

Abstract
This review article summarizes the last few decades of research on nickel hydroxide, an important material in physics and chemistry, that has many applications in engineering including, significantly, batteries. First, the structures of the two known polymorphs, denoted as α-Ni(OH)2 and β-Ni(OH)2, are described. The various types of disorder, which are frequently present in nickel hydroxide materials, are discussed including hydration, stacking fault disorder, mechanical stresses and the incorporation of ionic impurities. Several related materials are discussed, including intercalated α-derivatives and basic nickel salts. Next, a number of methods to prepare, or synthesize, nickel hydroxides are summarized, including chemical precipitation, electrochemical precipitation, sol–gel synthesis, chemical ageing, hydrothermal and solvothermal synthesis, electrochemical oxidation, microwave-assisted synthesis, and sonochemical methods. Finally, the known physical properties of the nickel hydroxides are reviewed, including their magnetic, vibrational, optical, electrical and mechanical properties. The last section in this paper is intended to serve as a summary of both the potentially useful properties of these materials and the methods for the identification and characterization of ‘unknown’ nickel hydroxide-based samples.
Keywords: nickel hydroxide, structure, synthesis, properties
1. Introduction
When we began to write this review on nickel hydroxides, we considered what information about these industrially significant materials would provide the most benefit to the scientific community. It has been 33 years since Oliva et al.'s monumental review [1]. The work covered in that review had its origins in the 1950s and 1960s when there was an interest in the water/hydroxyl/oxy–hydroxy content of thin/bulk nickel oxide films, leading to concepts such as bound water, water of composition, structural water and adsorbed water, and their importance for corrosion and battery performance. However, this was long before suitable analysis/measurement techniques were available. The advances made in the last number of years have been most significant and has led to the point where we know a lot more today. From the thousands of literature results that a simple search on the Internet for ‘nickel hydroxide’ turns up, it is obvious that these are important materials. However, this vast body of literature can quickly become overwhelming and confusing, especially to new students or researchers who are just beginning to work with nickel hydroxides and who may not yet be familiar with all the experimental techniques discussed in the literature. Moreover, nickel hydroxide materials find diverse applications in the fields of chemistry, physics and engineering. Since the first half of the twentieth century, nickel hydroxide has been used as an electrode material for battery technologies (e.g. [2,3]). Nickel hydroxides are also important components of the surface layers that form either electrochemically or by the corrosion of nickel metal and nickel-based alloys. In the late 1900s, research into nickel hydroxides was principally focused on these two areas. In the late 1960s, Bode et al. proposed a simple scheme to explain the reported behaviour of the electrochemical oxidation of nickel hydroxides to nickel(III) oxyhydroxide and the subsequent reduction back to nickel(II) hydroxide [4]. Their scheme involves two phases of nickel hydroxide, α- and β-Ni(OH)2, and two phases of the oxidized material, β- and γ-NiOOH (figure 1). Although the complete picture is somewhat more nuanced, the original scheme, sometimes with one or two more arrows added (such as the -Ni(OH)2 transition in figure 1), remains a good overall description of the processes that occur at nickel hydroxide battery electrodes.
Figure 1.
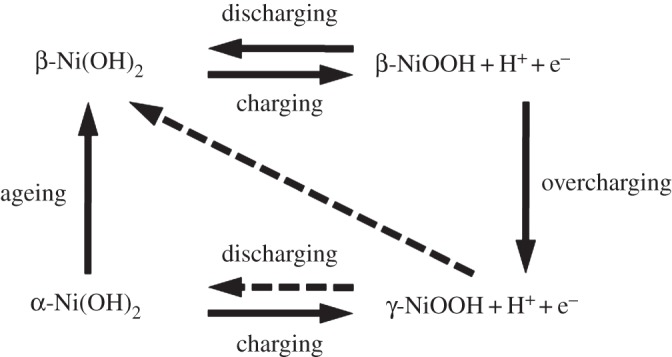
A general scheme of the chemical and electrochemical processes that occur at a nickel hydroxide battery electrode [1,4]. Copyright this work.
Today, nickel-based batteries are ubiquitous, including nickel–cadmium (NiCd) and nickel metal hydride (NiMH) technologies [5,6]. Recently, McBreen reviewed the applications of nickel hydroxides in modern battery technologies [6]. But nickel hydroxides are no longer limited to laboratories investigating batteries and corrosion processes. Today, these materials are involved in a diversity of practical applications, which include photocatalysis (e.g. [7–9]), electrocatalysis and electrosynthesis (e.g. [10–12]), supercapacitors (e.g. [13–18]), electrochromic devices (e.g. [12,19–21]), electrochemical sensors (e.g. [22–25]) and more. Decades of work has determined a much more complete description of the nickel hydroxides than the simple two-phase model shown above. There have been numerous advances in the preparation and characterization of nickel hydroxide materials. Nanotechnology has become widespread and complex morphologies that once defied imagination, such as nanoflowers and nanoribbons, are prepared and described on a regular basis. There are now literally hundreds of reported ways to synthesize nickel hydroxide materials, including newer methods such as microwave-assisted synthesis and sonochemistry.
This all amounts to an exciting and deeply interesting material that has a very rich history. However, this also means that understanding all the nuances of its structures and properties can be challenging. Moreover, finding an appropriate method to prepare nickel hydroxide with particular chemical or physical properties and at a suitable scale can be an overwhelming task. Therefore, in this review, we provide an up-to-date guide to nickel hydroxide materials and their modern applications. This review is aimed at an interdisciplinary audience and does not assume a strong background in any one field. We begin with a discussion of the different structures of nickel hydroxide, including the various types of structural disorder that the material tends to adopt (§2). Next, we organize the synthesis of nickel hydroxides into its most common methods, with some discussion of when to use each approach (§3). Finally, the physical properties and analytical characterization of nickel hydroxides are summarized and assessed (§4).
2. Structures
(a). The two fundamental crystal forms
Bode et al. [4] first identified the two known pseudopolymorphs of Ni(OH)2, denoted as α- and β-Ni(OH)2. The β-phase material (figure 2), which is isostructural with brucite, Mg(OH)2 [28], occurs naturally as the mineral theophrastite [29,30]. Note that the a- and b-axes in figure 2 are not orthogonal because this material has trigonal symmetry. Therefore, the angle between these axes, γ, is 120°. For a full explanation of unit cell symmetry and crystallographic notation, see [31]. The unit cell parameters, measured by X-ray diffraction (XRD) [26,28] and neutron diffraction [26,27], are listed in table 1. A sample XRD pattern is shown in figure 3 and the XRD parameters are summarized in table 2.
Figure 2.
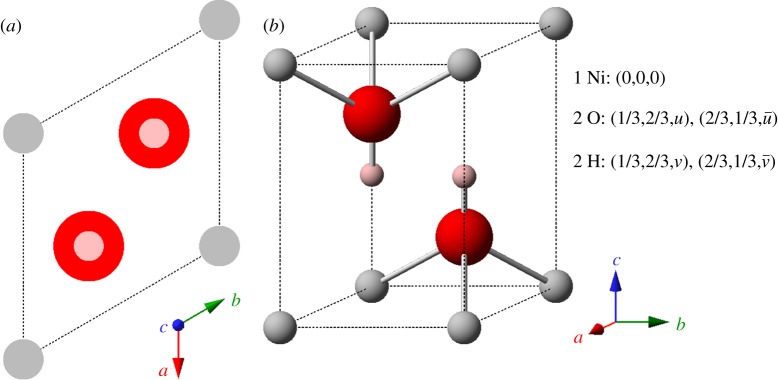
The crystal structure of β-Ni(OH)2 represented by (a) unit cell projection and (b) ball-and-stick unit cell (u=0.24 and v=0.47 [26]; or u=0.2221 and v=0.4275 [27]). Medium size (grey) spheres, Ni2+; large (red) spheres, O2−; small (pink) spheres, H+. Unit cell parameters are listed in table 1. Copyright this work. (Online version in colour.)
Table 1.
Unit cell parameters for the two fundamental phases of Ni(OH)2.
| β-Ni(OH)2 | β-Ni(OD)2 | α-Ni(OH)2⋅xH2O | ||
|---|---|---|---|---|
| [28] | [27] | [32] | ||
| space group | ml/No. 164 | lm/No. 162a | ||
| α=β | 90° | 90° | ||
| γ | 120° | 120° | ||
| a=b | 3.126 Å | 3.126 Å | dbNi−Ni | 3.08 Å |
| c | 4.605 Å | 4.593 Å | 8.0 Åc,d | |
aThe space group is for the idealized structure shown in figure 4. However, the H2O molecules do not actually occupy fixed lattice points and α-Ni(OH)2 materials are always turbostratic. Therefore, this is only a representative symmetry.
bThe a-parameter of α-Ni(OH)2 depends on the definition of the unit cell. The Ni–Ni spacing, however, may be directly compared to the a-parameter of β-Ni(OH)2.
cThe c-parameter may vary from this value if there are anionic impurities incorporated into the interlayer space.
Figure 3.
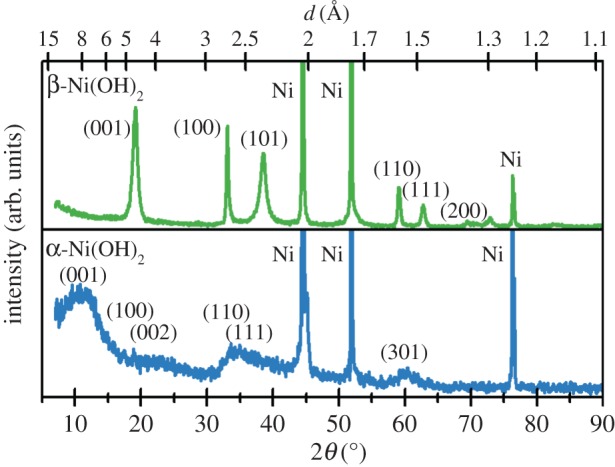
X-ray diffraction patterns of Ni(OH)2 films on Ni substrates collected using a Cu Kα X-ray source. Upper: β-Ni(OH)2 with stacking fault disorder (βsf) displays selective line broadening of the peaks that involve the crystallographic c-axis. Lower: α-Ni(OH)2 has low XRD intensities owing to high general structural disorder. Copyright this work. (Online version in colour.)
Table 2.
X-ray diffraction parameters of β-Ni(OH)2 calculated using the lattice parameters reported by McEwen [28]. Diffraction angles are listed for Cu Kα (λ=1.542 Å) and Co Kα (λ=1.789 Å) X-ray sources.
| Miller indices (hkl) | d(Å) | 2θCuKα(°) | 2θCoKα(°) | broadened by stacking faults |
|---|---|---|---|---|
| (001) | 4.605 | 19.3 | 22.4 | • |
| (100) | 2.707 | 33.1 | 38.6 | |
| (101) | 2.334 | 38.6 | 45.1 | • |
| (110) | 1.563 | 59.1 | 69.8 | |
| (111) | 1.480 | 62.8 | 74.4 | • |
| (200) | 1.354 | 69.4 | 82.7 | |
| (201) | 1.299 | 72.8 | 87.1 | • |
| (202) | 1.167 | 82.7 | 100.1 | • |
| (113) | 1.095 | 89.5 | 109.5 | • |
The α-Ni(OH)2⋅xH2O polymorph of nickel hydroxide consists of layers of β-Ni(OH)2, oriented parallel to the crystallographic ab-plane, intercalated by water molecules (figure 4) [4]. The degree of hydration varies within the range 0.41≤x≤0.7 [28,35]. Although the material is intrinsically hydrated, the water molecules are typically omitted from the written formula, i.e. the material is denoted α-Ni(OH)2. Note that the representation of α-Ni(OH)2 in figure 4 is not strictly correct or, at the very least, is somewhat misleading [1,28,36]. The intercalated water molecules do not occupy fixed sites, but rather they have some freedom to rotate and translate within the ab-plane. If the water molecules were close-packed with the hydroxide ions, as originally proposed by Bode et al. [4], the c-parameter would be approximately 7.6 Å. Actually, c≥7.8 Å, and water acts as an ‘amorphous glue’ that holds together the Ni(OH)2 layers [28]. As a result, adjacent layers have little, or no, tendency to orient relative to one another. This random layer orientation motif is known as a ‘turbostratic’ structure [28,37].
Figure 4.
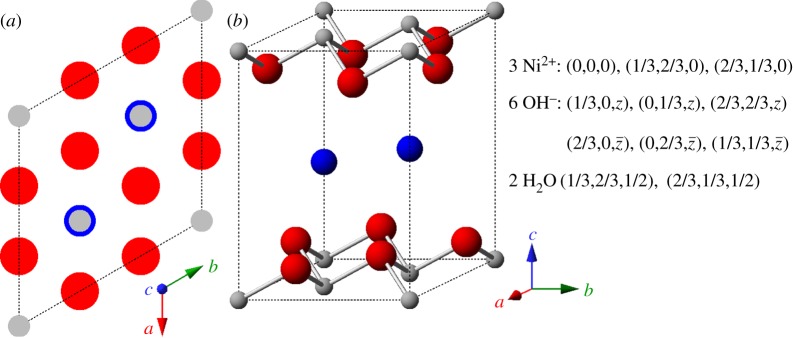
The idealized crystal structure of α-Ni(OH)2⋅xH2O represented by (a) unit cell projection and (b) ball-and-stick unit cell for x=0.67 (actual value varies, 0.41≤x≤0.7). Small (grey) spheres, Ni2+; large (red) spheres, OH−; medium size (blue) spheres, H2O. Unit cell parameters are listed in table 1. Copyright this work. (Online version in colour.)
Extended X-ray absorption fine structure (EXAFS) measurements show that there is a 0.05 Å contraction in the Ni–Ni distance, dNi−Ni, in α-Ni(OH)2 relative to β-Ni(OH)2 [32]. Therefore, the unit cell parameters for the idealized stoichiometry (3 Ni : 2 H2O) shown in figure 4 are a=b=5.335 Åand c=8.0 Å. Note that the a-parameter depends on the way in which the unit cell is defined, which in turn, is determined by the degree of hydration. dNi−Ni, however, may be compared directly with the a-parameter of β-Ni(OH)2 (table 1). Furthermore, the c-parameter is greater if the interlayer space contains anionic impurities (see §2b(iii)). A representative XRD pattern is shown in figure 3 and the XRD parameters are listed in table 3.
Table 3.
X-ray diffraction parameters of α-Ni(OH)2 calculated using the unit cell shown in figure 4 and the lattice parameters reported by Pandya et al. [32]. Fewer significant digits are provided for peaks involving the crystallographic c-axis [(001), (111), etc.]. Diffraction angles are listed for Cu Kα (λ=1.542 Å) and Co Kα (λ=1.789 Å) X-ray sources.
| Miller indices (hkl) | d (Å) | 2θCuKα(°) | 2θCoKα(°) |
|---|---|---|---|
| (001)a | 8.0 | 11.1 | 12.8 |
| (100) | 4.62 | 19.2 | 22.3 |
| (002) | 4.0 | 22.2 | 25.8 |
| (110) | 2.67 | 33.6 | 39.2 |
| (111) | 2.5 | 35.5 | 41.4 |
| (200) | 2.31 | 39.0 | 45.6 |
| (004) | 2.0 | 45.3 | 53.1 |
| (210) | 1.75 | 52.4 | 61.6 |
| (301) | 1.5 | 61.3 | 72.5 |
aWe have used the unit cell definition and Miller indices reported by Bode et al. and McEwen, in which the intersheet distance, dIS=c[4,28]. However, some authors use a different unit cell definition, such that dIS=c/3 (e.g. [33,34]). In these sources, the Miller indices also vary by a factor of three (e.g. where we report (001), they would report (003), etc.).
(b). Structural disorder
In addition to the two fundamental phases of nickel hydroxide, there are several possible types of structural disorder, including the incorporation of foreign ions, variable hydration and crystal defects including stacking faults [38]. Numerous methods have been used to denote the structural disorder, including adopting labels such as αam, α*, α′, IS and βbc [1,39–41], in part, because the nature of the disorder was not always fully understood. However, it is now possible to establish a less ambiguous method to identify and communicate nickel hydroxide structures. Quite simply, we recommend that the parent structure is first identified, followed by the observed type, or types, of disorder. For example, some βbc samples have since been identified as β-Ni(OH)2 with stacking fault disorder [34,38]. Such clearly defined short-hand notations may be useful, provided they actually are clearly defined. In this example, one could write βsf, where sf = stacking fault disorder.
The effects of structural disorder can have very important practical consequences. For example, well-crystallized β-Ni(OH)2 has lower electrochemical activity than disordered β-Ni(OH)2 materials [6]. However, the relationship between structural disorder and the measured properties is not always clear. The substitution of nickel sites with cobalt atoms improves the proton conductivity of β-Ni(OH)2. It is unclear, however, whether this is attributable to increased proton vacancies [42] or to increased stacking fault disorder, which is also known to affect the electrochemical activity of nickel hydroxide electrodes [43–45]. Therefore, it is important that all possible forms of disorder should be identified. These are briefly reviewed below.
(i). Hydration
Hydrated β-Ni(OH)2 materials are very important for electrochemical applications. The hydrated, electrochemically active form of the material has been given the representation [Ni(H2O)x](OH)2, where 0.1≤x≤0.4 [46,47]. The water molecules are weakly associated with the nickel cations and do not form hydrogen bonds with the lattice hydroxide [6,47–49]. One way to assess the amount of hydration is through thermogravimetric analysis or thermal gravimetric analysis (TGA) that involves the measurement of a sample's mass as its temperature, T, is slowly increased (ΔT/Δt is fixed and the sample mass is plotted against T) or as a function of time (T is fixed and mass is plotted against time). This method is very useful for hydrated materials because the removal of water may be measured as a sample's temperature is increased [50,51]. For β-Ni(OH)2, TGA has shown that surface water may be removed at relatively low temperatures, T≈80–90°C, and that the incorporated water can be fully removed at high temperatures, T≈160°C [35,46,47,52–54]. Thermal dehydration decreases the charge capacity of β-Ni(OH)2 battery electrodes by approximately 10–14% [47,49]. The presence of incorporated water can also increase the interlayer spacing by Δc≈0.1 Å[27], although because the role of stacking faults (see §2b(ii)) in these measurements is unknown, this expansion may not be wholly attributable to hydration effects. Incorporated water may be conclusively detected by the presence of additional O−H libration (bending) modes in the Raman and IR spectra [27,38,48,49,52] (see §4b).
α-Ni(OH)2⋅xH2O materials are always hydrated because water is intrinsic to their structure. TGA measurements show that the interlayer water in α-Ni(OH)2 is only removed at very high temperatures, T≈240–300°C [35,46,55]. The removal of the interlayer water may occur together with the removal of water from the decomposition of the hydroxide to NiO [46]. This is quite different from the TGA behaviour of β-Ni(OH)2 materials, which shows a dehydration process at T≈160°C from incorporated water, followed by a very broad feature at 170°C<T<525°C from decomposition to NiO [47]. Moreover, the mass loss of α-Ni(OH)2 from dehydration is significantly greater than that of the β-phase. The ‘pure’ material is generally reported within the range 0.41≤x≤0.7 [28,35], although the exact degree of hydration is quite variable and sometimes lies outside this range. The incorporation of foreign anions into the region between Ni(OH)2 layers strongly affects hydration (see §2b(iii)). In addition to the water limited to within this interlayer region, it has been suggested that water may associate with the lattice Ni, similar to what is observed for hydrated β-Ni(OH)2 materials [46,48]. TGA measurements show that a small amount of weakly bound water may be removed at approximately 110°C. This has been attributed to water adsorbed on the surface, but could also originate from bulk hydration of the Ni atoms [35]. EXAFS evidence that α-Ni(OH)2 materials can have Ni–O coordination numbers greater than six supports this possibility [32].
(ii). Stacking fault disorder
Stacking faults, which are common in layered double hydroxides (LDHs) [56], may be understood by examining the unit cell of β-Ni(OH)2 (figure 2). There is strong ionic bonding between Ni2+, O2− and H+ ions within each Ni(OH)2 sheet, whereas the interactions between adjacent layers are relatively weak. Therefore, Ni(OH)2 materials consist of several ‘stacked’ layers. Consequently, in addition to the aligned thermodynamic product, illustrated in figure 5a, one can propose three additional stacking motifs. The first arises from rotation about the c-axis by 60° (figure 5b), the second from translation within the ab-plane (figure 5c), and the last from both rotation and translation. One may alternatively consider stacking fault disorder by examining the oxygen anion stacking sequence, which is ideally AC AC AC AC, etc. In the presence of a stacking fault, this sequence is altered, e.g. to AC BA CB AC, etc. [57]. For more information on stacking fault disorder in nickel hydroxides, the reader is referred to Tessier et al. [45] or, for a detailed and general discussion of stacking fault disorder, Sebastian & Krishna [58]. Stacking faults cause selective line broadening in XRD patterns, because the disorder is only along the direction of the crystallographic c-axis (i.e. order within the ab-plane is unaffected by stacking faults) [43,45,57]. The relationship between stacking fault frequency and powder XRD peak widths and intensities has been simulated computationally [44,59]. Stacking faults in β-Ni(OH)2 materials also cause additional O−H stretching modes in their Raman and infrared (IR) spectra (see §4) [38]. One can therefore estimate the relative frequency of stacking fault disorders between two samples from the relative peak intensities in the Raman or IR spectra. However, spectral differences arising from the different stacking fault motifs, e.g. translation (figure 5b) versus rotation (figure 5c), have not been reported. It is meaningless to discuss stacking faults in α-Ni(OH)2 because of its turbostratic structure (the layer orientation is always random).
Figure 5.
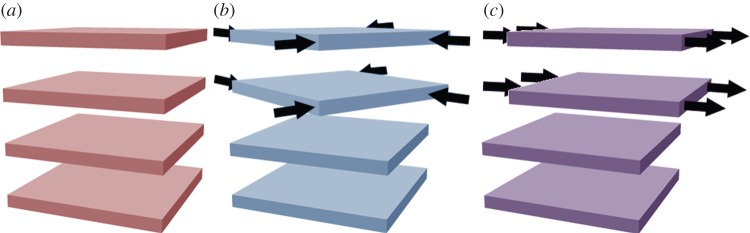
Schematic of stacking fault disorder between two adjacent layers. (a) No stacking faults are present and adjacent layers are aligned. (b) Rotation about the crystallographic c-axis. (c) Translation within the ab-plane. Copyright this work. (Online version in colour.)
(iii). Ionic substitution and foreign ion incorporation
Substitutional point defects involve the substitution of a lattice atom (or polyatomic ion) with another. Numerous cationic substitutions of nickel hydroxides have been prepared from bivalent metal cations and are given the general formula Ni1−xMx(OH)2, where M is the substituted metal atom. Substitution may be minor; for example, reagent impurities led to the preparation of Ni1−xCax(OH)2, x≤0.02 in [38]. More extensive cationic substitution may also occur, especially if there are isostructural materials. For example, solid solutions of β-Ni(OH)2 and Mg(OH)2 (brucite) of any composition are possible and have been used to correlate the vibrational modes of the two materials [60].
β-Ni(OH)2 materials have been prepared with cationic substitutions of the lattice Ni with Mg [60,61], Al [16], Ca [38,61], Co [42,43,52,61–64], Cu [61], Zn [61,65,66] and Cd [42,64]. All of these materials maintain the space group of the parent β-phase material, although generally with slightly modified unit cell parameters. For example, the interplanar spacing, c, decreases with increasing Co content [63]. This may not purely result from the different cationic radii, as the preparation of these substituted materials is often accompanied by other structural changes, such as decreased stacking fault disorder [43].
α-Ni(OH)2 materials have been prepared with cationic substitutions of the lattice Ni with Al [16,67–75], Mn [76,77], Fe [78,79], Co [14,69,74,75,80–82], Cu [67,83], Zn [65,66,70,84,85], Y [86] and Yb [87]. In some of these examples, the bivalent Ni2+ cation is replaced with a trivalent species (e.g. Fe3+). Therefore, the resulting materials must be hydroxide-rich, proton-deficient or there is a corresponding degree of anionic incorporation, which is further discussed below, to maintain charge balance (e.g. carbonate anions in [79]).
Ni(OH)2 materials can be isotopically enriched by treatment with D2O, which is another form of cationic substitution. Deuterated materials have been used for crystallographic studies (β-Ni(OD)2 [27] and α-Ni(OD)2 [88]) and to definitively identify the O−H and O−D stretching modes in IR spectra (β-Ni(OH)2/β-Ni(OD)2 [49,88,89] and α-Ni(OH)2/α-Ni(OD)2 [88]).
Foreign anions are commonly incorporated into α-Ni(OH)2. During precipitation from a nickel(II) salt, it is quite common for some of the counterions to be incorporated into the structure (see §3 for the methods of nickel hydroxide synthesis). The amount incorporated can be considerable and the resultant material is represented by the general formula α-Ni(OH)2−xAyBz ⋅ nH2O, where A and B are monovalent and divalent anions, respectively, and x=y+2z [90]. Because Ni(NO3)2 is widely used as a precursor to prepare nickel hydroxide materials, the most common foreign anion is nitrate [1,91], although chloride, sulfate, carbonate, cyanate, acetate, succinate, glutarate and adipate anions have also been reported [38,90,92,93]. The incorporated anions may either occupy lattice hydroxide sites or they may sit within the interlayer region [38,94]. The former tends to be less stable, probably owing to mechanical stresses from the size mismatch. By contrast, anions in the intercalation region tend to be more stable and have very similar spectroscopic properties to the free solution species. In the field of intercalation chemistry, the thickness of the interlayer space is known as the ‘gallery height’ [95].The presence of foreign ions in the interlayer space can increase the gallery height and, consequently, the interlayer spacing. For example, the interlayer spacing of α-Ni(OH)2 containing nitrate or carbonate is typically 7.8 Å ≤c≤8.2 Å , whereas the spacing of α-Ni(OH)2 that contains the larger anion adipate [] has been reported as high as c=13.2 Å. This is a 65% expansion from the parent structure [33,90]. The increased gallery height is presumably accompanied by the incorporation of additional water.
The incorporation of foreign anions into β-Ni(OH)2 materials is far less common because adjacent layers are close-packed. Furthermore, foreign anions are quickly removed during the preparation of β-Ni(OH)2 by chemical ageing, i.e. the conversion of α- to β-Ni(OH)2 (see §3d) [38,94].
(iv). α/β-Interstratification
One can consider a single grain or crystal of nickel hydroxide in which some of the layers are separated by intercalated water (α), whereas the rest are not (β). Such materials have been described as ‘interstratified phases consisting of α- and β-type structural motifs’ and not simple mixtures of α- and β-Ni(OH)2 crystals [33,41,96,97]. Rather, the presence of interstratification allows α- and β-Ni(OH)2 domains to coexist within a single crystal. This material, which we denote α/βIS-Ni(OH)2, is illustrated in figure 6. These domains may hypothetically be very large or as little as a single layer. Generally, the measured XRD pattern of materials containing primarily one phase (i.e. when the interstratification is minimal), is quite similar to that of the pure material. However, for mixed samples such as the α/βIS-Ni(OH)2 shown pictorially in figure 6, the diffraction lines appear shifted and broadened. This is because the long-range structural order required for diffraction measurements is diminished in interstratified materials. If each layer is solely α- or β-phase, the disorder is only oriented along the c-axis and the (hkl), l≠0, XRD lines will be selectively broadened or even absent from the measured pattern [59,97]. However, it is also possible to have both phases within a single layer, as shown in figure 6, in which case the structural disorder will be more generalized. The variation of the diffraction peak widths and relative intensity versus the diffraction angle in interstratified layered structures has been examined theoretically and experimentally for various compositions and structural motifs [59,96,98]. A sample diffraction pattern shown in figure 7.
Figure 6.

Schematic of α/β-interstratification, in which both phases coexist within a single crystal. In this example, the α-Ni(OH)2 contains intercalated carbonate anion impurities. Adapted from [96], copyright 1994, with permission from Elsevier.
Figure 7.
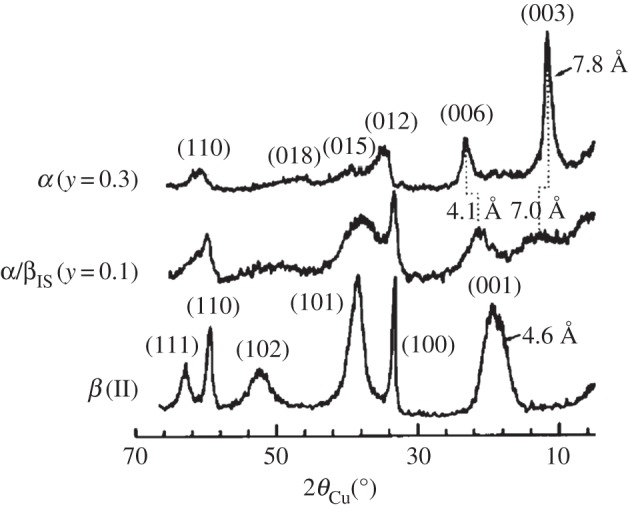
XRD patterns collected from α-Ni0.7Mn0.3(OH)2, α/βIS-Ni0.9Mn0.1(OH)2, and β-Ni(OH)2 using a Cu Kα X-ray. The α- and α/βIS-phase materials contained intercalated carbonate anions (figure 6). Adapted from [96], copyright 1994, with permission from Elsevier.
It should be noted that vibrational spectra collected from interstratified materials contain the characteristic O−H internal stretching modes of both the α- and β-phase materials (see §4b) [33,41,96]. Moreover, it is possible to have interstratification and other types of structural disorder, such as the mixed metal, carbonate-intercalated α/βIS-Ni0.9Mn0.1(OH)2 materials reported in Guerlou-Demourgues et al. [96] or the carbonate-intercalated α/βIS-Ni1−xCox(OH)2 in [33]. Guerlou-Demourgues et al. suggested that the small amount of carbonate in their samples was insufficient to homogeneously distribute within the interlayer space, which caused segregation, as represented in figure 6.
