

Abstract
The management of anemia in patients with chronic kidney disease (CKD) is difficult. The availability of erythropoiesis-stimulating agents (ESAs) has increased treatment options for previously transfusion-requiring patients, but the recent evidence of ESA side effects has prompted the search for complementary or alternative approaches. Next to ESA, parenteral iron supplementation is the second main form of anemia treatment. However, as of now, no systematic approach has been proposed to balance the concurrent administration of both agents according to individual patient's needs. Furthermore, the potential risks of excessive iron dosing remain a topic of controversy. How, when and whether to monitor CKD patients for potential iron overload remain to be elucidated. This review addresses the question of risk and benefit of iron administration in CKD, highlights the evidence supporting current practice, provides an overview of standard and potential new markers of iron status and outlines a new pharmacometric approach to physiologically compatible individualized dosing of ESA and iron in CKD patients.
Keywords: anemia, erythropoiesis, iron, iron deficiency, iron overload, kidney disease
INTRODUCTION
Anemia in chronic kidney disease (CKD) is primarily a consequence of insufficient erythropoietin (Epo) production. Prior to the introduction of erythropoiesis-stimulating agents (ESAs), patients with end-stage renal disease (ESRD) received blood transfusions to maintain hemoglobin (Hb) levels. Since then, clinical practice guidelines for using ESA evolved from preventing transfusions to normalizing Hb [1]. Recent reports identified adverse cardiovascular effects of Hb normalization with overly aggressive ESA treatment in ESRD patients [2, 3]. Consequently, the current Food and Drug Administration-approved ESA package insert emphasizes using the minimum necessary ESA dose [4]. Several studies have shown that supplementing ESA with aggressive intravenous (IV) iron treatment improves Hb compared with using ESA alone [5]. Although concurrent administration of ESA and IV iron is considered the standard of care in ESRD anemia management, insufficient evidence exists to support recurrent dose iron administration, with its potential for long-term risks of iron overload. This manuscript reviews the current state of our understanding of iron utilization in erythropoiesis and the potential risks of iron overload in ESRD. Although this topic applies to all CKD patients, we focus predominantly on hemodialysis-dependent ESRD patients, most affected due to ongoing blood loss.
ANEMIA AND IRON DEFICIENCY IN RENAL FAILURE
Anemia is a consequence of a lower than normal Hb concentration. The general definition of anemia includes clinical signs and symptoms in patients with Hb concentrations below a cut-off value for a given population. Clinically, symptomatic anemia presents as shortness of breath, lethargy, fatigue, skin pallor, palpitations, tachycardia, systolic flow murmurs and angina, among many others. Compensatory mechanisms may mask the severity of symptoms, if the Hb decreases gradually. Thus, the goal of anemia management is to reverse patient symptoms attributable to decreased Hb.
Patients with advanced kidney disease are frequently anemic. Multiple mechanisms may be responsible for the decrease in Hb in hemodialysis-dependent ESRD patients: (i) insufficient erythropoietin production and ESA dose, (ii) blood loss and resultant iron deficiency, (iii) accelerated destruction of red blood cells (RBCs) or (iv) bone marrow suppression. Erythropoietin is an essential stimulus for bone marrow production of new RBCs under normal and anemic conditions. Multiple factors, including uremic toxins, vitamin deficiencies and elevated inflammatory cytokines, play a role in bone marrow suppression contributing to erythropoietin's inability to compensate for normal RBC senescence (Figures 1 and 2A) [6]. As a physiological adaptation to a relative decrease in Hb in ESRD patients, a right shift in the oxygen–Hb dissociation curve results in reducing Hb requirements without impaired oxygen delivery [7].
FIGURE 1:
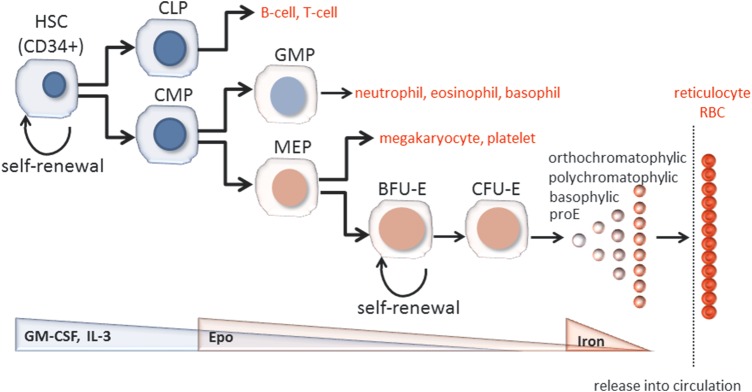
Model of hematopoiesis and terminal erythroid differentiation.
FIGURE 2:
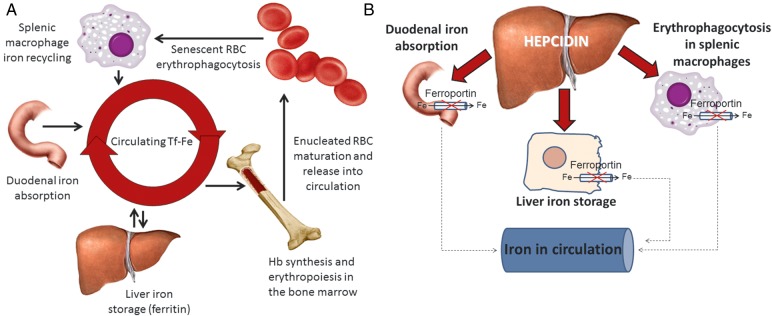
(A) Interaction between iron homeostatic and erythropoietic systems. (B) Hepcidin negatively regulates iron efflux from duodenal enterocytes and splenic macrophages, reducing the concentration of available iron in circulation.
Iron deficiency in CKD is multifactorial and is typically classified as absolute or functional. Absolute iron deficiency results from depleted body iron stores, frequently a result of blood loss. The hemodialysis procedure itself contributes due to unavoidable dialyzer blood loss, clotted dialysis membranes and frequent blood sampling, all totaling an average of 2–5 L of blood per year [8], equivalent to ∼0.5–1.5 g of iron annually. Functional iron deficiency is caused by insufficient iron availability at the site of erythroblast production, despite adequate body iron stores. Thus, diagnosing iron deficiency may be a challenge, especially in CKD patients. Although iron is required predominantly for adequate Hb synthesis, iron deficiency may also affect DNA synthesis, immune response, oxidative metabolism and mitochondrial electron transport. Iron deficiency has been linked to decreased muscle function in experimental animals [9, 10], cognitive impairment and fatigue in non-anemic women [11]. As a consequence, deciphering the roles of iron deficiency relative to the role of anemia in symptoms associated with a chronic illness such as CKD is difficult.
Elevated hepcidin levels in CKD patients may contribute to functional iron deficiency. Hepcidin is a peptide hormone that regulates absorption of dietary iron and iron recycled from senescent RBCs. It is thought to be the main regulator of iron homeostasis (Figure 2B). Hepcidin exerts its function by binding to the only known iron export protein, ferroportin (FPN-1), found on all cells involved in iron homeostasis. Hepcidin binds FPN-1, causes its internalization and degradation and results in impaired iron release [12]. Hepcidin is cleared by the kidneys and thus elevated in patients with CKD [13–16]. Increased hepcidin levels due to CKD result in a relative decrease in iron absorption, availability of recycled iron from macrophages and ultimately functional iron deficiency [17].
Platelet function is impaired in uremia and increases the likelihood of bleeding [18]. Concurrently, recent and old data indicate that iron deficiency is associated with reactive thrombocytosis [19], may contribute to increased risk for thromboembolic events [20, 21] and mortality in CKD patients treated with ESAs [22, 23] but is potentially reversible with the administration of supplemental iron [24, 25]. Thus, management of iron availability for erythropoiesis is essential to prevent ill consequences of both too much and too little iron.
