria |
| Pip5k1b | Chr19 24602322 | Genebody | 72%** | 4.06 | Phosphorylation |
| Pnkd | Chr1 74336698 | Genebody | 65%** | 2.00 | Hydrolase activity, Plays an aggregative role in the development of cardiac hypertrophy via NF‐kappa‐B signaling |
| Pou2f1 | Chr1 167866210 | Promoter | 70%** | −2.01 | Regulates gene expression in response to stress and metabolic signals |
| Ppm1 h | Chr10 122245574 | Genebody | 67% | 4.05 | Phosphatase activity, drug response in cancer, associated with systemic lupus erythematosus |
| Ppm1 h | Chr10 122144175 | Genebody | 19%** | 4.05 | |
| Prdm16 | Chr4 153999781 | Genebody | 68%** | 2.02 | Transcriptional regulation, Functions as a repressor of TGF‐beta signaling |
| Ppwd1 | Chr13 104995653 | Genebody | 67%** | 2.02 | Putative peptidylprolyl isomerase, may be involved in pre‐mRNA splicing |
| Ptn | Chr6 36663240 | Enhancer | 75%** | 4.07 | Angiogenesis, tumorigenesis, regulation of hematopoietic stem cell self renewal, mammary gland development |
| Prmt8 | Chr6 127665685 | Genebody | 79%* | −2.07 | Arginine methyltransferase, embryonic and neural development, regulated by auto‐methylation |
| Prkca | Chr11 108120704 | Genebody | 86%** | 2.00 | Regulation of transcription, cell growth, immune response, negative regulation of cell proliferation, apoptosis, differentiation, cardiac hypertrophy and angiogenesis |
| Ranbp3 | Chr17 56833581 | Genebody | 78%* | 2.02 | Nuclear export, negative regulator of TGF‐beta signaling through SMAD |
| Ptpro | Chr6 137362114 | Genebody | 71%** | 2.00 | Wnt‐protein binding, Candidate tumor suppressor, aberrantly methylated in cancer |
| Ptpru | Chr4 131336827 | Genebody | 69%** | 1.84 | Cell proliferation and migration, maintenance of epithelial integrity, neural development and possible megakaryocytopoiesis |
| Rbfox3 | Chr11 118610022 | Genebody | 65%* | 1.97 | RNA‐binding, associated with neurocytoma and cerebral artery occlusion |
| Rhd | Chr4 134418089 | Promoter | 81%** | −8.00 | Encodes member of Rh blood group proteins |
| Rbm39 | Chr2 155977061 | Genebody | 75% | 4.05 | Transcriptional co‐activator for steroid nuclear receptors, involved in pre‐mRNA splicing |
| Rpia | Chr6 70726365 | Genebody | 67%** | 2.02 | Carbohydrate metabolism |
| Robo2 | Chr16 74182881 | Genebody | 72% | −2.00 | Heart Morphogenesis, linear hear tube formation, neuronal development |
| Rpn2 | Chr2 157147564 | Genebody | 55%** | 2.04 | Ribosome binding, Dolichyl‐diphosphooligosaccharide‐protein glycotransferase activity |
| Rsu1 | Chr2 13110936 | Genebody | 79%** | 2.01 | Ras signal transduction pathway |
| Rsu1 | Chr2 13153759 | Genebody | 50%* | 2.01 | |
| Rufy2 | Chr10 62447503 | Genebody | 65% | 2.03 | Alzheimer's disease |
| Sav1 | Chr12 71078443 | Genebody | 76%** | 2.03 | Transcription, cell proliferation, cell death, cell migration, cell cycle exit, protein degradation and RNA splicing |
| Slc24a2 | Chr4 86672633 | Genebody | 58%** | 2.03 | Calcium and potassium transport |
| Slmap | Chr14 27308224 | Genebody | 79% | 4.07 | Myoblast fusion |
| Slco5a1 | Chr1 12939917 | Genebody | 61% | −1.97 | Transporter activity |
| Sorbs2 | Chr8 46655637 | Genebody | 79%** | 2.02 | Cytoskeletal adaptor activity and structural constituent of cytoskeleton |
| Sorbs2 | Chr8 46649975 | Genebody | 20% | 2.02 | |
| Stxbp6 | Chr12 46076554 | Genebody | 79%** | 1.96 | Regulates SNARE complex formation |
| St8sia5 | Chr18 77402859 | Enhancer | 80%** | 2.02 | Synthesis of gangliosides |
| Srsf9 | Chr5 115781199 | Genebody | 73% | 2.03 | Splicing |
| Tex9 | Chr9 72307706 | Genebody | 67%** | 2.02 | — |
| Tbc1d16 | Chr11 119004863 | Genebody | 64%** | 1.99 | Rab GTPase activator activity |
| Tbc1d16 | Chr11 119017735 | Genebody | 76%** | 1.99 | |
| Taf4a | Chr2 179700219 | Genebody | 72%** | 2.01 | Basal transcription |
| Tet2 | Chr3 133187569 | Genebody | 85%** | 2.01 | DNA demethylation regulating transcription |
| Tmem38a | Chr8 75105110 | Genebody | 69%** | 4.06 | Potassium channel activity |
| Tnrc6a | Chr7 130306203 | Genebody | 73%** | 2.02 | Gene silencing by RNA and microRNA |
| Tmem135 | Chr7 96306156 | Genebody | 82%* | 2.02 | Peroxisome organization |
| Tmem135 | Chr7 96397839 | Genebody | 29% | 2.02 | |
| Tubg2 | Chr11 101015396 | Promoter | 19% | −2.01 | Major constituent of microtubules, structural molecule activity |
| Vmn1r73 | Chr7 12307910 | Enhancer | 63%** | 2.00 | — |
| Wnk1 | Chr6 119946851 | Genebody | 86%** | −1.98 | Heart development, regulations of cell signaling, survival and proliferations, electrolyte homeostasis, cytoskeletal reorganization and sodium and chloride ion transport |
| Zbtb20 | Chr16 43500263 | Genebody | 89%** | −2.01 | Transcription factor involved in hematopoiesis, oncogenesis and immune response |
| Zbtb20 | Chr16 43219787 | Enhancer | 95% | −2.01 | |
| Zbtb20 | Chr16 43343231 | Genebody | 98% | −2.01 | |
| Zbtb20 | Chr16 43455368 | Genebody | 97% | −2.01 | |
| Wnt6 | Chr1 74821915 | Genebody | 65% | −4.35 | Tissue development |
| Zdhhc8 | Chr16 18231749 | Genebody | 72% | 4.00 | Susceptibility to schizophrenia |
| Zfp385b | Chr2 77445243 | Genebody | 73%* | −2.02 | Metal ion, nucleic acid, p53, and zinc ion binding, Apoptotic processes |
| Zfp385b | Chr2 77629407 | Genebody | 21%* | −2.02 | |
| Enah | Chr1 183952945 | Promoter | 25% | −2.01 | |