(v). Internal mechanical stress
There are several causes of internal mechanical stress. For example, the insertion of large polyatomic anions, such as nitrate, into the lattice sites or the intercalation space of α-Ni(OH)2 is expected to introduce mechanical stress. Internal stress occurs during the chemical ageing of α- to β-Ni(OH)2 because of the changing unit cell c-parameter (8.0–4.6 Å), which causes compressive and tensile forces along the c-direction of the α- and β-phases, respectively [94]. Mechanical stress is also reported for dried nickel hydroxide films. The lattice vibrational modes are shifted when the films are visibly cracked, showing that the removal of water changes the density of the materials and causes internal stresses [38]. Mechanical stress also occurs during the redox cycling of nickel hydroxide electrodes in batteries, because of the differences in densities of nickel hydroxide and nickel oxyhydroxide [1,99].
(c). α-Derivative structures
In addition to α-Ni(OH)2, there are several related materials that may not truly belong to the α-Ni(OH)2 phase. Nevertheless, they are often given the ‘α’ label because of certain structural similarities. For example, Nevravathi et al. [82] used the term ‘surfactant-intercalated α-nickel hydroxide’ to designate a material composed of single sheets of Ni(OH)2 separated by intercalated dodecyl sulfate. This term clearly indicates that the surfactant molecules are between the nickel hydroxide sheets. However, one may argue that the ‘α’ label should be reserved for materials that are composed of brucite-like nickel hydroxide intercalated with water molecules (i.e. α-Ni(OH)2⋅xH2O). Essentially, this suggests two possible interpretations of the ‘α’ label: (i) brucite-like Ni(OH)2 sheets with intercalated water or (ii) brucite-like Ni(OH)2 sheets with any intercalated species. With the crystallographic and historic perspectives in mind, we shall choose the first of these over the latter. Therefore, we now introduce the term ‘α-derivative structures’ for brucite-like Ni(OH)2 sheets with any intercalated species other than water. This new term is quite broad and may be considered a class of materials, rather than an individual crystallographic phase. These materials may be generally described as partially ordered layered structures, as the individual Ni(OH)2 layers are well ordered, whereas there is disorder in the intercalation space. This is also true for α-Ni(OH)2 itself. Several examples of these α-derivative structures are provided below.
(i). Surfactant-intercalated α-derivative structures
α-Ni(OH)2-like materials in which the intercalated water is replaced with surfactant molecules have been prepared using cetyltrimethylammonium bromide [100], dodecyl sulfate [82,92,100–106], dodecylbenzene sulfonate [103], p-aminobenzoate [18] and Tween 80 [100]. The polar or ionic regions of the surfactant molecules interact with the Ni(OH)2 sheets and the non-polar regions fill the interlayer space. Therefore, the interlayer space is hydrophobic and water is consequently excluded from the region. Note that these materials are fundamentally different from the α-Ni(OH)2 materials with incorporated foreign anions described in §2b(iii). Those previous examples are hydrated structures that are closely related to the ideal parent α-phase, whereas these surfactant-intercalated α-derivative materials do not contain water.
(ii). Exfoliated α-derivatives
The individual layers of α-Ni(OH)2 may be separated, i.e. exfoliated, to produce a colloidal suspension. The exfoliation of layered structures, in general, such as perovskites, hydroxyl double salts and LDHs, has been studied in more detail [107]. Just as the properties of graphene are wholly different than those of graphite, the properties of exfoliated nickel hydroxide can be quite different from those of either α- or β-Ni(OH)2 [102].
Generally, there are two exfoliation methods. In the first, the interlayer region is rendered organophilic via surfactant intercalation, as described above. Then, the use of an organic solvent spontaneously exfoliates the material [107]. For example, a dodecyl sulfate-intercalated α-derivative may be exfoliated by the addition of formamide [102] or 1-butanol [82]. In the second method, exfoliated α-derivatives are prepared in an aqueous solution. An intercalated α-derivative is prepared with an amphoteric molecule, which is any chemical species that has both acidic and basic functional groups. For example, p-aminobenzoic acid has a carboxylic acid group and a basic amine group. At high pH, this species is anionic and may be incorporated into the interlayer space to produce a p-aminobenzoate-intercalated α-derivative. When the pH is decreased, the molecule becomes a zwitterion. Zwitterions are electrically neutral molecules that contain regions of positive (–NH) and negative (–CO) charge. The result is that the material becomes unstable and Coulombic repulsion between adjacent, positively charged amine groups drives exfoliation of the material [108].
The colloidal sheets maintain the octahedral coordination of the Ni cations. However, as they are no longer stacked sheets of Ni(OH)2, they cannot correctly be designated as α- or β-phase materials. Rather, we recommend the term ‘exfoliated α-derivative,’ to describe the preparation of the material. It is also noteworthy that changing the solution conditions of the suspension may result in reformation of the surfactant-intercalated α-derivative [82].
(iii). Basic nickel salts
Several basic salts of nickel exist that are related to the nickel hydroxides [34]. In particular, nickel hydroxynitrates (NHNs) may be prepared from Ni(NO3)2 and include Ni2(OH)3(NO3) [109], Ni3(OH)4(NO3)2 [110], and Ni(OH)(NO3) [111]. These materials, which can be hydrated, have the general formula Ni(OH)2−x(NO3)x⋅nH2O, the same given to nitrate-containing α-Ni(OH)2 [34]. NHNs have trigonal symmetry and the nitrate anions are directly bonded to the Ni cations [34], which is also true for some α-Ni(OH)2 samples [38]. However, NHNs may be prepared as anhydrous materials, which is fundamentally different from α-Ni(OH)2 [34]. One may wonder, then, what differences exist between these two related materials. First, the interlayer spacing is consistently less in NHNs (typically, cNHN<7.6 Å) than in α-Ni(OH)2 (cα≥7.8 Å). Second, NHN materials have considerably sharper diffraction peaks [34], because adjacent layers in NHNs are close-packed. This is in contrast to the broad diffraction peaks that arise from the turbostratic α-Ni(OH)2 structure [1,28]. Therefore, the main difference between nitrate-containing α-Ni(OH)2 and hydrated NHN is the relative spacing and alignment of the adjacent layers.
Other basic salts of nickel include nullaginite [Ni2(OH)2CO3] [112], paraotwayite [Ni(OH)2−x(SO4,CO3)0.5x, x≈0.5] [113] and otwayite [(Ni,Mg)2(OH)2CO3⋅H2O] [114]. The unit cell symmetries of these three minerals (monoclinic, orthorhombic and monoclinic, respectively) are different from those of the NHNs, α-Ni(OH)2 and β-Ni(OH)2 (all trigonal).
3. Synthesis
Countless methods to prepare nickel hydroxide materials exist. As a result, finding a method to prepare a sample with appropriate properties can be a time-consuming and overwhelming task. Desired properties may include the phase (α or β), crystallinity (stacking fault disorder, purity, etc.), product morphology (porosity, nanostructure, etc.) and general form (bulk powder, thin film on a metallic substrate, etc.). Therefore, this portion of the review is intended to serve as a guide to direct the reader toward appropriate references by outlining some of the more widely used methods that are available. By no means is this summary comprehensive or exhaustive, as the compilation of all the different published methods is simply outside the scope of a simple review article. Rather, examples have been selected for their popularity, for their ease, or to represent the diversity of published methods.
In general, α-Ni(OH)2 may be prepared in bulk quantities by chemical precipitation or as a thin layer on a high surface area substrate by impregnation-precipitation (§3a). α-Ni(OH)2 may be deposited as a thin layer on a conductive substrate by electrochemical precipitation (§3b). A wide variety of porous and micro-/nanoscale structured materials may be prepared by sol–gel synthesis, as a bulk material or deposited onto a substrate by spin- or dip-coating (α or β, §3c). High-purity β-Ni(OH)2 materials may be produced by chemically ageing α-Ni(OH)2 (§3d) or purchased as a powder from a commercial supplier. Bulk quantities of micro- or nanostructured α- or β-Ni(OH)2 may be produced by hydrothermal or solvothermal synthesis (§3e). Nickel hydroxides are common components of corrosion deposits and electrochemically formed surface layers on nickel metal and nickel-based alloys (§3f). These six synthetic methods are schematically represented in figure 8. Finally, several less common methods, including microwave-assisted, sonochemical and solid-state synthesis, are briefly presented (§3g). Examples of six possible morphologies for α- and β-Ni(OH)2 prepared by various synthetic methods are shown in figure 9.
Figure 8.

Six methods to prepare Ni(OH)2. (a) The basification of an aqueous nickel(II) salt solution (e.g. addition of KOH(aq) to NiCl2(aq)). (b) Electrochemical precipitation onto a conductive substrate. The reduction of water produces hydroxide anions at the cathode surface, which react with the nickel(II) cations in solution. In this example, the anode material is a Ni sheet. (c) Sol–gel methods. A solution of a nickel(II) salt and a surfactant (e.g. sodium dodecylsulfate) is basified to form a Ni(OH)2 sol. The sol is treated (e.g. by centrifugation or evaporation) to produce a gel. (d) Chemical ageing converts α-Ni(OH)2 to β-Ni(OH)2. This is typically performed in concentrated alkaline media at high temperatures but also proceeds slowly in room temperature water. (e) The hydrothermal method. α/β-Ni(OH)2 precursor is dissolved in a pressure vessel at high temperatures. The temperature is subsequently decreased and β-Ni(OH)2 precipitates. (f) α/β-Ni(OH)2 surface layers form on nickel and nickel-based alloys as corrosion products or electrochemically formed surface layers, often underlaid by non-stoichiometric nickel oxide. Reproduced with permission from [115]. (Online version in colour.)
Figure 9.

Representative images of the morphologies discussed in this review: (a) Light micrograph of an electrochemically precipitated α-Ni(OH)2 film. The surface visibly cracked when the film was dried. Adapted with permission from [38]. (b) SEM image of β-Ni(OH)2 nanoflowers prepared by hydrothermal synthesis. Inset: high magnification shows the nanoflowers are composed of nanosheets. Adapted by permission from [22]. (c) TEM image of a ‘dandelion-like’ hollow β-Ni(OH)2 microsphere prepared by a sol–gel method. Reprinted with permission from [116], © 2009 American Chemical Society. (d) SEM of an ‘unusually large β-Ni(OH)2 ‘pseudo-single crystal’ prepared by a chemical precipitation method. Inset: high magnification of the particle basal plane and an edge. Reprinted with permission from [117], © 2011 American Chemical Society. (e) TEM image of α-Ni(OH)2 nanoribbons prepared by hydrothermal synthesis. Ribbons are 10–20 nm thick. Adapted with permission from [118]. © 2008 WILEY-VCH Verlag GmbH & Co. (f) SEM image of β-Ni(OH)2 nanoflowers prepared by microwave-assisted hydrothermal synthesis. Inset: high magnification of a single nanoflower. Reprinted from [119], © 2010, with permission from The Society of Powder Technology. (Online version in colour.)
Keen-eyed readers will notice that some of the product phases and descriptions do not exactly match those in the original articles. This is because, to the best of our ability, we have made efforts to re-evaluate crystallographic and spectroscopic results using the most recently available interpretations and assignments. It is not that we believe or wish to imply that previous researchers made errors. Rather, the community's understanding of some experimental results (e.g. vibrational mode assignments) has improved with time and one may now better understand decades-old experimental results. For example, selective line broadening in XRD patterns is now understood to usually arise from stacking fault disorder, rather than particle size effects [43].
(a). Chemical precipitation
The chemical precipitation of Ni(OH)2 is a one-step method, in which a basic solution (NaOH, NH3, etc.) is added, typically dropwise, to a nickel(II) salt solution (Ni(NO3)2, NiCl2, etc.), or vice versa. At sufficiently high pH, the hydroxide anion concentration exceeds the solubility limit and Ni(OH)2 forms as a precipitate
| 3.1 |
The phase and the crystallinity of the product are determined by the solution concentrations, the nickel(II) salt used, the base solution used (NaOH, NH4OH, etc.), the temperature and the pH of the solution during precipitation [40,120]. In general, precipitation at room temperature favours the formation of α-Ni(OH)2 and precipitation at elevated temperatures produces mixed-phase α/β- or pure β-Ni(OH)2 materials [35]. It has been suggested that the precipitation process always produces α-Ni(OH)2 initially, but that the initial product rapidly chemically ages (sometimes referred to as ‘ripening’) at high temperatures to the more crystalline β-phase [121]. Chemical ageing, the spontaneous transformation from α to β, is further discussed in §3d. The formation of pure β-Ni(OH)2 by chemical precipitation generally requires temperatures near or above the boiling point of water. These high temperature and pressure methods are described in §3e, which discusses hydrothermal synthesis. Precipitation at high temperatures in ambient pressure may be performed if very concentrated alkaline solutions are used (e.g. β-Ni(OH)2 was precipitated by reflux at 170°C in 63–75% (w/w) NaOH(aq) [122]). The choice of the nickel(II) salt and the basification agent also affects the product structure. For example, the basification of Ni(NO3)2 with urea at 90°C yields α-Ni(OH)2 precipitate, despite the high temperature [121]. This is presumably because of the incorporation of nitrate and cyanate anions into the interlayer space during precipitation. Through careful control of the experimental conditions, it is also possible to control the crystallite size. For example, figure 9d shows a large hexagonal β-Ni(OH)2 crystal prepared by Gourrier et al. [117].
Mixed hydroxides (e.g. Ni1−xMnx(OH)2), which may be used in NiCd or NiMH battery anodes, are easily prepared by chemical precipitation [6]. This is a common step in the synthesis of materials for Li-ion battery electrodes (e.g. LiNi1−xMnxO2) [77,123,124]. More complex mixed hydroxides, such as core–shell Ni1−xMnx(OH)2 spherical nanoparticles, where x varies as a function of the particle radius, may be prepared by gradually changing the composition of the precipitation solution [125]. Several representative precipitation methods are listed in table 4.
Table 4.
Representative methods from the literature for chemical precipitation of bulk Ni(OH)2 materials.
| Ni(II) salt | base | other reagents | product phase | comments | refs. |
|---|---|---|---|---|---|
| Ni(NO3)2 | NaOH | α-Ni(OH)2 | article discusses the effect of solution pH | [40] | |
| Ni(NO3)2 | KOH | α-Ni(OH)2 | [126] | ||
| Ni(NO3)2 | KOH | α-Ni(OH)2 | T=22°C | [35] | |
| contains some β-Ni(OH)2 | |||||
| Ni(NO3)2 | KOH | β-Ni(OH)2 | T=90°C | [35] | |
| stacking fault disorder | |||||
| contains some α-Ni(OH)2 | |||||
| Ni(NO3)2 | NH4OH | α-Ni(OH)2 | [127] | ||
| Ni(NO3)2 | NaOH | Na2CO3 | α-Ni(OH)2 | product contains Zn2+ | [84] |
| Zn(NO3)2 | |||||
| Ni(NO3)2 | KOH | sodium citrate | α/β-Ni(OH)2 | [126] | |
| NiCl2 | NH4OH | β-Ni(OH)2 | dried at 100°C for 12 h | [128] | |
| stacking fault and hydration disorder | |||||
| NiCl2 | NH4OH | graphene | β-Ni(OH)2 | dried at 100°C for 12 h | [128] |
| stacking fault, hydration and general disorder | |||||
| contains some α-Ni(OH)2 | |||||
| NiCl2 | NH4OH | CoCl2 | α/β-Ni(OH)2 | dried at 100°C for 12 h | [128] |
| product contains Co | |||||
| stacking fault, hydration and general disorder | |||||
| NiCl2 | NH4OH | Al2(SO4)3 | α-Ni(OH)2 | dried at 100°C for 12 h | [128] |
| product contains Al | |||||
| hydration and general disorder | |||||
| NiCl2 | NH4OH | Al2(SO4)3 | α-Ni(OH)2 | dried at 100°C for 12 h | [128] |
| CoCl2 | product contains Co and Al | ||||
| hydration and general disorder | |||||
| NiSO4 | NaOH | α-Ni(OH)2 | [129] | ||
| NiSO4 | NaOH | Fe2(SO4)3 | α/β-Ni(OH)2 | mostly, the α-phase material is produced | [79] |
| Na2CO3 | |||||
| product contains Fe3+ and CO | |||||
| NiSO4 | NaOH | β-Ni(OH)2 | T=20°C, dried at 60°C | [43] | |
| stacking fault and general disorder | |||||
| Ni(NO3)2 | NaOH | α/β-Ni(OH)2 | T=60°C | [101] | |
| product contains NO | |||||
| Ni(NO3)2 | OC(NH2)2 | α-Ni(OH)2 | T=70–90°C | [121] | |
| product contains OCN− and CO | |||||
| Ni(NO3)2 | Na2O2 | β-Ni(OH)2 | yields ‘coral-like’ nanoporous materials | [130] | |
| stacking fault disorder | |||||
| NiSO4 | NaOH | β-Ni(OH)2 | T=70°C, dried at 120°C | [43] | |
| stacking fault and some general disorder | |||||
| NiSO4 | NaOH | α/β-Ni(OH)2 | T=60°C | [101] | |
| product contains SO | |||||
| NiSO4 | NaOH | NH4OH | α/β-Ni(OH)2 | T=60°C | [131] |
| article discusses the effect of solution pH | |||||
| stacking fault disorder | |||||
| Ni(ClO4)2 | NaOH | β-Ni(OH)2 | stir at 110≤T≤170°C for 24 h | [122] | |
| yields hexagonal crystals (≤0.1 mm) | |||||
| Ni(ClO4)2 | NaOH | β-Ni(OH)2 | stir at T=140°C for 24 h, remove mother liquor, add water, then stir at T=170°C for 24 h | [117] | |
| yields hexagonal crystals (approx. 50 μm) |
The impregnation-precipitation method is used to prepare high surface area materials, commonly for battery anodes. A porous or highly textured conductive substrate is first impregnated by immersion in a nickel(II) salt solution. The substrate is then immersed in an alkaline solution to elicit the precipitation of Ni(OH)2. By repeating this cycle, one may obtain the desired electrode loading, i.e. the desired Ni(OH)2 layer thickness [39]. The deposited Ni(OH)2 maintains the morphology of the substrate [39,132]. This method has also been called ‘dip-coating’ [132], a term that also refers to other techniques. Battery electrodes are also prepared by the suspension-impregnation method, in which an Ni(OH)2 suspension is deposited onto a porous substrate [6,72,133–135]. Ni(OH)2 may also be deposited onto a substrate by slow precipitation from a single solution [16,136].
(b). Electrochemical precipitation
A thin layer of α-, α/β- or β-Ni(OH)2 may be deposited on a conductive substrate by electrochemical precipitation (cathodic electrodeposition, electro-precipitation, etc.). An example of electrochemically precipitated α-Ni(OH)2 is shown in figure 9a. In this process, shown schematically in figure 8b, the substrate is placed in a nickel(II) salt solution and is held at a negative potential to elicit a cathodic current from the reduction of water
| 3.2 |
If the solution contains nickel(II) nitrate, the reduction of nitrate anions may contribute to the production of the hydroxide anions [28]. Because Ni(OH)2 has very low solubility, chemical precipitation immediately occurs at the electrode (substrate) surface
| 3.3 |
The structure of the product (phase, crystallinity, etc.) is heavily dependent on the nature of the nickel(II) salt solution (concentration, counterion) and the cathodic current density employed. It is emphasized that only the formation of hydroxide is electrochemical. The precipitation step is identical to the chemical precipitation methods described in §3a. During this process, the oxidation state of the nickel ions does not change. Hence, this method is dissimilar to the oxidation of nickel metal or a nickel-based alloy, which is discussed in §3f.
At low Ni(NO3)2 concentrations, the Faradaic efficiency of the deposition is nearly 100%, e.g. [Ni(NO3)2]<0.2 M at 0.5 mA cm−2 [91]. That is, approximately 1 mol of Ni(OH)2 is produced for every 2 mol of hydroxide produced at the electrode surface. At higher concentrations, the deposition efficiency can be much lower, e.g. 20% in 1 M Ni(NO3)2 at 0.5 mA cm−2. This inefficiency in concentrated solutions is attributed to the formation of soluble species, such as [NiOH]+ or [Ni4(OH)4]4+, that diffuse away from the substrate.
In general, deposition currents on the order of a few mA cm−2 yield α-Ni(OH)2, whereas larger currents produce mixed α/β-phase materials [28,137]. However, it is difficult to predict the product phase or crystallinity because of the joint effects of salt concentration, deposition current, temperature, etc. (e.g. cf. [13,138]). Even the nature of the substrate has an influence; for example, deposition onto Fe is more facile than on Ni or Pt electrodes [137]. Several representative electrochemical precipitation methods are listed in table 5.
Table 5.
Representative methods from the literature for electrochemical precipitation of Ni(OH)2 films onto electrically conducting substrates. Depositions are performed galvanostatically (at constant current), except where noted.
| Ni(II) salt and reagents | conc. (mol L−1) | current density (mA cm−2) | deposition time min | temp. (°C) | substrate | product phase | comments | refs |
|---|---|---|---|---|---|---|---|---|
| Ni(NO3)2 | 0.1 | -2.5 | 10 | 22 | nickel sheet | α-Ni(OH)2 | product contains NO | [38] |
| Ni(NO3)2 | 0.005 | -0.1 | 60 | 25 | stainless steel sheet (316L) | α-Ni(OH)2 | product contains NO | [13] |
| produces nanoparticles (approx. 5 nm) | ||||||||
| Ni(NO3)2 | 0.005 | -2.0 | 30 | 40 | stainless steel sheet (316L) | β-Ni(OH)2 | stacking fault disorder and very hydrated | [138] |
| produces nanoparticles (approx. 100 nm) | ||||||||
| NiCl2 | 0.1 | -2.5 | 10 | 22 | nickel sheet | α-Ni(OH)2 | product contains Cl− | [38] |
| NiCl2 | 0.005 | -1.0 | —a | 10 | stainless steel sheet (316L) | β-Ni(OH)2 | stacking fault disorder and very hydrated | [139] |
| produces nanoparticles (less than 5 nm) | ||||||||
| NiSO4 | 0.1 | -2.5 | 2 | 22 | nickel sheet | α-Ni(OH)2 | very highly disordered material | [38] |
| product contains SO | ||||||||
| NiCl2 | 0.01 | -1.0 | 12.5 | 70 | graphite sheet | β-NixCo1−x(OH)2 | x=0.32 | [14] |
| CoCl2 | 0.02 | product contains some α-phase material | ||||||
| NaNO3 | 0.06 | |||||||
| Ni(NO3)2 | 0.3 | -6.25 | 30 | 25 | nickel sheet | α-Ni(OH)2 | product is highly disordered | [35] |
| KNO2 | 0.1 | unclear what the role KNO2 or KNO3 serve | ||||||
| KNO3 | 0.1 | |||||||
| NiCl2 | 0.01 | -1.0 | 12.5 | 70 | graphite sheet | α-Ni(OH)2 | NaNO3 is used to increase solution conductivity | [14] |
| NaNO3 | 0.06 | |||||||
| Ni(NO3)2 | 1.8 | −0.7 VbSCE | — | — | titanium sheet | α-Ni(OH)2 | Mix Brij 56 and aqueous solution, 50% (w/w) | [140] |
| NaNO3 | 0.075 | remove surfactant template at 100°C for 1.5 h | ||||||
| Brij 56 | pure | hexagonal pore array, dp=2.5 nm, dp−p=7.0 nm | ||||||
| Ni(NO3)2 | 0.01 | −1.1 VbAg/AgCl | 1–100 s | 25 | boron-doped diamond | — | nanoparticle size determined by deposition time | [141] |
| (1 s, d=12±3 nm; 15 s, d=39±9 nm) | ||||||||
| longer times result in agglomerates |
a ‘—’ Indicates experimental details that were not provided.
bDeposition performed potentiostatically (at constant potential).
(c). Sol–gel synthesis
The sol–gel method encompasses several related synthetic procedures. These always begin with the preparation of a sol, followed by treatment of the sol to prepare a gel. A sol is a colloidal suspension of very small particles, with a dimension, in at least one direction, between 1 nm and 1 μm, in a liquid [142]. The term ‘gel’ includes several related substances: (i) covalent polymer networks; (ii) polymer networks formed through physical aggregation; (iii) polymer networks formed through glassy junction points (e.g. gels based on block copolymers); (iv) lamellar structures (e.g. soap gels, phospholipids, clays); or (v) particulate disordered structures (e.g. particles with large geometrical anisotropy, such as in V2O5 gels) [142]. Sol–gel synthesis methods vary by how the sol is prepared and how it is treated to produce the gel.
A Ni(OH)2 sol may be prepared from chemically precipitated powder by repeatedly washing it with water [143,144]. This produces a suspension of nanoparticles. A gel may then be formed by network growth as an array of these discrete colloidal particles (e.g. by centrifuging the suspension as shown in figure 8c) [145]. Alternatively, one may form an interconnected, three-dimensional network by the simultaneous hydrolysis and polycondensation of an organometallic precursor. Metal alkoxide precursors are quite common [145]. For example, metathesis (ion exchange) may be used to prepare nickel ethoxide [146]
| 3.4 |
The hydrolysis of the metal alkoxide produces nickel hydroxide
| 3.5 |
The gelation can begin before the hydrolysis is complete (i.e. the sol and the gel steps can be concurrent). The product may be aged and dried under ambient atmosphere to produce a xerogel with high porosity and high specific surface area. Alternatively, the gel may be supercritically dried to produce an aerogel [146]. Aerogels have extremely high porosity and specific surface area [145]. NHN aerogels, which are related to α-Ni(OH)2 (see §2c(iii)), may also be prepared [147].
Sol–gel methods may be used to prepare α- and β-Ni(OH)2 with a variety of densities and porosities either as a bulk solid or a thin film on a substrate. An example of hollow microspheres of β-Ni(OH)2 prepared by Zhang & Zeng using a sol–gel method is shown in figure 9c. Representative sol–gel methods for preparing α- and β-Ni(OH)2 materials are listed in table 6.
Table 6.
Representative methods from the literature for sol–gel preparation of Ni(OH)2 materials. a-PEO, amorphous polyethylene oxide; CTAB, cetyltrimethyl ammonium bromide; DEA, diethanolamine; EDA, 1,2-ethanediamine; FTO, fluorine-doped tin oxide; OAc, [O2CCH3]− (acetate); OEt, [OCH2CH3]− (ethoxide); PVA, polyvinyl alcohol; SDS, sodium dodecylsulfate.
| Ni(II) salt | solvent(s) and reagents | method | product phase | comments | refs |
|---|---|---|---|---|---|
| NiCl2 | OC(NH2)2 | stir reactants at room temperature for 2 h | α-Ni(OH)2 | product contains some β-Ni(OH)2 | [148] |
| SDS | heat at 80°C for 6 h | yields nanoparticle (approx. 250 nm) xerogel | |||
| H2O | centrifuge to obtain product | product is mesoporous (dpore=3.6 nm) | |||
| dry overnight at 100°C | |||||
| NiCl2 | NaOEt | stir NiCl2, NaOEt and HOEt at 70–80°C for 3–5 h | α-Ni(OH)2 | dried with supercritical CO2 to yield an aerogel | [146] |
| HOEt | add aqueous NH4OH to obtain a sol | ||||
| NH4OH | leave overnight to obtain a hydrogel | ||||
| H2O | |||||
| NiCl2 | ethylene glycol | stir NiCl2 in ethylene glycol and n-butanol 30 min | α-Ni(OH)2 | film contains NiO | [21] |
| n-butanol | add HOAc and reflux at 70°C for 3 h | yields nanoparticle (d∼30 nm) xerogel | |||
| HOAc | add H2O and a-PEO/DEA then reflux for 1 h | ||||
| H2O | cool to room temperature to obtain sol | ||||
| a-PEO | dip-coat onto SnO2 glass | ||||
| DEA | dry at 100°C for 10–30 min | ||||
| NiCl2 | EtOH | stir NiCl2 with water and EtOH | β-Ni(OH)2 | stacking fault disorder | [149] |
| N2H4 | stir N2H4 and NaOH in water | yields clusters of nanoparticles (d∼65 nm) | |||
| NaOH | add second solution to first dropwise until pH≈10 | ||||
| centrifuge the sol to obtain a gel (powder) | |||||
| Ni(NO3)2 | H2O | stir Ni(NO3) and NH4OH, in water | β-Ni(OH)2 | Some stacking fault disorder | [150] |
| NH4OH | stir 1 h at 70°C | yields nanoparticle (d = 50–80 nm) xerogel | |||
| centrifuge, rinse and dry at 80°C for 12 h | |||||
| Ni(NO3)2 | H2O | stir Ni(NO3), NH4OH and PEG-400 in water | β-Ni(OH)2 | stacking fault disorder | [150] |
| NH4OH | stir 1 h at 70°C | yields nanoflakes (d=20–60 nm) and needles (d=10–20 nm, l=100–200 nm) | |||
| PEG-400 | centrifuge, rinse and dry at 80°C for 12 h | ||||
| Ni(NO3)2 | H2O | stir Ni(NO3), NH4OH and Tween-80 in water | β-Ni(OH)2 | stacking fault and hydration disorder | [150] |
| NH4OH | stir 1 h at 70°C | low crystallinity | |||
| Tween-80 | centrifuge, rinse and dry at 80°C for 12 h | yields needles (d∼10 nm, l≈100 nm) | |||
| Ni(NO3)2 | OC(NH2)2H2O | stir Ni(NO3)2 and urea in water | α-Ni(OH)2 | very low crystallinity | [151] |
| H2O | add maleic acid and stir 10 min to obtain sol | yields nanoparticle (d∼500 nm) xerogel | |||
| maleic acid | heat at 100°C for 24 h | product is mesoporous (dpore=16 nm) | |||
| centrifuge to obtain product | |||||
| Ni(NO3)2 | NaOH | stir Ni(NO3)2, NaOH and EDA in water | β-Ni(OH)2 | yields hollow, ‘dandelion-like’ microspheres | [116] |
| H2O | Dry at 100°C for 0–3 h | (d∼5 μm) composed of nanosheets | |||
| EDA | hydration disorder | ||||
| NiSO4 | H2O | combine NiSO4 and NaOH solutions | β-Ni(OH)2 | stacking fault disorder | [152] |
| NaOH | centrifuge the sol to obtain a gel and dry at 80°C | yields nanoparticle (d∼10 nm) xerogel | |||
| NiSO4 | H2O | stir all reagents except KOH in water | β-Ni(OH)2 | high surface area, mesoporous xerogel | [153] |
| sodium citrate | add KOH solution dropwise | ||||
| NaOAc | centrifuge, rinse and dry at 80°C | ||||
| KOH | |||||
| NiSO4 | CTAB | dissolve NiSO4 in CTAB, butanol, cyclohexane | α/β-Ni(OH)2 | mostly, the α-phase materials is produced | [154] |
| n-butanol | add NaOH solution dropwise | product contains SO | |||
| cyclohexane | transfer to ice water | ||||
| NaOH | centrifuge, rinse and dry | ||||
| NiSO4 | H2O | combine Ni(II) and LiOH solutions | α-Ni(OH)2 | film contains SO and CO foreign anions | [155] |
| LiOH | wash precipitate with water | ||||
| HOAc | acidify with acetic acid | ||||
| formamide | stir with formamide, glycerol, PVA | ||||
| glycerol | wet substrate with Teloksid A-30 | ||||
| PVA | dip-coat onto Pt, CdTe or Si substrate | ||||
| Teloksid A-30 | |||||
| Ni(OAc)2 | H2O | stir Ni(OAc)2 and water for 2 days | β-Ni(OH)2 | film may contain OAc− foreign anions | [156] |
| dip-coat onto Ni foil substrate | hydration disorder | ||||
| film was dried to yield NiO xerogels | |||||
| Ni(OAc)2 | glycerine | stir all reagents except water | unknown | clear gelatinous mass | [143] |
| KOH | let stand several days | ||||
| HOEt | dilute with water then partially dry | ||||
| H2O | |||||
| Ni(OAc)2 | glycerine | stir reagents | α-Ni(OH)2 | Ref. also prepared Ni1−xCox(OH)2, 0≤x≤0.5 | [157] |
| KOH | spin- or dip-coat onto FTO glass | ||||
| n-butanol | |||||
| Ni(OAc)2 | glycerine | spread sol onto glass, quartz, FTO- or Pt-coated Si substrate α-Ni(OH)2 | α-Ni(OH)2 | yields nanoparticles (d<10 nm) | [158] |
| KOH | dry under vacuum then remove glycerine at 240°C | ||||
| n-butanol |
(d). Chemical ageing
It has long been known that α-Ni(OH)2 spontaneously transforms to β-Ni(OH)2 in aqueous KOH [4]. This process is widely referred to as chemical ageing, or simply ageing, and is typically performed at high temperatures (e.g. 70–80°C) in concentrated alkaline media (e.g. 5 M, 30% KOH) [1,159]. The ageing process has also been studied at room temperature (22–25°C) in the mother liquor after chemical precipitation [160], in concentrated alkaline solutions [35,85] and in pure water [94,161]. These studies support the widespread belief that ageing occurs faster at higher temperatures. Furthermore, one may conclude by comparing [35,85,94] that ageing proceeds faster in concentrated alkaline media (7 M–8 M KOH, 25°C) than in pure water (18.2 MΩ cm, 22°C). β-Ni(OH)2 that is newly formed from α-Ni(OH)2 usually has stacking fault disorder. Through further chemical ageing, the stacking faults are slowly removed and the overall crystallinity increases [35,85,120,162]. Tessier et al. [85] found that chemically ageing β-Ni1−xZnx(OH)2 both increases the structural order and gradually leaches the Zn from the material. It is noted that β-Ni(OH)2 with superior crystallinity may be prepared by hydrothermal recrystallization ([43], see §3e) or via the formation and subsequent decomposition of Ni(NH3)6(OH)2 ([35], see §3f).