CURRENT CLINICAL STANDARDS IN THE TREATMENT OF ANEMIA IN CHRONIC KIDNEY DISEASE
Until 1987, ESRD patients with anemia required an average of six transfusions annually, adding 1.5 g of iron, 250 mg/unit, to the patient's iron stores [8, 26]. Currently, ESRD patients are treated with ESAs, decreasing their transfusion requirements [27]. The concurrent use of ESAs and IV iron has become standard therapy, driven by ongoing iron losses and by the desire to reduce exposure to ESAs [28]. In addition, financial incentives have driven care-givers to administer ‘cheaper’ IV iron in an attempt to decrease the use of ‘expensive’ ESAs [29]. Several studies demonstrate that ESRD patients treated with concurrent ESAs and IV iron have a greater increase in Hb concentration or the ability to reduce the ESA dose to maintain stable Hb level, relative to those treated with ESAs alone [30, 31]. The most frequently sited randomized controlled trial, the DRIVE study, tested the effect of IV iron in ESRD patients with ferritin levels between 500 and 1200 ng/mL [5]. In this study, after increasing their baseline ESA dose by 25%, patients treated with IV iron exhibited a faster (P = 0.035) and greater increase in Hb (1.6 ± 1.3 versus 1.1 ± 1.4 g/dL; P = 0.028) after 6 weeks than those who did not receive IV iron. Whether similar effects would be observed if no change in ESA dose was initiated at the start of the study, or whether the effect can be sustained longer than 6 weeks and whether the benefit of this approach outweighs the risks of clinically relevant iron overload remain to be determined. Thus, the DRIVE and other studies have not resolved reservations about the long-term safety of the widely practiced concurrent use of IV iron with ESAs in ESRD patients. Practice guidelines define a threshold ferritin of ≤500 mg/L and transferrin saturation (TSat) of ≤30% to initiate IV iron in anemic CKD patients [Kidney Disease Improving Global Outcomes (KDIGO)] [32] although the evidence to support them is weak [33].
Definitions of iron deficiency and iron overload in patients with CKD are complex. They require the expertise of multiple specialists and methods of assessment to properly evaluate the potential clinical risks and impact of therapy. The result is a delay in reaching consensus on how to identify CKD patients who would benefit from iron supplementation. Attempts to set thresholds using ferritin have been hampered by conflicting evidence of its responsiveness during inflammation. CKD patients with ferritin levels >500 ng/dL have also demonstrated increased Hb responsiveness to IV iron [5], suggesting that ferritin alone may not predict responsiveness to IV iron. Furthermore, even in ESRD patients with ferritin <500 ng/mL, hepatic iron overload was diagnosed as determined by magnetic susceptometry (SQUID) [34]. Patients with ferritin >1000 ng/mL had multi-organ iron deposition [35], and those receiving ESAs and IV iron according to the KDIGO and Kidney Disease Outcomes Quality Initiative guidelines had evidence of hepatic iron deposition [36]. Furthermore, discontinuing IV iron for 1 year in patients with hepatic iron overload did not result in changes in Hb at a stable ESA dose, despite a concomitant significant decrease in the initially high level of ferritin and TSat [35, 36]. This finding indicates that storage iron can be made available to maintain a steady Hb level with ESA alone.
Determining the critical ferritin level sufficient to avoid iron-deficient erythropoiesis and systemic iron overload is a challenge. The ability to set recommendations for initiation of treatment and for goals of iron therapy is limited by the absence of reliable methods to evaluate iron requirements, as well as, prospective, controlled, long-term clinical trials of supplemental iron safety and efficacy. Furthermore, the optimal timing and dose of ESA and iron are unresolved. Although physiological release of these agents into circulation is virtually continuous, exogenous administration occurs at non-physiologic intervals using supra-physiologic doses. This paradigm is driven by practicalities of patient management as higher doses prolong bioavailability of the drugs and enable extended dosing intervals. However, large peak concentrations may lead to the unintended and unpredictable side effects observed with ESAs. For example, large ESA doses increase the demand for iron in the bone marrow beyond the maximum capacity of transferrin to deliver it. Although the Hb concentration increases, resultant iron-deficient erythropoiesis leads to the production of microcytic RBCs and an increased red cell distribution width, which is an independent predictor of cardiovascular events [37].
Similarly, controversy continues regarding the use of large, single iron doses, the so-called load and hold approach, compared with the use of frequent, small doses of iron, commonly referred to as maintenance iron dosing. The former approach is frequently used with very large molecular weight parenteral iron products whereas the latter is required for smaller molecules to prevent generation of reactive oxygen species (ROS). Recently, Brookhart et al. reported a large, retrospective cohort study suggesting that the maintenance approach results in fewer infections than bolus dosing [38]. As pointed out in an accompanying editorial by Rhee and Kalantar-Zadeh, conclusions from this observational report are weakened by confounding by indication, even when sophisticated statistical approaches are applied [39].
We anticipate that as novel methods for measuring multiple erythropoiesis- and iron-related parameters become standardized and more readily available, a more robust understanding of how patients differ, one from another, will enable clinicians to apply expertise beyond the current trial-and-error approach and ultimately aid in the management of this complicated issue in CKD patients.
AVAILABLE NOVEL METHODS TO ASSESS IRON STATUS
Ferritin is the main iron storage protein, regulated by the intracellular iron concentration [40]. Although very low levels enable physicians to diagnose iron deficiency in otherwise healthy individuals, ferritin measurement has not demonstrated sufficient power to predict responsiveness to IV iron in patients receiving ESAs [5, 41, 42] despite a greater increase in Hb in those treated concurrently with ESAs and IV iron [5]. Due to the broad range of responses to iron therapy in CKD patients, elevated ferritin levels may be due to causes other than increased iron stores, requiring the use of additional diagnostic tools. Central to the broad range of response is the wide variability and individuality in how much iron is needed for adequate erythropoiesis, as well as, how much iron is stored and accessible relative to the ferritin concentration.
Soluble transferrin receptor 1 (sTfR1) concentration and sTfR1/log ferritin have been proposed as estimates of iron-deficient erythropoiesis since circulating sTfR1 concentration is proportional to cellular expression of the membrane-associated TfR1 and increases with increased cellular iron requirements and cellular proliferation [43–45].
Ferritin can vary in its iron content, and methods to measure iron-loaded ferritin have been developed. Each ferritin molecule is a complex of 24 subunits forming a large stable cylinder with the capacity to contain >4000 iron atoms in its core [46]. Ferritin iron content would thus enable a quantitative approach of the relationship of total ferritin to iron-loaded ferritin in different conditions of concurrent inflammation and aberrant iron regulation [47, 48]. An analysis of ferritin iron content in hemodialysis patients thus far has been difficult to interpret [49].
TSat has not demonstrated sufficient power to predict responsiveness of ESRD patients on ESAs to IV iron [5, 41]. When the capacity of the iron-binding protein transferrin is exceeded, non-transferrin-bound iron (NTBI) is produced. NTBI is typically undetectable until TSat exceeds 80% [50, 51]. NTBI levels can be measured by spectrophotometry. In a cross-sectional randomized study of patients with β-thalassemia, NTBI was evaluated as an index of iron overload, demonstrating a significant positive correlation between mean NTBI, liver iron concentration (LIC) and ferritin [52].
Labile plasma iron is the fraction of the NTBI that is redox active. Labile plasma iron levels >0.4 μm are highly correlated with iron overload [53]. Several methods for measuring labile plasma iron have been proposed. However, at present, few laboratories worldwide have established the methodology for accurate and reproducible NTBI and labile plasma iron measurements. Analysis of these parameters was performed in hemodialysis patients receiving IV iron, demonstrating an increase in non-apotransferrin moieties and NTBI relative to untreated patients [54].