| Pkd2 | Chr5 104885245 | Promoter | 22% | 1.99 | Tubular morphogenesis, associated with autosomal dominant polycystic kidney disease |
| Mylk2 | Chr2 152734667 | Promoter | 20% | −1.91 | Cardiac function and global muscle contraction |
| Ppp1r1c | Chr2 79544612 | Promoter | 25% | −1.93 | Promotes cell cycle progression and increases cell susceptibility to TNF‐induced apoptosis |
| Spns2 | Chr11 72304383 | Promoter | 59%** | −4.03 | Migration of myocardial precursors; cardiovascular, immunological and neural development |
| Elf1 | Chr14 79879478 | Promoter | 31% | −4.05 | Endothelial transcription factor |
| Ppp1r3c | Chr19 36813263 | Promoter | 44% | −2.01 | Glycogen synthase, Regulated by Hif1α |
| Gucy1a3 | Chr3 81943306 | Genebody | 23%** | −2.02 | Cardiac function, vascular smooth muscle function |
| Gucy1a3 | Chr3 81993497 | Enhancer | 23%** | −2.02 | |
| Ank2 | Chr3 126729907 | Enhancer | 97% | −2.01 | Expression and targeting of SPTBN1 in neonatal cardiomyocytes and regulation of neonatal cardiomyocyte contraction rate |
| Sfrp2 | Chr3 83534574 | Enhancer | 30% | 8.05 | Cell growth and differentiation, Wnt signaling, myogenesis and eye retinal development, methylation of gene is a potential marker for colorectal cancer |
| Erbb4 | Chr1 68142043 | Genebody | 23% | −4.04 | Heart development, cardiac muscle differentiation and postnatal cardiomyocyte differentiation, CNS development, neural crest cell migration, gene transcription, cell proliferation, differentiation, migration and apoptosis |
| Erbb4 | Chr1 68846422 | Genebody | 20% | −4.04 | |
| Erbb4 | Chr1 68938673 | Genebody | 38% | −4.04 | |
| Erbb4 | Chr1 68954441 | Genebody | 20% | −4.04 | |
| Erbb4 | Chr1 69128731 | Genebody | 24% | −4.04 | |
| Mysm1 | Chr4 94660067 | Enhancer | 28%** | −2.00 | Histone modification and transcriptional co‐activation |
| Foxp1 | Chr6 98892345 | Genebody | 19% | −2.04 | Cardiomyocyte proliferation |
| Foxp1 | Chr6 99008217 | Genebody | 22%* | −2.04 | |
| Foxp1 | Chr6 99018432 | Genebody | 34% | −2.04 | |
| Foxp1 | Chr6 99163145 | Genebody | 20% | −2.04 | |
| Foxp1 | Chr6 99197140 | Genebody | 23% | −2.04 | |
| Foxp1 | Chr6 99357158 | Genebody | 22% | −2.04 | |
| Foxp1 | Chr6 99391693 | Enhancer | 43%* | −2.04 | |
| Unc5c | Chr3 141321392 | Genebody | 19% | −8.07 | Cell migration in neural development, axon extension and induction of apoptosis |
| Unc5c | Chr3 141342275 | Genebody | 26% | −8.07 | |
| Unc5c | Chr3 141359065 | Genebody | 20% | −8.07 | |
| Sox5 | Chr6 143990289 | Genebody | 20% | −4.01 | Embryonic development, cell fate determination, transcriptional regulation |
| Sox5 | Chr6 144141969 | Genebody | 19% | −4.01 | |
| Psd3 | Chr8 70278799 | Genebody | 19%* | −2.03 | Guanine nucleotide exchange factor for ARF6 |
| Psd3 | Chr8 70338543 | Genebody | 96% | −2.03 | |
| Psd3 | Chr8 70459139 | Genebody | 23%* | −2.03 | |
| Dock1 | Chr7 142059905 | Genebody | 28% | −2.01 | Cytoskeletal rearrangements necessary for phagocytosis of apoptotic cells and in cell motility Guanine nucleotide exchange factor |
| Dock1 | Chr7 142061686 | Genebody | 19% | −2.01 | |
| Dock1 | Chr7 142073658 | Genebody | 30% | −2.01 | |
| Dock1 | Chr7 142115224 | Genebody | 24% | −2.01 | |
| Dock1 | Chr7 142136783 | Genebody | 24% | −2.01 | |
| Dock1 | Chr7 142190260 | Genebody | 53% | −2.01 | |
| Wwox | Chr8 117142250 | Genebody | 31% | −2.03 | Apoptosis, TGFB1 signaling and TGFB1‐mediated cell death. Inhibits Wnt signaling |
| Wwox | Chr8 117354691 | Genebody | 26% | −2.03 | |
| Wwox | Chr8 117632214 | Genebody | 22% | −2.03 | |
| Vegfc | Chr8 55148115 | Enhancer | 21% | 2.02 | Angiogenesis and endothelial cell growth |
| Vegfc | Chr8 55239360 | Genebody | 18.5%* | 2.02 | |
| Vegfc | Chr8 55247617 | Genebody | 22% | 2.02 | |
| Pard3 | Chr8 129591554 | Genebody | 28%* | −2.00 | Adaptor protein, asymmetrical cell division and cell polarization, plays a role in epithelial tight junctions |
| Pard3 | Chr8 129640346 | Genebody | 96%** | −2.00 | |
| Pard3 | Chr8 129830952 | Genebody | 96% | −2.00 | |
| Pard3 | Chr8 129845627 | Genebody | 21%** | −2.00 | |
| Pard3 | Chr8 129875488 | Genebody | 22% | −2.00 | |
| Pard3 | Chr8 129914177 | Genebody | 24% | −2.00 | |
| Thsd4 | Chr9 59846658 | Genebody | 19%* | −2.00 | Attenuates TGFB signaling |
| Thsd4 | Chr9 59968525 | Genebody | 22% | −2.00 | |
| Thsd4 | Chr9 60041974 | Genebody | 19%* | −2.00 | |
| Thsd4 | Chr9 60098887 | Genebody | 96%* | −2.00 | |
| Rora | Chr9 68624882 | Genebody | 19%** | −2.01 | Regulated genes involved in lipid metabolism |
| Rora | Chr9 68726640 | Genebody | 24%** | −2.01 | |
| Rora | Chr9 68865723 | Genebody | 19% | −2.01 | |
| Utrn | Chr10 12126998 | Genebody | 20%* | 2.02 | Anchors cytoskeleton to plasma membrane |
| Utrn | Chr10 12213501 | Genebody | 20% | 2.02 | |
| Utrn | Chr1012395503 | Genebody | 19% | 2.02 | |
| Mef2c | Chr13 83687636 | Genebody | 96% | −1.99 | Transcription activator controls cardiac morphogenesis and myogenesis, plays a role in vascular development |
| Mef2c | Chr13 83755977 | Genebody | 30%** | −1.99 | |
| Mef2c | Chr13 83797333 | Genebody | 25% | −1.99 | |
| Odz2 | Chr11 35940645 | Genebody | 35%* | −1.98 | Neural development |
| Odz2 | Chr11 36164904 | Genebody | 20%* | −1.98 | |
| Odz2 | Chr11 36400641 | Genebody | 32%** | −1.98 | |