Chemical ageing has long been proposed to occur via a dissolution-precipitation mechanism. At sufficiently high pH, this process may be facilitated by [Ni(OH)3]− ions [1,85,121]. As a result, chemical ageing has also been referred to as ‘ripening’, in that the unstable α-Ni(OH)2 particles slowly dissolve and the thermodynamically favoured β-Ni(OH)2 particles slowly grow. This is somewhat analogous to Ostwald ripening in solids and sols, for example, in which a dissolution-precipitation process leads to the formation of larger particle sizes. However, the solubility of Ni(OH)2 is very low in neutral and slightly alkaline media (7<pH<14), which would make such a mechanism improbable under milder conditions. In particular, this mechanism does not explain the spontaneous transformation of α- to β-Ni(OH)2 in pure water at room temperature [94,161]. Chemical ageing may alternatively be considered analogous to a zipper with hydroxide as the teeth, supported on nickel backbones (figure 10) [94]. The water molecule next to the α/β-boundary (dark blue circle) blocks the opposing hydroxyl groups from ‘zipping’ into place, analogous to the zipper's slider (i.e. the part one pulls to operate a zipper). As the water is removed, the molecules move stochastically (to the right in figure 10), which allows the hydroxide ‘teeth’ to interdigitate (i.e. to zip closed). The ageing process actually involves stacked, two-dimensional sheets of Ni(OH)2 and, hence, a full representation of the process would be considerably more complex. Nevertheless, this simple model provides informative predictions. The process is apparently mass-transfer limited, which may explain why ageing is faster at higher temperatures [94]. Moreover, it predicts an additional water vibrational mode, corresponding to the ‘slider’ molecule, and red- and blue-shifts of the β- and α-phase O−H stretching modes from tensile and compressive forces, respectively (figure 10). These have all been observed in the Raman spectra of partially aged samples [94]. However, this model suffers the major setback that it does not explain the observed kinetic effect of using an alkaline ageing solution. Therefore, it is possible that the two pathways compete kinetically, with dissolution-precipitation favoured in concentrated alkaline solutions and the ‘zipper’ model favoured in lower pH conditions. The rate of both mechanisms is predicted to increase at elevated temperatures.
Figure 10.
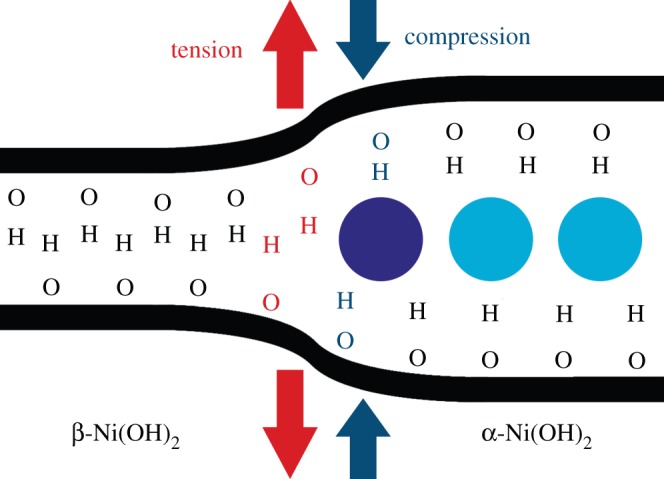
Representative schematic of the boundary between the α- and β-Ni(OH)2 phases during chemical ageing showing the mechanical forces near the α/β boundary. These forces correspond to observed blue- and red-shifts in the frequencies of various lines in the Raman spectra of partially aged samples. Figure reproduced with permission from [94]. (Online version in colour.)
Chemical ageing also removes intercalated species, such as nitrate or chloride anions, from the interlayer region. Raman spectroscopy measurements show that the removal of intercalated nitrate anions is much faster than the dehydration process [94,159]. This may be due to mechanical stresses from the size mismatch of the incorporated nitrate anions.
The term ‘ageing’ also refers to the transformation of α-Ni(OH)2 electrode materials to β-Ni(OH)2 via prolonged electrochemical cycling between Ni(OH)2 and NiOOH [71,85,163–167]. In the field of rechargeable battery anodes, this transformation is often accompanied by decreased electrode conductivity and charge capacity [6]. In order to obtain more consistent charge–discharge cycling, commercial batteries use β-Ni(OH)2 anodes [6].
(e). Hydrothermal and solvothermal synthesis
Hydrothermal (aqueous) and solvothermal (non-aqueous) synthesis methods are represented in figure 8d. The precursor materials and the solvent are placed in a pressure vessel and then the reaction mixture is heated. Typical reaction temperatures for Ni(OH)2 synthesis are 150–200°C, well above the boiling point of water. Because the vessel has a fixed volume, as the solution is heated, the pressure increases. Therefore, the boiling point increases and the solution remains liquid at such high temperatures that the precursors fully dissolve [168]. The scale of this method is limited by the size of the pressure vessel. In the laboratory, this is usually in the range of hundreds of millilitres to a few litres. Larger pressure vessels, on the order of hundreds of litres, are commonly used for industrial applications. Therefore, hydrothermal and solvothermal methods may be used for large-scale production of Ni(OH)2 materials.
In general, the thermodynamic product, β-Ni(OH)2, tends to form at the high temperatures used in hydrothermal synthesis. However, it is possible to produce α-Ni(OH)2 materials by introducing an intercalation agent into the reaction mixture [169], by using a non-aqueous solution [170], by limiting the reaction time or by using a low reaction temperature [118]. For example, the addition of urea to a reaction mixture results in the formation of NH3-intercalated α-Ni(OH)2 [169].
Hydrothermal and solvothermal syntheses may be used to produce Ni(OH)2 nanoscale materials with unique morphologies. For example, one may prepare micrometre-scale ‘nanoflower’ aggregates composed of nanosheets (figure 9b) [17,22,170]. The morphology is maintained when the nanoflowers are calcinated, to form NiO [22], or chemically aged, to form crystalline β-Ni(OH)2 [17]. However, prolonged ageing can flatten the nanoflowers. One can also produce ‘nanoribbons’ or ‘nanobelts’ (figure 9e). Several representative literature methods are summarized in table 7.
Table 7.
Representative methods from the literature for hydrothermal and solvothermal preparation of Ni(OH)2 materials. CG, cyanoguanidine; CNT, carbon nanotube; CTAB, cetyltrimethyl ammonium bromide; DEG, diethylene glycol; EG, ethylene glycol; GO, graphene oxide; OAc, acetate (H3CCO); PSS, sodium poly(4-styrenesulfonate).
| precursors | solvent | temperature (°C) | time h | product phase | comments | refs |
|---|---|---|---|---|---|---|
| α-Ni(OH)2 | H2O | 150–200 | —a | β-Ni(OH)2 | very crystalline product | [43] |
| α-Ni(OH)2 | H2O | 200 | — | β-Ni(OH)2 | yields ‘non-porous, hexagonal thin platelets’ | [127] |
| Ni(NO3)2 | H2O | 180 | 18 | β-Ni(OH)2 | yields nanoflowers (dflower≈10–15 μm) composed of nanosheets approximately 15 nm thick some stacking fault disorder | [22] |
| CG | ||||||
| NiCl2 | H2O | 180 | 6 | β-Ni(OH)2 | hollow microspheres (d≈2 μm) | [171] |
| PSS | ||||||
| NH4OH | ||||||
| further treatment yields ‘hollow microspheres wrapped in graphene oxide’ | ||||||
| NiOAc | H2O | 200 | 5–30 | β-Ni(OH)2 | nanosheets (d≈200–300 nm, h≈12–20 nm) | [172] |
| NH4OH | ||||||
| 5 h: hexagonal sheets; 30 h: round sheets | ||||||
| good crystallinity (some stacking fault disorder) | ||||||
| NiSO4 | H2O | 120–200 | 24 | α/β-Ni(OH)2 | studied the effect of reactant ratio and temp. on product morphology and phase | [118] |
| NaOH | ||||||
| α- ‘nanobelts’/nanoribbons (d=10–20 nm) or nanowires (d=20–30 nm, l=1–3 μm) | ||||||
| β- hexagonal or rectangular ‘nanoplates’/ nanosheets | ||||||
| Ni(NO3)2 | H2O | 120 | 1.7 | α-Ni(OH)2 | contains intercalated NH3 | [169] |
| OC(NH2)2 | ‘Fibrillar’ nanoscale morphology | |||||
| Ni(NO3)2 | H2O | 200 | 24 | β-Ni(OH)2 | nanosheets (d≈100–120 nm) | [173] |
| NH4OH | CH3OH | stacking fault and hydration disorder | ||||
| NiOAc | H2O | 200 | 1–10.5 | α/β-Ni(OH)2 | nanosheets and nanoflowers | [170] |
| EG | α-Ni(OH)2 forms at shorter times and higher EG concentrations | |||||
| crystallinity increases with reaction time stacking fault disorder | ||||||
| NiCl2 | EG | 170 | 1–4 | β-Ni(OH)2 | porous network of nanoparticles | [174] |
| CTAB | ||||||
| NaBH4 | ||||||
| particle and pore sizes depend on temp and time | ||||||
| product contains Ni metal and β-Ni(OH)2 | ||||||
| highly disordered material | ||||||
| Ni foam | H2O | 150 | 6 | β-Ni(OH)2 | nanoflakes form on the Ni foam substrate | [175] |
| H2O2 | ||||||
| NaOH | ||||||
| stacking fault disorder | ||||||
| NiCl2 | DEG | 180 | 10 | α-Ni(OH)2 | yields nanoflowers (dflower≈400 nm) | [17] |
| NaOAc | ||||||
| may be prepared with GO and CNTs | ||||||
| α-Ni(OH)2 nanoflowers | H2O | 180 | 1 | β-Ni(OH)2 | maintains nanoflower morphology | [17] |
| (from prev. method) | ||||||
| stacking fault disorder | ||||||
| 10 | β-Ni(OH)2 | nanoflowers become flattened | ||||
| less stacking fault disorder than after 1 h |
a ‘—’ Indicates experimental details that were not provided.
(f). Surface layers on nickel
An air-formed oxide layer, 6–8 Åthick, rapidly forms on Ni metal that has been mechanically or electro-polished. Ellipsometry measurements [176] and angle-resolved X-ray photoelectron spectroscopy (XPS) [177] show that this is a bilayer, with a Ni(OH)2 layer underlaid by NiO. The underlayer is non-stoichiometric NiOx, x≥1, with approximately 2% lattice parameter expansion [178]. On the basis of the XPS peak shapes and positions, the hydroxide layer is assessed to be α-Ni(OH)2 [179]. Ni(OH)2 is also a common component of the passive layers and corrosion deposits on Ni metal [176,180,181] and Ni-containing alloys [182,183] that form in alkaline (e.g. [176]) and neutral media (e.g. [183]).
Ni(OH)2 also forms electrochemically on Ni electrodes in alkaline media, which has importance for the development of Ni-based electrocatalysts [23,105,179,184–187]. Consequently, the surface electrochemistry has been extensively studied using atomic force microscopy [188], ellipsometry [181,189–191], IR spectroscopy [192], Raman spectroscopy [166,193,194], UV–Vis spectroscopy [192,195], voltammetry [179,185,196–201], X-ray scattering [176], XPS [179,202] and gravimetry with an electrochemical quartz crystal microbalance [203,204]. During a forward voltammetric sweep, a surface bilayer of α-Ni(OH)2 underlaid by NiOx forms initially [179]. This corresponds to a voltammetric peak at approximately 0.3 V versus the reversible hydrogen electrode (RHE). At higher potentials, the surface layer thickens and some β-Ni(OH)2 forms. It is unknown whether the β-Ni(OH)2 is formed electrochemically or by the ageing of the α-Ni(OH)2. The relative amount of β-Ni(OH)2 tends to be greater at slower potential scan rates.
The voltammetric peak at approximately 0.3 V versus RHE has been used to estimate the surface area of nickel electrodes [197]. The method approximates that the peak corresponds to the formation of a monolayer consisting entirely of α-Ni(OH)2. Recently, this method has been refined by adding an oxalate salt to the electrolyte, which is proposed to limit the surface hydroxide to a single layer [205]. Moreover, to avoid the problem of the concurrent formation of NiOx, this new method instead measures the Ni(OH)2/NiOOH redox pair.
(g). Other preparation methods
Several remaining methods to prepare Ni(OH)2 materials that do not fall neatly into the six previous categories are briefly presented here. Presently, these methods represent only a small portion of the vast literature on Ni(OH)2 materials. However, their adoption and usage may well increase with time. As with the methods in §§3a–3e, each method uses a nickel source, already in the nickel(II) oxidation state, and a source of hydroxide anions.
(i). Electrodialysis
The electrodialysis method is related to electrochemical precipitation. The method uses a two-electrode electrochemical cell with two compartments that are separated by an anionic membrane. When a current is applied, Ni2+ cations are generated by the oxidative dissolution of the Ni metal anode and OH− anions are produced by the reduction of water molecules at the cathode. The Ni(OH)2 deposits onto the anode side of the membrane [206]. The product is a mixture of α- and β-Ni(OH)2 [162]. The α-phase component contains incorporated anions and the β-phase component has considerable stacking fault disorder.
(ii). Microwave-assisted synthesis
Ni(OH)2 may be prepared by microwave-assisted synthesis, in which a microwave oven is used to heat a solution more homogeneously than conventional heating [93,119,121,207,208]. Microwave-assisted methods produce very uniform nanoparticle shapes and narrow size distributions, but otherwise mirror the results of homogeneous precipitation or hydrothermal synthesis. For example, Soler-Illia et al. [121] used microwave heating and aged α-Ni(OH)2 samples at 150°C, in a pressure vessel, to produce β-Ni(OH)2. Microwave heating may be used to produce unique nanoscale morphology. For example, Ni(NO3)2 and urea dissolved in water and ethanol and heated in a microwave oven at 90°C for 15 min produces nanoflowers (d=0.7–1.0 μm) composed of nanoflakes [93]. The flakes are, in turn, composed of nanocrystals d=2–3 nm). In another example, a Ni(NO3)2, urea, NH4OH and PEG-20 000 solution placed in a pressure vessel and heated in a microwave oven at 160°C for 30 min produces nanoflowers (d=2.5–4.0 μm) composed of nanosheets 10–20 nm thick and 0.2–1.5 μm wide (figure 9f) [119]. The XRD pattern contains peaks from β-Ni(OH)2. Several other peaks show a material with c=6.2 Å , which may be ascribed to an NHN component (see §2c(iii)) on the basis of the d-spacing (cα≥7.8 Å, cNHN≤7.6 Å) and the absence of any clear α-Ni(OH)2 modes in the Fourier-transform infrared spectroscopy (FTIR) spectra.
(iii). Sonochemistry
Sonochemical methods, which use the application of powerful ultrasound radiation (20 kHz–10 MHz) to drive chemical reactions, may be used to prepare Ni(OH)2 materials with unique nanostructures [55,67,83,87,209]. In general, sonochemistry has significant advantages for the preparation amorphous products, the insertion of nanoscale materials into mesoporous materials, the deposition of nanoparticles on ceramic and polymeric surfaces, and the formation of proteinaceous micro- and nanospheres [210]. For example, the application of 20 kHz ultrasound to a solution of Ni(NO3)2 and urea produces very disordered, nanoscale α-Ni(OH)2 fibres of approximately 200 nm long and 15 nm wide [55]. The application of 20 kHz ultrasound at a lower power to a solution of Ni(NO3)2 and NH4OH produces nanoparticles of nitrate-intercalated α-Ni(OH)2 (d≈5 nm) [209]. One may similarly prepare α-Ni1−x−yCoxCdy(OH)2 by adding Co(NO3)2 and/or Cd(NO3)2 to the reaction flask during the synthesis.
(iv). Solid-state synthesis
Liu & Yu [211] report a two-step solid-state method for preparing β-Ni(OH)2 at room temperature. First, Ni(CH3CO2) (nickel acetate) and H2C2O4 (oxalic acid) are ground together in a 1 : 1 molar ratio with a mortar. The resulting NiC2O4 (nickel oxalate) is similarly ground together with NaOH in a 1 : 2 molar ratio. The green powder is rinsed with water and dried at 80°C to obtain β-Ni(OH)2 with stacking fault and general structural disorder. The product is composed of clusters of nanoscale fibres (d=3–9 nm, l=10–50 nm).
(v). Purification by ‘recrystallization’
Barnard et al. [35] report a unique method of β-Ni(OH)2 purification. A Ni(OH)2 precursor is dissolved in an 8 M NH3 aqueous solution. This resulting blue solution of Ni(NH3)6(OH)2 is then added to concentrated H2SO4 and stored in a desiccator for several days. The NH3 is gradually removed from the material and green, glassy flakes of β-Ni(OH)2 are left behind. The authors describe their process as recrystallization, although it does not fit the conventional meaning of the term. Barnard et al.'s method yields very high purity, highly crystalline β-Ni(OH)2 with minimal stacking fault disorder. However, a slightly more crystalline product may be prepared by hydrothermal recrystallization [43].
4. Physical properties and analytical characterization
So far, this review has focused primarily on the chemical and structural properties of the various forms of nickel hydroxide. However, this material system possesses many interesting physical properties, such as in electricity and magnetism, that have potential applications and others, such as vibrational and optical spectroscopy, including XPS, that can be very useful in characterizing nickel hydroxide materials for research and developmental investigations. In this section, we provide an overview of the large amount of work on the physical properties of Ni(OH)2. We also highlight how this knowledge can be usefully applied in characterizing ‘unknown’ materials. In general, we find that vibrational spectroscopy is perhaps the quickest and simplest method for the identification and characterization of α- and β-Ni(OH)2, either as bulk materials or thin films. Moreover, it is evident that the stable β form of Ni(OH)2 is by now well characterized, but that α-Ni(OH)2 still requires a lot more work. Such characterization work is made particularly difficult by the multitudinous forms of α-Ni(OH)2, owing to its inherent turbostratic disorder, its tendency to incorporate variable amounts of water and/or impurities and its poor long-term stability.
(a). Magnetic properties
(i). Bulk magnetic properties
Compounds containing Ni2+ ions typically exhibit magnetism owing to the particular electronic properties of the transition metal ions. The layered structure of Ni(OH)2, and β-Ni(OH)2 in particular, is analogous or identical to that of many transition metal halides including for example NiCl2, all of which exhibit ferromagnetism within their layers of metal ions in the crystallographic a–b plane, with their parallel electron spins aligned either along or perpendicular to the crystallographic c-axis, but overall they form antiferromagnets with the spins in one layer pointing in the opposite direction to those in the adjacent layers [212]. This type of layered-structure magnetism is classified as being metamagnetic, because the external magnetic field-induced transition to the paramagnetic phase can occur without the existence of an intermediate spin-flop phase.
The early reports on the magnetic properties of β-Ni(OH)2 showed that at low temperatures it was, indeed, an antiferromagnet with the magnetic ordering occurring at a Néel temperature TN of 25.8 K [26,213,214]. The magnetic unit cell shown in figure 11 is twice as long along the c-axis as the chemical unit cell [26] and the magnetic moments in each a–b plane are aligned parallel or antiparallel to the c-axis [213]. Thus, β-Ni(OH)2 forms a quasi-two-dimensional antiferromagnet with a strong ferromagnetic intralayer exchange interaction (labelled J1 in figure 11) and two different weak antiferromagnetic interlayer exchange interactions (labelled J2 and J3 in figure 11). These three different exchange interactions between the pairs of Ni ions at different separations are mediated by the different arrangements of adjacent oxygen ions [26] and have the values J1/k=+2.70 K, J2/k=−0.28 K and J3/k=−0.09 K [213] where the plus (minus) sign indicates ferromagnetic (antiferromagnetic) exchange. Theoretical calculations from first principles methods based on density functional theory [215] confirm that the magnetic ground state of β-Ni(OH)2 is the A-type antiferromagnetic one, i.e. the a–b plane Ni layers are ordered ferromagnetically and with antiferromagnetic coupling between layers along the c-axis [26]. Using a high-spin (S=1) state on the Ni atoms, Hermet et al. calculated the ferromagnetic exchange constant J1/k to be 2.46 K, which is in good agreement with the experimental value [213] given above. They calculated the Ni atom magnetic moment to be 1.8 μB (the Bohr magneton), also in good agreement with experiment [26] (μ=2.0±0.2 μB).
Figure 11.
The magnetic structure of β-Ni(OH)2 showing the three principal exchange interactions J1, J2 and J3, and the hexagonal arrangement of spins within the a–b plane. The spin directions within each plane are shown by the arrows aligned along the c-axis at the site of each Ni atom (small (grey) spheres). The O and H atoms are denoted by large (red) and small (pink) spheres, respectively. Copyright this work. (Online version in colour.)
Hermet et al. [215] remeasured the magnetic susceptibility of β-Ni(OH)2 using a large pseudo-single crystal containing a low defect density. From the peak in the magnetic susceptibility they found TN = 26 K (compared with 30 K in [214]) in agreement with other kinds of measurements of TN [26,213] and observed a Curie–Weiss behaviour for temperatures above 150 K. They determined a Curie temperature of −22.3 K and a Curie constant of 1.15 emu K mol−1, corresponding to an effective paramagnetic moment of 3.04 μB consistent with earlier reports [216] and with the expected value for a high-spin Ni ion. Specific heat measurements [213] show that β-Ni(OH)2 exhibits a crossover from two-dimensional Ising behaviour to a three-dimensional Ising one with temperature approaching TN.
In contrast to β-Ni(OH)2 (referred to simply as Ni(OH)2 in many of the early papers), very little research has been carried out into the magnetic properties of α-Ni(OH)2. We know of only the study by Rall et al. [217,218] on α-Ni(OH)2⋅3.3H2O in the form of flowers with a petal thickness of approximately 5 nm and with lattice constants of a=3.0 Åand c=8.6 Å. Comparative studies of the magnetic susceptibility of powdered samples of α-Ni(OH)2 and β-Ni(OH)2 yielded maxima at 13–16 K and 25 K, respectively, and a 40 times greater value for the field-cooled susceptibility in α-Ni(OH)2 versus that in β-Ni(OH)2, indicating differences in their magnetic ordering. A Curie–Weiss behaviour was observed above the transition temperature for α-Ni(OH)2 (β-Ni(OH)2), giving a Curie temperature of 35 K (20.5 K) and a Curie constant of 87.4 (112) 10−4 emu K g−1 Oe−1, corresponding to an effective paramagnetic moment of 3.13 (2.95) μB and high-spin S=1.00 (0.93). The magnetic field dependence of the magnetization for α-Ni(OH)2 at 2 K was found to be characteristic of a ferromagnetic phase. Rall et al. concluded that turbostratic α-Ni(OH)2 was a hard ferromagnet below TC≈13 K as a consequence of the large separation between a and b plane layers of Ni ions considerably reducing any potential interplanar antiferromagnetic interaction. This conclusion was confirmed from a study of nickel hydroxide layers spaced even further apart (c=3.05 nm) than in α-Ni(OH)2 by intercalation with dodecyl sulfate [219]. Again, ferromagnetic ordering is observed with a TC of 23 K and such intercalated systems are essentially two-dimensional Ising-like ferromagnets with the value of TC mainly determined by the in-plane exchange interaction (see [219,220] and references therein).
Recently, studies of the magnetic properties of β-Ni(OH)2 nanoparticles have been reported [152,221,222]. Liu et al. [152] found that just as in the bulk material there is a transition from paramagnetism to antiferromagnetism in the nanoparticles, but it occurs at a lower temperature (22 K for 10 nm size nanoparticles) than in the bulk. Tiwari & Rajeev [222] reported an even lower transition temperature of 18 K for their approximately 8 nm size nanoparticles of arbitrary shapes and sizes. Rall et al. [221] compared the magnetic properties of samples with a nanoplate morphology (33 nm across and 4.3 nm thick) with those of bulk-like β-Ni(OH)2. Nanosize effects are again shown to lower TN and from the field dependence of the magnetization are also found to introduce a two-step mechanism to produce ferromagnetism with the first transition at 28 kOe owing to flipping of the Ni ion spins at the nanoplate surface and the second at 55 kOe. For comparison, the field-induced transition to ferromagnetism occurs at 53.5 kOe in bulk material.
(ii). Nuclear magnetic resonance spectroscopy
There is very little available information on the use of nuclear magnetic resonance (NMR) spectroscopy to characterize the nickel hydroxides. There are no known reports that used NMR to study β-Ni(OH)2.
Proton (1H) NMR spectra of α-Ni(OH)2 materials contain two distinct peaks. The first is a broad absorption line that is unchanged by thermal dehydration, which arises from hydroxyl groups bonded to the nickel atoms [1]. The chemical shifts that are measured by NMR spectroscopy arise from the shielding interaction, σ, which is a rank 2 tensor, i.e. a three-dimensional value. Hence, NMR chemical shifts measured from anisotropic solid materials are orientation-dependent and NMR peaks measured from polycrystalline materials are generally broad. Hence, the observed breadth of this first hydrogen peak is expected. The second chemical environment yields a narrow absorption line that decreases when samples are dehydrated by heating [1]. This second environment is consistent with loosely bound intersheet water molecules. The free rotation of these water molecules causes σ to adopt an averaged isotropic value.
(b). Vibrational properties
The vibrational properties of chemical compounds are unique to each compound and thus form ‘fingerprints’ that may be used for identification and analytical characterization. The principal techniques for measuring vibrational spectra are optical ones, including IR spectroscopy, conventional Raman (inelastic light scattering) spectroscopy and surface-enhanced Raman spectroscopy (SERS) [223,224]. These techniques have the advantage of ease of use and wide availability, but are generally restricted to measuring optical modes of vibration at zero-wave vector in solids. The activities of vibrations in these techniques are governed by symmetry considerations, which are discussed below. Inelastic neutron scattering (INS), on the other hand, allows measurement of all acoustic and optical vibrations (or phonons) at wave vectors throughout the Brillouin zone [225] at the disadvantage of being a specialized technique available at only a few locations worldwide.
(i). Vibrational modes of β-Ni(OH)2
Factor group analysis [223] can be used to ascertain the vibrational modes of a crystalline solid from its space group symmetry and atomic positions within the primitive unit cell. From the D3d symmetry of β-Ni(OH)2, the factor group theory when performed at the Brillouin zone centre predicts four Raman-active vibrations with even parity, two Eg and two A1g modes, and four IR-active vibrations with odd parity, two Eu and two A2u modes [226]. All optical modes are either IR- or Raman-active, i.e. there are no silent modes. This means that both techniques are required to identify all the vibrational modes. Group theory also predicts the displacements of the atoms in each of these so-called normal modes, which allows a further subdivision of them into lattice and internal (molecular-like) modes of vibration [226].