Labile plasma iron permeates cell membranes and, as part of the labile iron pool (LIP), causes cellular damage by production of ROS [53, 55]. The LIP likely represents the redox active cellular iron that results in oxidative stress and cytotoxicity [56]. Although cellular iron is mostly bound to various components, such as Hb, heme, ferritin and various enzymes, some iron, namely the LIP, is hypothesized to be made up of iron ions bound to low-affinity ligands that vary in composition and quantity under different physiological settings. It is localized primarily, but not exclusively, in the cytosol [57]. Recent data suggest that markers of the LIP are increased in patients with iron overload [57, 58] and decreased in patients treated with iron chelators [59]. Flow cytometry assays of the LIP [60] demonstrate that cultured hepatocytes exposed to serum from IV iron-treated hemodialysis patients with increased concentrations of NTBI, resulted in an increased LIP relative to serum from untreated patients [54].
Hepcidin is regulated by iron, inflammation and erythropoiesis. These factors and reduced renal clearance are implicated mechanisms responsible in elevated circulating hepcidin in CKD patients [14, 15]. In a small sample of ESRD patients, serum hepcidin concentration was reduced following hemodialysis, correlating well with ferritin, inversely with reticulocyte count and not at all with markers of inflammation [16]. Furthermore, hepcidin concentration decreased following ESA administration in some [14, 16, 61], but not all studies [42]. Hepcidin concentration did not correlate with responsiveness to ESAs [62]. Other studies show that serum hepcidin concentration was not predictive of response to IV iron in patients receiving ESAs, although all patients responded to IV iron by increasing Hb [63]. Studies are ongoing to determine whether and how hepcidin measurement can elucidate the multiple potential factors resulting in anemia in patients with CKD. To date, no consensus has been reached on a methodology to measure hepcidin. The collective interpretation of multiple factors, all exerting competing influences on hepcidin, is both complicated and necessary. Proteins related to hepcidin regulation are being evaluated for their utility in informing iron status in ESRD patients [64]. Recently, hepcidin-lowering drugs have been shown to improve iron utilization in an animal model of CKD [65]. In this study, lowering hepcidin levels resulted in decreased spleen iron content, increased reticulocyte count and higher EPO levels. Hepcidin correlates with mortality in dialysis patients [66, 67].
Liver iron concentration is regarded as the reference standard to estimate body iron stores. Studies demonstrate an association between LIC levels above 15–20 mg/g dry weight and hepatic dysfunction [68], hepatic fibrosis [69] and worse prognosis [70]. Noninvasive methods for measuring LIC include super-conducting quantum interference device biomagnetic liver susceptometry (SQUID-BLS) and magnetic resonance imaging (MRI). These tools have mostly replaced invasive liver biopsy for measuring organ iron deposition. SQUID-BLS is a very accurate and well-validated noninvasive method for measuring liver iron [71] and has been used to evaluate LIC in hemodialysis patients [34]. However, clinical availability of SQUID is restricted in light of the high purchase and maintenance costs of the machine and the requirement of dedicated trained staff. MRI, on the other hand, is widely available and less expensive. The MRI R2 and R2* techniques demonstrate an average sensitivity of >85% and specificity of >92% up to an LIC of 15 mg/g dry weight and can be applied with little training and at any center with a reasonably new MRI machine. It provides a rapid and accurate method for estimating hepatic iron concentration suitable for diagnosis and management of iron overload [72]. Several studies have already shown abnormally high iron stores and severe iron overload in the liver using these noninvasive methods in hemodialysis patients treated with ESAs and IV iron [36, 73, 74].
Because evidence from diseases associated with iron overload demonstrates significant morbidity and mortality from heart failure, tools to evaluate cardiac iron are relevant for optimal patient care. Myocardial T2* MRI provides an accurate assessment of cardiac iron status and correlates well with left ventricular function [75, 76]. Histological and MRI studies have previously demonstrated that myocardial iron distribution is heterogeneous. Thus, further evolution of this technique, using a multi-slice multi-echo T2* approach, enables the evaluation of myocardial iron from the mid-ventricular septum and the whole left ventricle [77, 78]. Preliminary studies using myocardial T2* MRI reveal no evidence of iron overload in a small cohort of ESRD patients on hemodialysis treated concurrently with standard doses of ESAs and IV iron [28].
Table 1 presents a summary of iron markers currently used as a clinical standard in ESRD, as well as other available and novel iron markers.
Table 1.
Markers of iron status in ESRD
| Current clinical standard | Available but not routinely used for iron monitoring in ESRD | Novel |
|---|---|---|
|
|
|
IRON OVERLOAD AND ITS POTENTIAL CLINICAL CONSEQUENCES IN ESRD
Iron overload may result from disease or from disease management, as no physiologic means of excreting iron exist in humans. For example, hereditary hemochromatosis results from abnormally increased iron absorption, whereas repetitive transfusions for hereditary anemia may result in similar tissue iron deposition. Data from transfusion-dependent β-thalassemia patients suggest that transfusion iron overload results in significant morbidity and mortality [51, 59, 79–85]. As iron accumulation progresses, exceeding transferrin binding capacity, iron is deposited in organs and can result in cirrhosis, hepatocellular carcinoma, diabetes, skin pigmentation, destructive osteoarthritis and cardiomyopathy. If untreated, iron overload in these diseases results in heart failure and death in 10–30 years.
Although clinical evidence of harm due to iron overload is undisputed, the primary vehicle of tissue iron overload and ensuing toxicity has not been clearly delineated. Indirect but compelling evidence suggests that NTBI leads to organ iron deposition [86]. The pathologically relevant fraction of NTBI, labile plasma iron, is translocated across cell membranes in an unregulated manner [87]. The ROS produced by these reactions have the potential to oxidize lipids, proteins and nucleic acids, resulting in premature apoptosis, cell death and tissue and organ damage. These reactions are implicated as the cause of the clinical manifestations observed in iron overload syndromes [88]. Labile plasma iron also results in increased asymmetric dimethylarginine, inhibiting endothelial nitric oxide, and causes endothelial dysfunction [89] and impairs neutrophil, macrophage and lymphocyte immune functions [54, 89–94].
Inhibition of nitric oxide and resultant endothelial dysfunction has been associated with impaired cardiovascular function, the major cause of death in ESRD patients [95]. In fact, a correlation between carotid artery medial thickness and IV iron dose in ESRD patients has already been demonstrated [96]. In addition, increased incidence of infections and related mortality correlate with iron overload in this patient population [97–99]. Although a recent retrospective study in ESRD patients did not find an increase in infectious complications after instituting a more aggressive IV iron protocol in their dialysis unit [100], a meta-analysis of trials evaluating the use of IV iron did find a 33% increased risk of infection compared with patients receiving oral or no iron [101].
In ESRD patients treated with a combination of ESAs and IV iron, a significant portion of the IV iron dose administered deposits in organs [102]. Furthermore, because a high dose of IV iron is given rapidly, the binding capacity of transferrin is easily exceeded, resulting in the generation of NTBI and labile plasma iron [94]. Because only transferrin-bound iron can deliver iron for erythropoiesis, most of IV iron may be unavailable for erythropoiesis and may be directly shuttled to iron stores in parenchymal cells where it accumulates and is expected to cause cellular injury (Figure 3).
FIGURE 3:
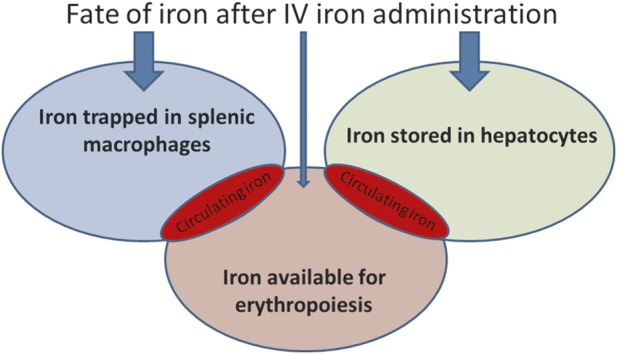
Clinical evidence suggests that the majority of IV iron administered to patients is unavailable for erythropoiesis.