| Odz2 | Chr11 36725324 | Genebody | 27%** | −1.98 | |
| Ptprk | Chr10 27825617 | Genebody | 20%* | −2.01 | Regulation of cell contact and adhesions, tumor invasion/metastasis, Negative regulator of EGFR signaling |
| Ptprk | Chr10 28084547 | Genebody | 24% | −2.01 | |
| Ptprk | Chr10 28226602 | Genebody | 24%** | −2.01 | |
| Enox1 | Chr14 77538059 | Enhancer | 97% | −2.00 | Oxidoreductase activity, nucleotide binding |
| Enox1 | Chr14 77771534 | Genebody | 23% | −2.00 | |
| Enox1 | Chr14 77876556 | Genebody | 20%* | −2.00 | |
| Enox1 | Chr14 77977395 | Genebody | 20%** | −2.00 | |
| Zfpm2 | Chr15 40787554 | Genebody | 29% | −2.01 | Transcription regulator important in heart morphogenesis and coronary vessel development from epicardium |
| Zfpm2 | Chr15 40824705 | Genebody | 95%** | −2.01 | |
| Zfpm2 | Chr15 40848971 | Genebody | 19% | −2.01 | |
| Dach1 | Chr14 98263585 | Genebody | 97%* | −1.99 | Transcription factor important in organogenesis |
| Dach1 | Chr14 98304412 | Genebody | 26% | −1.99 | |
| Dach1 | Chr14 98510700 | Genebody | 96% | −1.99 | |
| Erc2 | Chr14 28475844 | Genebody | 97% | −1.99 | Cytomatrix organization at nerve terminal active zones regulating release of neurotransmitters |
| Erc2 | Chr14 28668685 | Genebody | 30% | −1.99 | |
| Erc2 | Chr14 28822634 | Genebody | 95% | −1.99 | |
| Erc2 | Chr14 28845440 | Genebody | 24% | −1.99 | |
| Vps13b | Chr15 35600466 | Genebody | 23%** | −1.99 | Protein sorting in post Golgi membrane traffic, may play a role in development and function of the eye, hematological system and the CNS |
| Vps13b | Chr15 35777972 | Genebody | 19% | −1.99 | |
| Vps13b | Chr15 35817931 | Genebody | 23%* | −1.99 | |
| 2810403A07Rik | Chr3 88506464 | Genebody | 84% | −2.01 | RNA binding |
| Tle4 | Chr19 14528880 | Genebody | 20%* | −2.01 | Transcriptional co‐repressor of members in Wnt signaling |
| Tle4 | Chr19 14621827 | Genebody | 24% | −2.01 | |
| Prkg1 | Chr19 30823222 | Genebody | 19%* | −2.02 | Regulates cardiac function, smooth muscle contraction, platelet activation and adhesion |
| Prkg1 | Chr19 31528132 | Genebody | 24% | −2.02 | |
| Prkg1 | Chr19 31595749 | Genebody | 20% | −2.02 | |
| Hif1a | Chr12 75032797 | Genebody | 25% | −2.00 | Hypoxia response, Master transcriptional regulator |
| Malt1 | Chr18 65586771 | Promoter | 19%* | −2.00 | NF‐kappaB activation |
| Igf2r | Chr17 12965268 | Promoter | 44% | −2.02 | Activation of TGF‐β. Intracellular trafficking of lysosomal enzymes and degradation of IGF2, tumorigenesis, Paternally imprinted |
| Krtap9‐1 | Chr11 99731190 | Promoter | 27% | 8.00 | Hair shaft formation |
| Ccdc40 | Chr11 119085948 | Promoter | 20% | −4.03 | Motile cilia function. Ciliary dyskinesia type 15 |
| Cftr | Chr6 18119186 | Promoter | 20% | −2.00 | Chloride channel and enzyme binding, associated with cystic fibrosis |
| Cftr | Chr6 18242079 | Genebody | 42% | −2.00 | |
| Pdzd2 | Chr15 12315935 | Genebody | 23% | 2.02 | Prostate tumorigenesis |
| Pdzd2 | Chr15 12342544 | Genebody | 27% | 2.02 | |
| Pdzd2 | Chr15 12521967 | Promoter | 19% | 2.02 | |
| Elovl2 | Chr13 41317598 | Promoter | 23% | −4.04 | Atherosclerosis, protein binding and fatty acid elongase activity |
| Gpr18 | Chr14 122316980 | Promoter | 96% | 1.98 | Regulation of immune system, bipolar disorder |
| Rpn1 | Chr6 88030514 | Promoter | 24% | −2.01 | Dolichyl‐diphosphooligosaccharide‐protein glycotransferase activity |
| Mosc1 | Chr1 186637543 | Promoter | 19% | −4.12 | — |
| Tmem150c | Chr5 100589921 | Promoter | 25% | −2.04 | — |
| Shroom3 | Chr5 93236380 | Promoter | 27% | −2.01 | Regulation of cell shape in neuroepithelium |
| Calb1 | Chr4 15806105 | Promoter | 24% | −1.95 | Functions in purkinje cells |
| Gpr61 | Chr3 107962781 | Promoter | 22%** | 8.01 | G‐protein coupled receptor signaling |
| Cd40 | Chr2 164871483 | Enhancer | 96% | −2.02 | Immune and inflammatory response |
| Ephx1 | Chr1 182951005 | Promoter | 23% | 2.02 | Cis‐stilbene‐oxide hydrolase activity, epoxide hydrolase activity. Plays a role in preeclampsia |
| Arhgap29 | Chr3 121628392 | Enhancer | 96% | −2.00 | Rho GTPase activator activity, essential role in blood vessel tubulogenesis |
| Fbxl4 | Chr4 22244260 | Enhancer | 21% | −2.02 | Cell cycle control |
| Fbxl4 | Chr4 22264607 | Enhancer | 21% | −2.02 | |
| Zmat3 | Chr3 32233835 | Genebody | 96%** | −1.96 | TP53‐dependent growth regulatory pathway and TP53‐mediate apoptosis, inhibits tumor cell growth |
| Zmat3 | Chr3 32278426 | Enhancer | 22% | −1.96 | |
| Fam125b | Chr2 33790838 | Enhancer | 23%** | −2.03 | Vesicular trafficking |
| Serpinb2 | Chr1 109370955 | Enhancer | 31% | −4.08 | Serine‐type endopeptidase inhibitor activity |
| Rims1 | Chr1 22615396 | Genebody | 97%* | −2.02 | Exocytosis, maintenance of neurotransmitter release and regulation of release during short‐term synaptic plasticity |
| Rims1 | Chr1 22763512 | Genebody | 96% | −2.02 | |
| Dst | Chr1 34249899 | Genebody | 20% | −2.01 | Cytoskeletal linker protein, Regulation of keratinocyte polarity and mobility |
| Dst | Chr1 34315083 | Genebody | 20% | −2.01 | |
| Dst | Chr1 34322642 | Genebody | 26% | −2.01 | |
| Etl4 | Chr2 20373784 | Genebody | 23% | 2.00 | Intervertebral disk development |
| Etl4 | Chr2 20576360 | Genebody | 95%* | 2.00 | |
| Esrrg | Chr1 189527991 | Genebody | 46% | −2.00 | Transcriptional activator via estrogen response elements |