Raman-active vibrational modes of β-Ni(OH)2. The Raman spectrum of β-Ni(OH)2 is shown in figure 12a. Factor group theory predicts four Raman-active transitions [226]. It is well established that three of these modes are at 310–318, 445–453 and 3581 cm−1 (figure 13). The symmetry of these transitions has been disputed, however, because the position of the fourth mode had not been clearly determined until recently [38,228]. However, it is worth noting that the Raman-active lattice modes of brucite [Mg(OH)2], which is isomorphic with β-Ni(OH)2, are at similar positions: 280, 443, 725 and 3652 cm−1 [229]. A detailed theoretical analysis of brucite vibrational modes revealed the atomic displacements corresponding to each mode and thus allowed symmetry assignments of the observed Raman features [226]. Thus, by analogy with brucite, a Raman peak measured at approximately 880 cm−1 [38] has been ascribed to the final lattice mode of β-Ni(OH)2 and the symmetries of the four Raman-active transitions were assigned.
Figure 12.
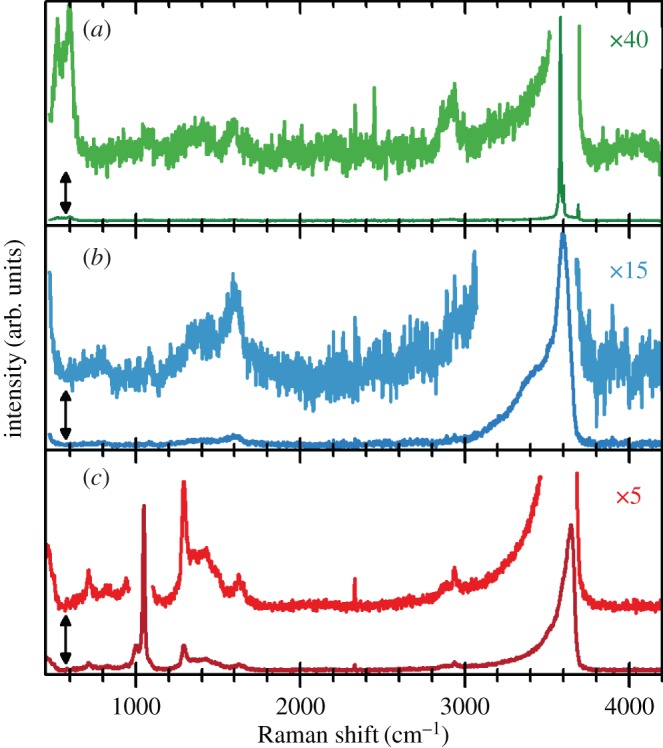
Raman spectra of (a) β-Ni(OH)2, (b) α-Ni(OH)2 and (c) nitrate-intercalated α-Ni(OH)2. Plots are enlarged and offset to show low intensity features. Copyright this work. (Online version in colour.)
Figure 13.
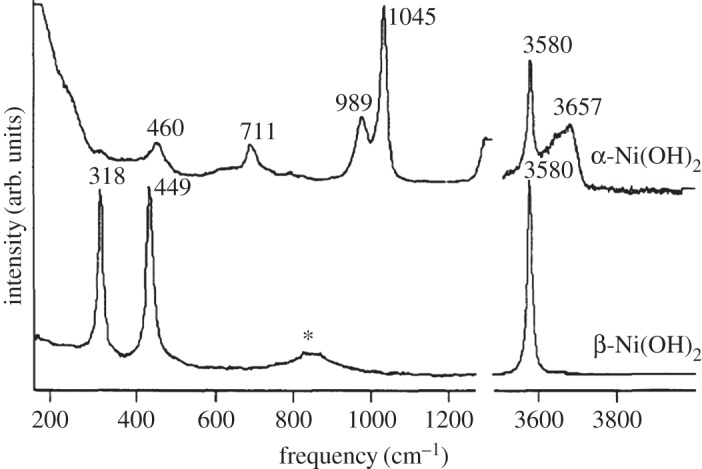
Raman spectra of nitrate-intercalated α-Ni(OH)2 and β-Ni(OH)2 showing lower frequency modes. The α-phase sample also contained some β-Ni(OH)2, as evidenced from the strong peak at 3580 cm−1, owing to the difficulty of preparing high purity α-Ni(OH)2. The peaks annotated with an asterisk (*) are spurious peaks arising from the instrument objective. Reproduced with permission from Johnston C & Graves PR, 1990 [227], In situ Raman spectroscopy study of the nickel oxyhydroxide electrode (NOE) system. Appl. Spectrosc. 44, 105–115.
In addition to the four principal Raman peaks, there are several additional modes that correspond to combinations, overtones and stacking fault disorder [38]. Combination modes and harmonic overtones are formally forbidden under the quantum selection rules [230]. However, these rules tend to relax when there is general structural disorder, because the symmetry of the crystal deviates from the ideal case. Thus, second-order modes are typically very weak and increase in intensity for disordered samples. Consequently, these additional modes are useful for identifying structural disorder in a sample. Because β-Ni(OH)2 has an inversion centre, the vibrational modes are designated as even (subscript g for the German ‘gerade’) or odd (subscript u for ‘ungerade’). Generally, even modes are Raman-active and odd modes are IR-active, which also applies for second-order modes. However, this rule is also relaxed if there is sufficient structural disorder.
A Raman band at 508–519 cm−1 is sometimes reported. The intensity of this feature varies between samples and is generally more intense for less crystalline samples [228]. Although it has sometimes been ascribed to a lattice mode, the frequency is much lower than expected for the Eg mode [226] and its intensity can vary independently of the other lattice modes [42]. Therefore, this band is assigned to the harmonic overtone of an acoustic vibrational mode at approximately 250–270 cm−1 that is present in INS measurements [38,228].
A very weak Raman band is observed at 601 cm−1 that is not a predicted lattice mode. This peak is taken to be the harmonic overtone of the Eg transition at 306–318 cm−1 [38].
At intermediate frequencies, there is a very weak feature at approximately 1600 cm−1 superimposed with a weak peak at approximately 1630 cm−1. The latter is ascribed to the O−H bend of H2O that has adsorbed on the material surface or has been trapped within the material. The former is similar in shape to free water but is in the wrong position. Thus, this is taken to be the O−H bend for a small degree of hydration in the structure (formally termed a libration mode for lattice species [226]). A small amount of water is often present in β-Ni(OH)2 and may improve the electrochemical activity for battery electrodes [46,47]. The water is weakly associated with the nickel cations and does not form hydrogen bonds with the lattice hydroxide [6,47–49].
A sharp band at 3601 cm−1 is often present in measured Raman spectra. This peak has previously been variously assigned to adsorbed H2O, crystal defects, impurities, surface OH groups and the O−H stretch for a new crystal phase, βbc-Ni(OH)2 [52,159,228]. However, it is also associated with selective XRD line broadening, which affects only the diffraction peaks that involve the crystallographic c-axis [38,52]. Because selective line broadening is not a crystallite size effect, the Scherrer equation should not be applied. Rather, this peak indicates the presence of stacking fault disorder (see §2b(ii)). The presence of this peak has been associated with superior electrochemical [52] and photocatalytic [9] properties, which suggests that stacking fault disorder may be a positive feature for certain applications [38,115].
A weak but sharp peak has been reported at 3655 cm−1 that is assigned to either the IR-active stacking fault O−H stretching mode or a localized mode around Ca point defects from the starting material used to prepare the β-Ni(OH)2 samples. In the former case, this out-of-phase peak should be quite weak in the Raman spectrum. In the latter explanation, the A1g for bulk Ca(OH)2 is observed at 3620 cm−1 [229]. Hall et al. [38] only detected this peak in less-ordered samples that contained Ca in their XPS spectra, which makes either explanation equally plausible.
One additional Raman peak is observed at 3688 cm−1. The intensity has been reported to be linked to that of the peak at 3601 cm−1, but has have also been observed to vary independently [42,52]. Bernard et al. [42] observed that dry samples, which initially show this peak, that are rinsed with water and re-analysed while still wet, lose the peak completely. This indicates that it is a surface-related phenomenon, although the exact origin is unclear. Bernard et al. proposed that the O−H stretch for surface hydroxide groups is at higher energy than for those opposed by adjacent layers. Upon hydration the transition becomes similar to either the bulk O−H stretching mode, A1g, or that of free H2O [42]. This hypothesis is supported by a more recent Raman study of wet and dry Ni(OH)2 samples [94].
The reported Raman-active β-Ni(OH)2 vibrational peaks are summarized in table 8, where we also show the results of the lattice dynamical calculations of Hermet et al. [215] based on their spin generalized gradient approximation (SGGA) model. Their calculated values are in very good agreement with the observed ones for the Raman-active modes.
Table 8.
Comparison of literature values for the vibrational modes in β-Ni(OH)2 and equivalent modes in Mg(OH)2 (brucite). ‘sh’ indicates the peak was observed as a shoulder.
| peak assignment | [38] | mode frequency (cm−1) literature | theory of [215] | Mg(OH)2 |
|---|---|---|---|---|
| Raman | ||||
| Eg lattice mode | — | 310 [42], 310 [52], 315 [117], 309 [194], 313 [228], 318 [227], 319 [231], 312–313 [232], 306 [233], 315 [234] | 316 | 280 [229] |
| A1g lattice mode | — | 445 [42], 450 [52], 450 [117], 446 [194], 447 [228], 449 [227], 458 [231], 448–453 [232], 445 [233], 450 [234] | 445 | 443 [229] |
| second-order acoustic mode | 516 | 510 [42], 515 [52], 510 [194], 508 [228], 519 [231], 519 [232], 510 [233], 517 [234] | — | |
| 2 Eg second-order lattice mode | 601 | — | — | |
| Eg O−H bend | ∼880 | ∼850 [227], 880 [234] | 868 | 725 [229] |
| trapped H2O, O−H bend | ∼1600 | — | — | |
| free H2O, O−H bend | ∼1635 | — | — | |
| A1g O−H stretch | 3581 | 3570 [42], 3585 [52], 3581 [117], 3565 [159], 3579 [194], 3581 [228], 3580 [227], 3599 [231], 3580–3583 [232], 3579 [233], 3580 [234], 3580 [235] | 3600 | 3652 [229] |
| disordered, O−H stretch | 3601 | 3580 [42], 3605 [52], 3601 [117], 3600 [159], 3599 [194], 3601 [228], 3600 [232], 3600 [234] | — | |
| surface, O−H stretch | 3688 | 3680 [42], 3687 [159], 3687 [232] | — | |
| infrared | ||||
| Eu (TO)/A2u (TO) lattice modes | — | 345 [40], 350 [48], 350 [49], 348 [60], 354 [89], 350 [161], 332 [228], 350 [227], 340 [231] | 354/418 | 368 [236] |
| Eu (LO)/A2u (LO) lattice modes | — | 460 [40], 450 [48], 450 [49], 460 [52], 451 [60], 475 [89], ∼450 [139], 460 [161], 440 [228], 452 [227], 458 [231] | 366/469 | 562 [236] |
| Eu (TO) O−H bend | — | 520 [40], 530 [48], 545 [49], 525 [52], 549 [60], 530 [89], 517 [139], 510 [161], 530 [228], 553 [227], 525 [231] | 496 | 462 [236] |
| Eu (LO) O−H bend | 1030 | 1030 sh [52] | 665 | — |
| free H2O, O−H bend | ∼1630 | 1620 [48], 1630 [52] | ||
| A2u (TO) O−H stretch | 3629 | 3650 [40], 3650 [48], 3650 [49], 3635 [60], 3650 [89], 3640 [139], 3650 [161], 3630 [227], 636[228], 3639 [235], 3650 [237] | 3643 | 3698 [236] |
| disordered, (TO) O−H stretch | 3634 | — | — | |
| A2u (LO) O−H stretch | 3643 | — | 3673 | — |
| disordered, (LO) O−H stretch | 3652 | — | — | |
| A1g+A2u (LO)/A1g+Eu (LO) | 4035 | 4028 [235] | — | |
| third-order combination | ∼4275 | 4274 [235] | — | |
| A1g+A2u (TO/LO) | 7020 | 7016 [235] | — |
Infrared-active vibrational modes of β-Ni(OH)2. The IR spectrum of β-Ni(OH)2 is shown in figure 14a and factor group theory predicts four IR-active vibrational modes for β-Ni(OH)2 [226]. There are twice as many IR peaks as there are Raman peaks for β-Ni(OH)2 because of the splitting of each mode into transverse optical (TO)/longitudinal optical (LO) components owing to the nature of these vibrations [226]. The magnitude of this splitting effect has been calculated for brucite and can be used to assist peak identification for β-Ni(OH)2 [226].
Figure 14.
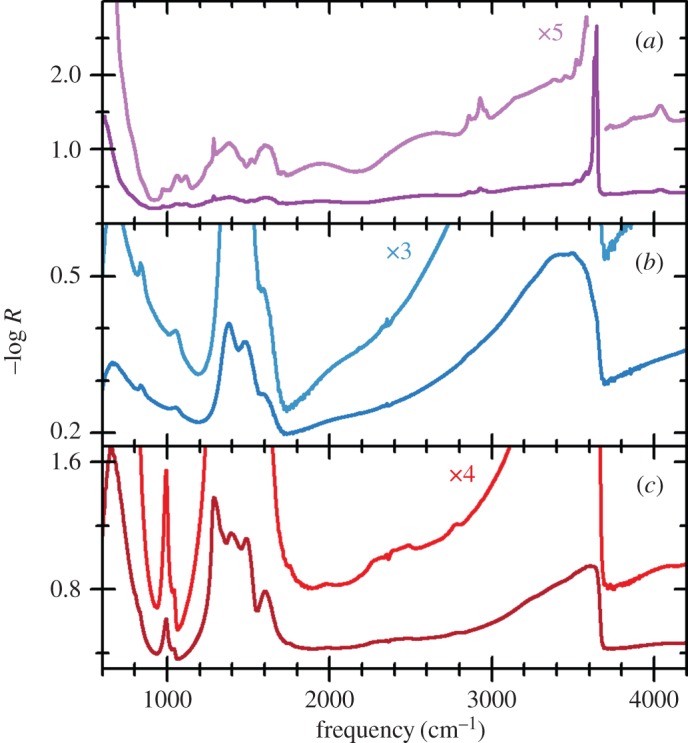
Infrared reflectance spectra of (a) β-Ni(OH)2, (b) α-Ni(OH)2 and (c) nitrate-intercalated α-Ni(OH)2. Plots are enlarged and offset to show low absorbance features. Copyright this work. (Online version in colour.)
Until recently, only four IR lattice modes had been previously identified: 332–354, 440–475, 510–553 and 3630–3650 cm−1 [228]. These four peaks are clearly visible in figure 15. However, in [38], the lattermost peak is reported to consist of overlapping peaks at 3629 and 3643 cm−1. This matches the expected LO/TO splitting for the A2u mode at approximately 3635 cm−1 (cf. 14 cm−1 for Mg(OH)2 [226]). Therefore, these peaks are assigned to the A2u (TO) and A2u (LO) modes, respectively. An IR study of β-Ni(OD)2 showed that the two apparent peaks at 350 and 450 cm−1 are actually each composed of two overlapping peaks: one each that red-shifts on deuteration and one each that does not observably shift. The peak at 550 cm−1 is also observed to red-shift on deuteration [49]. From brucite, the two remaining A2u modes are not predicted to significantly shift on deuteration, whereas all four of the Eu modes are [226]; hence, these peaks can be assigned accordingly. This conclusion is supported by a study that observed the shift in position of three lattice modes for Mg1−xNix(OH)2, where 0≤x≤1 [60]. The remaining Eu mode is predicted at a frequency greater than 550 cm−1 and has a very large TO/LO splitting (cf. 370 cm−1 for brucite), thus this mode is assigned to a broad band at approximately 1030 cm−1 [38]. This peak has very low intensity and was unreported until recently, although close examination reveals that it may be visible as a weak shoulder in older results [52]. Thus, the eight IR-active lattice modes have now been identified and assigned (table 8).
Figure 15.
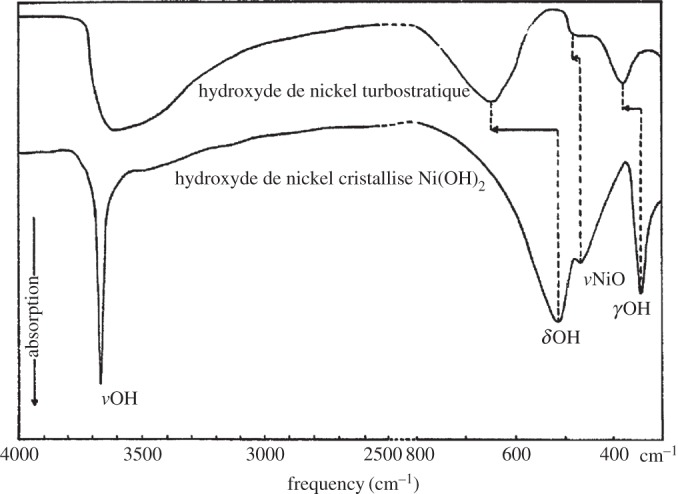
Infrared reflectance spectra of α-Ni(OH)2 (upper) and β-Ni(OH)2 (lower). Reprinted from [161], Copyright 1972, with permission from Elsevier.
A broad peak at approximately 1630 cm−1 arises from the O−H bending mode of H2O that is either adsorbed on the material surface or trapped within the structure. The peaks at approximately 3380 cm−1 and approximately 3450 cm−1 are from the O−H stretch of the H2O.
A sharp but weak peak measured at 3581 cm−1 corresponds exactly to the Raman-active A1g transition. This is formally forbidden in IR spectroscopy; however, structural disorder allows a very weak peak to be seen in the IR spectra. Conversely, a weak A2u mode should also be allowed in the Raman spectrum. However, this mode overlaps with other features of the sample and, hence, cannot be observed in the Raman spectrum. A shoulder at 3601 cm−1 corresponds to the in-phase O−H stretch of disordered β-Ni(OH)2 discussed in the Raman section. The corresponding out-of-phase O−H stretch (LO) is a strong, sharp peak at 3652 cm−1. The corresponding TO mode is visible in the spectra for sample β1 as a shoulder at 3634 cm−1. Note that disorder shifts both the TO and LO modes to higher frequency by 5–9 cm−1, which reaffirms that these two peaks are connected.
The peak at 2658 cm−1 is sharp, very weak and close to the positions reported for the A2u mode of β-Ni(OD)2 (cf. 2705 cm−1 [49], 2680 cm−1 [89]). The difference in position could arise because this study observes localized modes from the naturally present D (0.0115%), whereas the reference materials were highly enriched.
Several previously unreported weak features are observed in the IR spectra for β-Ni(OH)2 that can be ascribed to combination bands. As this crystal structure has an inversion centre, the overall symmetry must be odd (ungerade) for a transition to be IR-active. Thus, all overtone transitions are even and therefore Raman-active, whereas an IR mode or an acoustic mode combined with a Raman mode is odd and therefore IR-active. The peak assignments were created by considering the IR and Raman lattice modes (table 8) and the acoustic transitions observed by INS [228]. One particularly interesting combination is the sharp peak at 3520 cm−1 assigned to the combination of the A1g mode and a subtractive mode, i.e. a hot band, for the acoustic transition at approximately 60 cm−1 [228]. Hot bands are not typically observed in IR spectra at room temperature. However, the acoustic transition is sufficiently low in energy that a Boltzmann distribution predicts a population ratio of 57 to 43 for the ground state and first excited state, respectively. Another combination of the A1g and a subtractive mode is observed at 3566 cm−1.
The reported IR-active β-Ni(OH)2 peaks are summarized in table 8. It can be seen there that the lattice dynamical calculations of Hermet et al. [215] are not in agreement with experimental values for some of the IR-active modes, which suggests that more work needs to be done on these particular IR modes.
(ii). Vibrational modes of α-Ni(OH)2
Most generally, α-Ni(OH)2 has trigonal symmetry consisting of planes of β-Ni(OH)2 intercalated with water, but the irregularities in the structure precludes a group theory analysis, similar to that performed for β-Ni(OH)2 in §4b(i), of the vibrational modes of α-Ni(OH)2.
Raman-active vibrational modes of α-Ni(OH)2. The Raman spectra of α-Ni(OH)2 and nitrate-intercalated α-Ni(OH)2 are shown in figure 12b and c, respectively. There are Raman-active lattice modes at 460 cm−1 (figure 13) and 495 cm−1 [38]. Weak features, assigned to second-order transitions, are observed at approximately 790 cm−1 and approximately 1075 cm−1. Some authors report additional Raman-active lattice modes [194,232]. However, these are not observed in the most recent work [38] and can all be ascribed to β-Ni(OH)2 impurities.
Structural disorder causes the internal O−H bending mode of lattice OH to split and broaden into a weak feature at approximately 1400 cm−1 (figure 12). As the Raman intensity is quite weak it is necessary to use IR, discussed below, to observe fine details for these peaks. Furthermore, this broad feature underlies several nitrate bands in the nitrate-intercalated α-Ni(OH)2 and is therefore often not clearly seen. A second internal O−H bending mode at 1590–1620 cm−1 is similar in width and only slightly shifted from the position of free H2O. Thus, this can be assigned to the O−H bending mode of the intercalated H2O. A third internal O−H bending mode is observed at 1630 cm−1, which arises from adsorbed H2O on the surface or H2O trapped within the structure.
There are several O−H stretching modes from 3240 to 3515 cm−1 that arise from free H2O. The internal O−H stretching modes from lattice OH and intersheet H2O are visible from 3590 to 3650 cm−1. This region is quite different between samples and such differences have previously been suggested to arise from the presence of intermediate phases [233]. However, the lattice modes do not vary as significantly between samples and these differences probably arise because this phase tends to adopt disorder by the incorporation of foreign ions, variable hydration and turbostratic structure [1]. The reported Raman-active α-Ni(OH)2 peaks are summarized in table 9.
Table 9.
Comparison of literature values for the vibrational modes in α-Ni(OH)2.
| mode frequency (cm−1) |
||
|---|---|---|
| peak assignment | [38] | literature frequency |
| Raman | ||
| lattice mode | 460 | 461 [194], 451–460 [227], 462–464 [232], 460 [233] |
| lattice mode | 495 | — |
| second-order lattice mode | ∼790 | — |
| second-order lattice mode | ∼1075 | — |
| O−H bend, lattice OH | ∼1400 | — |
| O−H bend, layer H2O | 1590–1620 | — |
| O−H bend, free H2O | 1630 | — |
| O−H stretch, free H2O | ∼3240, ∼3380, 3515 | — |
| O−H stretch, lattice OH/layer H2O | 3590, 3620, 3642–3650 | 3625 [159], 3637 [194], 3657 [227], 3650–3653 [232], 3656 [233] |
| infrared | ||
| lattice mode | — | 400 [40], 380 [161] |
| lattice mode | — | 460 [40], 461 [139], 480 [161], 474 [173], 480 [238] |
| lattice mode | ≤650−670 | 625 [40], 662 [139], 650 [161], 645 [173], 650–655 [238] |
| combination lattice mode | 835 | — |
| combination lattice mode | 1055 | 1055 [139] |
| O−H bend, lattice OH | 1380–1390 | 1400 [139] |
| O−H bend, lattice OH | 1480–1490 | 1540 [139] |
| O−H bend, layer H2O | 1600 | 1650 [40], 1640 [139], 1600 [161], 1650 [237], 1615–1620 [238] |
| O−H bend, free H2O | 1630 | — |
| O−H stretch, free H2O | 3220/3240, 3380/3390, 3500/3515 | 3350 [40], ∼3500 [161], ∼3400 [173], 3350 [237], ∼3500 [238] |
| O−H stretch, lattice OH/layer H2O | 3570, 3600–3640 | ∼3600 [161], ∼3600 [173] |
Infrared-active vibrational modes of α-Ni(OH)2. There are three IR-active α-Ni(OH)2 modes at 380–400, 460–480 and 625–670 cm−1 (figure 15). Weakly visible combination bands are observed at 835 and 1055 cm−1 (figure 14). The latter is not visible in the spectra of nitrate-intercalated α-Ni(OH)2 samples because it is obscured by nitrate bands (see ‘Nitrate ions’ in §4b(iii)).
Two peaks from the O−H bending modes of lattice OH are observed at 1380–1390 and 1480–1490 cm−1. These peaks tend to vary slightly between samples because of structural disorder and the mechanical stress induced when the materials are dried. The O−H bending mode from intercalated H2O is measured at 1600 cm−1, which matches the Raman spectra. The absorbance of these first three O−H bending modes is too great to be assigned to free H2O that is trapped within the material, whereas a weaker peak is observed at 1630 cm−1 that is consistent with this assignment.
As in the Raman spectra, several O−H stretching modes are observed in the IR spectra of the α-phase samples. Broad features from free H2O are measured at 3220–3515 cm−1 and peaks corresponding to lattice OH and intersheet H2O are observed at 3570–3640 cm−1. The variability from structural disorder is the same as in the Raman spectra.
A combination band of the O−H stretch and a lattice mode is measured at 4260–4280 cm−1. A combination of O−H stretching modes is measured at 7140 cm−1. Additional features are difficult to distinguish by FTIR because of periodic interference fringes arising from the Fourier transformation. The reported IR-active α-Ni(OH)2 peaks are summarized in table 9.
(iii). Vibrational signatures of disorder and ad-ions
Disorder. The current body of literature can be simplified by considering two phases, α and β, with varying types and degrees of disorder [38]. α-Ni(OH)2 samples are turbostratic, where adjacent layers are separated by amorphous H2O. The anions in the Ni2+ salt used for sample preparation often incorporate into this interlayer region. β-Ni(OH)2 materials often contain stacking faults, incorporated during preparation. Moreover, different degrees of hydration can occur, which is observed by large H2O bands in the Raman and IR spectra. Finally, other factors such as crystallite size, point defects, stacking faults, etc., are expected to affect the spectroscopic properties of the materials, such as whether second-order transitions appear in Raman measurements. Using the most recent vibrational peak assignments, one may simultaneously, but separately, monitor structural disorder from the incorporation of foreign ions, hydration and crystal defects such as stacking faults. Thus, any changes in the performance or stability of nickel hydroxide catalysts and electrodes can now be linked with their true origin. This will enable systematic and directed development of materials for real applications.
Nitrate ions. Nitrate anions are often incorporated in samples prepared chemically or electrochemically from Ni(NO3)2. Free nitrate (i.e. nitrate anions in solution) has D3h symmetry and four vibrational modes: ν1 at 1050 cm−1 (A′1 IR-inactive), ν2 at 831 cm−1 ( Raman-inactive), ν3 at 1390 cm−1 (E′) and ν4 at 720 cm−1 (E′) [239]. In solution, the ν1 band becomes weakly IR-active and the ν3 band splits into a doublet at 1356 and 1410 cm−1, both of which are weakly Raman-active. This split results from rotation and is therefore only expected if the chemical environment allows free rotation. Furthermore, these bands can shift and split into additional modes when nitrate interacts with cations to form ion pairs [240].
The Raman and IR spectra of nitrate-intercalated α-Ni(OH)2 may contain all four nitrate modes (figure 16), although the relative intensities differ between the two techniques. The most intense Raman peak is at 1047 cm−1 (ν1) and the strongest IR peak is at 1340 cm−1 (ν2). The positions and relative intensities closely match those of free nitrate, which supports that the nitrate ions are located in the interlayer space and that this region is chemically similar to free solution [38,94]. The positions of the nitrate modes are even closer to those of free nitrate in Raman spectra that were collected in situ from an α-Ni(OH)2 sample immersed in water [94].
Figure 16.
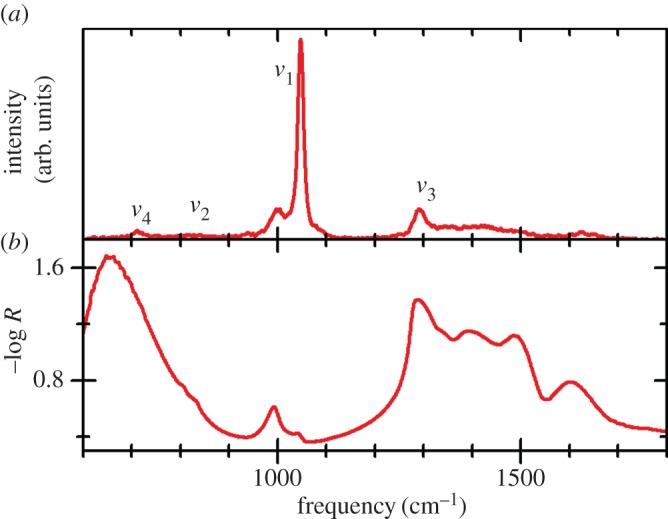
(a) Raman and (b) IR reflectance spectra of nitrate-intercalated α-Ni(OH)2. Copyright this work. (Online version in colour.)
In the spectra shown in figure 16, there is an additional set of nitrate peaks, which implies that nitrate can occupy two distinct chemical environments. The relative Raman and IR intensities of these peaks do not match the corresponding vibrational modes of free nitrate. This indicates that there are two sets of selection rules and confirms the presence of two types of nitrate, rather than peak splitting from ion-pairing effects [38,240]. The change in selection rules also demonstrates that the second position in the crystal reduces the symmetry below D3h. It is unclear whether the second type of nitrate has a different orientation within the interlayer space or whether it substitutes lattice hydroxide. Therefore, vibrational spectroscopy may be used to rapidly identify the presence of nitrate in two distinct chemical environments by examining the Raman and IR spectra.
Anion-intercalated α-Ni(OH)2 materials are quite similar to the basic nickel salts. For example, nitrate-intercalated α-Ni(OH)2 is closely related to the NHNs (see §2c(iii)). However, the NHNs are close-packed structures, whereas α-Ni(OH)2 is a turbostratic structure in which the intercalated water layer acts as an ‘amorphous glue’. Therefore, the nitrate peaks in the NHNs are sharper and do not closely match the free nitrate peak positions. For example, the reported IR spectrum of a NHN contains a sharp peak at 990 cm−1 (ν1), but does not contain the free nitrate-like peak at approximately 1050 cm−1 [34]. Therefore, it can be concluded that the second chemical environment in the α-Ni(OH)2 sample discussed above is more like the close-packed environment in a NHN.