Before the use of ESAs, when transfusion was the mainstay of anemia management in ESRD patients, evidence of parenchymal iron deposition was ubiquitous. In an autopsy study from that period, 82% of patients were found to have significant iron deposition in the liver and spleen and 23% in the heart [103]. Since the availability of ESAs, evidence of iron deposition has been demonstrated by MRI in ESRD patients concurrently treated with IV iron [34–36, 74]. The Normal HCT Trial was a randomized controlled trial to evaluate the benefit of normalizing hematocrit in ESRD patients. The results of this trial demonstrated that patients assigned to the high Hb target group exhibited a significant increase in mortality from cardiovascular events. Post hoc analysis revealed that among patients assigned to the high Hb target group, survival was strongly inversely correlated with the dose of IV iron [1]. Several unanswered questions about these data remain: (i) was iron deposition a result of RBC transfusion-derived iron, IV iron or a combination? (ii) Is there evidence that iron deposition is reversible with available treatments? And (iii) does reversal of iron deposition have clinically relevant consequences of decreased morbidity and mortality? Furthermore, it remains unknown whether resistance to ESAs is a marker of disease severity or if excess iron causes increased morbidity and mortality. Taken together, although significantly more evidence is needed to alter patient management, currently available data suggest the need for caution on the part of physicians who treat ESRD patients with IV iron.
COMPUTATIONAL APPROACH TO BALANCING ERYTHROPOIETIN AND IRON
The complex interactions between Epo and iron, as well as the long- and short-term side effects of these agents are not completely understood. The dynamic nature of erythropoiesis and its inherent within- and between-subject variability pose a well-appreciated challenge to physicians. The standard ‘one-size-fits-all’ ESA and IV iron dosing is not optimal. To address this challenge, we propose new dosing algorithms for ESA and IV iron based on mathematical modeling and feedback control design. This approach enables personalized dosing of these agents, aimed at achieving desired therapeutic outcomes while avoiding adverse side effects. The algorithms described below can be easily implemented on a computer as part of a clinical decision support system integrated with electronic health records.
For decades, pharmacometric approaches and mathematical modeling have been applied to better understand erythropoiesis and ferrokinetics [104–106], using experimental and clinical data to create equations representing relationships between stimuli and responses. Such equations are built through estimates of how blood parameters and iron markers respond to ESA and IV iron dosing over time (Figure 4). Here, we illustrate this concept with a simple example, based on currently used laboratory parameters.
FIGURE 4:

Mathematical models of ESA and/or IV iron dosing require an understanding of the relationships between surrogates of erythropoiesis and iron metabolism.
To describe ferritin change over time, we can write the following equation:
Similarly, to describe TSat change over time:
Finally, to delineate a relationship between Hb, ESA dose and iron status:
Mathematical modeling quantifies the right-hand-side terms in these equations on a patient-by-patient basis using engineering methods called ‘system identification’. In our example, we can use longitudinal data on ferritin, TSat, Hb, as well as, IV iron and ESA dose to quantify some of these terms [107, 108]. Our goal is to maximize the model's predictive ability by minimizing the error between calculated and measured outcomes. Once validated, a mathematical model will be used to systematically derive patient-specific dosing rules for ESA and IV iron. We propose ‘feedback control design’ to derive these rules. Based on measured patient response (e.g. Hb), feedback control computes the proper corrective action, like the ESA dose adjustment, such that the future response behaves in a desired manner, even in situations when the mathematical model is not perfect (uncertainty). For example, feedback control based ESA dosing rules should lead to reaching target Hb within a specified time limit, without under- and over-shooting.
To illustrate this concept, consider the ESA dose adjustment to achieve a desired target Hb. The achieved Hb in a real patient can be affected over time by many unpredictable factors, e.g. consequences of blood loss. This leads to a difference between the achieved and desired Hb (error). The fundamental principle of feedback control is to minimize this error:
A feedback control rule for ESA dose adjustment could be:
in which ‘change in Hb error’ represents how Hb error is changing over time. ESA dose adjustment based not only the current Hb error but also its trend ‘anticipates’ the corrective action and accounts for the dynamic nature of the process. Further details of mathematical modeling, system identification and feedback control design are beyond the scope of this review [109].
We and others have demonstrated that feedback control can successfully support ESA dosing in anemia management of ESRD patients [110–113], and additional studies are underway to provide further evidence of its utility (ClinicalTrials.gov: NCT01719146, NCT01975844). Based on our experience, we postulate that feedback control can also be successful in the more complicated management of concurrent ESA and IV iron dosing.
CONCLUSIONS AND RECOMMENDATIONS
The administration of ESAs with IV iron is practiced widely in hemodialysis centers worldwide. Noninvasive measures in IV iron-treated ESRD patients demonstrate tissue iron deposition. However, thus far, long-term, deleterious consequences of iron overload have not yet been proven in the ESRD population. Conflicting evidence is likely related to the time required for clinical consequences of iron overload to become evident. Experience with RBC transfusion-related iron overload in β-thalassemic patients suggests that it will take years before informed conclusions can be drawn about iron overload in ESRD patients. This effort is further hampered by the relatively short life expectancy of many ESRD patients. What remains is the nagging suspicion that iron overload contributes to shortened survival in hemodialysis patients. Universally acceptable recommendations for the diagnosis and treatment of iron deficiency or iron overload in CKD and ESRD patients are questionable. Both the clinical and basic science communities agree that IV iron administration to complement ESA administration should be used judiciously, anticipating the potential need to evaluate iron status with the use of multiple modalities outlined in this review.
We suggest that the definition of iron deficiency as the cause of anemia and iron overload in ESRD patients be based on a complement of evidence obtained from changes in RBC parameters, ferritin and TSat, and tissue iron deposition measured by standardized, noninvasive imaging. Reaching consensus on the definition of iron overload and iron deficiency would be an important step toward helping physicians determine whether they are present in patients and provide homogenous, reasonable and obtainable end points for future studies. Rigorous randomized controlled clinical trials are required to evaluate any potential harm from extended use of IV iron in ESRD patients, particularly those without clear evidence of iron deficiency. These studies are qualitatively different from those already published, since their primary objective would not aim to assess whether ESRD patients with elevated ferritin respond to IV iron with increasing Hb, but rather ask the question of optimal management to balance the risks and benefits of IV iron. Although randomized controlled clinical trials are the gold standard in evidence-based medicine, such long-term studies would be hampered by logistical, financial and ethical complications and provide only partial resolution of this issue. We assert that, at best, such studies can lead to population-wide guidelines not accounting for specific therapeutic needs of individual patients.
We propose a complementary paradigm of anemia management that derives a treatment strategy based on mathematical modeling of human physiology in ESRD and feedback control design. Mathematical models are derived from patient data to understand the complex interactions between parameter surrogates of physiological processes. Once such models are built, individualized ESA and IV iron dosing algorithms can be designed using feedback control. This systematic application of modeling and control concepts are poised to empower physicians to provide more individualized and clinically optimal results.
CONFLICT OF INTEREST STATEMENT
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
ACKNOWLEDGEMENT
This work was supported by the National Institute of Diabetes And Digestive And Kidney Diseases of the National Institutes of Health under Award Numbers R01DK093832 and K25DK09600601.
