

Abstract
Techniques largely used for protein interaction studies and discovery of intracellular receptors, such as affinity capture complex purification and yeast two-hybrid, may produce inaccurate datasets due to protein insolubility, transient or weak protein interactions, or irrelevant intracellular context. A versatile tool to overcome these limitations as well as to potentially create vaccines and engineer peptides and antibodies as targeted diagnostic and therapeutic agents, is the phage display technique. We have recently developed a new technology for screening internalizing phage (iPhage) vectors and libraries utilizing a ligand/receptor-independent mechanism to penetrate eukaryotic cells. iPhage particles provide a unique discovery platform for combinatorial intracellular targeting of organelle ligands along with their corresponding receptors and to fingerprint functional protein domains in living cells. Here we explain the design, cloning, construction, and production of iPhage-based vectors and libraries, along with basic ligand-receptor identification and validation methodologies for organelle receptors. An iPhage library screening can be performed in ~8 weeks.
INTRODUCTION
Over almost three decades, phage selection performed in vitro and in vivo has provided insight into the biology at the cell surface through the identification of novel functions for known proteins, novel multi-protein complexes, and targetable expression patterns in normal and pathologic settings1–15. Recently, we have shown that intracellular ligand-directed delivery can be accomplished by the use of cell-penetrating peptides that induce receptor-independent crossing of eukaryotic cell membranes. These peptides are usually arginine-rich cationic sequences of 10–30 residues in length. One of the best characterized peptides is penetratin (pen), which is derived from the third helix of the homeodomain of the Drosophila antennapedia protein16–18. Pen has been used to transport peptides, recombinant proteins, antibodies, small interfering RNAs, and micro RNAs into cells19–26.
The concept of an intracellular “ZIP code” system has long been well-established27–31. However, very few, if any, high-throughput discovery tools are currently available for the identification of intracellular ZIP codes. Here we use a recently introduced class of filamentous bacteriophage-based reagents that integrate recombinant pen as a fusion protein with a recombinant major coat protein (rpVIII), thereby enabling receptor-independent phage particle entry into, and intracellular distribution within, mammalian cells32. Moreover, either random peptide libraries or specific individual motifs can be displayed simultaneously on the minor coat protein (pIII), a feature allowing intracellular library selection and organelle-targeting. We have named this new family of reagents “internalizing phage” (iPhage) vectors and libraries32. This integrated technology platform permits ligand-directed combinatorial selection and targeting of cell organelles and intracellular signaling/metabolic pathways.
Peptide-based molecular probes identified by phage display technology expand the peptide repertoire for in vivo diagnosis and therapy of obesity, cancer, and eye diseases. Numerous peptides that bind cancer-associated antigens have been discovered by affinity selection of ligands from phage display random libraries. A few of the peptides selected by phage display are currently being developed toward preclinical and clinical applications33–38. The success of phage-derived peptides depends on the quality of the library screen, functional assays, and receptor validation. Here we describe the method for generating and screening the iPhage display system, and explain how to select and validate candidate internalizing homing peptide (iHoPe) ligands (Figure 1).
Figure 1.
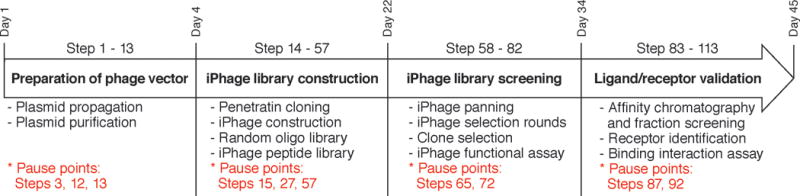
Flowchart of the iPhage combinatorial library technology. Steps involved in iPhage construction, library cloning, and ligand/receptor validation are shown in bold. Times required to complete these steps are depicted in the time line (days). Pause points are indicated in red.
The iPhage methodology described here can also be integrated with the adeno-associated virus/phage (AAVP) constructs39–45 to generate internalizing AAVP (iAAVP) vectors and libraries, which have been used for the isolation of blocking peptides for apoptosis-inducing drugs46 and for RNA interference (RNAi) library screens47, 48. This new class of phage vector is under development in our laboratory and is based on a two-step cloning strategy: generation of an iPhage vector containing a multicloning site (iPhage-MCS vector), and subsequent cloning of pAAV containing the eukaryotic expression cassette of interest between flanking Inverted Terminal Repeats (ITR) sequences into the iPhage-MCS. Furthermore, it may be possible to combine receptor-targeting peptides with intracellular bioactive peptides discovered by iPhage, thus creating a potential to modulate cell function in a tissue- and organ-specific fashion. The new resource introduced here can be used to target intracellular ZIP codes, interrogate signal transduction pathways, and participate in the discovery and development of an organelle-targeted cell biology and pharmacology in mammalian cells.