| Esrrg | Chr1 189976300 | Genebody | 21%* | −2.00 | |
| Dnm3 | Chr1 163949959 | Genebody | 22% | −1.99 | Megakaryocyte development, likely involved in endocytosis |
| Dnm3 | Chr1 164108264 | Genebody | 40%** | −1.99 | |
| Rbms1 | Chr2 60615270 | Genebody | 25% | −2.02 | Cell cycle progression, apoptosis, DNA replication and gene transcription. |
| Rbms1 | Chr2 60789032 | Genebody | 19% | −2.02 | |
| Lrba | Chr3 86163560 | Genebody | 23% | −2.01 | Signal transduction and vesicle trafficking |
| Lrba | Chr3 86267521 | Genebody | 21% | −2.01 | |
| Plcb1 | Chr2 134819839 | Genebody | 23% | −1.99 | Intracellular transduction of extracellular signals |
| Plcb1 | Chr2 135145732 | Genebody | 25% | −1.99 | |
| Meis2 | Chr2 115688950 | Genebody | 96% | −2.01 | Transcriptional regulation |
| Meis2 | Chr2 115750462 | Genebody | 96% | −2.01 | |
| Bach2 | Chr4 32560314 | Genebody | 21%* | −2.00 | Transcriptional regulation |
| Bach2 | Chr4 32629036 | Genebody | 21% | −2.00 | |
| Kcnd3 | Chr3 105447527 | Genebody | 16%* | −1.84 | Smooth muscle contraction, heart rate, insulin secretion, neuronal excitability and cell volume |
| Fam19a1 | Chr6 96068370 | Genebody | 31% | −1.91 | Regulators of immune cells and cells of the nervous system |
| Fam19a1 | Chr6 96235296 | Genebody | 19%** | −1.91 | |
| Drosha | Chr15 12715566 | Enhancer | 22% | −2.03 | Cleaves ds‐RNA in micro RNA processing |
| Drosha | Chr15 12817558 | Genebody | 27% | −2.03 | |
| Gsk3b | Chr16 38062422 | Enhancer | 20% | −2.01 | Negative regulator in hormonal control of glucose homeostasis, Wnt signaling and the regulation of transcription factors and microtubules. Regulates NFatc1 expression. Mediates development of insulin resistance |
| Gsk3b | Chr16 38138300 | Genebody | 20%** | −2.01 | |
| Gsk3b | Chr16 38218276 | Genebody | 96% | −2.01 | |
| Pmm2 | Chr16 8627532 | Enhancer | 33% | −2.02 | Glycoprotein biosynthesis |
| Osta | Chr16 32515415 | Enhancer | 31% | −2.00 | Transporter activity |
| Krt8 | Chr15 101867475 | Enhancer | 23% | −4.02 | Signal transduction and cellular differentiation |
| Slc38a4 | Chr15 96905406 | Enhancer | 96% | −2.00 | Sodium‐dependent amino acid transporter |
| Slc38a2 | Chr15 96516885 | Enhancer | 20%* | −2.03 | Supply of maternal nutrients to fetus through placenta, transport of amino acids at blood‐brain barrier |
| Adra2a | Chr19 54118496 | Promoter | 26% | 1.99 | Mediates the catecholamine‐induced inhibition of adenylate cyclase |
Table provides differentially expressed genes with corresponding differential methylation (DM) sites. Bolded DM values indicate decreased methylation at E14.5 whilst unbolded DM values indicates increased methylation at E14.5.
*Notes P<0.05 in methylation changes and **notes P<0.01 in methylation changes. Gene functions are summarized.
Recent studies of the correlation between DNA methylation and gene expression have found that increased promoter and enhancer methylation often lead to gene silencing while DNA methylation at gene bodies corresponds with gene activation.53 We therefore examined the functional correlation between differential gene expression and changes in DNA methylation. We found that among the top 15 downregulated genes at E14.5, 4 of them contained an increase in methylation of sites located within promoter or enhancer regions and an additional 4 showed decreased methylation in their gene bodies (Figure 5B). Of the top 15 upregulated genes, 4 had decreased methylation in their enhancers and 3 exhibited increased methylation in their gene bodies. We found that, while 12.7% (23) of the 181 differentially expressed genes contained differentially methylated sites within the promoter region, methylation of only 60.8% (14) of those genes was predictive of their expression difference between E11.5 and E14.5, and overall 43.6% (79) of genes had differentially methylated sites predictive of expression change. The findings suggest that not all DNA methylation is functional; most genes are regulated independent of methylation.54 Nevertheless, the observed correlations between gene expression and DNA methylation during heart development do support that DNA methylation regulates expression of a subset of genes during heart development.
Increased DNA Methylation at Enhancers is Associated With Decreased Expression of the Cardiac‐Essential Gene Has2 in the Developing Heart
These correlations suggest a regulatory relationship between DNA methylation and cardiac‐important genes. Notably, Has2 is essential for endocardial to mesenchymal transformation and heart valve formation.51,55–56,6 Gene network analysis using the Genemania open freeware (http://www.genemania.org) further revealed potential genetic and/or physical interactions among genes or pathways involved in heart development. The top 20 genes that were identified to interact (either genetically or physically) with Has2 by the network analysis were significantly enriched with functions involved in heart development. These genes included Cdh2, Epo, Kcna5, Myocd, Tbx20, Hand1, Mef2c, and Nfatc4 (Figure 6).49,57–59 Regulation of these genes by DNA methylation to influence their expression will ultimately affect their pathway and downstream functions, which are essential for heart development.
Figure 6.