REFERENCES
- 1.Besarab A, Bolton WK, Browne JK, et al. The effects of normal as compared with low hematocrit values in patients with cardiac disease who are receiving hemodialysis and epoetin. N Engl J Med. 1998;339:584–590. doi: 10.1056/NEJM199808273390903. [DOI] [PubMed] [Google Scholar]
- 2.Singh AK, Szczech L, Tang KL, et al. Correction of anemia with epoetin alfa in chronic kidney disease. N Engl J Med. 2006;355:2085–2098. doi: 10.1056/NEJMoa065485. [DOI] [PubMed] [Google Scholar]
- 3.Pfeffer MA, Burdmann EA, Chen CY, et al. A trial of darbepoetin alfa in type 2 diabetes and chronic kidney disease. N Engl J Med. 2009;361:2019–2032. doi: 10.1056/NEJMoa0907845. [DOI] [PubMed] [Google Scholar]
- 4.Kliger AS, Fishbane S, Finkelstein FO. Erythropoietic stimulating agents and quality of a patient's life: individualizing anemia treatment. Clin J Am Soc Nephrol. 2012;7:354–357. doi: 10.2215/CJN.11961111. [DOI] [PubMed] [Google Scholar]
- 5.Coyne DW, Kapoian T, Suki W, et al. Ferric gluconate is highly efficacious in anemic hemodialysis patients with high serum ferritin and low transferrin saturation: results of the Dialysis Patients' Response to IV Iron with Elevated Ferritin (DRIVE) Study. J Am Soc Nephrol. 2007;18:975–984. doi: 10.1681/ASN.2006091034. [DOI] [PubMed] [Google Scholar]
- 6.Eschbach JW. The anemia of chronic renal failure: pathophysiology and the effects of recombinant erythropoietin. Kidney Int. 1989;35:134–148. doi: 10.1038/ki.1989.18. [DOI] [PubMed] [Google Scholar]
- 7.Hertig A, Ferrer-Marin F. Correction of anaemia on dialysis: did we forget physiology? Nephrol Dial Transplant. 2011;26:1120–1122. doi: 10.1093/ndt/gfr001. [DOI] [PubMed] [Google Scholar]
- 8.Sargent JA, Acchiardo SR. Iron requirements in hemodialysis. Blood Purif. 2004;22:112–123. doi: 10.1159/000074931. [DOI] [PubMed] [Google Scholar]
- 9.Ackrell BA, Maguire JJ, Dallman PR, et al. Effect of iron deficiency on succinate- and NADH-ubiquinone oxidoreductases in skeletal muscle mitochondria. J Biol Chem. 1984;259:10053–10059. [PubMed] [Google Scholar]
- 10.McLane JA, Fell RD, McKay RH, et al. Physiological and biochemical effects of iron deficiency on rat skeletal muscle. Am J Physiol. 1981;241:C47–C54. doi: 10.1152/ajpcell.1981.241.1.C47. [DOI] [PubMed] [Google Scholar]
- 11.Krayenbuehl PA, Battegay E, Breymann C, et al. Intravenous iron for the treatment of fatigue in nonanemic, premenopausal women with low serum ferritin concentration. Blood. 2011;118:3222–3227. doi: 10.1182/blood-2011-04-346304. [DOI] [PubMed] [Google Scholar]
- 12.Nemeth E, Tuttle MS, Powelson J, et al. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science. 2004;306:2090–2093. doi: 10.1126/science.1104742. [DOI] [PubMed] [Google Scholar]
- 13.Tomosugi N, Kawabata H, Wakatabe R, et al. Detection of serum hepcidin in renal failure and inflammation by using ProteinChip System. Blood. 2006;108:1381–1387. doi: 10.1182/blood-2005-10-4043. [DOI] [PubMed] [Google Scholar]
- 14.Ashby DR, Gale DP, Busbridge M, et al. Plasma hepcidin levels are elevated but responsive to erythropoietin therapy in renal disease. Kidney Int. 2009;75:976–981. doi: 10.1038/ki.2009.21. [DOI] [PubMed] [Google Scholar]
- 15.Zaritsky J, Young B, Wang HJ, et al. Hepcidin—a potential novel biomarker for iron status in chronic kidney disease. Clin J Am Soc Nephrol. 2009;4:1051–1056. doi: 10.2215/CJN.05931108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Weiss G, Theurl I, Eder S, et al. Serum hepcidin concentration in chronic haemodialysis patients: associations and effects of dialysis, iron and erythropoietin therapy. Eur J Clin Invest. 2009;39:883–890. doi: 10.1111/j.1365-2362.2009.02182.x. [DOI] [PubMed] [Google Scholar]
- 17.Babitt JL, Lin HY. Molecular mechanisms of hepcidin regulation: implications for the anemia of CKD. Am J Kidney Dis. 2010;55:726–741. doi: 10.1053/j.ajkd.2009.12.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ple H, Maltais M, Corduan A, et al. Alteration of the platelet transcriptome in chronic kidney disease. Thromb Haemost. 2012;108:605–615. doi: 10.1160/TH12-03-0153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kadikoylu G, Yavasoglu I, Bolaman Z, et al. Platelet parameters in women with iron deficiency anemia. J Natl Med Assoc. 2006;98:398–402. [PMC free article] [PubMed] [Google Scholar]
- 20.Gurbel PA. The relationship of platelet reactivity to the occurrence of post-stenting ischemic events: emergence of a new cardiovascular risk factor. Rev Cardiovasc Med. 2006;7(Suppl 4):S20–S28. [PubMed] [Google Scholar]
- 21.Aljaroudi WA, Halabi AR, Harrington RA. Platelet inhibitor therapy for patients with cardiovascular disease: looking toward the future. Curr Hematol Rep. 2005;4:397–404. [PubMed] [Google Scholar]
- 22.Streja E, Kovesdy CP, Greenland S, et al. Erythropoietin, iron depletion, and relative thrombocytosis: a possible explanation for hemoglobin-survival paradox in hemodialysis. Am J Kidney Dis. 2008;52:727–736. doi: 10.1053/j.ajkd.2008.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vaziri ND. Understanding iron: promoting its safe use in patients with chronic kidney failure treated by hemodialysis. Am J Kidney Dis. 2013;61:992–1000. doi: 10.1053/j.ajkd.2012.10.027. [DOI] [PubMed] [Google Scholar]
- 24.Kurekci AE, Atay AA, Sarici SU, et al. Effect of iron therapy on the whole blood platelet aggregation in infants with iron deficiency anemia. Thromb Res. 2000;97:281–285. doi: 10.1016/s0049-3848(99)00150-4. [DOI] [PubMed] [Google Scholar]
- 25.Dahl NV, Henry DH, Coyne DW. Thrombosis with erythropoietic stimulating agents—does iron-deficient erythropoiesis play a role? Semin Dial. 2008;21:210–211. doi: 10.1111/j.1525-139X.2008.00435.x. [DOI] [PubMed] [Google Scholar]
- 26.Milman N. Iron therapy in patients undergoing maintenance hemodialysis. Acta Med Scand. 1976;200:315–319. doi: 10.1111/j.0954-6820.1976.tb08238.x. [DOI] [PubMed] [Google Scholar]
- 27.Eschbach JW, Egrie JC, Downing MR, et al. Correction of the anemia of end-stage renal disease with recombinant human erythropoietin. Results of a combined phase I and II clinical trial . N Engl J Med. 1987;316:73–78. doi: 10.1056/NEJM198701083160203. [DOI] [PubMed] [Google Scholar]
- 28.Szczech LA, Barnhart HX, Inrig JK, et al. Secondary analysis of the CHOIR trial epoetin-alpha dose and achieved hemoglobin outcomes. Kidney Int. 2008;74:791–798. doi: 10.1038/ki.2008.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Freburger JK, Ng LJ, Bradbury BD, et al. Changing patterns of anemia management in US hemodialysis patients. Am J Med. 2012;125:906–914. doi: 10.1016/j.amjmed.2012.03.011. e909. [DOI] [PubMed] [Google Scholar]