Other anions. Similar to nitrate, other anions may be incorporated into α-Ni(OH)2 samples during preparation. This includes monatomic anions, such as chloride, and polyatomic anions, such as sulfate and carbonate. In the case of monatomic impurities, there are no internal modes to detect by vibrational spectroscopy. In the case of sulfate anions, there are weak IR peaks reported at 980 and 1120 cm−1 [38] that are similar to the positions of the ν1 and ν3 modes of free sulfate (cf. 981 cm−1 and 1104 cm−1, respectively [239]). By analogy to the nitrate bands discussed above, this may indicate that sulfate is in the hydrated interlayer space. A second set of bands is more strongly observed in the Raman and IR spectra at 520, 685, 1060 and 1170 cm−1. The first two peaks are assigned to ν2 and ν4 modes (cf. 451 cm−1 and 613 cm−1, respectively [239]). The Raman spectra also show a combination of ν1 and ν2 at 1590 cm−1, however, this is not observed by IR. Thus, sulfate ions also adopt some chemical environment that shifts the peaks to higher frequency [38]. Similarly, incorporated carbonate anions have been observed in the Raman and IR spectra of carbonate-intercalated α-Ni(OH)2 materials [80,241]. The reported modes correspond to the four principal modes of free carbonate ions: ν1 at 1063 cm−1 (A′1, IR-inactive), ν2 at 879 cm−1 (, Raman-inactive), ν3 at 1415 cm−1 (E′) and ν4 at 680 cm−1 (E′) [239].
Therefore, one can readily determine the presence of polyatomic anions, including nitrate, sulfate and carbonate, in α-Ni(OH)2 using Raman or IR spectroscopy. Such measurements may be used to examine the effects of incorporated anions on the behaviour of an electrode or catalyst.
(iv). Surface-enhanced Raman spectroscopy
It is often necessary to examine ultrathin surface layers, such as the air-formed film on Ni, which is 6–8 Åthick [178], or the passive films that form in solution, which vary with experimental conditions, but can be as thin as approximately 1 nm for Ni electrodes [242]. These surface layers are often below the detection limit of conventional Raman spectroscopy. For example, Nan et al. collected Raman spectra in situ during voltammetry on Ni electrodes in strong alkaline media. Although they did measure some weak lattice modes for oxides and hydroxides, the only strong peaks were from β-NiOOH. Furthermore, they were unable to obtain peaks in the characteristic O−H stretching region (3300–3800 cm−1), presumably because the surface layer was too thin and the background water dominated their results [194]. For this reason, we consider the practical use of SERS, which can offer as much as a 106 signal enhancement over conventional Raman spectroscopy. The SERS effect may be achieved by using a suitably roughened Cu, Ag or Au substrate. Alternatively, by coating a sample surface with Ag or Au nanoparticles, one can measure the SERS spectra of ultrathin surface layers without the need for a specialized substrate [243,244]. However, the relative intensities of SERS peaks are difficult to predict and can vary considerably from conventional Raman spectra [244]. Thus, proper interpretation of SERS results is often challenging. We recently reported a clarified model of the structure of Ni(OH)2 and identified the Raman and IR modes of the α and β phases [38] and subsequently applied these findings to the in situ Raman spectroscopy of nickel hydroxides [94]. We now apply these results to re-interpret literature SERS results, to summarize the known SERS-active modes and to assess the applicability of the three vibrational optical spectroscopy techniques for the analysis of Ni(OH)2 materials and surface layers.
In past work, we recommend the use of the O−H stretching region (approx. 3200–3700 cm−1) as a ‘fingerprint’ for identifying the presence and relative amounts of the α- and β-Ni(OH)2 phases in a sample [38,94]. This avoids spectral overlap with lattice modes from several materials (NiOx, NiOOH, AgO, etc.) that may be present. Kostecki and McLarnon included a high-quality SERS spectrum in this frequency range of precipitated α-Ni(OH)2 measured in 1 M NaOH at −0.73 V versus the RHE. The inset of figure 17 shows the re-plotted, original data (cf. fig. 9 in [233]). We removed the fluorescence and water background components using the same methodology illustrated in [94]. The relative intensities of the bulk solution bands are expected to be similar to those of our reference spectrum of water because the water component is predominantly from conventional Raman, rather than SERS. Some mismatch is expected from the differences in solution compositions and instrumental set-ups. However, the advantages of deconvoluting the water and sample bands far outweigh these minor sources of error. Qualitatively, the resultant SERS spectrum is quite different from either the Raman or the IR ex situ spectra of α-Ni(OH)2, which are also shown in figure 17. The SERS spectrum contains an O−H stretching mode of α-Ni(OH)2 at approximately 3610 cm−1. This peak is consistent with literature references of Raman- and IR-active lattice hydroxyl modes for α-Ni(OH)2, which are quite variable [38]. The spectrum is, however, dominated by two broad peaks at 3450 and 3300 cm−1 that correspond to the IR-active O−H stretching modes of the interlayer water. There is also a sharp but weak peak at approximately 3520 cm−1, which may be attributed to water molecules at the boundary between α- and β-Ni(OH)2, although there are no β-Ni(OH)2 peaks visible in the spectrum, possibly because the spectrum is quite noisy. Significantly, we find that the relative intensities of the O−H stretching modes vary considerably between IR, SERS and Raman spectroscopy, as shown in figure 17. Nevertheless, by examining the characteristic ‘fingerprint’ region, α-Ni(OH)2 may be identified from SERS results.
Figure 17.
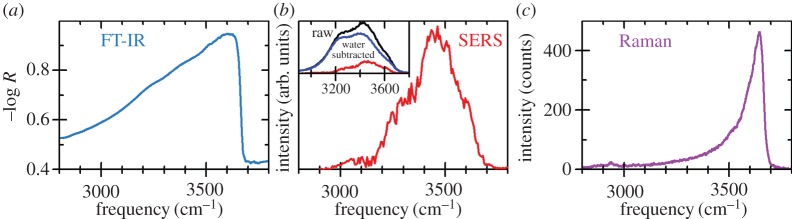
A comparison of the O−H stretching region of α-Ni(OH)2 as measured by FT-IR, SERS (data taken from fig. 9, −0.2 V Hg/HgO in [233]), inset shows data treatment to remove the contribution of water using the procedure in [94]. Copyright this work. (Online version in colour.)
Desilvestro et al. deposited thin Ni(OH)2 films on Au substrates and measured SERS during voltammetry in alkaline media. Their in situ SERS spectra of freshly deposited α-Ni(OH)2 contain a dominant peak at 455 cm−1 [245] or at 460 cm−1 [166] and contain a shoulder at approximately 385 cm−1 [166,245]. We can now ascribe these peaks to IR-active α-Ni(OH)2 lattice modes. The presence of IR-active modes and the absence of any Raman-active lattice peaks reaffirm the unpredictable nature of the SERS selection rules. The authors also measured SERS on precipitated Ni(OH)2, in air, which contains broad peaks at 3610 and 3525 cm−1 that probably arise from the α-Ni(OH)2 O−H stretching modes of lattice hydroxyl and interlayer water, respectively. However, their data are too weak to interpret further [166].
Kostecki & McLarnon [233] measured SERS during long-term voltammetric cycling between Ni(OH)2 and NiOOH. During such cycling, it is known that the composition of the reduced state is initially α-Ni(OH)2 and, after cycling, gradually contains a greater β-Ni(OH)2 component [179]. The SERS spectra of the initial state contains the same α-Ni(OH)2 lattice modes present in the results of Desilvestro et al. After cycling, the spectra contain additional SERS peaks at 445 and 522 cm−1. We attribute the first peak to the β-Ni(OH)2 A2u (LO), Eu (LO) and/or A1g lattice modes and the second peak to the β-Ni(OH)2 Eu (TO) lattice mode.
Melendres & Pankuch [193] conducted in situ SERS measurements on Ag-nanoparticle-coated Ni electrodes in alkaline media. Figure 18 shows the re-plotted, original data (cf. fig. 5 in [193]) before and after the removal of the fluorescence and water background components. The peak shapes and frequencies of the O−H stretching modes in the resultant spectra closely match the literature values for Ni(OH)2 and the α and β phase components were readily identified. Consistent with Kostecki and McLarnon's results, the intensities of the α-phase interlayer water modes are greater than for Raman or IR spectroscopy. Our re-examination of their spectra reveals that during 1 h potentiostatic treatments, α-Ni(OH)2 formed at the Ni surface at approximately 0.2 VRHE (figure 18a). We ascribe the weak shoulders at 3575 cm−1 and 3620 cm−1 to the β-Ni(OH)2 A1g and A2u (TO) peaks, respectively, which indicates that the film contains a very small β-Ni(OH)2 component. The amount of β-Ni(OH)2 at the surface increases at 0.6 VRHE (figure 18b) and at 1.0 VRHE (figure 18c), the β-Ni(OH)2 A1g and A2u (TO) peaks are clearly present. The SERS activity of the IR-active A2u (TO) peak is greater than that of the Raman-active A1g peak.
Figure 18.
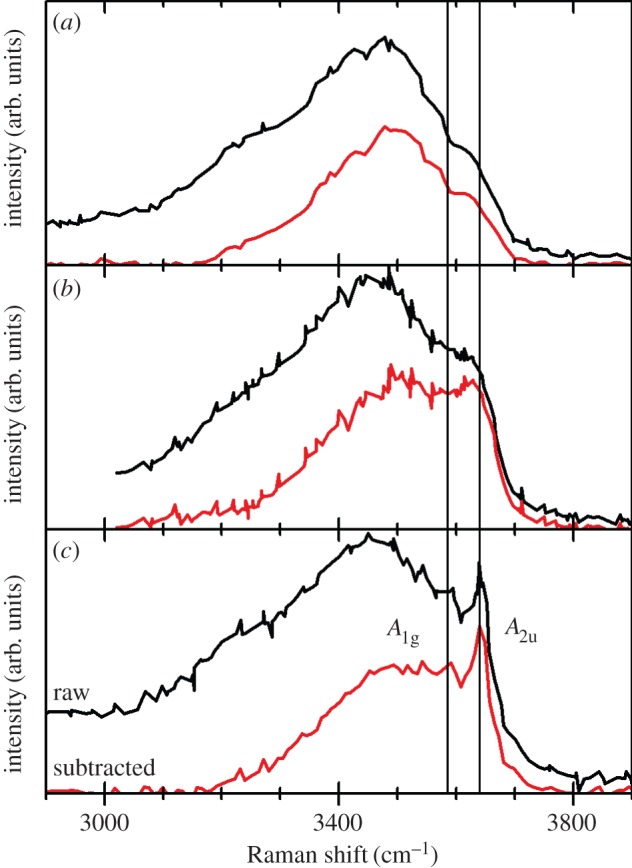
The in situ SERS measurements of Melendres & Pankuch (fig. 5(a)–(c) in [193]) were first digitized (upper curves, black) and then the background fluorescence and water components were removed (lower curves, red) using the method illustrated in figure 17. Copyright this work. (Online version in colour.)
Melendres & Pankuch also presented spectra showing the lattice mode frequencies (200–1000 cm−1, fig. 4 in [193]). From the electrodes held at or above approximately 0.2 VRHE, the authors labelled a peak at 450–456 cm−1. Upon reassessing their results, we attribute this feature to a convolution of two IR-active α-Ni(OH)2 lattice modes, at approximately 380 and at 460 cm−1. An additional peak at 510–512 cm−1 is present from electrodes held at 0.6–1.0 VRHE. Both NiOx and β-Ni(OH)2 surface components are expected under these conditions. However, the Raman-active NiO peak would be much broader than the observed SERS peak, although we expect that it is, to some extent, convoluted in the spectrum. Thus, we attribute the small, sharp SERS peak at approximately 510 cm−1 to the β-Ni(OH)2 Eu (TO) lattice mode. Presumably, the additional SERS-active β-Ni(OH)2 peak at 445 cm−1 is convoluted with the α-Ni(OH)2 peak at 460 cm−1 in the spectra, which reinforces the benefits of examining the O−H stretching ‘fingerprint’ region. Our interpretation of these results support recent voltammetric peak assignments of Ni in alkaline media; during a forward potential sweep a surface layer of mostly α-Ni(OH)2 forms over a non-stoichiometric NiOx underlayer [179]. However, the SERS results suggest that β-Ni(OH)2 is a surface layer component at lower potentials than previously thought. It is noted that there is no indication of stacking fault disorder in the SERS spectra.
There are a few other examples in which Ni(OH)2-containing surface layers have been examined using SERS. Berchmans et al. [246] studied catalytic Ni(OH)2 layers on modified Au substrates, with the goal of developing electrocatalysts for the oxidation of glucose. They did not, however, show the O−H stretching frequencies and their spectra were convoluted with several vibrational modes from AuO and some organic compounds. Hence, interpretation of their results is difficult. Oblonsky & Devine [247,248] examined passive films on Ni electrodes, coated with either Ag or Au particles, in borate buffer solutions. Their spectra of electrodes held at 0.84 VRHE contain peaks from the IR-active α-Ni(OH)2 lattice modes at 375 and 460 cm−1 [248]. There is a broad peak at approximately 3520 cm−1 that we attribute to the O−H stretching mode of α-Ni(OH)2 interlayer water. At 1.34 VRHE, there is an additional peak at approximately 520 cm−1 that may arise from either NiO or β-Ni(OH)2. However, the data is noisy in the O−H stretching frequency range and has an upper limit of 3600 cm−1, which prevents a conclusive analysis of their results. These studies not only demonstrate potential applications of SERS for characterizing Ni(OH)2 surface layers but also that further work is necessary to determine and summarize the SERS-active modes.
All the known SERS-active vibrational modes and their activities in IR and Raman spectroscopy are listed in table 10. Curiously, the most intense SERS peaks for both α- and β-Ni(OH)2 correspond to IR-active modes. Although we did not find an example of SERS from pure β-Ni(OH)2, these results should provide sufficient information to interpret SERS results for ultrathin surface layers that contain α- or β-Ni(OH)2 film components.
Table 10.
Known SERS-active vibrational modes of α- and β-Ni(OH)2 and their activities in IR and conventional Raman (R) spectroscopy.
| SERS shift (cm−1) | mode assignment | activity | refs | |
|---|---|---|---|---|
| α-Ni(OH)2 | ||||
| 375–385 | lattice mode | IR | [193,233,245,248] | |
| 460 | lattice mode | IR | [166,193,233,245,248] | |
| 3300–3320 | layer H2O, O−H stretch | IR | [193,233] | |
| 3480–3520 | layer H2O, O−H stretch | IR | [166,193,233,248] | |
| 3610–3640 | lattice OH, O−H stretch | R | IR | [166,193,233] |
| β-Ni(OH)2 | ||||
| 445 | A2u (LO)/Eu (LO)/A1g lattice | R | IR | [193,233], |
| 510–520 | Eu (TO) O−H bend | IR | [193,233] | |
| 3575–3585 | A1g O−H stretch | R | [193] | |
| 3620–3630 | A2u (TO) O−H stretch | IR | [193] |
We now assess the viability of the three vibrational optical spectroscopy techniques for characterizing Ni(OH)2-based materials. The greater availability and relatively lower cost of FTIR instruments make this perhaps the most accessible method of the three. Furthermore, measurement times are typically quite short and no sample preparation is required. However, the very strong IR absorbance of water makes in situ measurements very difficult. Sharper peaks and greater spectral resolution may be attained by using Raman spectroscopy. Conventional Raman spectroscopy may be used to rapidly identify and characterize Ni(OH)2 bulk materials, for sample thicknesses down to approximately 0.1 μm and perhaps even lower for β-Ni(OH)2. Furthermore, we have demonstrated in this article that Raman spectroscopy is a suitable method for monitoring structural changes in situ. However, it is necessary to use SERS in order to characterize ultrathin surface layers. The strength of SERS is its sensitivity at the surface, although this makes the technique unsuitable for considering the bulk structure. Hence, by choosing the appropriate analytical technique, vibrational spectroscopy may be used to characterize and monitor the structure of Ni(OH)2 materials or surfaces.
(v). Inelastic neutron scattering
There have been very few studies of Ni(OH)2 by INS, primarily because of the problem of obtaining large enough single crystals. This is unfortunate, as INS can provide valuable information on lattice and internal modes of vibration at specific wavevectors throughout the Brillouin zone. The two INS studies to date [228,249] have used crystal powders, which means that INS provides information on the combined density of phonon states (DOS) obtained by integrating all vibrations throughout the Brillouin zone over all crystal orientations. Such studies do not allow conclusive assignments of the origin of the peaks in the DOS, but nevertheless, they can provide useful information that complements IR and Raman results.
In 1995, Baddour-Hadjean et al. [249] reported on an INS investigation of proton dynamics in β-Ni(OH)2 and their results are shown in figure 19. Peaks in the DOS spectrum correspond in most cases to modes involving H atom vibrations, as INS is particularly sensitive to H atoms [249]. For example, the two OH bending modes are evident at 680 and 880 cm−1. They assigned these modes to the expected Eu and Ag zone centre symmetry vibrations, respectively, in agreement with Raman but not IR results (table 11). Peaks seen at energy transfers above 1000 cm−1 were attributed to vibrational overtones, combinations and at high energy transfers, the OH stretching mode. In their results, this mode peaks at approximately 3800 cm−1, which is considerably higher than the 3580–3650 cm−1 range where the Brillioun zone centre modes are seen in Raman and IR spectroscopy. This indicates that in INS the DOS results for the OH stretching mode are dominated by contributions from phonons at the Brillioun zone boundary. The low energy peaks (figure 19) are assigned to acoustic (90 and 115 cm−1) and lattice modes (313–294, 358, 394 and 452–421 cm−1). The INS lattice mode results are compared with Raman and IR data in table 11.
Figure 19.
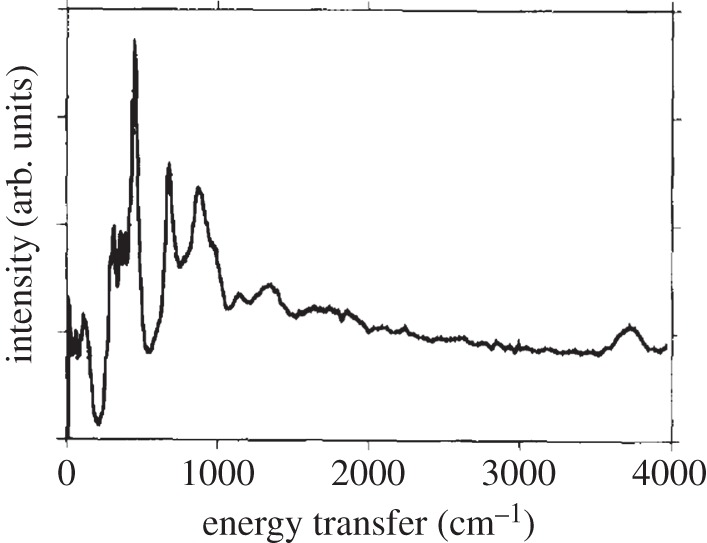
INS spectrum of powdered β-Ni(OH)2 at 25 K. Reprinted from [249], © 1995, with permission from Elsevier.
Table 11.
INS peak positions and mode assignments for β-Ni(OH)2, as reported by Baddour-Hadjean et al. [249]. The vibrational mode assignments from [38] and the range of Raman and IR peak positions obtained at room temperature (approx. 295 K) are provided for comparison where applicable (for individual values, see [38] and references therein).
| vibrational mode assignment |
||||
|---|---|---|---|---|
| INS peak position (cm−1) | [249] | [38] | Raman peak position (cm−1) | IR peak position (cm−1) |
| 90 | acoustic | acoustic | ||
| 115 | acoustic | acoustic | ||
| 294/313 | A1g(T) | Eg | 306–319 | |
| 358 | Eu(T) | Eu (TO)/A2u (TO) | 332–354 | |
| 394 | A2u(T) | |||
| 421/452 | Eg(T) | A1g/Eu(LO)/A2u(LO) | 445–458 | 440–475 |
| 680 | Eu(R) | 2 Eg | 601 | |
| 880 | Eg(R) | Eg | 850/880 | |
Bantignies et al. [228] performed a comprehensive study of the vibrational properties of β-Ni(OH)2 and β-Ni(OD)2 employing INS as well as IR, and Raman spectroscopies. From INS measurements at 293 K, they report low-frequency peaks at 130, 310–315 and 430–460 cm−1 that are similar in both the H and D compounds, which they attribute to lattice vibrations. At higher frequencies, up to 1200 cm−1, they observed two other peaks at 670 and 870 cm−1, in agreement with Baddour-Hadjean et al. However, as for Baddour-Hadjean et al. case, it was generally difficult to be precise about the correlation of these peaks with the IR and Raman results, because of the differences in the frequencies of peaks obtained by INS compared with IR and Raman spectroscopy owing to the DOS effect mentioned above.
(vi). Considerations for the application of vibrational spectroscopy
Vibrational spectroscopy measurements provide information on the proportions of α- and β-Ni(OH)2 and their fine structural details with high sensitivity. The two phases are easily distinguished from their strong internal O−H stretching modes at 3581–3640 cm−1 in pure and mixed samples (figure 20), in situor ex situ. Moreover, vibrational spectroscopy can characterize sample disorder from: (i) the incorporation of foreign ions, (ii) hydration and (iii) crystal defects including stacking faults. See fig. 4 in [94] for an example of how in situ Raman spectroscopy has been used to analyse the chemical transformation from α-Ni(OH)2 to β-Ni(OH)2.
Figure 20.
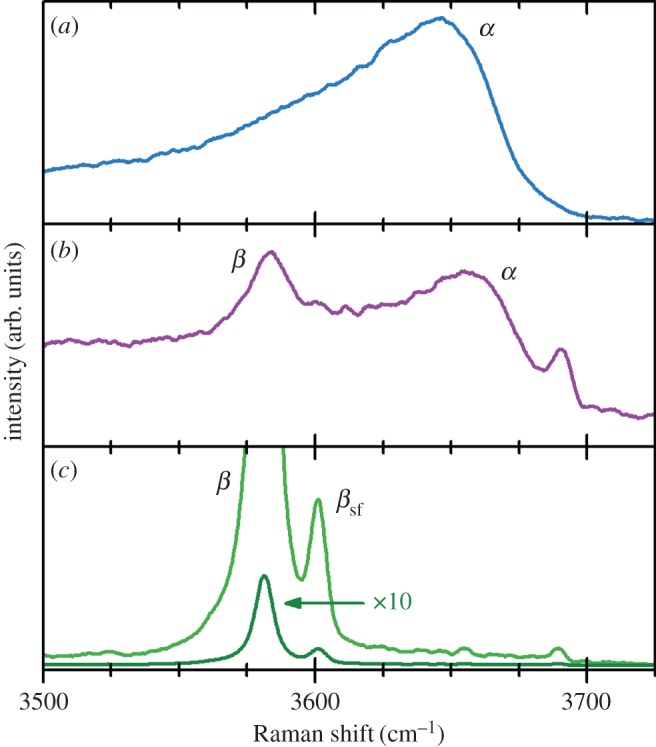
Raman spectra, plotted on the same intensity scale, showing the O−H internal modes of samples (a) α-Ni(OH)2, (b) mixed α/β-Ni(OH)2 and (c) β-Ni(OH)2. Copyright this work. (Online version in colour.)
With regard to choosing the most appropriate technique, IR spectroscopy tends to be the most readily available and may be used to rapidly characterize dry Ni(OH)2. Raman spectroscopy offers greater spectral resolution and is better suited for in situ measurements, owing to the lower intensity of the water modes in Raman spectra. Considerable information may be attained from only the most intense Raman modes, making these methods suitable even for very minute amounts of material or in the presence of high matrix interference. Finally, SERS may be applied to probe only the very surface of a sample or to characterize ultrathin samples.
(c). Electronic properties
(i). Dielectric properties
The dielectric response of β-Ni(OH)2 has been investigated experimentally and theoretically by Hermet et al. [215]. The static dielectric constant (ε0) experiments were performed on a disc-shaped pellet made from pressed powder with gold electrodes at frequencies between 1 Hz and 1 MHz at temperatures ranging from 150 K to room temperature. A value of 4.1 for ε0 was obtained at 150 K at 1 MHz. Because the sample was in powder form, this value represents an average of the two different tensor components of ε0 : (component zz, where z is aligned along the trigonal c-axis, //) and (component xx=yy, where x is aligned along the crystallographic a-axis, ⊥). The computed values were obtained from a consideration of the electronic and ionic (produced by the IR vibrational modes) contributions to ε0, and at 0 K yielded and . Their average is ε0=5.96, which is an overestimate of the experimental value of 4.1 at 150 K. The discrepancy between experiment and theory is attributed to differences in the measured and calculated IR oscillator strengths [215]. The calculated results show that the dielectric response of ε0 is mainly electronic along the optical c-axis, but with nearly equal ionic and electronic contributions in the a–b plane, and that this difference arises primarily from just the Eu(TO) vibrational mode at 496 cm−1 (frequency value obtained from their theory).
Using a linear response formalism, Hermet et al. [215] have also evaluated the electronic dielectric permittivity tensor , which is related to the second derivative of the electronic energy with respect to an applied electric field. Their calculated values of the tensor components are and , with corresponding refractive indices of n⊥=1.88 and n//=1.82. Thus, β-Ni(OH)2 is a negative uniaxial crystal with a fairly isotropic electronic response.
(ii). Electronic properties
Using plane-wave pseudopotential density-functional-theory, Hermet et al. [215] predicted that β-Ni(OH)2 is a semiconductor with a direct band gap of around 2.9 eV; the experimental value for the band gap, from the UV–Vis absorption spectrum, appears to lie in the range 3–3.5 eV [9,215]. For a detailed discussion of the UV–Vis spectrum of β-Ni(OH)2, see §4d.
(iii). Electrical conductivity
Measurements of the electrical conductivity of Ni(OH)2 samples are very worthwhile for assessing their usefulness in various energy storage applications including, most importantly, batteries and also supercapacitors. Such measurements require the attachment of electrodes to the sample and, as this is not easily done for powders of typically high resistance, only a few such studies have been performed so far (e.g. [250–255]). Motori et al. [252] carried out the first systematic study of the electrical properties of nickel hydroxide for alkaline cell systems. They found that a pure uncycled electrode of α-Ni(OH)2 exhibited a low conductivity that ranged from 10−13 to 10−17 S cm−1 depending on the temperature (298–423 K) and time (1–120 min) after the application of a step voltage (figure 21). The dielectric loss factor and dielectric constant were measured over the frequency range 10−2–106 Hz and both showed an increase with decreasing frequency and increasing temperature. From their studies Motori et al. deduced that the low conductivity was the result of ionic conduction in the α-phase. After electrochemical cycling between the Ni(II) and Ni(III) oxidation states, the electrode was determined to be the β-hydroxide. This β-Ni(OH)2 (denoted as the ‘reduced’ material in [252]) behaved as an insulator, like the α-phase material, but with higher corresponding conductivity, loss factor and dielectric constant values. Interestingly, the absorption of water in the uncycled (α) and also reduced (β) electrodes resulted in a very strong increase in the electrical conductivity to as much as 10−4 S cm−1 at 2% absorbed water content and 1 min after the voltage application. This is consistent with reports that some structural water is necessary for ‘active’ electrode materials [6]. The highly resistive properties of nickel hydroxide were confirmed by Deabate et al. [251], who also proposed a schematic model incorporating protonic conduction that could account for the observed electrical conductivity and dielectric properties of Ni(OH)2. Palencsár & Scherson [253,254] have performed in situ measurements of a single spherical Ni(OH)2 microparticle electrode just approximately 30 μm in diameter. The results they obtained from immersion in the 9 M KOH electrolyte showed that the effective resistance of their ‘activated’ Ni(OH)2 microparticle, which may be assumed to be hydrated β-Ni(OH)2, increased by a factor of approximately 50 on oxidation to NiOOH. This is in qualitative agreement with the ex situ results obtained by Motori et al. [252]. Other microstructural details have also been proposed to affect the electrical and proton conductivity of nickel hydroxide materials [228]. However, without detailed studies of the different types of structural disorder, such as stacking fault disorder and interstratification, it remains unknown how the conductivity is affected by various types and extents of disorder.
Figure 21.
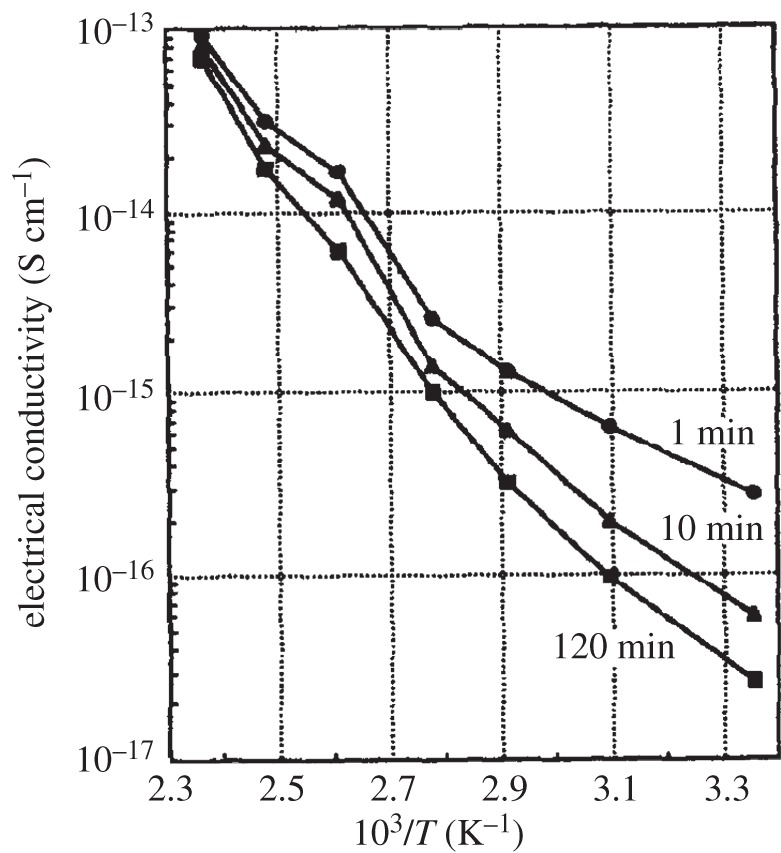
Electrical conductivity at 1, 10 and 120 min after application of a step voltage as a function of reciprocal temperature for a pure uncycled electrode of α-Ni(OH)2. Reprinted from [252], Copyright 1994, with permission from Elsevier.