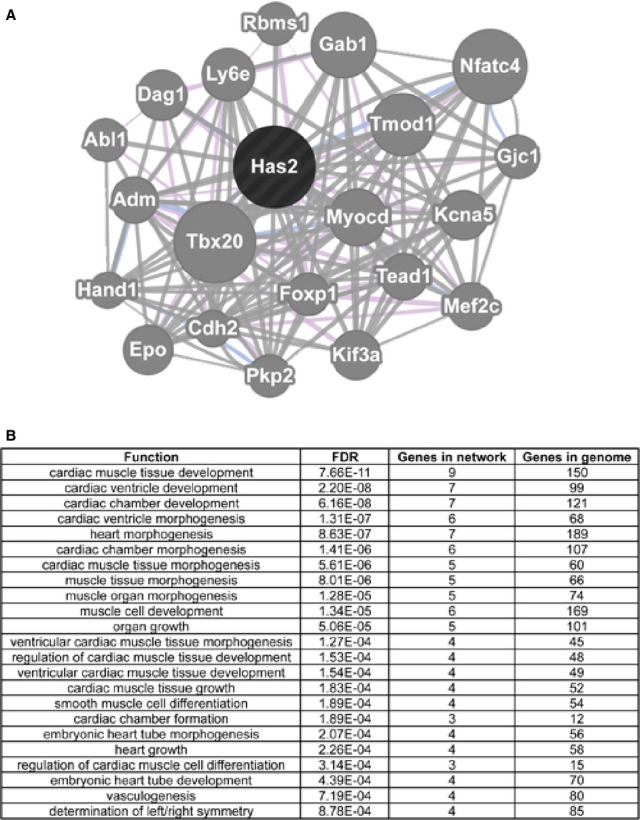
Network analysis for DNA methylation‐regulated Has2. Top: Genemania analysis reveals multiple relationships between Has2 and multiple cardiac genes including Myocd, Kcna5, Mef2c, Hand1, Tbx20 and Nfatc4. Bottom: Top functions of genes in the Has2 network.
Previous knockout studies in mice have shown that Has2 is essential for development of cardiac valves and septa.51,56 Here, our DNA methylation analysis indicated that an ACGT site located within 1 kb of a previously determined enhancer of Has2, marked by enriched H3K27ac at E14.5,44 exhibited an increase in methylation as confirmed by Sequenom's MassArray (Figure 7A). Based on what is known about DNA methylation, we expected to see a decrease in Has2 expression at E14.5, with confirmation by qPCR analysis (Figure 7B). To further characterize its expression in the developing hearts, we carried out RNA in situ hybridization. The results showed that Has2 expression was predominately expressed in the endocardial cells and their mesenchymal progeny cells that form the primitive heart valves at E11.5, but its expression markedly diminished by E14.5 (Figure 7C), consistent with its function in endocardial to mesenchymal transformation around E11.5 for heart valve development.
Figure 7.
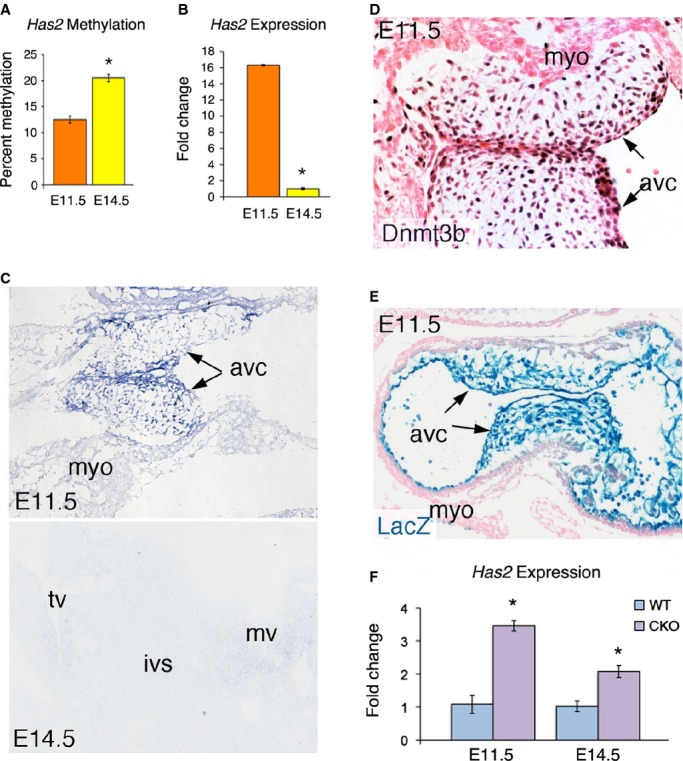
Has2 expression is regulated by Dnmt3b. A, MassArray showing increased Has2 enhancer methylation at E14.5 (n=2). B, RT‐qPCR showing decreased Has2 expression at E14.5 (n=3). C, RNA in situ hybridization showing that Has2 expression is predominantly in the atrioventricular canal (avc), with less expression in the myocardium (myo) at E11.5; and the expression is diminished by E14.5. mv/tv, mitral/tricuspid valve; ivs, interventricular septum. D, IHC showing Dnmt3b is predominantly expressed in AVC at E11.5. E, X‐gal staining showing the Nfatc1‐Cre mediated LacZ expression in the AVC at E11.5. F, RT‐qPCR showing that deletion of Dnmt3b resulted in increased Has2 expression at E11.5 and E14.5 (n=3). Error bars represent standard error. *Marks statistical significance (P<0.001, 2 factor ANOVA in [F]). ANOVA indicates analysis of variance; CKO, conditional knockout; IHC, immunohistochemistry; RT‐qPCR, quantitative real‐time polymerase chain reaction; WT, wild type.
Dnmt3b Suppresses Expression of the Cardiac‐Essential Gene Has2 in the Developing Heart
We next chose to determine experimentally the function of DNA methylation in Has2 expression in the developing heart valves. To overcome the difficulty of not being able to directly assay the in vivo role of methylation of the Has2 enhancer on the expression of the gene in the developing heart, we inactivated the activity of DNA methyltransferase 3b (Dnmt3b), which is responsible for de novo methylation during embryonic development.22 First, we showed that, like Has2, Dnmt3b was expressed predominately in the endocardial cells, precursor cells for the heart valves (Figure 7D). The expression pattern of Dnmt3b suggests that it has a role in DNA methylation in the endocardial cell lineages and may therefore regulate Has2 expression in the developing heart valves. Indeed, deletion of Dnmt3b in the endocardial cells and their valve progeny, using the endocardial‐specific Nfatc1Cre mice (Figure 7E),9 resulted in significantly increased Has2 expression in the developing heart valves at E11.5 and E14.5 (Figure 7F). The results support that DNA methylation of the Has2 enhancer plays a role in repressing its expression during heart development.
Discussion
In this study, we generated a developmental profile of DNA methylation using methyl sensitive tiny fragment enrichment coupled with massively parallel sequencing (MSFE/MPS). Greater coverage and increased sensitivity for the detection of methylation are achieved using this method compared with microarray‐based techniques. The method also provides more detailed information specific for ACGT sites and is capable of detecting intermediate levels of methylation, allowing for detection of modest changes in methylation.60
Our technique was adapted from the HpaII tiny fragment enrichment by ligation mediated PCR (HELP)‐tagging assay developed by Suzuki et al32–33 In the original HELP‐tagging assay a methylation sensitive restriction enzyme, HpaII, is used to assess methylation of CpG dinucleotides located within its recognition site 5′‐CCGG‐3′. We modified this technique by using a different methylation‐sensitive restriction enzyme, HpyCH4IV, whose recognition site is 5′‐ACGT‐3′ even though it lacks a methylation‐insensitive isoschizomer. Previous studies have confirmed that sequencing reads/tags from a single methylation sensitive restriction enzyme without its isoschizomer are highly correlated with methylation status.32,61 Furthermore, we used independent validation methods, such as LUMA and MassArray to confirm methylation levels determined by the MSFE/MPS.