- 30.Rozen-Zvi B, Gafter-Gvili A, Paul M, et al. Intravenous versus oral iron supplementation for the treatment of anemia in CKD: systematic review and meta-analysis. Am J Kidney Dis. 2008;52:897–906. doi: 10.1053/j.ajkd.2008.05.033. [DOI] [PubMed] [Google Scholar]
- 31.Provenzano R, Schiller B, Rao M, et al. Ferumoxytol as an intravenous iron replacement therapy in hemodialysis patients. Clin J Am Soc Nephrol. 2009;4:386–393. doi: 10.2215/CJN.02840608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kidney Disease: Improving Global Outcomes (KDIGO) CKD Work Group. KDIGO 2012 clinical practice guideline for the evaluation and management of chronic kidney disease. Kidney Int Suppl. 2013;3:1–150. [Google Scholar]
- 33.Guyatt GH, Oxman AD, Kunz R, et al. Going from evidence to recommendations. BMJ. 2008;336(7652):1049–1051. doi: 10.1136/bmj.39493.646875.AE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Canavese C, Bergamo D, Ciccone G, et al. Validation of serum ferritin values by magnetic susceptometry in predicting iron overload in dialysis patients. Kidney Int. 2004;65:1091–1098. doi: 10.1111/j.1523-1755.2004.00480.x. [DOI] [PubMed] [Google Scholar]
- 35.Ghoti H, Rachmilewitz EA, Simon-Lopez R, et al. Evidence for tissue iron overload in long-term hemodialysis patients and the impact of withdrawing parenteral iron. Eur J Haematol. 2012;89:87–93. doi: 10.1111/j.1600-0609.2012.01783.x. [DOI] [PubMed] [Google Scholar]
- 36.Rostoker G, Griuncelli M, Loridon C, et al. Hemodialysis-associated hemosiderosis in the era of erythropoiesis-stimulating agents: a MRI study. Am J Med. 2012;125:991–999. doi: 10.1016/j.amjmed.2012.01.015. e991. [DOI] [PubMed] [Google Scholar]
- 37.Vaya A, Hernandez JL, Zorio E, et al. Association between red blood cell distribution width and the risk of future cardiovascular events. Clin Hemorheol Microcirc. 2012;50:221–225. doi: 10.3233/CH-2011-1428. [DOI] [PubMed] [Google Scholar]
- 38.Brookhart MA, Freburger JK, Ellis AR, et al. Infection risk with bolus versus maintenance iron supplementation in hemodialysis patients. J Am Soc Nephrol. 2013;24:1151–1158. doi: 10.1681/ASN.2012121164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rhee CM, Kalantar-Zadeh K. Is iron maintenance therapy better than load and hold? J Am Soc Nephrol. 2013;24:1028–1031. doi: 10.1681/ASN.2013050456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Torti FM, Torti SV. Regulation of ferritin genes and protein. Blood. 2002;99:3505–3516. doi: 10.1182/blood.v99.10.3505. [DOI] [PubMed] [Google Scholar]
- 41.Singh AK, Coyne DW, Shapiro W, et al. Predictors of the response to treatment in anemic hemodialysis patients with high serum ferritin and low transferrin saturation. Kidney Int. 2007;71:1163–1171. doi: 10.1038/sj.ki.5002223. [DOI] [PubMed] [Google Scholar]
- 42.Ford BA, Coyne DW, Eby CS, et al. Variability of ferritin measurements in chronic kidney disease; implications for iron management. Kidney Int. 2009;75:104–110. doi: 10.1038/ki.2008.526. [DOI] [PubMed] [Google Scholar]
- 43.Cazzola M, Beguin Y. New tools for clinical evaluation of erythron function in man. Br J Haematol. 1992;80:278–284. doi: 10.1111/j.1365-2141.1992.tb08133.x. [DOI] [PubMed] [Google Scholar]
- 44.Skikne BS, Flowers CH, Cook JD. Serum transferrin receptor: a quantitative measure of tissue iron deficiency. Blood. 1990;75:1870–1876. [PubMed] [Google Scholar]
- 45.R'Zik S, Beguin Y. Serum soluble transferrin receptor concentration is an accurate estimate of the mass of tissue receptors. Exp Hematol. 2001;29:677–685. doi: 10.1016/s0301-472x(01)00641-5. [DOI] [PubMed] [Google Scholar]
- 46.Harrison PM, Arosio P. The ferritins: molecular properties, iron storage function and cellular regulation. Biochim Biophys Acta. 1996;1275:161–203. doi: 10.1016/0005-2728(96)00022-9. [DOI] [PubMed] [Google Scholar]
- 47.Herbert V, Jayatilleke E, Shaw S, et al. Serum ferritin iron, a new test, measures human body iron stores unconfounded by inflammation. Stem Cells. 1997;15:291–296. doi: 10.1002/stem.150291. [DOI] [PubMed] [Google Scholar]
- 48.Yamanishi H, Iyama S, Yamaguchi Y, et al. Relation between iron content of serum ferritin and clinical status factors extracted by factor analysis in patients with hyperferritinemia. Clin Biochem. 2002;35:523–529. doi: 10.1016/s0009-9120(02)00380-6. [DOI] [PubMed] [Google Scholar]
- 49.Spada PL, Rossi C, Alimonti A, et al. Ferritin iron content in haemodialysis patients: comparison with septic and hemochromatosis patients. Clin Biochem. 2008;41:997–1001. doi: 10.1016/j.clinbiochem.2008.05.003. [DOI] [PubMed] [Google Scholar]
- 50.von Bonsdorff L, Lindeberg E, Sahlstedt L, et al. Bleomycin-detectable iron assay for non-transferrin-bound iron in hematologic malignancies. Clin Chem. 2002;48:307–314. [PubMed] [Google Scholar]
- 51.Piga A, Longo F, Duca L, et al. High nontransferrin bound iron levels and heart disease in thalassemia major. Am J Hematol. 2009;84:29–33. doi: 10.1002/ajh.21317. [DOI] [PubMed] [Google Scholar]
- 52.Taher A, Musallam KM, El Rassi F, et al. Levels of non-transferrin-bound iron as an index of iron overload in patients with thalassaemia intermedia. Br J Haematol. 2009;146:569–572. doi: 10.1111/j.1365-2141.2009.07810.x. [DOI] [PubMed] [Google Scholar]
- 53.Pootrakul P, Breuer W, Sametband M, et al. Labile plasma iron (LPI) as an indicator of chelatable plasma redox activity in iron-overloaded beta-thalassemia/HbE patients treated with an oral chelator. Blood. 2004;104:1504–1510. doi: 10.1182/blood-2004-02-0630. [DOI] [PubMed] [Google Scholar]
- 54.Scheiber-Mojdehkar B, Lutzky B, Schaufler R, et al. Non-transferrin-bound iron in the serum of hemodialysis patients who receive ferric saccharate: no correlation to peroxide generation. J Am Soc Nephrol. 2004;15:1648–1655. doi: 10.1097/01.asn.0000130149.18412.56. [DOI] [PubMed] [Google Scholar]
- 55.Cabantchik ZI, Breuer W, Zanninelli G, et al. LPI-labile plasma iron in iron overload. Best Pract Res Clin Haematol. 2005;18:277–287. doi: 10.1016/j.beha.2004.10.003. [DOI] [PubMed] [Google Scholar]
- 56.Jacobs A. Low molecular weight intracellular iron transport compounds. Blood. 1977;50:433–439. [PubMed] [Google Scholar]
- 57.Prus E, Fibach E. The labile iron pool in human erythroid cells. Br J Haematol. 2008;142:301–307. doi: 10.1111/j.1365-2141.2008.07192.x. [DOI] [PubMed] [Google Scholar]