(d). Optical absorbance and reflectance
(i). β-Ni(OH)2
The reflectance spectrum of β-Ni(OH)2 powder has been measured over the wavelength range 340–2000 nm by Ikeda & Vedanand [256]. These authors did not state the temperature of their measurements, but room temperature seems to be implied by their experimental conditions. They also did not explicitly identify the phase of their sample, but from their reported XRD results, it was in the β phase. The optical absorption spectrum, as calculated from the sample reflectance [256], is shown in figure 22. This spectrum was interpreted by crystal field theory assuming the local symmetry of the Ni(II) ions cubic from the octahedral (Oh) arrangement of oxygen atoms. This assumption was justified on the grounds that the expected trigonal site symmetry becomes nearly cubic in appearance owing to the averaging effects of a powdered sample combined with the lack of polarization analysis [256]. The standard cubic (octahedral) crystal field parameters were determined: 10Dq (Δo)=9000 cm−1, B (Racah B parameter)=925 cm−1 and C=10 800 cm−1 [257]. These values were calculated from the 3A2g to 3T2g(F) band transition at 9000 cm−1 (1100 nm), 3A2g to 3T1g(P) band at 26 000 cm−1 (385 nm) and the 3A2g to 1Eg(D) band at 11 400 cm−1 (877 nm); these transitions are marked in figure 22. The Racah B parameter exhibits a small (14%) reduction from that of free Ni(II) ions (1080 cm−1), which implies that the Ni–OH bonds are mostly ionic. Calculations of the Born effective charge tensors for β-Ni(OH)2 confirm this conclusion [215].
Figure 22.
The UV–Vis reflectance spectra measured by Ikeda & Vedenand [256] and Yu et al. [9] were digitized and replotted. Three electronic transitions, discussed in the text and in [256] and the estimated band-gap energy (Eg) are indicated on the figure. Copyright this work. (Online version in colour.)
Yu et al. measured reflectance data from precipitated Ni(OH)2 and reported peaks at approximately 670 nm (reported at 15 300 cm−1 (654 nm) in [256]) and approximately 385 nm (figure 22) [9], which correspond to the and transitions, respectively, calculated for the β phase [256]. Two additional peaks can be seen at approximately 740 nm and approximately 320 nm that are likely from α-Ni(OH)2, although, the study does not provide conclusive evidence regarding this assignment [9]. An additional transition is observed above 33 000 cm−1 (below 300 nm) [9,80] that, based on its high absorbance, may be from a charge transfer transition. Alternatively, this steep absorbance edge may correspond to the material's band-gap transition. By extrapolation, as shown by the dashed grey lines in figure 22, the X-intercept is found to be approximately 350 nm. This corresponds to a band-gap energy of Eg∼3.5 eV. Hermet et al. [215] measured the diffuse reflectance of β-Ni(OH)2 at room temperature between 200 and 800 nm. They reported a band gap in the ultraviolet of energy Eg∼3.0–3.5 eV. This is consistent with the value estimated from Yu et al.'s spectrum. A plane-wave pseudopotential density-functional-theory calculation [215] predicts that β-Ni(OH)2 is a semiconductor with a direct band gap of Eg=2.9 eV, which is slightly below the experimental value.
(ii). α-Ni(OH)2 and in situ reflectance spectroscopy
Hahn et al. [192,195] have demonstrated the feasibility of using UV–Vis reflectance spectroscopy at fixed wavelengths λ for in situ characterization of the spectrum of surface layers on metal electrodes during electrochemical experiments. When applied to the nickel electrode-alkaline solution interface they were able to obtain the reflectance-absorption spectrum [−(ΔR/R),λ]E of a thin layer of α-Ni(OH)2 that is shown in figure 23. Two absorption maxima are evident at 360 and 500 nm, respectively, demonstrating the power of the technique. The origin, or origins, of these peaks was not discussed by Hahn et al. However, based on the optical absorption spectrum of β-Ni(OH)2, it seems that they are associated with optical transitions within the Ni ion, although the peak at 360 nm could also involve some electronic transition of the α-Ni(OH)2 phase.
Figure 23.
Reflection–absorption spectrum of α-Ni(OH)2 at room temperature obtained by section of the tridimensional diagram in [195] at electrode potential E=−0.82 V versus the mercuric sulfate electrode. Adapted from [195], with permission from Elsevier.
(iii). Fluorescence
The strong background signal commonly observed in the Raman spectra of α and β films (e.g. [38]) indicates that the samples are fluorescent. Fluorescence is also observed from the air-formed NiOx on the substrate, but it is relatively weak. The fluorescence arises from the electronic structure of the Ni(II) cations, which have 3d8 valence configurations and octahedral coordination of OH− anions. Ideally, in the ground state (3A2g) the three highest occupied orbitals have t2g symmetry and there are unpaired, spin-aligned valence electrons in two, singly occupied eg orbitals. However, these orbitals can be greatly affected by small distortions of the octahedral geometry. Thus, the fluorescent background present in measured Raman spectra may not be intrinsic to the β-Ni(OH)2 material. Rather, it could be a result of structural disorder. Moreover, it is possible that the presumed fluorescence is in fact phosphorescence, which requires excitation to a triplet state. Such processes tend to be slower in crystalline samples and faster with increasing structural disorder [38,115].
In the case of α-Ni(OH)2, fluorescence is only sometimes observed in the measured Raman spectra, which signifies that α-Ni(OH)2 is not inherently fluorescent [38,115]. This also suggests that when there is a fluorescent background in measured Raman spectra, it may arise from structural disorder or changes in the electronic structure owing to the incorporation of impurities (e.g. intercalated chloride or nitrate anions) [38,115].
(iv). Ellipsometry
Spectroscopic ellipsometry is widely used to characterize the optical properties of thin films [258]. By varying the angle of incidence of the incoming light and analysing the reflected light for various polarizations, the resulting set of reflectance spectra for a known film thickness can be used to elucidate the optical constants of the film. Conversely, if the optical constants of the film are known, then an ellipsometric measurement at a single wavelength provides sufficient information to deduce the film thickness. The technique can also be used to analyse such properties in multi-layer structures.
Thus, ellipsometry is a useful technique for investigating thin film development in dynamic systems such as Ni(OH)2 films grown on Ni, where there have been several studies (e.g. [189–191] and references therein). de Souza et al. [189] used spectroscopic ellipsometry and linear sweep voltammetry to study the oxidation of nickel in alkaline solutions in situ. They found that during a positive scan in 1 M KOH starting at 0 V versus RHE, a thin film of Ni(OH)2 is formed at E∼0.5 V, followed by the appearance of nickel oxyhydroxides at approximately 1.4 V. A considerable number of optical models were investigated in order to best reproduce the observed spectroscopic ellipsometry measurements in the different potential regions. de Souza et al.'s conclusions are generally consistent with the present understanding of the voltammetry of nickel in alkaline media [179].
As an example, we show in figure 24 the measured and fitted spectra in terms of the ellipsometry parameters Ψ (amplitude) and Δ (phase difference) obtained at a potential of E=−0.4V versus Hg/HgO (approx. 0.5 V versus RHE) in 1 M KOH. The values of the optical constants n (real part of the complex refractive index) and k (imaginary part) for Ni(OH)2 and NiOOH used in the model predictions were those of Crocker [259], as shown in figure 25, where it can be seen that the refractive index is almost constant at n≈1.6 throughout the visible wavelength region. For the underlying Ni substrate, de Souza et al. used their own measurements of n and k. The best interpretation of the experimental data was obtained using a 0.2-nm-thick film of Ni(OH)2, consistent with a first anodically formed film containing mostly Ni(OH)2. This example demonstrates how sensitive this technique can be in resolving a film thickness and its composition. However, further work is necessary to establish the different optical parameters for α- and β-Ni(OH)2 materials.
Figure 24.
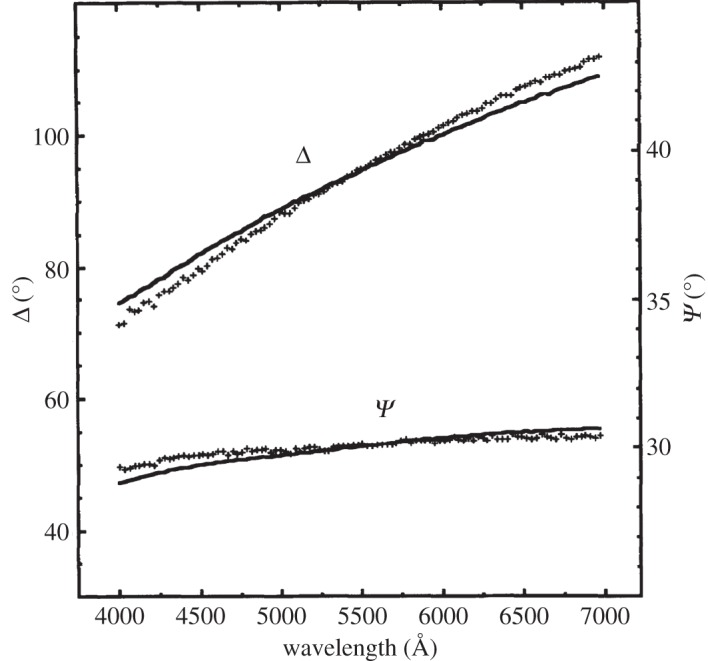
Comparison of the best model fit to experimental ellipsometry spectra for a Ni electrode in 1.0 M NaOH solution polarized at −0.4 V DS Hg/HgO/1.0 M NaOH. The compact film was concluded to consist of 0.2 nm of Ni(OH)2: model (—) and experimental data (+) [189]. Adapted from [189], with permission from Elsevier.
Figure 25.
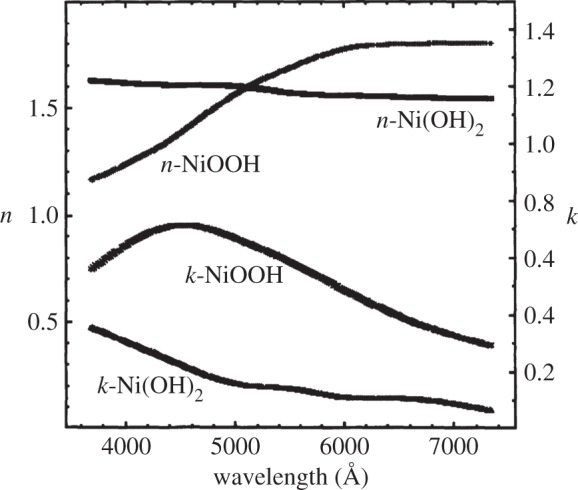
Spectral dependence of the optical constants of nickel hydroxide and nickel oxyhydroxide films components electrochemically formed on Ni in 1 M NaOH(aq) [189,259]. Adapted from [189], with permission from Elsevier.
The combined ellipsometry and reflectance study by Paik & Szklarska-Smialowska [191] of the anodic film formed on nickel in sodium hydroxide solutions revealed variations of n and k with time and potential that showed similar to de Souza et al. [189] that there are gradual changes occurring in the composition and structure, such as from Ni(OH)2 to NiO. Thus, the film composition at any given time will not be uniform, but their calculation using the single film model gives the average values of n and k as long as there are no sharp boundaries between different layers in the film. They found at their working wavelength of 546.1 nm that n varied from about 2.0 to about 2.5 with increasing potential, which are slightly higher values than those found by de Souza et al. [189]. Values obtained for k were small (0–0.5) when the film was potentiostatically aged or when the potential was raised gradually. The thickness of the film ranged from about 1.0 to 2.0 nm within 30 min of anodic polarization. Based on these optical constants and other coulometric data, the anodically created film was estimated to comprise mostly Ni(OH)2 with some NiO. Using the same wavelength of 564.1 nm, Visscher & Barendrecht [190] in their ellipsometry study found slightly lower values for n (1.45–1.55), but equally small values for k (0–0.03).
(v). Photoelectron spectroscopy
XPS has been used to study β-Ni(OH)2 [260,261] and, to a lesser extent, α-Ni(OH)2 [38]. In the XPS spectrum, the oxidation state of the Ni atoms may be assessed from the binding energy (BE) of the Ni 2p3/2 electrons (BENi(0)≈852.6 eV; BENi(II)=853.7–854.9 eV [260]). The determination of exact positions of measured XPS peaks requires a ‘charge correction’ to account for the buildup of electrostatic charge at the surface. Typically, this is done by adjusting the position of the C 1s peak from adventitious hydrocarbons on the sample surface [262]. Note that not all authors use the same reference value (typically 284.8–285 eV [262]). Unfortunately, this correction is not always exact, such as when a sample is covered with multiple surface layers that have different electrical conductivities. Therefore, an exact analysis of XPS data should use peak fitting to comprehensively analyse the various types of peak splitting that occur (figure 26). All Ni 2p peaks are subject to spin-orbit coupling, which produces 2p3/2 and 2p1/2 peaks. In the case of Ni(OH)2, the other most important effect is multiplet splitting, which occurs because the unpaired core electron (2p5) couples with the unpaired Ni(II) valence electrons (3d8). The area ratio of each multiplet peak is consistent between samples [262]. Thus, peak fitting using established parameters allows for a quantitative analysis of mixed samples that contain Ni, NiO, β-Ni(OH)2, γ-NiOOH and/or β-NiOOH. The reader is referred to the work of Biesinger et al., who report a complete set of fitting parameters for these materials, including peak line-shapes, area ratios, background treatment and BE splitting [260].
Figure 26.
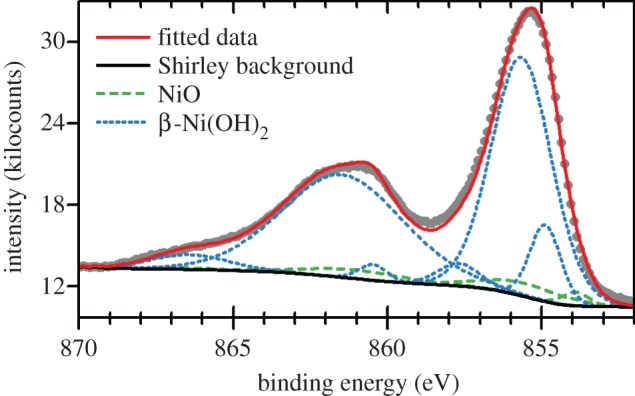
Ni 2p X-ray photoelectron spectrum (XPS) of β-Ni(OH)2 on a metallic Ni substrate. Experimental methods and parameters are described in [38]. The spectrum was fitted employing parameters and procedures described by Biesinger et al. [260]. (Online version in colour.)
There are only two known reports that discuss the XPS spectra of α-Ni(OH)2 [38,179]. The peak fitting parameters for this material have not been established. However, the XPS spectrum of α-Ni(OH)2 may instead be approximated using the parameters for β-Ni(OH)2. Characteristically, the fitted curve will not exactly match the data at 858–859 eV; the experimental spectrum of α-Ni(OH)2 has slightly greater intensity in this BE region than the fitted spectrum of β-Ni(OH)2 [38]. This may be used in either pure [38] or mixed (e.g. Ni/NiO/α-Ni(OH)2 in fig. 9a-i in [179]) samples. It is presently not possible, however, to quantify the components in a mixed α/β-Ni(OH)2 sample from XPS results.
Ultraviolet photoelectron spectroscopy (UPS) has been used to calculate work functions for surface layers on Ni electrodes [177,263]. The results support that the passive surface layer that forms on Ni in alkaline solutions consists of Ni(OH)2 underlaid by NiO [177]. Furthermore, the electrochemical behaviour of such surface Ni(OH)2 films may be analysed by UPS [263]. UPS is not, however, a useful technique for the identification of unknown surface components. It also cannot be used to differentiate between the α- and β-phases.
(e). Mechanical properties
(i). Elastic constants
The elastic constant, C, of a material characterizes its response to applied stress. For trigonal (D3d) symmetry materials such as β-Ni(OH)2, there are six independent constants within a fourth-rank tensor describing all 21 components of C. No measurements of these constants have been reported to our knowledge, but they have been calculated by Hermet et al. [215] for both the clamped-ion and relaxed-ion cases. The relaxed-ion constants are generally smaller than the fixed-ion ones. These calculated constants satisfy the Born mechanical stability conditions, indicating that β-Ni(OH)2 is mechanically stable. The bulk and shear moduli are reported from theory, based on the Voigt approximation to be B=55.70 GPa and G=35.09 GPa, respectively, giving a calculated value of G/B=0.63 for their ratio [215]. Such a ratio is typical of an ionic material.
5. Concluding remarks
This review, although quite detailed, has only touched just the surface of a vast sea of papers on the subject of nickel hydroxides and their applications. By restricting the coverage to what we feel are the essentials of the subject we hope to give pointers to useful papers in the literature and at the same time provide an up-to-date compendium of known properties and preparation methods. Readers requiring further details of a particular aspect of nickel hydroxides are encouraged to start first with the references provided here and then proceed to search through the literature by using references within these selected articles and via the Internet.
References
- 1.Oliva P, Leonardi J, Laurent JF, Delmas C, Braconnier JJ, Figlarz M, Fievet F, de Guibert A. 1982. Review of the structure and the electrochemistry of nickel hydroxides and oxy-hydroxides. J. Power Sources 8, 229–255 (doi:10.1016/0378-7753(82)80057-8) [Google Scholar]
- 2.Hauel AP. 1939. The cadmium–nickel storage battery. Trans. Electrochem. Soc. 76, 435–452 (doi:10.1149/1.3500304) [Google Scholar]
- 3.Robert AK. 1954. Alkaline electric storage battery cell. Patent no. US2677006 A. [Google Scholar]
- 4.Bode H, Dehmelt K, Witte J. 1966. Zur Kenntnis der Nickelhydroxidelektrode-I. Über das Nickel(II)-Hydroxidhydrat. Electrochim. Acta 11, 1079–1087 (doi:10.1016/0013-4686(66)80045-2) [Google Scholar]
- 5.Reddy TB, Linden D. 2011. Linden's handbook of batteries, p. 1200 4th edn New York, NY: McGraw-Hill. [Google Scholar]
- 6.McBreen J. 2011. Nickel hydroxides. In Handbook of battery materials (eds Daniel C, Besenhard JO.), pp. 149–168, 2nd edn Weinheim, Germany: Wiley-VCH Verlag GmbH & Co. KGaA. [Google Scholar]
- 7.Ran J, Yu J, Jaroniec M. 2011. Ni(OH)2 modified CdS nanorods for highly efficient visible-light-driven photocatalytic H2 generation. Green Chem. 13, 2708–2713 (doi:10.1039/C1GC15465F) [Google Scholar]
- 8.Yan Z, Yu X, Zhang Y, Jia H, Sun Z, Du P. 2014. Enhanced visible light-driven hydrogen production from water by a noble-metal-free system containing organic dye-sensitized titanium dioxide loaded with nickel hydroxide as the cocatalyst. Appl. Catal. B 160–161, 173–178 (doi:10.1016/j.apcatb.2014.05.017) [Google Scholar]
- 9.Yu J, Hai Y, Cheng B. 2011. Enhanced photocatalytic H2-production activity of TiO2 by Ni(OH)2 cluster modification. J. Phys. Chem. C 115, 4953–4958 (doi:10.1021/jp111562d) [Google Scholar]
- 10.Gao M, Sheng W, Zhuang Z, Fang Q, Gu S, Jiang J, Yan Y. 2014. Efficient water oxidation using nanostructured α-nickel–hydroxide as an electrocatalyst. J. Am. Chem. Soc. 136, 7077–7084 (doi:10.1021/ja502128j) [DOI] [PubMed] [Google Scholar]
- 11.Lyons MEG, Cakara A, O'Brien P, Godwin I, Coyle RL. 2012. Redox, pH sensing and electrolytic water splitting properties of electrochemically generated nickel hydroxide thin films in aqueous alkaline solution. Int. J. Electrochem. Sci. 7, 11768–11795 [Google Scholar]
- 12.Vidotti M, Torresi R, De Torresi SIC. 2010. Nickel hydroxide modified electrodes: a review study concerning its structural and electrochemical properties aiming the application in electrocatalysis, electrochromism and secondary batteries. Quim. Nova 33, 2176–2186 (doi:10.1590/S0100-40422010001000030) [Google Scholar]
- 13.Aghazadeh M, Ghaemi M, Sabour B, Dalvand S. 2014. Electrochemical preparation of α-Ni(OH)2 ultrafine nanoparticles for high-performance supercapacitors. J. Solid State Electrochem 18, 1569–1584 (doi:10.1007/s10008-014-2381-7) [Google Scholar]
- 14.Chen J-C, Hsu C-T, Hu C-C. 2014. Superior capacitive performances of binary nickel–cobalt hydroxide nanonetwork prepared by cathodic deposition. J. Power Sources 253, 205–213 (doi:10.1016/j.jpowsour.2013.12.073) [Google Scholar]
- 15.Cheng H, Su AD, Li S, Nguyen ST, Lu L, Lim CYH, Duong HM. 2014. Facile synthesis and advanced performance of Ni(OH)2/CNTs nanoflake composites on supercapacitor applications. Chem. Phys. Lett. 601, 168–173 (doi:10.1016/j.cplett.2014.03.071) [Google Scholar]
- 16.Huang J, Lei T, Wei X, Liu X, Liu T, Cao D, Yin J, Wang G. 2013. Effect of Al-doped β-Ni(OH)2 nanosheets on electrochemical behaviors for high performance supercapacitor application. J. Power Sources 232, 370–375 (doi:10.1016/j.jpowsour.2013.01.081) [Google Scholar]
- 17.Ma X, Liu J, Gong X, Che R. 2014. A facile phase transformation method for the preparation of 3D flower-like β-Ni(OH)2/GO/CNTs composite with excellent supercapacitor performance. J. Mater. Chem. A 2, 12692–12696 (doi:10.1039/C4TA02221A) [Google Scholar]
- 18.Sebastian M, Nethravathi C, Rajamathi M. 2013. Interstratified hybrids of α-hydroxides of nickel and cobalt as supercapacitor electrode materials. Mater. Res. Bull. 48, 2715–2719 (doi:10.1016/j.materresbull.2013.03.029) [Google Scholar]
- 19.Cordoba-Torresi SI, Gabrielli C, Goff AH-L, Torresi R. 1991. Electrochromic behavior of nickel oxide electrodes I. Identification of the colored state using quartz crystal microbalance. J. Electrochem. Soc. 138, 1548–1553 (doi:10.1149/1.2085830) [Google Scholar]
- 20.Mortimer R, Sialvi M, Varley T, Wilcox G. 2014. An in situ colorimetric measurement study of electrochromism in the thin-film nickel hydroxide/oxyhydroxide system. J. Solid State Electrochem. 18, 3359–3367 (doi:10.1007/s10008-014-2618-5) [Google Scholar]
- 21.Sharma PK, Fantini MCA, Gorenstein A. 1998. Synthesis, characterization and electrochromic properties of NiOxHy thin film prepared by a sol–gel method. Solid State Ionics 113–115, 457–463 (doi:10.1016/S0167-2738(98)00310-5) [Google Scholar]
- 22.Fan Y, Yang Z, Cao X, Liu P, Chen S, Cao Z. 2014. Hierarchical macro-mesoporous Ni(OH)2 for nonenzymatic electrochemical sensing of glucose. J. Electrochem. Soc. 161, B201–B206 (doi:10.1149/2.0251410jes) [Google Scholar]
- 23.Miao Y, Ouyang L, Zhou S, Xu L, Yang Z, Xiao M, Ouyang R. 2014. Electrocatalysis and electroanalysis of nickel, its oxides, hydroxides and oxyhydroxides toward small molecules. Biosensors Bioelectr. 53, 428–439 (doi:10.1016/j.bios.2013.10.008) [DOI] [PubMed] [Google Scholar]
- 24.Rastgar S, Shahrokhian S. 2014. Nickel hydroxide nanoparticles-reduced graphene oxide nanosheets film: layer-by-layer electrochemical preparation, characterization and rifampicin sensory application. Talanta 119, 156–163 (doi:10.1016/j.talanta.2013.10.047) [DOI] [PubMed] [Google Scholar]
- 25.Yang H, Gao G, Teng F, Liu W, Chen S, Ge Z. 2014. Nickel hydroxide nanoflowers for a nonenzymatic electrochemical glucose sensor. J. Electrochem. Soc. 161, B216–B219 (doi:10.1149/2.0521410jes) [Google Scholar]
- 26.Szytula A, Murasik A, Balanda M. 1971. Neutron diffraction study of Ni(OH)2. Phys. Status Solidi B 43, 125–128 (doi:10.1002/pssb.2220430113) [Google Scholar]
- 27.Greaves C, Thomas MA. 1986. Refinement of the structure of deuterated nickel hydroxide, Ni(OD)2, by powder neutron diffraction and evidence for structural disorder in samples with high surface area. Acta Crystallogr. Sect. B 42, 51–55 (doi:10.1107/S0108768186098592) [Google Scholar]
- 28.McEwen RS. 1971. Crystallographic studies on nickel hydroxide and the higher nickel oxides. J. Phys. Chem. 75, 1782–1789 (doi:10.1021/j100681a004) [Google Scholar]
- 29.Livingstone A, Bish D. 1982. On the new mineral theophrastite, a nickel hydroxide, from Unst, Shetland, Scotland. Mineral. Mag. 46, 1–5 (doi:10.1180/minmag.1982.046.338.01) [Google Scholar]
- 30.Marcopoulos T, Economou M. 1981. Theophrastite, Ni(OH)2, a new mineral from Northern Greece. Am. Mineral. 66, 1020–1021. [Google Scholar]
- 31.Giacovazzo C, et al. 2011. Fundamentals of crystallography, p. 864, 3rd edn Oxford, UK: Oxford University Press. [Google Scholar]
- 32.Pandya KI, O'Grady WE, Corrigan DA, McBreen J, Hoffman RW. 1990. Extended X-ray absorption fine structure investigations of nickel hydroxides. J. Phys. Chem. 94, 21–26 (doi:10.1021/j100364a005) [Google Scholar]
- 33.Delmas C, Faure C, Borthomieu Y. 1992. The effect of cobalt on the chemical and electrochemical behaviour of the nickel hydroxide electrode. Mater. Sci. Eng. 13, 89–96 (doi:10.1016/0921-5107(92)90147-2) [Google Scholar]
- 34.Rajamathi M, Kamath PV. 1998. On the relationship between α-nickel hydroxide and the basic salts of nickel. J. Power Sources 70, 118–121 (doi:10.1016/S0378-7753(97)02656-6) [Google Scholar]
- 35.Barnard R, Randell CF, Tye FL. 1981. Studies concerning the ageing of α and β-Ni(OH)2 in relation to nickel–cadmium cells. In Proc. 12th Int. Power Sources Symp Brighton, September 1980 (ed. Thompson J.), pp. 401–425 London, UK: Academic Press. [Google Scholar]
- 36.Le Bihan S, Figlarz M. 1973. Note sur l’article: ‘Zur kenntnis der Nickel-hydroxidelektrode—III. Thermogravimetrische Untersuchungen an Nickel(II)-hydroxigen’ par W. Dennstedt et W. Löser. Electrochim. Acta 18, 123–124 (doi:10.1016/0013-4686(73)87021-5) [Google Scholar]
- 37.Le Bihan S, Guenot J, Figlarz M. 1970. Sur la cristallogénèse de l'hydroxyde de nickel Ni(OH)2. C. R. de l'Académie des Science: Paris 270, 2131–2133. [Google Scholar]
- 38.Hall DS, Lockwood DJ, Poirier S, Bock C, MacDougall BR. 2012. Raman and Infrared spectroscopy of α and β phases of thin nickel hydroxide films electrochemically formed on nickel. J. Phys. Chem. A 116, 6771–6784 (doi:10.1021/jp303546r) [DOI] [PubMed] [Google Scholar]
- 39.Barnard R, Randell CF, Tye FL. 1980. Studies concerning charged nickel hydroxide electrodes. I. Measurement of reversible potentials. J. Appl. Electrochem. 10, 109–125 (doi:10.1007/BF00937345) [Google Scholar]
- 40.Faure C, Delmas C, Fouassier M. 1991. Characterization of a turbostratic α-nickel hydroxide quantitatively obtained from an NiSO4 solution. J. Power Sources 35, 279–290 (doi:10.1016/0378-7753(91)80112-B) [Google Scholar]
- 41.Rajamathi M, Vishnu Kamath P, Seshadri R. 2000. Polymorphism in nickel hydroxide: role of interstratification. J. Mater. Chem. 10, 503–506 (doi:10.1039/a905651c) [Google Scholar]