HpyCH4IV provides comparable genome coverage to HpaII, having 1.7 million recognition sites located throughout the genome. Future studies using both enzymes will not only double the coverage but also examine and compare DNA methylation in both CG rich and non‐CG rich regions. In addition, the use of HpyCH4IV will, in future studies, allow us to directly examine, in a genome‐wide manner, the effect of methylation on transcription factor binding sites such as Hif1α, whose consensus binding sequence is 5′‐ACGTG‐3′ and has been shown to be regulated by DNA methylation.62
The original analytical pipeline was also modified to analyze our MPS data generated by using HpyCH4IV.37 Within the HELP‐tagging protocol an internal experimental control is used in which contaminating fragments are recognized based on the absence of the digested restriction site.60 Additionally, to better identify differentially methylated sites between developmental stages, we generated a threshold to determine levels of methylation at individual loci. We employed these modifications to generate a genome wide developmental profile of methylation at ACGT sites in the developing heart. The results showed no significant global change in methylation over mid‐stage heart development, although our study did not include the repetitive regions of the genome as they could not be aligned and thus discarded in the pipeline. We further confirmed that there is no significant change in global methylation patterns using LUMA.
Although drastic changes in global DNA methylation are not present during heart development, differential methylation was detected at a small subset of individual loci throughout the genome in this study. Furthermore, we detected a number of differentially methylated sites in which the change in methylation between E11.5 and E14.5 corresponds with the observed change in expression of the nearby gene, suggesting that there is a regulatory relationship between DNA methylation and the expression of cardiac‐important genes including Has2.
Has2 has previously been identified to be essential for heart development, playing a role in epicardial cell differentiation, heart valve development, and septation.51,55 A DNA methylation‐regulated cardiac gene program was generated by performing a network analysis for Has2 using Genemania. Network analysis revealed multiple relationships between Has2 and other cardiac‐important gene products, including previously mentioned Tbx20, involved in endocardial cushion formation and heart valve remodeling,57 Hand 1 involved in ventricle morphogenesis,58 and Nfatc4, a member of the nuclear factor of activated T‐cell family that are known to be essential for heart development.59 Additional connections have been identified between Has2 and Gjc1 (Connexin45), known to play an important role in cardiac morphogenesis and conduction,63 and Cdh2, Epo, Kcna5, Mef2c, and Myocd, which are essential for heart development and function.
We further studied methylation of Has2 and its expression in the developing heart in detail, as it has an increase in enhancer methylation that corresponds with a decrease in its expression over mid‐stage heart development in the developing heart valves. We showed by qPCR analysis, RNA in situ hybridization, and genetic knockout that Dnmt3b regulates Has2 expression, possibly through its enhancer methylation.
In this study we were able to assay 1.64 million ACGT sites for potential changes in DNA methylation during mid‐stage cardiac development, identifying 2901 differentially methylated sites, and determined a number of developmentally important cardiac genes that are likely to be regulated by DNA methylation. However, current methods for studying functionality of DNA methylation at specific sites in the developing heart are limited. We circumvented this limitation by revealing the dependence of Has2 expression on Dnmt3b expression in the developing heart valves. Our results are mainly discovery and by necessity preliminary, and will require a much larger sample size to detect more sites with more subtle DNA methylation changes between the 2 developmental stages.
In conclusion, our results support an essential role for DNA methylation in the regulation of cardiac essential genes during heart development and suggest abnormal DNA methylation may contribute to the pathogenesis of congenital heart disease. Using this study as a starting point, we plan to investigate further candidate genes as well as the role of additional epigenetic modifications that may play a role in heart development and disease.
Sources of Funding
Disclosures
None.
Acknowledgments
The authors thank Wendy Lui, Pratistha Koirala, Shahina Maqbool, David Reynolds, and Bernice Morrow for helpful discussions or technique support.
References
- 1.Webb S, Brown NA, Anderson RH. Formation of the atrioventricular septal structures in the normal mouse. Circ Res. 1998; 82:645-656. [DOI] [PubMed] [Google Scholar]
- 2.Savolainen SM, Foley JF, Elmore SA. Histology atlas of the developing mouse heart with emphasis on E11.5 to E18.5. Toxicol Pathol. 2009; 37:395-414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.De la Cruz MV, Gimenez‐Ribotta M, Saravalli O, Cayre R. The contribution of the inferior endocardial cushion of the atrioventricular canal to cardiac septation and to the development of the atrioventricular valves: study in the chick embryo. Am J Anat. 1983; 166:63-72. [DOI] [PubMed] [Google Scholar]
- 4.Lincoln J, Alfieri CM, Yutzey KE. Development of heart valve leaflets and supporting apparatus in chicken and mouse embryos. Dev Dyn. 2004; 230:239-250. [DOI] [PubMed] [Google Scholar]
- 5.Wang Y, Wu B, Chamberlain AA, Lui W, Koirala P, Susztak K, Klein D, Taylor V, Zhou B. Endocardial to myocardial notch‐wnt‐bmp axis regulates early heart valve development. PLoS One. 2013; 8:e60244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Camenisch TD, Schroeder JA, Bradley J, Klewer SE, McDonald JA. Heart‐valve mesenchyme formation is dependent on hyaluronan‐augmented activation of ErbB2‐ErbB3 receptors. Nat Med. 2002; 8:850-855. [DOI] [PubMed] [Google Scholar]
- 7.Ramsdell AF, Moreno‐Rodriguez RA, Wienecke MM, Sugi Y, Turner DK, Mjaatvedt CH, Markwald RR. Identification of an autocrine signaling pathway that amplifies induction of endocardial cushion tissue in the avian heart. Acta Anat. 1998; 162:1-15. [DOI] [PubMed] [Google Scholar]