- 58.Ghoti H, Amer J, Winder A, et al. Oxidative stress in red blood cells, platelets and polymorphonuclear leukocytes from patients with myelodysplastic syndrome. Eur J Haematol. 2007;79:463–467. doi: 10.1111/j.1600-0609.2007.00972.x. [DOI] [PubMed] [Google Scholar]
- 59.Ghoti H, Fibach E, Merkel D, et al. Changes in parameters of oxidative stress and free iron biomarkers during treatment with deferasirox in iron-overloaded patients with myelodysplastic syndromes. Haematologica. 2010;95:1433–1434. doi: 10.3324/haematol.2010.024992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Prus E, Fibach E. Flow cytometry measurement of the labile iron pool in human hematopoietic cells. Cytometry A. 2008;73:22–27. doi: 10.1002/cyto.a.20491. [DOI] [PubMed] [Google Scholar]
- 61.van der Putten K, Jie KE, van den Broek D, et al. Hepcidin-25 is a marker of the response rather than resistance to exogenous erythropoietin in chronic kidney disease/chronic heart failure patients. Eur J Heart Fail. 2010;12:943–950. doi: 10.1093/eurjhf/hfq099. [DOI] [PubMed] [Google Scholar]
- 62.Kato A, Tsuji T, Luo J, et al. Association of prohepcidin and hepcidin-25 with erythropoietin response and ferritin in hemodialysis patients. Am J Nephrol. 2008;28:115–121. doi: 10.1159/000109968. [DOI] [PubMed] [Google Scholar]
- 63.Tessitore N, Girelli D, Campostrini N, et al. Hepcidin is not useful as a biomarker for iron needs in haemodialysis patients on maintenance erythropoiesis-stimulating agents. Nephrol Dial Transplant. 2010;25:3996–4002. doi: 10.1093/ndt/gfq321. [DOI] [PubMed] [Google Scholar]
- 64.Malyszko J, Malyszko JS, Levin-Iaina N, et al. Is hemojuvelin a possible new player in iron metabolism in hemodialysis patients? Int Urol Nephrol. 2012;44:1805–1811. doi: 10.1007/s11255-011-0084-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sun CC, Vaja V, Chen S, et al. A hepcidin lowering agent mobilizes iron for incorporation into red blood cells in an adenine-induced kidney disease model of anemia in rats. Nephrol Dial Transplant. 2013;28:1733–1743. doi: 10.1093/ndt/gfs584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wagner M, Ashby D. Hepcidin—a well-known iron biomarker with prognostic implications in chronic kidney disease. Nephrol Dial Transplant. 2013;28:2936–2939. doi: 10.1093/ndt/gft330. [DOI] [PubMed] [Google Scholar]
- 67.van der Weerd NC, Grooteman MP, Bots ML, et al. Hepcidin-25 is related to cardiovascular events in chronic haemodialysis patients. Nephrol Dial Transplant. 2013;28:3062–3071. doi: 10.1093/ndt/gfs488. [DOI] [PubMed] [Google Scholar]
- 68.Jensen PD, Jensen FT, Christensen T, et al. Relationship between hepatocellular injury and transfusional iron overload prior to and during iron chelation with desferrioxamine: a study in adult patients with acquired anemias. Blood. 2003;101:91–96. doi: 10.1182/blood-2002-06-1704. [DOI] [PubMed] [Google Scholar]
- 69.Angelucci E, Baronciani D, Lucarelli G, et al. Needle liver biopsy in thalassaemia: analyses of diagnostic accuracy and safety in 1184 consecutive biopsies. Br J Haematol. 1995;89:757–761. doi: 10.1111/j.1365-2141.1995.tb08412.x. [DOI] [PubMed] [Google Scholar]
- 70.Telfer PT, Prestcott E, Holden S, et al. Hepatic iron concentration combined with long-term monitoring of serum ferritin to predict complications of iron overload in thalassaemia major. Br J Haematol. 2000;110:971–977. doi: 10.1046/j.1365-2141.2000.02298.x. [DOI] [PubMed] [Google Scholar]
- 71.Agarwal MB. Advances in management of thalassemia. Indian Pediatr. 2004;41:989–992. [PubMed] [Google Scholar]
- 72.Voskaridou E, Douskou M, Terpos E, et al. Magnetic resonance imaging in the evaluation of iron overload in patients with beta thalassaemia and sickle cell disease. Br J Haematol. 2004;126:736–742. doi: 10.1111/j.1365-2141.2004.05104.x. [DOI] [PubMed] [Google Scholar]
- 73.Ghoti H, Fibach E, Westerman M, et al. Increased serum hepcidin levels during treatment with deferasirox in iron-overloaded patients with myelodysplastic syndrome. Br J Haematol. 2011;153:118–120. doi: 10.1111/j.1365-2141.2011.08587.x. [DOI] [PubMed] [Google Scholar]
- 74.Ferrari P, Kulkarni H, Dheda S, et al. Serum iron markers are inadequate for guiding iron repletion in chronic kidney disease. Clin J Am Soc Nephrol. 2011;6:77–83. doi: 10.2215/CJN.04190510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Anderson LJ, Holden S, Davis B, et al. Cardiovascular T2-star (T2*) magnetic resonance for the early diagnosis of myocardial iron overload. Eur Heart J. 2001;22:2171–2179. doi: 10.1053/euhj.2001.2822. [DOI] [PubMed] [Google Scholar]
- 76.Westwood M, Anderson LJ, Firmin DN, et al. A single breath-hold multiecho T2* cardiovascular magnetic resonance technique for diagnosis of myocardial iron overload. J Magn Reson Imaging. 2003;18:33–39. doi: 10.1002/jmri.10332. [DOI] [PubMed] [Google Scholar]
- 77.Pepe A, Positano V, Santarelli MF, et al. Multislice multiecho T2* cardiovascular magnetic resonance for detection of the heterogeneous distribution of myocardial iron overload. J Magn Reson Imaging. 2006;23:662–668. doi: 10.1002/jmri.20566. [DOI] [PubMed] [Google Scholar]
- 78.Positano V, Pepe A, Santarelli MF, et al. Standardized T2* map of normal human heart in vivo to correct T2* segmental artefacts. NMR Biomed. 2007;20:578–590. doi: 10.1002/nbm.1121. [DOI] [PubMed] [Google Scholar]
- 79.Nielsen P, Engelhardt R, Duerken M, et al. Using SQUID biomagnetic liver susceptometry in the treatment of thalassemia and other iron loading diseases. Transfus Sci. 2000;23:257–258. doi: 10.1016/s0955-3886(00)00101-6. [DOI] [PubMed] [Google Scholar]
- 80.Kletzmayr J, Horl WH. Iron overload and cardiovascular complications in dialysis patients. Nephrol Dial Transplant. 2002;17(Suppl 2):25–29. doi: 10.1093/ndt/17.suppl_2.25. [DOI] [PubMed] [Google Scholar]
- 81.Drueke TB, Massy ZA. Intravenous iron: how much is too much? J Am Soc Nephrol. 2005;16:2833–2835. doi: 10.1681/ASN.2005080804. [DOI] [PubMed] [Google Scholar]
- 82.Borgna-Pignatti C, Rugolotto S, De Stefano P, et al. Survival and complications in patients with thalassemia major treated with transfusion and deferoxamine. Haematologica. 2004;89:1187–1193. [PubMed] [Google Scholar]
- 83.Hershko CM, Link GM, Konijn AM, et al. Iron chelation therapy. Curr Hematol Rep. 2005;4:110–116. [PubMed] [Google Scholar]
- 84.Hershko C, Link G, Cabantchik I. Pathophysiology of iron overload. Ann N Y Acad Sci. 1998;850:191–201. doi: 10.1111/j.1749-6632.1998.tb10475.x. [DOI] [PubMed] [Google Scholar]