- 42.Bernard MC, Cortes R, Keddam M, Takenouti H, Bernard P, Senyarich S. 1996. Structural defects and electrochemical reactivity of β-Ni(OH)2. J. Power Sources 63, 247–254 (doi:10.1016/s0378-7753(96)02482-2) [Google Scholar]
- 43.Delmas C, Tessier C. 1997. Stacking faults in the structure of nickel hydroxide: a rationale of its high electrochemical activity. J. Mater. Chem. 7, 1439–1443 (doi:10.1039/a701037k) [Google Scholar]
- 44.Ramesh TN, Kamath PV. 2008. The effect of stacking faults on the electrochemical performance of nickel hydroxide electrodes. Mater. Res. Bull. 43, 2827–2832 (doi:10.1016/j.materresbull.2008.06.010) [Google Scholar]
- 45.Tessier C, Haumesser PH, Bernard P, Delmas C. 1999. The structure of Ni(OH)2: from the ideal material to the electrochemically active one. J. Electrochem. Soc. 146, 2059–2067 (doi:10.1149/1.1391892) [Google Scholar]
- 46.Dennstedt W, Löser W. 1971. Zur kenntnis der nickelhydroxid-elektrode—III. Thermogravimetrische untersuchungen an Nickel(II)-hydroxiden. Electrochim. Acta 16, 429–435 (doi:10.1016/0013-4686(71)85015-6) [Google Scholar]
- 47.Kober FP. On the structure of electrochemically active hydrated nickel-oxide electrodes. In Power Sources: Proceedings for the 5th International Power Sources Symposium (ed. Collins DH.), pp. 257–268 Brighton, London: Academic Press. [Google Scholar]
- 48.Kober FP. 1965. Analysis of the charge–discharge characteristics of nickel-oxide electrodes by infrared spectroscopy. J. Electrochem. Soc. 112, 1064–1067 (doi:10.1149/1.2423361) [Google Scholar]
- 49.Kober FP. 1967. Infrared spectroscopic investigation of charged nickel hydroxide electrodes. J. Electrochem. Soc. 114, 215–218 (doi:10.1149/1.2426549) [Google Scholar]
- 50.Brown ME. 2001. Introduction to thermal analysis: techniques and applications, p. 264, 2nd edn Boston, MA: Kluwer Academic Publishers. [Google Scholar]
- 51.Nicholls JR. 1954. Thermogravimetric analysis. Nature 173, 1011–1012 (doi:10.1038/1731011b0) [Google Scholar]
- 52.Audemer A, Delahaye A, Farhi R, Sac-Epée N, Tarascon J-M. 1997. Electrochemical and Raman studies of beta-type nickel hydroxides Ni1−xCox(OH)2 electrode materials. J. Electrochem. Soc. 144, 2614–2620 (doi:10.1149/1.1837873) [Google Scholar]
- 53.Freitas MBJG. 2001. Nickel hydroxide powder for NiOċOH/Ni(OH)2 electrodes of the alkaline batteries. J. Power Sources 93, 163–173 (doi:10.1016/S0378-7753(00)00570-X) [Google Scholar]
- 54.Mani B, de Neufville JP. 1988. Dehydration of chemically and electrochemically impregnated (CI and EI) nickel hydroxide electrodes. J. Electrochem. Soc. 135, 800–803 (doi:10.1149/1.2095777) [Google Scholar]
- 55.Jeevanandam P, Koltypin Y, Gedanken A. 2001. Synthesis of nanosized α-nickel hydroxide by a sonochemical method. Nano Lett. 1, 263–266 (doi:10.1021/nl010003p) [Google Scholar]
- 56.Radha AV, Kamath PV, Shivakumara C. 2007. Order and disorder among the layered double hydroxides: combined Rietveld and DIFFaX approach. Acta Crystallogr. Section B 63, 243–250 (doi:10.1107/S010876810700122X) [DOI] [PubMed] [Google Scholar]
- 57.Ramesh TN. 2009. Crystallite size effects in stacking faulted nickel hydroxide and its electrochemical behaviour. Mater. Chem. Phys. 114, 618–623 (doi:10.1016/j.matchemphys.2008.10.017) [Google Scholar]
- 58.Sebastian MT, Krishna P. 1994. Random, non-random, and periodic faulting in crystals, p. 383 Langhorne, PA: Gordon and Breach Science Publishers. [Google Scholar]
- 59.Ramesh TN, Jayashree RS, Kamath PV. 2003. Disorder in layered hydroxides: DIFFaX simulation of the X-ray powder diffraction patterns of nickel hydroxide. Clays Clay Miner. 51, 570–576 (doi:10.1346/ccmn.2003.0510511) [Google Scholar]
- 60.de Oliveira EF, Hase Y. 2003. Infrared study of magnesium–nickel hydroxide solid solutions. Vib. Spectrosc. 31, 19–24 (doi:10.1016/s0924-2031(02)00049-8) [Google Scholar]
- 61.Zhang W, Jiang W, Yu L, Fu Z, Xia W, Yang M. 2009. Effect of nickel hydroxide composition on the electrochemical performance of spherical Ni(OH)2 positive materials for Ni–MH batteries. Int. J. Hydrogen Energy 34, 473–480 (doi:10.1016/j.ijhydene.2008.07.129) [Google Scholar]
- 62.Begum SN, Muralidharan VS, Ahmed Basha C. 2009. The influences of some additives on electrochemical behaviour of nickel electrodes. Int. J. Hydrogen Energy 34, 1548–1555 (doi:10.1016/j.ijhydene.2008.11.074) [Google Scholar]
- 63.Watanabe K, Koseki M, Kumagai N. 1996. Effect of cobalt addition to nickel hydroxide as a positive material for rechargeable alkaline batteries. J. Power Sources 58, 23–28 (doi:10.1016/0378-7753(95)02272-4) [Google Scholar]
- 64.Watanabe K, Kumagai N. 1998. Thermodynamic studies of cobalt and cadmium additions to nickel hydroxide as material for positive electrodes. J. Power Sources 76, 167–174 (doi:10.1016/S0378-7753(98)00150-5) [Google Scholar]
- 65.Chen H, Wang JM, Zhao YL, Zhang JQ, Cao CN. 2005. Electrochemical performance of Zn-substituted Ni(OH)2 for alkaline rechargeable batteries. J. Solid State Electrochem. 9, 421–428 (doi:10.1007/s10008-004-0578-x) [Google Scholar]
- 66.Hu B, Chen S-F, Liu S-J, Wu Q-S, Yao W-T, Yu S-H. 2008. Controllable synthesis of zinc-substituted α- and β-nickel hydroxide nanostructures and their collective intrinsic properties. Chem. Eur. J. 14, 8928–8938 (doi:10.1002/chem.200800458) [DOI] [PubMed] [Google Scholar]
- 67.Bao J, Zhu Y, Xu QS, Zhuang YH, Zhao RD, Zeng YY, Zhong HL. 2012. Structure and electrochemical performance of Cu and Al codoped nanometer α-nickel hydroxide. Adv. Mater. Res. 479, 230–233 (doi:10.4028/www.scientific.net/AMR.479-481.230) [Google Scholar]
- 68.Chen H, Wang JM, Pan T, Zhao YL, Zhang JQ, Cao CN. 2005. The structure and electrochemical performance of spherical Al-substituted α-Ni(OH)2 for alkaline rechargeable batteries. J. Power Sources 143, 243–255 (doi:10.1016/j.jpowsour.2004.11.041) [Google Scholar]
- 69.Chen X, Long C, Lin C, Wei T, Yan J, Jiang L, Fan Z. 2014. Al and Co co-doped α-Ni(OH)2/graphene hybrid materials with high electrochemical performances for supercapacitors. Electrochim. Acta 137, 352–358 (doi:10.1016/j.electacta.2014.05.151) [Google Scholar]
- 70.Jayashree RS, Vishnu Kamath P. 2001. Suppression of the α → β-nickel hydroxide transformation in concentrated alkali: role of dissolved cations. J. Appl. Electrochem. 31, 1315–1320 (doi:10.1023/a:1013876006707) [Google Scholar]
- 71.Kamath PV, Dixit M, Indira L, Shukla AK, Kumar VG, Munichandraiah N. 1994. Stabilized α-Ni(OH)2 as electrode material for alkaline secondary cells. J. Electrochem. Soc. 141, 2956–2959 (doi:10.1149/1.2059264) [Google Scholar]
- 72.Li YW, Yao JH, Liu CJ, Zhao WM, Deng WX, Zhong SK. 2010. Effect of interlayer anions on the electrochemical performance of Al-substituted α-type nickel hydroxide electrodes. Int. J. Hydrogen Energy 35, 2539–2545 (doi:10.1016/j.ijhydene.2010.01.015) [Google Scholar]
- 73.Qi J, Xu P, Lv Z, Liu X, Wen A. 2008. Effect of crystallinity on the electrochemical performance of nanometer Al-stabilized α-nickel hydroxide. J. Alloys Compd. 462, 164–169 (doi:10.1016/j.jallcom.2007.07.102) [Google Scholar]
- 74.Wang CY, Zhong S, Bradhurst DH, Liu HK, Dou SX. 2002. Ni/Al/Co-substituted α-Ni(OH)2 as electrode materials in the nickel metal hydride cell. J. Alloys Compd. 330–332, 802–805 (doi:10.1016/S0925-8388(01)01515-8) [Google Scholar]
- 75.Zhao YL, Wang JM, Chen H, Pan T, Zhang JQ, Cao CN. 2004. Different additives-substituted α-nickel hydroxide prepared by urea decomposition. Electrochim. Acta 50, 91–98 (doi:10.1016/j.electacta.2004.07.016) [Google Scholar]
- 76.Guerlou-Demourgues L, Delmas C. 1996. Electrochemical behavior of the manganese-substituted nickel hydroxides. J. Electrochem. Soc. 143, 561–566 (doi:10.1149/1.1836480) [Google Scholar]
- 77.Zhou F, Zhao X, van Bommel A, Rowe AW, Dahn JR. 2009. Coprecipitation synthesis of NixMn1−x(OH)2 mixed hydroxides. Chem. Mater. 22, 1015–1021 (doi:10.1021/cm9018309) [Google Scholar]
- 78.Demourgues-Guerlou L, Delmas C. 1993. Structure and properties of precipitated nickel–iron hydroxides. J. Power Sources 45, 281–289 (doi:10.1016/0378-7753(93)80017-J) [Google Scholar]
- 79.Liu C, Huang L, Li Y, Sun D. 2010. Synthesis and electrochemical performance of amorphous nickel hydroxide codoped with Fe3+ and CO2−3. Ionics 16, 215–219 (doi:10.1007/s11581-009-0383-8) [Google Scholar]
- 80.Delmas C, Braconnier JJ, Borthomieu Y, Hagenmuller P. 1987. New families of cobalt substituted nickel oxyhydroxides and hydroxides obtained by soft chemistry. Mater. Res. Bull. 22, 741–751 (doi:10.1016/0025-5408(87)90027-4) [Google Scholar]
- 81.Martins PR, Ferreira LMC, Araki K, Angnes L. 2014. Influence of cobalt content on nanostructured alpha-phase-nickel hydroxide modified electrodes for electrocatalytic oxidation of isoniazid. Sensors Actuators B 192, 601–606 (doi:10.1016/j.snb.2013.11.029) [Google Scholar]
- 82.Nethravathi C, Ravishankar N, Shivakumara C, Rajamathi M. 2007. Nanocomposites of α-hydroxides of nickel and cobalt by delamination and co-stacking: enhanced stability of α-motifs in alkaline medium and electrochemical behaviour. J. Power Sources 172, 970–974 (doi:10.1016/j.jpowsour.2007.01.098) [Google Scholar]
- 83.Bao J, Zhu Y, Zhang Z, Xu Q, Zhao W, Chen J, Zhang W, Han Q. 2013. Structure and electrochemical properties of nanometer Cu substituted α-nickel hydroxide. Mater. Res. Bull. 48, 422–428 (doi:10.1016/j.materresbull.2012.10.059) [Google Scholar]
- 84.Dixit M, Kamath PV, Gopalakrishnan J. 1999. Zinc-substituted α-nickel hydroxide as an electrode material for alkaline secondary cells. J. Electrochem. Soc. 146, 79–82 (doi:10.1149/1.1391567) [Google Scholar]
- 85.Tessier C, Guerlou-Demourgues L, Faure C, Basterreix MT, Nabias G, Delmas C. 2000. Structural and textural evolution of zinc-substituted nickel hydroxide electrode materials upon ageing in KOH and upon redox cycling. Solid State Ionics 133, 11–23 (doi:10.1016/S0167-2738(00)00690-1) [Google Scholar]
- 86.Ren J, Zhou Z, Gao XP, Yan J. 2006. Preparation of porous spherical α-Ni(OH)2 and enhancement of high-temperature electrochemical performances through yttrium addition. Electrochim. Acta 52, 1120–1126 (doi:10.1016/j.electacta.2006.07.028) [Google Scholar]
- 87.Xu Q, Zhu Y, Han Q, Zhao R, Zhuang Y, Liu Y, Zhang S, Miao C. 2014. Preparation of Yb-substituted α-Ni(OH)2 and its physicochemical properties. J. Alloys Compd. 584, 1–6 (doi:10.1016/j.jallcom.2013.08.097) [Google Scholar]
- 88.Bardé F, Palacin MR, Chabre Y, Isnard O, Tarascon JM. 2004. In situ neutron powder diffraction of a nickel hydroxide electrode. Chem. Mater. 16, 3936–3948 (doi:10.1021/cm0401286) [Google Scholar]
- 89.Minkova N, Krusteva M, Nikolov G. 1984. Spectroscopic study of nickel hydroxyde, nickel carbonate-hexahydrate and nickel hydroxocarbonate. J. Mol. Struct. 115, 23–26 (doi:10.1016/0022-2860(84)80006-x) [Google Scholar]
- 90.Delahaye-Vidal A, Beaudoin B, Sac-Epée N, Tekaia-Elhsissen K, Audemer A, Figlarz M. 1996. Structural and textural investigations of the nickel hydroxide electrode. Solid State Ionics 84, 239–248 (doi:10.1016/0167-2738(96)00030-6) [Google Scholar]
- 91.Streinz CC, Hartman AP, Motupally S, Weidner JW. 1995. The effect of current and nickel nitrate concentration on the deposition of nickel hydroxide films. J. Electrochem. Soc. 142, 1084–1089 (doi:10.1149/1.2044134) [Google Scholar]
- 92.Lee JW, Ko JM, Kim J-D. 2011. Hierarchical microspheres based on α-Ni(OH)2 nanosheets intercalated with different anions: synthesis, anion exchange, and effect of intercalated anions on electrochemical capacitance. J. Phys. Chem. C 115, 19445–19454 (doi:10.1021/jp206379h) [Google Scholar]
- 93.Xu L, Ding Y-S, Chen C-H, Zhao L, Rimkus C, Joesten R, Suib SL. 2007. 3D flowerlike α-nickel hydroxide with enhanced electrochemical activity synthesized by microwave-assisted hydrothermal method. Chem. Mater. 20, 308–316 (doi:10.1021/cm702207w) [Google Scholar]
- 94.Hall DS, Lockwood DJ, Poirier S, Bock C, MacDougall BR. 2014. Applications of in situ Raman spectroscopy for identifying nickel hydroxide materials and surface layers during chemical aging. ACS Appl. Mater. Interfaces 6, 3141–3149 (doi:10.1021/am405419k) [DOI] [PubMed] [Google Scholar]
- 95.Alberti G, Bein T. 1996. Layered solids and their intercalation chemistry. In Solid-state supramolecular chemistry: two- and three-dimensional inorganic networks (eds Alberti G, Bein T.), pp. 1–23 New York, NY: Pergamon Press. [Google Scholar]
- 96.Guerlou-Demourgues L, Denage C, Delmas C. 1994. New manganese-substituted nickel hydroxides: Part 1. Crystal chemistry and physical characterization. J. Power Sources 52, 269–274 (doi:10.1016/0378-7753(94)02023-X) [Google Scholar]
- 97.Rajamathi MN, Subbanna G, Vishnu Kamath P. 1997. On the existence of a nickel hydroxide phase which is neither α nor β. J. Mater. Chem. 7, 2293–2296 (doi:10.1039/a700390k) [Google Scholar]
- 98.Hendricks S, Teller E. 1942. X-ray interference in partially ordered layer lattices. J. Chem. Phys. 10, 147–167 (doi:10.1063/1.1723678) [Google Scholar]
- 99.Häring P, Kötz R. 1995. Nanoscale thickness changes of nickel hydroxide films during electrochemical oxidation/reduction monitored by in situ atomic force microscopy. J. Electroanal. Chem. 385, 273–277 (doi:10.1016/0022-0728(94)03869-5) [Google Scholar]
- 100.Giarola D, Catarini da Silva P, Urbano A, Oliveira F, Texeira Tarley C, Dall'Antonia L. 2014. Surfactant effect on electrochemical-induced synthesis of α-Ni(OH)2. J. Solid State Electrochem. 18, 497–504 (doi:10.1007/s10008-013-2280-3) [Google Scholar]
- 101.Coudun C, Grillon F, Hochepied J-F. 2006. Surfactant effects on pH-controlled synthesis of nickel hydroxides. Colloids Surfaces A 280, 23–31 (doi:10.1016/j.colsurfa.2006.01.018) [Google Scholar]
- 102.Ida S, Shiga D, Koinuma M, Matsumoto Y. 2008. Synthesis of hexagonal nickel hydroxide nanosheets by exfoliation of layered nickel hydroxide intercalated with dodecyl sulfate ions. J. Am. Chem. Soc. 130, 14038–14039 (doi:10.1021/ja804397n) [DOI] [PubMed] [Google Scholar]
- 103.Nethravathi C, Rajamathi M, Ravishankar N, Basit L, Felser C. 2010. Synthesis of graphene oxide-intercalated α-hydroxides by metathesis and their decomposition to graphene/metal oxide composites. Carbon 48, 4343–4350 (doi:10.1016/j.carbon.2010.07.047) [Google Scholar]
- 104.Tan Y, Srinivasan S, Choi K-S. 2005. Electrochemical deposition of mesoporous nickel hydroxide films from dilute surfactant solutions. J. Am. Chem. Soc. 127, 3596–3604 (doi:10.1021/ja0434329) [DOI] [PubMed] [Google Scholar]
- 105.Wang D, Yan W, Botte GG. 2011. Exfoliated nickel hydroxide nanosheets for urea electrolysis. Electrochem. Commun. 13, 1135–1138 (doi:10.1016/j.elecom.2011.07.016) [Google Scholar]
- 106.Wang M, Ren W, Zhao Y, Liu Y, Cui H. 2013. One-pot synthesis of powder-form β-Ni(OH)2 monolayer nanosheets with high electrochemical performance. J. Nanoparticle Res. 15, 1–8 (doi:10.1007/s11051-013-1849-1) [Google Scholar]
- 107.Jacobson AJ. 1996. Colloidal dispersion of compounds with layer and chain structures. In Solid-state supramolecular chemistry: two- and three-dimensional inorganic networks (eds Alberti G, Bein T.), pp. 315–335 New York, NY: Pergamon Press. [Google Scholar]
- 108.Nethravathi C, Viswanath B, Sebastian M, Rajamathi M. 2010. Exfoliation of α-hydroxides of nickel and cobalt in water. J. Colloid Interface Sci. 345, 109–115 (doi:10.1016/j.jcis.2010.01.047) [DOI] [PubMed] [Google Scholar]
- 109.Petrov K, Zotov N, Mirtcheva E, Garcia-Martinez O, Rojas RM. 1994. Effect of composition on the lattice parameters and thermal behaviour of nickel(II)–cobalt(II) hydroxide nitrate solid solutions. J. Mater. Chem. 4, 611–614 (doi:10.1039/JM9940400611) [Google Scholar]
- 110.Gallezot P, Prettre M. 1969. Étude structurale d'un nitrate basique de nickel non stœchiométrique. Bull. Soc. Chim. Fr. 407, 407–411. [Google Scholar]
- 111.Louër M, Louër D, Grandjean D. 1973. Etude structurale des hydroxynitrates de nickel et de zinc. I. Classification structurale. Acta Crystallogr. Sect. B 29, 1696–1703 (doi:10.1107/S0567740873005285) [Google Scholar]
- 112.Nickel EH, Berry LG. 1981. The new mineral nullaginite and additional data on the related minerals rosasite and glaukosphaerite. Can. Mineral. 19, 315–324. [Google Scholar]
- 113.Nickel EH, Graham J. 1987. Paraotwayite, a new nickel hydroxide mineral from Western Australia. Can. Mineral. 25, 409–411. [Google Scholar]
- 114.Nickel EH, Robinson BW, Davis C, MacDonald RD. 1977. Otwayite, a new nickel mineral from Western Australia. Am. Mineral. 62, 999–1002. [Google Scholar]
- 115.Hall DS. 2014. An electrochemical and spectroscopic investigation of nickel electrodes in alkaline media for applications in electro-catalysis. PhD thesis, University of Ottawa, Ottawa, Ontario, Canada. [Google Scholar]
- 116.Zhang S, Zeng HC. 2009. Self-assembled hollow spheres of β-Ni(OH)2 and their derived nanomaterials. Chem. Mater. 21, 871–883 (doi:10.1021/cm8028593) [Google Scholar]
- 117.Gourrier L, Deabate S, Michel T, Paillet M, Hermet P, Bantignies J-L, Henn F. 2011. Characterization of unusually large ‘pseudo-single crystal’ of β-nickel hydroxide. J. Phys. Chem. C 115, 15067–15074 (doi:10.1021/jp203222t) [Google Scholar]
- 118.Dong L, Chu Y, Sun W. 2008. Controllable synthesis of nickel hydroxide and porous nickel oxide nanostructures with different morphologies. Chem. Eur. J. 14, 5064–5072 (doi:10.1002/chem.200701627) [DOI] [PubMed] [Google Scholar]
- 119.Zhu Z, Wei N, Liu H, He Z. 2011. Microwave-assisted hydrothermal synthesis of Ni(OH)2 architectures and their in situ thermal convention to NiO. Adv. Powder Technol. 22, 422–426 (doi:10.1016/j.apt.2010.06.008) [Google Scholar]
- 120.Ramesh TN, Kamath PV. 2006. Synthesis of nickel hydroxide: effect of precipitation conditions on phase selectivity and structural disorder. J. Power Sources 156, 655–661 (doi:10.1016/j.jpowsour.2005.05.050) [Google Scholar]
- 121.Soler-Illia GJDAA, Jobbágy M, Regazzoni AE, Blesa MA. 1999. Synthesis of nickel hydroxide by homogeneous alkalinization. Precipitation mechanism. Chem. Mater. 11, 3140–3146 (doi:10.1021/cm9902220) [Google Scholar]
- 122.Wallner H, Gatterer K. 2002. Growth of pure Ni(OH)2 single crystals from solution—control of the crystal size. Z. Anorg. Allg. Chem. 628, 2818–2820 (doi:10.1002/1521-3749(200213)628:13<2818::AID-ZAAC2818>3.0.CO;2-J) [Google Scholar]
- 123.Rodrigues I, Wontcheu J, MacNeil D. 2012. A novel coprecipitation method towards the synthesis of NixMnxCo(1-2x)(OH)2 for the preparation of lithium metal oxides. J. Solid State Electrochem. 16, 1121–1132 (doi:10.1007/s10008-011-1502-9) [Google Scholar]
- 124.Sun Y-K, Lee D-J, Lee YJ, Chen Z, Myung S-T. 2013. Cobalt-free nickel rich layered oxide cathodes for lithium-ion batteries. ACS Appl. Mater. Interfaces 5, 11434–11440 (doi:10.1021/am403684z) [DOI] [PubMed] [Google Scholar]
- 125.Camardese J, McCalla E, Abarbanel DW, Dahn JR. 2014. Determination of shell thickness of spherical core-shell NixMn1−x(OH)2 particles via absorption calculations of X-ray diffraction patterns. J. Electrochem. Soc. 161, A814–A820 (doi:10.1149/2.084405jes) [Google Scholar]
- 126.Kamath PV, Subbanna GN. 1992. Electroless nickel hydroxide: synthesis and characterization. J. Appl. Electrochem. 22, 478–482 (doi:10.1007/BF01077552) [Google Scholar]
- 127.Fievet F, Figlarz M. 1975. Preparation and study by electron microscopy of the development of texture with temperature of a porous oxhydroxide nickel oxide. J. Catal. 39, 350–356 (doi:10.1016/0021-9517(75)90300-0) [Google Scholar]
- 128.Chen X, Long C, Lin C, Wei T, Yan J, Jiang L, Fan Z. 2014. Al and Co co-doped α-Ni(OH)2/graphene hybrid materials with high electrochemical performance for supercapacitors. Electrochim. Acta. 137, 352–358 (doi:10.1016/j.electacta.2014.05.151) [Google Scholar]
- 129.Yang D, Wang R, He M, Zhang J, Liu Z. 2005. Ribbon- and boardlike nanostructures of nickel hydroxide: synthesis, characterization, and electrochemical properties. J. Phys. Chem. B 109, 7654–7658 (doi:10.1021/jp050083b) [DOI] [PubMed] [Google Scholar]
- 130.Li J, Luo F, Tian X, Lei Y, Yuan H, Xiao D. 2013. A facile approach to synthesis coral-like nanoporous β-Ni(OH)2 and its supercapacitor application. J. Power Sources 243, 721–727 (doi:10.1016/j.jpowsour.2013.05.172) [Google Scholar]
- 131.Song Q, Tang Z, Guo H, Chan SLI. 2002. Structural characteristics of nickel hydroxide synthesized by a chemical precipitation route under different pH values. J. Power Sources 112, 428–434 (doi:10.1016/S0378-7753(02)00396-8) [Google Scholar]
- 132.Feng K, Li W, Xie S, Lu X. 2014. Nickel hydroxide decorated hydrogenated zinc oxide nanorod arrays with enhanced photoelectrochemical performance. Electrochim. Acta 137, 108–113 (doi:10.1016/j.electacta.2014.05.152) [Google Scholar]
- 133.Chen J, Bradhurst DH, Dou SX, Liu HK. 1999. Nickel hydroxide as an active material for the positive electrode in rechargeable alkaline batteries. J. Electrochem. Soc. 146, 3606–3612 (doi:10.1149/1.1392522) [Google Scholar]
- 134.Ferrando WA. 1985. Performance of suspension-impregnated sintered nickel composite electrodes. J. Electrochem. Soc. 132, 2417–2419 (doi:10.1149/1.2113588) [Google Scholar]
- 135.Oshitani M, Yufu H, Takashima K, Tsuji S, Matsumaru Y. 1989. Development of a pasted nickel electrode with high active material utilization. J. Electrochem. Soc. 136, 1590–1593 (doi:10.1149/1.2096974) [Google Scholar]
- 136.Wang Y, Cao D, Wang G, Wang S, Wen J, Yin J. 2011. Spherical clusters of β-Ni(OH)2 nanosheets supported on nickel foam for nickel metal hydride battery. Electrochim. Acta 56, 8285–8290 (doi:10.1016/j.electacta.2011.06.098) [Google Scholar]
- 137.Jayashree RS, Kamath PV. 1999. Factors governing the electrochemical synthesis of α-nickel (II) hydroxide. J. Appl. Electrochem. 29, 449–454 (doi:10.1023/A:1003493711239) [Google Scholar]
- 138.Tizfahm J, Safibonab B, Aghazadeh M, Majdabadi A, Sabour B, Dalvand S. 2014. Supercapacitive behavior of β-Ni(OH)2 nanospheres prepared by a facile electrochemical method. Colloids Surf. A 443, 544–551 (doi:10.1016/j.colsurfa.2013.12.024) [Google Scholar]
- 139.Aghazadeh M, Golikand AN, Ghaemi M. 2011. Synthesis, characterization, and electrochemical properties of ultrafine β-Ni(OH)2 nanoparticles. Int. J. Hydrogen Energy 36, 8674–8679 (doi:10.1016/j.ijhydene.2011.03.144) [Google Scholar]
- 140.Zhao D-D, Bao S-J, Zhou W-J, Li H-L. 2007. Preparation of hexagonal nanoporous nickel hydroxide film and its application for electrochemical capacitor. Electrochem. Commun. 9, 869–874 (doi:10.1016/j.elecom.2006.11.030) [Google Scholar]
- 141.Hutton LA, Vidotti M, Patel AN, Newton ME, Unwin PR, Macpherson JV. 2010. Electrodeposition of nickel hydroxide nanoparticles on boron-doped diamond electrodes for oxidative electrocatalysis. J. Phys. Chem. C 115, 1649–1658 (doi:10.1021/jp109526b) [Google Scholar]
- 142.McNaught AD, Wilkinson A, Nic M, Jirat J, Kosata B, Jenkins A. 2014. IUPAC compendium of chemical terminology (the ‘gold book’) on-line corrected version. Oxford, UK: Blackwell Scientific Publications. [Google Scholar]
- 143.Tower OF. 1923. Note on colloidal nickel hydroxide. J. Phys. Chem. 28, 176–178 (doi:10.1021/j150236a009) [Google Scholar]
- 144.Tower OF, Cooke MC. 1921. On the preparation of colloidal solutions of nickel and cobalt hydroxides and some other compounds of these metals. J. Phys. Chem. 26, 728–735 (doi:10.1021/j150224a003) [Google Scholar]
- 145.Hench LL, West JK. 1990. The sol-gel process. Chem. Rev. 90, 33–72 (doi:10.1021/cr00099a003) [Google Scholar]
- 146.Wu M, Gao J, Zhang S, Chen A. 2006. Synthesis and characterization of aerogel-like mesoporous nickel oxide for electrochemical supercapacitors. J. Porous Mater. 13, 407–412 (doi:10.1007/s10934-006-8038-x) [Google Scholar]
- 147.Gash AE, Satcher JH, Jr, Simpson RL. 2004. Monolithic nickel(II)-based aerogels using an organic epoxide: the importance of the counterion. J. Non-Cryst. Solids 350, 145–151 (doi:10.1016/j.jnoncrysol.2004.06.030) [Google Scholar]
- 148.Xing W, Li F, Yan Z-F, Lu GQ. 2004. Synthesis and electrochemical properties of mesoporous nickel oxide. J. Power Sources 134, 324–330 (doi:10.1016/j.jpowsour.2004.03.038) [Google Scholar]
- 149.Tientong J, Garcia S, Thurber CR, Golden TD. 2014. Synthesis of nickel and nickel hydroxide nanopowders by simplified chemical reduction. J. Nanotechnol. 2014, 6 (doi:10.1155/2014/193162) [Google Scholar]