- 8.Zhang Z, Zhou B. Accelerated coronary angiogenesis by vegfr1‐knockout endocardial cells. PLoS One. 2013; 8:e70570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wu B, Wang Y, Lui W, Langworthy M, Tompkins KL, Hatzopoulos AK, Baldwin HS, Zhou B. Nfatc1 coordinates valve endocardial cell lineage development required for heart valve formation. Circ Res. 2011; 109:183-192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wu B, Zhang Z, Lui W, Chen X, Wang Y, Chamberlain AA, Moreno‐Rodriguez RA, Markwald RR, O'Rourke BP, Sharp DJ, Zheng D, Lenz J, Baldwin HS, Chang CP, Zhou B. Endocardial cells form the coronary arteries by angiogenesis through myocardial‐endocardial VEGF signaling. Cell. 2012; 151:1083-1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Montgomery RL, Davis CA, Potthoff MJ, Haberland M, Fielitz J, Qi X, Hill JA, Richardson JA, Olson EN. Histone deacetylases 1 and 2 redundantly regulate cardiac morphogenesis, growth, and contractility. Genes Dev. 2007; 21:1790-1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lee Y, Song AJ, Baker R, Micales B, Conway SJ, Lyons GE. Jumonji, a nuclear protein that is necessary for normal heart development. Circ Res. 2000; 86:932-938. [DOI] [PubMed] [Google Scholar]
- 13.Naya FJ, Wu C, Richardson JA, Overbeek P, Olson EN. Transcriptional activity of MEF2 during mouse embryogenesis monitored with a MEF2‐dependent transgene. Development. 1999; 126:2045-2052. [DOI] [PubMed] [Google Scholar]
- 14.Goldberg AD, Allis CD, Bernstein E. Epigenetics: a landscape takes shape. Cell. 2007; 128:635-638. [DOI] [PubMed] [Google Scholar]
- 15.Lee DY, Hayes JJ, Pruss D, Wolffe AP. A positive role for histone acetylation in transcription factor access to nucleosomal DNA. Cell. 1993; 72:73-84. [DOI] [PubMed] [Google Scholar]
- 16.Prokhortchouk E, Defossez PA. The cell biology of DNA methylation in mammals. Biochim Biophys Acta. 2008; 1783:2167-2173. [DOI] [PubMed] [Google Scholar]
- 17.Vilkaitis G, Merkiene E, Serva S, Weinhold E, Klimasauskas S. The mechanism of DNA cytosine‐5 methylation. Kinetic and mutational dissection of Hhai methyltransferase. J Biol Chem. 2001; 276:20924-20934. [DOI] [PubMed] [Google Scholar]
- 18.Jackson‐Grusby L, Beard C, Possemato R, Tudor M, Fambrough D, Csankovszki G, Dausman J, Lee P, Wilson C, Lander E, Jaenisch R. Loss of genomic methylation causes p53‐dependent apoptosis and epigenetic deregulation. Nat Genet. 2001; 27:31-39. [DOI] [PubMed] [Google Scholar]
- 19.Reik W. Stability and flexibility of epigenetic gene regulation in mammalian development. Nature. 2007; 447:425-432. [DOI] [PubMed] [Google Scholar]
- 20.Howlett SK, Reik W. Methylation levels of maternal and paternal genomes during preimplantation development. Development. 1991; 113:119-127. [DOI] [PubMed] [Google Scholar]
- 21.Olek A, Walter J. The pre‐implantation ontogeny of the H19 methylation imprint. Nat Genet. 1997; 17:275-276. [DOI] [PubMed] [Google Scholar]
- 22.Okano M, Bell DW, Haber DA, Li E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell. 1999; 99:247-257. [DOI] [PubMed] [Google Scholar]
- 23.Movassagh M, Choy MK, Goddard M, Bennett MR, Down TA, Foo RS. Differential DNA methylation correlates with differential expression of angiogenic factors in human heart failure. PLoS One. 2010; 5:e8564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shahrzad S, Bertrand K, Minhas K, Coomber BL. Induction of DNA hypomethylation by tumor hypoxia. Epigenetics. 2007; 2:119-125. [DOI] [PubMed] [Google Scholar]
- 25.Watson JA, Watson CJ, McCrohan AM, Woodfine K, Tosetto M, McDaid J, Gallagher E, Betts D, Baugh J, O'Sullivan J, Murrell A, Watson RW, McCann A. Generation of an epigenetic signature by chronic hypoxia in prostate cells. Hum Mol Genet. 2009; 18:3594-3604. [DOI] [PubMed] [Google Scholar]
- 26.Waterland RA, Michels KB. Epigenetic epidemiology of the developmental origins hypothesis. Annu Rev Nutr. 2007; 27:363-388. [DOI] [PubMed] [Google Scholar]
- 27.Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, Yamanaka S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007; 131:861-872. [DOI] [PubMed] [Google Scholar]
- 28.Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006; 126:663-676. [DOI] [PubMed] [Google Scholar]
- 29.Jeanpierre M, Turleau C, Aurias A, Prieur M, Ledeist F, Fischer A, Viegas‐Pequignot E. An embryonic‐like methylation pattern of classical satellite DNA is observed in ICF syndrome. Hum Mol Genet. 1993; 2:731-735. [DOI] [PubMed] [Google Scholar]
- 30.Ueda Y, Okano M, Williams C, Chen T, Georgopoulos K, Li E. Roles for Dnmt3b in mammalian development: a mouse model for the ICF syndrome. Development. 2006; 133:1183-1192. [DOI] [PubMed] [Google Scholar]
- 31.Khulan B, Thompson RF, Ye K, Fazzari MJ, Suzuki M, Stasiek E, Figueroa ME, Glass JL, Chen Q, Montagna C, Hatchwell E, Selzer RR, Richmond TA, Green RD, Melnick A, Greally JM. Comparative isoschizomer profiling of cytosine methylation: the help assay. Genome Res. 2006; 16:1046-1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Suzuki M, Jing Q, Lia D, Pascual M, McLellan A, Greally JM. Optimized design and data analysis of tag‐based cytosine methylation assays. Genome Biol. 2010; 11:R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Suzuki M, Greally JM. DNA methylation profiling using hpaii tiny fragment enrichment by ligation‐mediated PCR (HELP). Methods. 2010; 52:218-222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Karimi M, Johansson S, Stach D, Corcoran M, Grander D, Schalling M, Bakalkin G, Lyko F, Larsson C, Ekstrom TJ. LUMA (Luminometric Methylation Assay)—a high throughput method to the analysis of genomic DNA methylation. Exp Cell Res. 2006; 312:1989-1995. [DOI] [PubMed] [Google Scholar]
- 35.Karimi M, Johansson S, Ekstrom TJ. Using LUMA: a luminometric‐based assay for global DNA‐methylation. Epigenetics. 2006; 1:45-48. [DOI] [PubMed] [Google Scholar]
- 36.Thompson RF, Suzuki M, Lau KW, Greally JM. A pipeline for the quantitative analysis of CG dinucleotide methylation using mass spectrometry. Bioinformatics. 2009; 25:2164-2170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dubin R, Jing Q, O'Broin P, Calder B, Moskowitz D, Suzuki M, McLellan A, Greally J. WASP: wiki‐based automated sequence processor for epigenomics and genomics applications. J Biomol Tech. 2010; 21:S11 [Google Scholar]
- 38.Robinson MD, McCarthy DJ, Smyth GK. Edger: a bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010; 26:139-140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real‐time quantitative PCR and the 2(‐Delta Delta C(T)) method. Methods. 2001; 25:402-408. [DOI] [PubMed] [Google Scholar]