- 85.Rund D, Rachmilewitz E. Beta-thalassemia. N Engl J Med. 2005;353:1135–1146. doi: 10.1056/NEJMra050436. [DOI] [PubMed] [Google Scholar]
- 86.Hershko C, Graham G, Bates GW, et al. Non-specific serum iron in thalassaemia: an abnormal serum iron fraction of potential toxicity. Br J Haematol. 1978;40:255–263. doi: 10.1111/j.1365-2141.1978.tb03662.x. [DOI] [PubMed] [Google Scholar]
- 87.Hebbel RP. Auto-oxidation and a membrane-associated ‘Fenton reagent’: a possible explanation for development of membrane lesions in sickle erythrocytes. Clin Haematol. 1985;14:129–140. [PubMed] [Google Scholar]
- 88.Le Lan C, Loreal O, Cohen T, et al. Redox active plasma iron in C282Y/C282Y hemochromatosis. Blood. 2005;105:4527–4531. doi: 10.1182/blood-2004-09-3468. [DOI] [PubMed] [Google Scholar]
- 89.Gupta A, Zhuo J, Zha J, et al. Effect of different intravenous iron preparations on lymphocyte intracellular reactive oxygen species generation and subpopulation survival. BMC Nephrol. 2010;11:16. doi: 10.1186/1471-2369-11-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Deicher R, Ziai F, Cohen G, et al. High-dose parenteral iron sucrose depresses neutrophil intracellular killing capacity. Kidney Int. 2003;64:728–736. doi: 10.1046/j.1523-1755.2003.00125.x. [DOI] [PubMed] [Google Scholar]
- 91.Patruta SI, Edlinger R, Sunder-Plassmann G, et al. Neutrophil impairment associated with iron therapy in hemodialysis patients with functional iron deficiency. J Am Soc Nephrol. 1998;9:655–663. doi: 10.1681/ASN.V94655. [DOI] [PubMed] [Google Scholar]
- 92.Guo D, Jaber BL, Lee S, et al. Impact of iron dextran on polymorphonuclear cell function among hemodialysis patients. Clin Nephrol. 2002;58:134–142. doi: 10.5414/cnp58134. [DOI] [PubMed] [Google Scholar]
- 93.Zager RA. Parenteral iron compounds: potent oxidants but mainstays of anemia management in chronic renal disease. Clin J Am Soc Nephrol. 2006;1(Suppl 1):S24–S31. doi: 10.2215/CJN.01410406. [DOI] [PubMed] [Google Scholar]
- 94.Lim CS, Vaziri ND. The effects of iron dextran on the oxidative stress in cardiovascular tissues of rats with chronic renal failure. Kidney Int. 2004;65:1802–1809. doi: 10.1111/j.1523-1755.2004.00580.x. [DOI] [PubMed] [Google Scholar]
- 95.Zoccali C. Endothelial dysfunction and the kidney: emerging risk factors for renal insufficiency and cardiovascular outcomes in essential hypertension. J Am Soc Nephrol. 2006;17(4 Suppl 2):S61–S63. doi: 10.1681/ASN.2005121344. [DOI] [PubMed] [Google Scholar]
- 96.Reis KA, Guz G, Ozdemir H, et al. Intravenous iron therapy as a possible risk factor for atherosclerosis in end-stage renal disease. Int Heart J. 2005;46:255–264. doi: 10.1536/ihj.46.255. [DOI] [PubMed] [Google Scholar]
- 97.Zager RA. Parenteral iron treatment induces MCP-1 accumulation in plasma, normal kidneys, and in experimental nephropathy. Kidney Int. 2005;68:1533–1542. doi: 10.1111/j.1523-1755.2005.00565.x. [DOI] [PubMed] [Google Scholar]
- 98.Weiss G, Meusburger E, Radacher G, et al. Effect of iron treatment on circulating cytokine levels in ESRD patients receiving recombinant human erythropoietin. Kidney Int. 2003;64:572–578. doi: 10.1046/j.1523-1755.2003.00099.x. [DOI] [PubMed] [Google Scholar]
- 99.Boelaert JR, Daneels RF, Schurgers ML, et al. Iron overload in haemodialysis patients increases the risk of bacteraemia: a prospective study. Nephrol Dial Transplant. 1990;5:130–134. doi: 10.1093/ndt/5.2.130. [DOI] [PubMed] [Google Scholar]
- 100.Bansal A, Sandhu G, Gupta I, et al. Effect of aggressively driven intravenous iron therapy on infectious complications in end-stage renal disease patients on maintenance hemodialysis. Am J Ther. 2012 doi: 10.1097/MJT.0b013e31825425bd. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 101.Linton TK, Pacheco MA, McIntyre DO, et al. Development of bioassessment-based benchmarks for iron. Environ Toxicol Chem. 2007;26:1291–1298. doi: 10.1897/06-431.1. [DOI] [PubMed] [Google Scholar]
- 102.Beshara S, Lundqvist H, Sundin J, et al. Kinetic analysis of 52Fe-labelled iron(III) hydroxide-sucrose complex following bolus administration using positron emission tomography. Br J Haematol. 1999;104:288–295. doi: 10.1046/j.1365-2141.1999.01170.x. [DOI] [PubMed] [Google Scholar]
- 103.Gokal R, Weatherall DJ, Bunch C. Iron induced increase in red cell size in haemodialysis patients. Q J Med. 1979;48:393–401. [PubMed] [Google Scholar]
- 104.Boll IT. A quantitative kinetic model of human erythropoiesis. Hamatol Bluttransfus. 1972;10:9–12. [PubMed] [Google Scholar]
- 105.Franzone PC, Paganuzzi A, Stefanelli M. A mathematical model of iron metabolism. J Math Biol. 1982;15:173–201. doi: 10.1007/BF00275072. [DOI] [PubMed] [Google Scholar]
- 106.Stefanelli M, Bentley DP, Cavill I, et al. Quantitation of reticuloendothelial iron kinetics in humans. Am J Physiol. 1984;247(5 Pt 2):R842–R849. doi: 10.1152/ajpregu.1984.247.5.R842. [DOI] [PubMed] [Google Scholar]
- 107.Nichols B, Shrestha RP, Horowitz J, et al. Simplification of an erythropoiesis model for design of anemia management protocols in end stage renal disease. Conf Proc IEEE Eng Med Biol Soc. 2011;2011:83–86. doi: 10.1109/IEMBS.2011.6089902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Chait Y, Horowitz J, Nichols B, et al. Control-relevant erythropoiesis modeling in end-stage renal disease. IEEE Trans Biomed Eng. 2014;61:658–664. doi: 10.1109/TBME.2013.2286100. [DOI] [PubMed] [Google Scholar]
- 109.Ljung L. System Identification: Theory for the User. Prentice-Hall: Placed Published; 1987. [Google Scholar]
- 110.Brier ME, Gaweda AE, Dailey A, et al. Randomized trial of model predictive control for improved anemia management. Clin J Am Soc Nephrol. 2010;5:814–820. doi: 10.2215/CJN.07181009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Gaweda AE, Aronoff GR, Jacobs AA, et al. Individualized Anemia Management Reduces Hemoglobin Variability in Hemodialysis Patients. J Am Soc Nephrol. 2014;25:159–166. doi: 10.1681/ASN.2013010089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Gaweda AE, Jacobs AA, Aronoff GR, et al. Model predictive control of erythropoietin administration in the anemia of ESRD. Am J Kidney Dis. 2008;51:71–79. doi: 10.1053/j.ajkd.2007.10.003. [DOI] [PubMed] [Google Scholar]
- 113.Gaweda AE, Aronoff GR, Jacobs AA, et al. Individualized anemia management reduces hemoglobin variability in hemodialysis patients. J Am Soc Nephrol. 2014;25:159–166. doi: 10.1681/ASN.2013010089. [DOI] [PMC free article] [PubMed] [Google Scholar]