- 150.Xiao-yan G, Jian-cheng D. 2007. Preparation and electrochemical performance of nano-scale nickel hydroxide with different shapes. Mater. Lett. 61, 621–625 (doi:10.1016/j.matlet.2006.05.026) [Google Scholar]
- 151.Xing S, Wang Q, Ma Z, Wu Y, Gao Y. 2012. Synthesis of mesoporous α-Ni(OH)2 for high-performance supercapacitors. Mater. Lett. 78, 99–101 (doi:10.1016/j.matlet.2012.03.023) [Google Scholar]
- 152.Liu XH, Liu W, Lv XK, Yang F, Wei X, Zhang ZD, Sellmyer DJ. 2010. Magnetic properties of nickel hydroxide nanoparticles. J. Appl. Phys. 107, 083919 (doi:10.1063/1.3374468) [Google Scholar]
- 153.Cheng J, Cao G-P, Yang Y-S. 2006. Characterization of sol–gel-derived NiOx xerogels as supercapacitors. J. Power Sources 159, 734–741 (doi:10.1016/j.jpowsour.2005.07.095) [Google Scholar]
- 154.Liu C, Li Y. 2009. Synthesis and characterization of amorphous α-nickel hydroxide. J. Alloys Compd. 478, 415–418 (doi:10.1016/j.jallcom.2008.11.049) [Google Scholar]
- 155.Šurca A, Orel B, Pihlar B, Bukovec P. 1996. Optical, spectroelectrochemical and structural properties of sol-gel derived Ni-oxide electrochromic film. J. Electroanal. Chem. 408, 83–100 (doi:10.1016/0022-0728(96)04509-3) [Google Scholar]
- 156.Liu KC, Anderson MA. 1996. Porous nickel oxide/nickel films for electrochemical capacitors. J. Electrochem. Soc. 143, 124–130 (doi:10.1149/1.1836396) [Google Scholar]
- 157.Martins PR, Araújo Parussulo AL, Toma SH, Rocha MA, Toma HE, Araki K. 2012. Highly stabilized alpha-NiCo(OH)2 nanomaterials for high performance device application. J. Power Sources 218, 1–4 (doi:10.1016/j.jpowsour.2012.06.065) [Google Scholar]
- 158.Rocha MA, Winnischofer H, Araki K, Anaissi FJ, Toma HE. 2011. A New insight on the preparation of stabilized alpha-nickel hydroxide nanoparticles. J. Nanosci. Nanotechnol. 11, 3985–3996 (doi:10.1166/jnn.2011.3872) [DOI] [PubMed] [Google Scholar]
- 159.Bernard MC, Bernard P, Keddam M, Senyarich S, Takenouti H. 1996. Characterisation of new nickel hydroxides during the transformation of α Ni(OH)2 to β Ni(OH)2 by ageing. Electrochim. Acta 41, 91–93 (doi:10.1016/0013-4686(95)00282-J) [Google Scholar]
- 160.Osiñska M, Stefanowicz T, Paukszta D. 2004. Nickel hydroxide ageing time influence on its solubility in water acidified with sulphuric acid. J. Hazard. Mater. 112, 177–182 (doi:10.1016/j.jhazmat.2004.05.028) [DOI] [PubMed] [Google Scholar]
- 161.Le Bihan S, Figlarz M. 1972. Croissance de l'hydroxyde de nickel Ni(OH)2 à partird'un hydroxyde de nickel turbostratique. J. Cryst. Growth 13/14, 458–461 (doi:10.1016/0022-0248(72)90280-1) [Google Scholar]
- 162.Deabate S, Fourgeot F, Henn F. 2000. X-ray diffraction and micro-Raman spectroscopy analysis of new nickel hydroxide obtained by electrodialysis. J. Power Sources 87, 125–136 (doi:10.1016/S0378-7753(99)00437-1) [Google Scholar]
- 163.Briggs GWD, Snodin PR. 1982. Ageing and the diffusion process at the nickel hydroxide electrode. Electrochim. Acta 27, 565–572 (doi:10.1016/0013-4686(82)85041-X) [Google Scholar]
- 164.Briggs GWD, Wynne-Jones WFK. 1962. The nickel hydroxide electrode; the effects of ageing—I X-ray diffraction study of the electrode process. Electrochim. Acta 7, 241–248 (doi:10.1016/0013-4686(62)87001-7) [Google Scholar]
- 165.Briggs GWD, Wynne-Jones WFK. 1962. The nickel hydroxide electrode; the effect of ageing—II changes in electrochemical behaviour. Electrochim. Acta 7, 249–256 (doi:10.1016/0013-4686(62)87002-9) [Google Scholar]
- 166.Desilvestro J, Corrigan DA, Weaver MJ. 1988. Characterization of redox states of nickel hydroxide film electrodes by in situ surface Raman spectroscopy. J. Electrochem. Soc. 135, 885–892 (doi:10.1149/1.2095818) [Google Scholar]
- 167.Godwin IJ, Lyons MEG. 2013. Enhanced oxygen evolution at hydrous nickel oxide electrodes via electrochemical ageing in alkaline solution. Electrochem. Commun. 32, 39–42 (doi:10.1016/j.elecom.2013.03.040) [Google Scholar]
- 168.Rabenau A. 1985. The role of hydrothermal synthesis in preparative chemistry. Angew. Chemie Int. 24, 1026–1040 (doi:1002/anie.10261) [English edition] [Google Scholar]
- 169.Dixit M, Subbanna GN, Kamath PV. 1996. Homogeneous precipitation from solution by urea hydrolysis: a novel chemical route to the α-hydroxides of nickel and cobalt. J. Mater. Chem. 6, 1429–1432 (doi:10.1039/JM9960601429) [Google Scholar]
- 170.Yang L-X, Zhu Y-J, Tong H, Liang Z-H, Li L, Zhang L. 2007. Hydrothermal synthesis of nickel hydroxide nanostructures in mixed solvents of water and alcohol. J. Solid State Chem. 180, 2095–2101 (doi:10.1016/j.jssc.2007.05.009) [Google Scholar]
- 171.Wang Y, Gai S, Niu N, He F, Yang P. 2013. Fabrication and electrochemical performance of 3D hierarchical β-Ni(OH)2 hollow microspheres wrapped in reduced graphene oxide. J. Mater. Chem. A 1, 9083–9091 (doi:10.1039/c3ta11161j) [Google Scholar]
- 172.Liang Z-H, Zhu Y-J, Hu X-L. 2004. β-nickel hydroxide nanosheets and their thermal decomposition to nickel oxide nanosheets. J. Phys. Chem. B 108, 3488–3491 (doi:10.1021/jp037513n) [Google Scholar]
- 173.Gong L, Liu X, Su L. 2011. Facile solvothermal synthesis Ni(OH)2 nanostructure for electrochemical capacitors. J. Inorg. Organomet. Polym. Mater. 21, 866–870 (doi:10.1007/s10904-011-9519-1) [Google Scholar]
- 174.Wang M, Ni Y, Cao L, Zhao D, Ma X. 2013. Porous Ni/β-Ni(OH)2 superstructures: rapid solvothermal synthesis, characterization, and electrochemical property. J. Colloid Interface Sci. 401, 8–13 (doi:10.1016/j.jcis.2013.01.016) [DOI] [PubMed] [Google Scholar]
- 175.Xiong X-H, Wang Z-X, Guo H-J, Li X-H. 2015. Facile synthesis of ultrathin nickel hydroxides nanoflakes on nickel foam for high-performance supercapacitors. Mater. Lett. 138, 5–8 (doi:10.1016/j.matlet.2014.09.069) [Google Scholar]
- 176.Medway SL, Lucas CA, Kowal A, Nichols RJ, Johnson D. 2006. In situ studies of the oxidation of nickel electrodes in alkaline solution. J. Electroanal. Chem. 587, 172–181 (doi:10.1016/j.jelechem.2005.11.013) [Google Scholar]
- 177.Hoppe H-W, Strehblow HH. 1989. XPS and UPS examinations of the formation of passive layers on Ni in 1 M sodium hydroxide and 0.5 M sulphuric acid. Surf. Interface Anal. 14, 121–131 (doi:10.1002/sia.740140305) [Google Scholar]
- 178.MacDougall B, Cohen M. 1974. Anodic oxidation of nickel in neutral sulfate solution. J. Electrochem. Soc. 121, 1152–1159 (doi:10.1149/1.2402003) [Google Scholar]
- 179.Hall DS, Bock C, MacDougall BR. 2013. The electrochemistry of metallic nickel: oxides, hydroxides, hydrides and alkaline hydrogen evolution. J. Electrochem. Soc. 160, F235–F243 (doi:10.1149/2.026303jes) [Google Scholar]
- 180.Seyeux A, Maurice V, Klein LH, Marcus P. 2005. In situ scanning tunnelling microscopic study of the initial stages of growth and of the structure of the passive film on Ni(111) in 1 mM NaOH(aq). J. Solid State Electrochem. 9, 337–346 (doi:10.1007/s10008-004-0627-5) [Google Scholar]
- 181.Visscher W, Barendrecht E. 1983. Anodic oxide films of nickel in alkaline electrolyte. Surf. Sci. 135, 436–452 (doi:10.1016/0039-6028(83)90235-2) [Google Scholar]
- 182.Machet A, et al. 2004. XPS and STM study of the growth and structure of passive films in high temperature water on a nickel-base alloy. Electrochim. Acta 49, 3957–3964 (doi:10.1016/j.electacta.2004.04.032) [Google Scholar]
- 183.Zhang X, Zagidulin D, Shoesmith DW. 2013. Characterization of film properties on the NiCrMo alloy C-2000. Electrochim. Acta 89, 814–822 (doi:10.1016/j.electacta.2012.11.029) [Google Scholar]
- 184.Fleischmann M, Korinek K, Pletcher D. 1972. The kinetics and mechanism of the oxidation of amines and alcohols at oxide-covered nickel, silver, copper, and cobalt electrodes. J. Chem. Soc. Perkin Trans. 2, 1396–1403 (doi:10.1039/P29720001396) [Google Scholar]
- 185.Hall DS, Bock C, MacDougall BR. 2013. Surface layers in alkaline media: nickel hydrides on metallic nickel electrodes. ECS Trans. 50, 165–179 (doi:10.1149/05031.0165ecst) [Google Scholar]
- 186.Park K-W, Choi J-H, Kwon B-K, Lee S-A, Sung Y-E, Ha H-Y, Hong S-A, Kim H, Wieckowski A. 2002. Chemical and electronic effects of Ni in Pt/Ni and Pt/Ru/Ni alloy nanoparticles in methanol electrooxidation. J. Phys. Chem. B 106, 1869–1877 (doi:10.1021/jp013168v) [Google Scholar]
- 187.Siegert M, Yates MD, Call DF, Zhu X, Spormann A, Logan BE. 2014. Comparison of nonprecious metal cathode materials for methane production by electromethanogenesis. ACS Sust. Chem. Eng. 2, 910–917 (doi:10.1021/sc400520x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Kowal A, Niewiara R, czyk B, Haber J. 1996. In situ atomic force microscopy observation of change in thickness of nickel hydroxide layer on Ni electrode. Langmuir 12, 2332–2333 (doi:10.1021/la9515229) [Google Scholar]
- 189.de Souza LMM, Kong FP, McLarnon FR, Muller RH. 1997. Spectroscopic ellipsometry study of nickel oxidation in alkaline solution. Electrochim. Acta 42, 1253–1267 (doi:10.1016/S0013-4686(96)00298-8) [Google Scholar]
- 190.Visscher W, Barendrecht E. 1980. The anodic oxidation of nickel in alkaline solution. Electrochim. Acta 25, 651–655 (doi:10.1016/0013-4686(80)87072-1) [Google Scholar]
- 191.Paik W-K, Szklarska-Smialowska Z. 1980. Reflectance and ellipsometric study of anodic passive films formed on nickel in sodium hydroxide solution. Surf. Sci. 96, 401–412 (doi:10.1016/0039-6028(80)90316-7) [Google Scholar]
- 192.Hahn F, Floner D, Beden B, Lamy C. 1987. In situ investigation of the behaviour of a nickel electrode in alkaline solution by UV-vis and IR reflectance spectroscopies. Electrochim. Acta 32, 1631–1636 (doi:10.1016/0013-4686(87)90016-8) [Google Scholar]
- 193.Melendres CA, Pankuch M. 1992. On the composition of the passive film on nickel: a surface-enhanced Raman spectroelectrochemical study. J. Electroanal. Chem. 333, 103–113 (doi:10.1016/0022-0728(92)80384-g) [Google Scholar]
- 194.Nan J, Yang Y, Lin Z. 2006. In situ photoelectrochemistry and Raman spectroscopic characterization on the surface oxide film of nickel electrode in 30 wt.% KOH solution. Electrochim. Acta 51, 4873–4879 (doi:10.1016/j.electacta.2006.01.031) [Google Scholar]
- 195.Hahn F, Beden B, Croissant MJ, Lamy C. 1986. In situ UV visible reflectance spectroscopic investigation of the nickel electrode-alkaline solution interface. Electrochim. Acta 31, 335–342 (doi:10.1016/0013-4686(86)80087-1) [Google Scholar]
- 196.Machado SAS, Avaca LA. 1994. The hydrogen evolution reaction on nickel surfaces stabilized by H-absorption. Electrochim. Acta 39, 1385–1391 (doi:10.1016/0013-4686(94)e0003-i) [Google Scholar]
- 197.Alsabet M, Grden M, Jerkiewicz G. 2011. Electrochemical growth of surface oxides on nickel. Part 1: formation of α-Ni(OH)2 in relation to the polarization potential, polarization time, and temperature. Electrocatalysis 2, 317–330 (doi:10.1007/s12678-011-0067-9) [Google Scholar]
- 198.Alsabet M, Grdeñ M, Jerkiewicz G. 2014. Electrochemical growth of surface oxides on nickel. Electrocatalysis 1–12 (doi:10.1007/s12678-014-0214-1) [Google Scholar]
- 199.Grdeñ M, Alsabet M, Jerkiewicz G. 2012. Surface science and electrochemical analysis of nickel foams. ACS Appl. Mater. Interfaces 4, 3012–3021 (doi:10.1021/am300380m) [DOI] [PubMed] [Google Scholar]
- 200.Seghiouer A, Chevalet J, Barhoun A, Lantelme F. 1998. Electrochemical oxidation of nickel in alkaline solutions: a voltammetric study and modelling. J. Electroanal. Chem. 442, 113–123 (doi:10.1016/S0022-0728(97)00498-1) [Google Scholar]
- 201.Vukoviæ M. 1994. Voltammetry and anodic stability of a hydrous oxide film on a nickel electrode in alkaline solution. J. Appl. Electrochem. 24, 878–882 (doi:10.1007/bf00348775) [Google Scholar]
- 202.Casella IG, Guascito MR, Sannazzaro MG. 1999. Voltammetric and XPS investigations of nickel hydroxide electrochemically dispersed on gold surface electrodes. J. Electroanal. Chem. 462, 202–210 (doi:10.1016/S0022-0728(98)00413-6) [Google Scholar]
- 203.Grdeñ M, Klimek K. 2005. EQCM studies on oxidation of metallic nickel electrode in basic solutions. J. Electroanal. Chem. 581, 122–131 (doi:10.1016/j.jelechem.2005.04.026) [Google Scholar]
- 204.Grdeñ M, Klimek K, Czerwinski A. 2004. A quartz crystal microbalance study on a metallic nickel electrode. J. Solid State Electrochem. 8, 390–397 (doi:10.1007/s10008-003-0461-1) [Google Scholar]
- 205.Hall DS, Bock C, MacDougall BR. 2014. An oxalate method for measuring the surface area of nickel electrodes. J. Electrochem. Soc. 161, H787–H795 (doi:10.1149/2.0711412jes) [Google Scholar]
- 206.Lumbroso R. 1993. Process for obtaining metallic hydroxides. EP. Register Patent # EP0559590. [Google Scholar]
- 207.Mondal AK, Su D, Chen S, Zhang J, Ung A, Wang G. 2014. Microwave-assisted synthesis of spherical β-Ni(OH)2 superstructures for electrochemical capacitors with excellent cycling stability. Chem. Phys. Lett. 610–611, 115–120 (doi:10.1016/j.cplett.2014.07.025) [Google Scholar]
- 208.Yan J, Fan Z, Sun W, Ning G, Wei T, Zhang Q, Zhang R, Zhi L, Wei F. 2012. Advanced asymmetric supercapacitors based on Ni(OH)2/graphene and porous graphene electrodes with high energy density. Adv. Funct. Mater. 22, 2632–2641 (doi:10.1002/adfm.201102839) [Google Scholar]
- 209.Vidotti M, Salvador RP, Córdoba de Torresi SI. 2009. Synthesis and characterization of stable Co and Cd doped nickel hydroxide nanoparticles for electrochemical applications. Ultrason. Sonochem. 16, 35–40 (doi:10.1016/j.ultsonch.2008.07.006) [DOI] [PubMed] [Google Scholar]
- 210.Gedanken A. 2004. Using sonochemistry for the fabrication of nanomaterials. Ultrason. Sonochem. 11, 47–55 (doi:10.1016/j.ultsonch.2004.01.037) [DOI] [PubMed] [Google Scholar]
- 211.Liu X, Yu L. 2004. Synthesis of nanosized nickel hydroxide by solid-state reaction at room temperature. Mater. Lett. 58, 1327–1330 (doi:10.1016/j.matlet.2003.09.054) [Google Scholar]
- 212.Lockwood DJ. 1982. Light scattering from electronic and magnetic excitations in transition-metal halides. In Light scattering in solids III (eds Cardona M, Güntherodt G.), pp. 59–92 Berlin, Germany: Springer. [Google Scholar]
- 213.Enoki T, Tsujikawa I. 1978. Specific heat of a quasi-two-dimensional antiferromagnet Ni(OH)2. J. Phys. Soc. Jpn. 45, 1515–1519 (doi:10.1143/JPSJ.45.1515) [Google Scholar]
- 214.Takada T, Bando Y, Kiyama M, Miyamoto H, Sato T. 1966. The magnetic property of Ni(OH)2. J. Phys. Soc. Jpn. 21, 2745–2746 (doi:10.1143/JPSJ.21.2745) [Google Scholar]
- 215.Hermet P, Gourrier L, Bantignies JL, Ravot D, Michel T, Deabate S, Boulet P, Henn F. 2011. Dielectric, magnetic, and phonon properties of nickel hydroxide. Phys. Rev. B 84, 235211 (doi:10.1103/PhysRevB.84.235211) [Google Scholar]
- 216.Takada T, Bando Y, Kiyama M, Miyamoto H, Sato T. 1966. The magnetic property of β-Co(OH)2. J. Phys. Soc. Jpn. 21, 2726–2726 (doi:10.1143/JPSJ.21.2726) [Google Scholar]
- 217.Rall JD, Seehra MS. 2012. The nature of the magnetism in quasi-2D layered α-Ni(OH)2. J. Phys. 24, 076002 (doi:10.1088/0953-8984/24/7/076002) [DOI] [PubMed] [Google Scholar]
- 218.Rall JD, Seehra MS, Shah N, Huffman GP. 2010. Comparison of the nature of magnetism in α-Ni(OH)2 and β-Ni(OH)2. J. Appl. Phys. 107, 09B511 (doi:10.1063/1.3358015) [Google Scholar]
- 219.Seehra MS, Singh V. 2013. Magnetic ordering of nickel hydroxide layers 30 Å apart obtained by intercalating dodecyl sulfate. J. Phys. 25, 356001 (doi:10.1088/0953-8984/25/35/356001) [DOI] [PubMed] [Google Scholar]
- 220.Taibi M, Ammar S, Jouini N, Fievet F, Molinie P, Drillon M. 2002. Layered nickel hydroxide salts: synthesis, characterization and magnetic behaviour in relation to the basal spacing. J. Mater. Chem. 12, 3238–3244 (doi:10.1039/B204087E) [Google Scholar]
- 221.Rall JD, Seehra MS, Choi ES. 2010. Metamagnetism and nanosize effects in the magnetic properties of the quasi-two-dimensional system β-Ni(OH)2. Phys. Rev. B 82, 184403 (doi:10.1103/PhysRevB.82.184403) [Google Scholar]
- 222.Tiwari SD, Rajeev KP. 2008. Paramagnetic to ferromagnetic transition and superparamagnetic blocking in Ni(OH)2 nanoparticles. Phys. Rev. B 77, 224430 [Google Scholar]
- 223.Ferraro JR, Nakamoto K, Brown CW. 2003. Introductory Raman spectroscopy 2nd edn. Burlington, MA: Academic Press. [Google Scholar]
- 224.Schrader B. 2008. Infrared and Raman spectroscopy: methods and applications New York, NY: John Wiley & Sons. [Google Scholar]
- 225.Hudson BS. 2006. Vibrational spectroscopy using inelastic neutron scattering: overview and outlook. Vib. Spectrosc. 42, 25–32 (doi:10.1016/j.vibspec.2006.04.014) [Google Scholar]
- 226.Pascale F, Tosoni S, Zicovich-Wilson C, Ugliengo P, Orlando R, Dovesi R. 2004. Vibrational spectrum of brucite, Mg(OH)2: a periodic ab initio quantum mechanical calculation including OH anharmonicity. Chem. Phys. Lett. 396, 308–315 (doi:10.1016/j.cplett.2004.08.047) [Google Scholar]
- 227.Johnston C, Graves PR. 1990. In situ Raman spectroscopy study of the nickel oxyhydroxide electrode (NOE) system. Appl. Spectrosc. 44, 105–115 (doi:10.1366/0003702904085769) [Google Scholar]
- 228.Bantignies JL, Deabate S, Righi A, Rols S, Hermet P, Sauvajol JL, Henn F. 2008. New insight into the vibrational behavior of nickel hydroxide and oxyhydroxide using inelastic neutron scattering, far/mid-infrared and Raman spectroscopies. J. Phys. Chem. C 112, 2193–2201 (doi:10.1021/jp075819e) [Google Scholar]
- 229.Dawson P, Hadfield CD, Wilkinson GR. 1973. The polarized infra-red and Raman spectra of Mg(OH)2 and Ca(OH)2. J. Phys. Chem. Solids 34, 1217–1225 (doi:10.1016/s0022-3697(73)80212-4) [Google Scholar]
- 230.Laporte O, Meggers WF. 1925. Some rules of spectral structure. J. Opt. Soc. Am. 11, 459–460 (doi:10.1364/JOSA.11.000459) [Google Scholar]
- 231.Jackovits JF. 1982. In Proc Symposium on the Nickel Electrode, (eds Funther RG, Gross S.), pp. 48–68 Pennington, NJ: The Electrochemical Society. [Google Scholar]
- 232.Cornilsen BC, Karjala PJ, Loyselle PL. 1988. Structural models for nickel electrode active mass. J. Power Sources 22, 351–357 (doi:10.1016/0378-7753(88)80029-6) [Google Scholar]
- 233.Kostecki R, McLarnon F. 1997. Electrochemical and in situ Raman spectroscopic characterization of nickel hydroxide electrodes. J. Electrochem. Soc. 144, 485–493 (doi:10.1149/1.1837437) [Google Scholar]
- 234.Murli C, Sharma SM, Kulshreshtha SK, Sikka SK. 2001. High-pressure behavior of β-Ni(OH)2—a Raman scattering study. Physica B 307, 111–116 (doi:10.1016/s0921-4526(01)00646-9) [Google Scholar]
- 235.Weckler B, Lutz HD. 1996. Near-infrared spectra of M(OH)Cl (M=Ca, Cd, Sr), Zn(OH)F, γ-Cd(OH)2, Sr(OH)2, and brucite-type hydroxides M(OH)2 (M = Mg, Ca, Mn, Fe, Co, Ni, Cd). Spectrochim. Acta, Part A 52, 1507–1513 (doi:10.1016/0584-8539(96)01693-5) [Google Scholar]
- 236.de Oliveira EF, Hase Y. 2001. Infrared study and isotopic effect of magnesium hydroxide. Vib. Spectrosc. 25, 53–56 (doi:10.1016/s0924-2031(00)00107-7) [Google Scholar]
- 237.Torresi RM, Vázquez MV, Gorenstein A, Córdoba de Torresi SI. 1993. Infrared characterization of electrochromic nickel hydroxide prepared by homogeneous chemical precipitation. Thin Solid Films 229, 180–186 (doi:10.1016/0040-6090(93)90361-R) [Google Scholar]
- 238.Portemer F, Delahaye-Vidal A, Figlarz M. 1992. Characterization of active material deposited at the nickel hydroxide electrode by electrochemical impregnation. J. Electrochem. Soc. 139, 671–678 (doi:10.1149/1.2069283) [Google Scholar]
- 239.Herzberg G, Spinks J. 1966. Molecular spectra and molecular structure: II. Infrared and Raman spectra of polyatomic molecules 6th edn. Toronto, Canada: D. Van Nostrand Company, Inc. [Google Scholar]
- 240.Riddell JD, Lockwood DJ, Irish DE. 1972. Ion pair formation in NaNO3/D2O solutions: Raman and infrared spectra, partial molal volumes, conductance, and viscosity. Can. J. Chem. 50, 2951–2962 (doi:10.1139/v72-474) [Google Scholar]
- 241.Guerlou-Demourgues L, Delmas C. 1996. Electrochemical behavior of the manganese-substituted nickel hydroxides. J. Electrochem. Soc. 143, 561–566 (doi:10.1149/1.1836480) [Google Scholar]
- 242.MacDougall B, Cohen M. 1976. Mechanism of the anodic oxidation of nickel. J. Electrochem. Soc. 123, 1783–1789 (doi:10.1149/1.2132697) [Google Scholar]
- 243.Haynes CL, McFarland AD, Duyne RPV. 2005. Surface-enhanced Raman spectroscopy. Anal. Chem. 77, 338A–346A (doi:10.1021/ac053456d) [Google Scholar]
- 244.Smith E, Dent G, Wiley J. 2005. Modern Raman spectroscopy: a practical approach. Hoboken, NJ: J. Wiley. [Google Scholar]
- 245.Desilvestro J, Corrigan DA, Weaver MJ. 1986. Spectroelectrochemistry of thin nickel hydroxide films on gold using surface-enhanced Raman spectroscopy. J. Phys. Chem. 90, 6408–6411 (doi:10.1021/j100282a002) [Google Scholar]
- 246.Berchmans S, Yegnaraman V, Sandhyarani N, Murty KVGK, Pradeep T. 1999. Formation of a nickel hydroxide monolayer on Au through a self-assembled monolayer of 5,5′-dithiobis(2-nitrobenzoic acid): voltammetric, SERS and XPS investigations of the modified electrodes. J. Electroanal. Chem. 468, 170–179 (doi:10.1016/s0022-0728(99)00163-1) [Google Scholar]
- 247.Oblonsky LJ, Devine TM. 1995. A surface enhanced Raman spectroscopic study of the passive films formed in borate buffer on iron, nickel, chromium and stainless steel. Corros. Sci. 37, 17–41 (doi:10.1016/0010-938x(94)00102-c) [Google Scholar]
- 248.Oblonsky LJ, Devine TM. 1995. Surface enhanced Raman spectra from the films formed on nickel in the passive and transpassive regions. J. Electrochem. Soc. 142, 3677–3682 (doi:10.1149/1.2048398) [Google Scholar]
- 249.Baddour-Hadjean R, Fillaux F, Tomkinson J. 1995. Proton dynamics in β-Ni(OH)2 and β-NiOOH. Physica B 213–214, 637–639 (doi:10.1016/0921-4526(95)00235-2) [Google Scholar]
- 250.Bardé F, Taberna PL, Tarascon JM, Palacín MR. 2008. Evidence for electronic and ionic limitations at the origin of the second voltage plateau in nickel electrodes, as deduced from impedance spectroscopy measurements. J. Power Sources 179, 830–836 (doi:10.1016/j.jpowsour.2008.01.045) [Google Scholar]
- 251.Deabate S, Henn F, Devautour S, Giuntini JC. 2003. Conductivity and dielectric relaxation in various Ni(OH)2 samples. J. Electrochem. Soc. 150, 23 (doi:10.1149/1.1573203) [Google Scholar]
- 252.Motori A, Sandrolini F, Davolio G. 1994. Electrical properties of nickel hydroxide for alkaline cell systems. J. Power Sources 48, 361–370 (doi:10.1016/0378-7753(94)80032-4) [Google Scholar]
- 253.Palencsár A, Scherson D. 2005. In situ resistance measurements of single particle microelectrodes as a function of their state of charge. ECS Meeting Abstracts MA2005–01, 112 [Google Scholar]
- 254.Palencsár A, Scherson DA. 2005. In situ resistance measurements of single particle spherical Ni(OH)2 microelectrodes as a function of their state of charge. Electrochem. Solid-State Lett. 8, 328 (doi:10.1149/1.1904508) [Google Scholar]
- 255.Palencsár I, Scherson D. 2006. In situ interparticle resistance measurements for materials of relevance to battery applications. ECS Meeting Abstracts MA2006–01 258 See http://ma.ecsdl.org/content/MA2006-01/5/258. [Google Scholar]
- 256.Ikeda K, Vedanand S. 1999. Optical spectrum of synthetic theophrastite, Ni(OH)2. Neues Jahrb. Mineral., Monatsh 1, 21–26. [Google Scholar]
- 257.Tanabe Y, Sugano S. 1954. On the absorption spectra of complex ions. 9, 753–766 (doi:10.1143/JPSJ.9.753) [Google Scholar]
- 258.Tompkins HG, McGahan WA. 1999. Spectroscopic ellipsometry and reflectometry: a user's guide. New York, NY: Wiley. [Google Scholar]
- 259.Crocker RW. 1992. Structural transformation of nickel hydroxide films during anodic oxidation. PhD thesis, University of California and Lawrence Berkeley Laboratory, Berkeley, CA, USA. [Google Scholar]
- 260.Biesinger MC, Payne BP, Lau LWM, Gerson A, Smart RSC. 2009. X-ray photoelectron spectroscopic chemical state quantification of mixed nickel metal, oxide and hydroxide systems. Surf. Interface Anal. 41, 324–332 (doi:10.1002/sia.3026) [Google Scholar]
- 261.Grosvenor AP, Biesinger MC, Smart RSC, McIntyre NS. 2006. New interpretations of XPS spectra of nickel metal and oxides. Surf. Sci. 600, 1771–1779 (doi:10.1016/j.susc.2006.01.041) [Google Scholar]
- 262.Briggs D, Grant JT. 2003. Surface analysis by Auger and X-ray photoelectron spectroscopy Trowbridge, UK: IM Publications and SurfaceSpectra Limited. [Google Scholar]
- 263.Hoppe H-W, Strehblow H-H. 1990. XPS and UPS examinations of passive layers on Ni and FE53Ni alloys. Corros. Sci. 31, 167–177 (doi:10.1016/0010-938X(90)90105-E) [Google Scholar]