- 40.Heintzman ND, Stuart RK, Hon G, Fu Y, Ching CW, Hawkins RD, Barrera LO, Van Calcar S, Qu C, Ching KA, Wang W, Weng Z, Green RD, Crawford GE, Ren B. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat Genet. 2007; 39:311-318. [DOI] [PubMed] [Google Scholar]
- 41.Tie F, Banerjee R, Stratton CA, Prasad‐Sinha J, Stepanik V, Zlobin A, Diaz MO, Scacheri PC, Harte PJ. CBP‐mediated acetylation of histone H3 lysine 27 antagonizes Drosophila Polycomb silencing. Development. 2009; 136:3131-3141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Blow MJ, McCulley DJ, Li Z, Zhang T, Akiyama JA, Holt A, Plajzer‐Frick I, Shoukry M, Wright C, Chen F, Afzal V, Bristow J, Ren B, Black BL, Rubin EM, Visel A, Pennacchio LA. ChIP‐Seq identification of weakly conserved heart enhancers. Nat Genet. 2010; 42:806-810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stadler MB, Murr R, Burger L, Ivanek R, Lienert F, Scholer A, van Nimwegen E, Wirbelauer C, Oakeley EJ, Gaidatzis D, Tiwari VK, Schubeler D. DNA‐binding factors shape the mouse methylome at distal regulatory regions. Nature. 2011; 480:490-495. [DOI] [PubMed] [Google Scholar]
- 44.Shen Y, Yue F, McCleary DF, Ye Z, Edsall L, Kuan S, Wagner U, Dixon J, Lee L, Lobanenkov VV, Ren B. A map of the cis‐regulatory sequences in the mouse genome. Nature. 2012; 488:116-120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rada‐Iglesias A, Bajpai R, Swigut T, Brugmann SA, Flynn RA, Wysocka J. A unique chromatin signature uncovers early developmental enhancers in humans. Nature. 2011; 470:279-283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang Y, Morrisey E. Regulation of cardiomyocyte proliferation by FOXP1. Cell Cycle. 2010; 9:4251-4252. [DOI] [PubMed] [Google Scholar]
- 47.Alexandrovich A, Arno M, Patient RK, Shah AM, Pizzey JA, Brewer AC. Wnt2 is a direct downstream target of GATA6 during early cardiogenesis. Mech Dev. 2006; 123:297-311. [DOI] [PubMed] [Google Scholar]
- 48.Tian Y, Yuan L, Goss AM, Wang T, Yang J, Lepore JJ, Zhou D, Schwartz RJ, Patel V, Cohen ED, Morrisey EE. Characterization and in vivo pharmacological rescue of a Wnt2‐Gata6 pathway required for cardiac inflow tract development. Dev Cell. 2010; 18:275-287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lin Q, Schwarz J, Bucana C, Olson EN. Control of mouse cardiac morphogenesis and myogenesis by transcription factor MEF2C. Science. 1997; 276:1404-1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ghosh TK, Song FF, Packham EA, Buxton S, Robinson TE, Ronksley J, Self T, Bonser AJ, Brook JD. Physical interaction between TBX5 and MEF2C is required for early heart development. Mol Cell Biol. 2009; 29:2205-2218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Camenisch TD, Spicer AP, Brehm‐Gibson T, Biesterfeldt J, Augustine ML, Calabro A, Jr, Kubalak S, Klewer SE, McDonald JA. Disruption of hyaluronan synthase‐2 abrogates normal cardiac morphogenesis and hyaluronan‐mediated transformation of epithelium to mesenchyme. J Clin Invest. 2000; 106:349-360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Osborne JD, Flatow J, Holko M, Lin SM, Kibbe WA, Zhu LJ, Danila MI, Feng G, Chisholm RL. Annotating the human genome with disease ontology. BMC Genomics. 2009; 10suppl 1:S6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Aran D, Toperoff G, Rosenberg M, Hellman A. Replication timing‐related and gene body‐specific methylation of active human genes. Hum Mol Genet. 2011; 20:670-680. [DOI] [PubMed] [Google Scholar]
- 54.Lande‐Diner L, Zhang J, Ben‐Porath I, Amariglio N, Keshet I, Hecht M, Azuara V, Fisher AG, Rechavi G, Cedar H. Role of DNA methylation in stable gene repression. J Biol Chem. 2007; 282:12194-12200. [DOI] [PubMed] [Google Scholar]
- 55.Craig EA, Austin AF, Vaillancourt RR, Barnett JV, Camenisch TD. TGFbeta2‐mediated production of hyaluronan is important for the induction of epicardial cell differentiation and invasion. Exp Cell Res. 2010; 316:3397-3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Camenisch TD, Molin DG, Person A, Runyan RB, Gittenberger‐de Groot AC, McDonald JA, Klewer SE. Temporal and distinct TGFbeta ligand requirements during mouse and avian endocardial cushion morphogenesis. Dev Biol. 2002; 248:170-181. [DOI] [PubMed] [Google Scholar]
- 57.Cai X, Zhang W, Hu J, Zhang L, Sultana N, Wu B, Cai W, Zhou B, Cai CL. Tbx20 acts upstream of Wnt signaling to regulate endocardial cushion formation and valve remodeling during mouse cardiogenesis. Development. 2013; 140:3176-3187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.McFadden DG, Barbosa AC, Richardson JA, Schneider MD, Srivastava D, Olson EN. The Hand1 and Hand2 transcription factors regulate expansion of the embryonic cardiac ventricles in a gene dosage‐dependent manner. Development. 2005; 132:189-201. [DOI] [PubMed] [Google Scholar]
- 59.Chen Y, Yuen WH, Fu J, Huang G, Melendez AJ, Ibrahim FB, Lu H, Cao X. The mitochondrial respiratory chain controls intracellular calcium signaling and NFAT activity essential for heart formation in Xenopus laevis. Mol Cell Biol. 2007; 27:6420-6432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Oda M, Glass JL, Thompson RF, Mo Y, Olivier EN, Figueroa ME, Selzer RR, Richmond TA, Zhang X, Dannenberg L, Green RD, Melnick A, Hatchwell E, Bouhassira EE, Verma A, Suzuki M, Greally JM. High‐resolution genome‐wide cytosine methylation profiling with simultaneous copy number analysis and optimization for limited cell numbers. Nucleic Acids Res. 2009; 37:3829-3839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ball MP, Li JB, Gao Y, Lee JH, LeProust EM, Park IH, Xie B, Daley GQ, Church GM. Targeted and genome‐scale strategies reveal gene‐body methylation signatures in human cells. Nat Biotechnol. 2009; 27:361-368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wenger RH, Kvietikova I, Rolfs A, Camenisch G, Gassmann M. Oxygen‐regulated erythropoietin gene expression is dependent on a CpG methylation‐free hypoxia‐inducible factor‐1 DNA‐binding site. Eur J Biochem. 1998; 253:771-777. [DOI] [PubMed] [Google Scholar]
- 63.Kruger O, Maxeiner S, Kim JS, van Rijen HV, de Bakker JM, Eckardt D, Tiemann K, Lewalter T, Ghanem A, Luderitz B, Willecke K. Cardiac morphogenetic defects and conduction abnormalities in mice homozygously deficient for connexin40 and heterozygously deficient for connexin45. J Mol Cell Cardiol. 2006; 41:787-797. [DOI] [PubMed] [Google Scholar]