Applications of the iPhage technology
The iPhage technology can be applied to the unbiased delineation of intracellular pathways, intracellular protein-protein complexes, and organelle receptors in their native conformation. The expression of the pen protein on the recombinant pVIII allows, in a receptor-independent fashion, the penetration of a complete phage library into mammalian cells in vitro; well-established peptide motifs expressed on the minor capsid protein pIII can thereby be delivered to different sub-cellular locations. Note that for the purpose of this protocol we describe step-by-step the separation of the mitochondria fraction, and identification and validation of mitochondria-ligand peptides. However, separation of the nuclear and cytosolic fractions after iPhage library panning, bioactive iPhage particle identification, and peptide sequence selection can also be achieved following the same guidelines described herein. Ultimately, bioactive intracellular peptide ligands identified by iPhage can be tailored to other targeting entities for tissue selectivity.
Limitations of the iPhage technology
Identification of a target or protein receptor by phage display relies mostly on an effective way to separate specific from non-specific binders. In conventional in vitro phage display panning, removal of non-specific binders can be easily achieved by thorough wash steps. In the case of iPhage, however, the entire phage library enters the cell, and the identification of specifically bound phage requires distinct and systematic approaches. The proper isolation of organelles will determine enrichment of specific peptide sequences in sub-cellular fractions and will have an impact on the identification and validation of ligand/receptor pairs.
Our studies with iPhage suggest that mainly soluble proteins and the surface of organelles are targetable using this technology. The binding properties of iPhage towards single-pass or integral membrane proteins, or the ability of iPhage particles to target a given organelle’s internal space (lumen), such as the mitochondrial matrix, have yet to be evaluated. Thus, iPhage panning, identification, and receptor validation data must be analyzed carefully. Further and complementary experiments such as electron microscopy studies must be performed to ascertain whether iPhage can cross the membrane of the organelle of interest. Likewise, novel ligand peptides and receptors should be confirmed functionally.
Advantages of the iPhage technology
Several approaches for the discovery of peptides with intracellular functions have been developed in the last few years. For example, retroviral expression of peptide libraries in mammalian cells guided the isolation of blocking peptides for cell death-inducing drugs46; the same screening system coupled to fluorescent markers identified organelle-homing peptides with useful biological properties49. Similarly, many systematic approaches have been used to interrogate intracellular pathways and activities, including shRNA and RNAi library screening47, 48 and yeast (two) hybrid techniques50, 51. The former screen identifies genes involved in specific processes but does not place them in context, e.g., upstream, downstream, or at the nexus of the specific function assayed. The latter identifies putative binding moieties and interaction networks, and provides better context, but lacks direct functional insight because only binding partners are identified.
Conversely, in the past decade, combinatorial phage display technology has expanded in its application to decipher the molecular diversity of peptide binding specificity to isolated proteins, purified antibodies, cell surface receptors, intracellular/cyto-domains, and blood vessels in vivo52 in an unbiased functional manner without any preconceived notion of the nature of the target. Moreover, phage display technology, including iPhage, is a relatively inexpensive methodology, libraries can be easily obtained, and the technique is simple, rapid to set up, and requires no special equipment.
Our initial studies with iPhage have identified peptides that have the potential to modulate cell function in a tissue- and/or organ-specific manner after organelle-specific delivery, for example, via the triggering of apoptosis32. These data suggest that iPhage is a valuable tool for interrogation and evaluation of intracellular molecular interactions, enabling direct targeting of intracellular organelles and metabolic/signaling pathways in living cells, as well as allowing the selection of bioactive peptides from virtually any subcellular fraction and organelle.
Experimental design
Generation of the internalizing phage peptide library
The construction of iPhage random peptide libraries is based on cloning DNA fragments encoding peptide sequences into the phage genome fused to the pIII coat protein gene (Figure 2). Incorporation and expression of the gene fusion product results in the presentation of the peptide on the phage surface, on which it can interact with and bind to a potential target. A phage library can consist of up to 109 unique phage clones, each displaying a different peptide. The size of the peptide insert as well as its expression orientation (linear or cyclic), are two variations that can be adjusted to fit the purpose of the screen. In this protocol, we use a X4YX4 (X, any residue; Y, tyrosine) linear library to exemplify the potential of the iPhage technology. In addition, the success of the screening is integrally dependent on how well the library is constructed. Although insertless phage cannot be avoided and will be eliminated in the phage selection biopanning procedures, it is important to limit the presence of insertless phage in the library to maintain library diversity.
Figure 2.
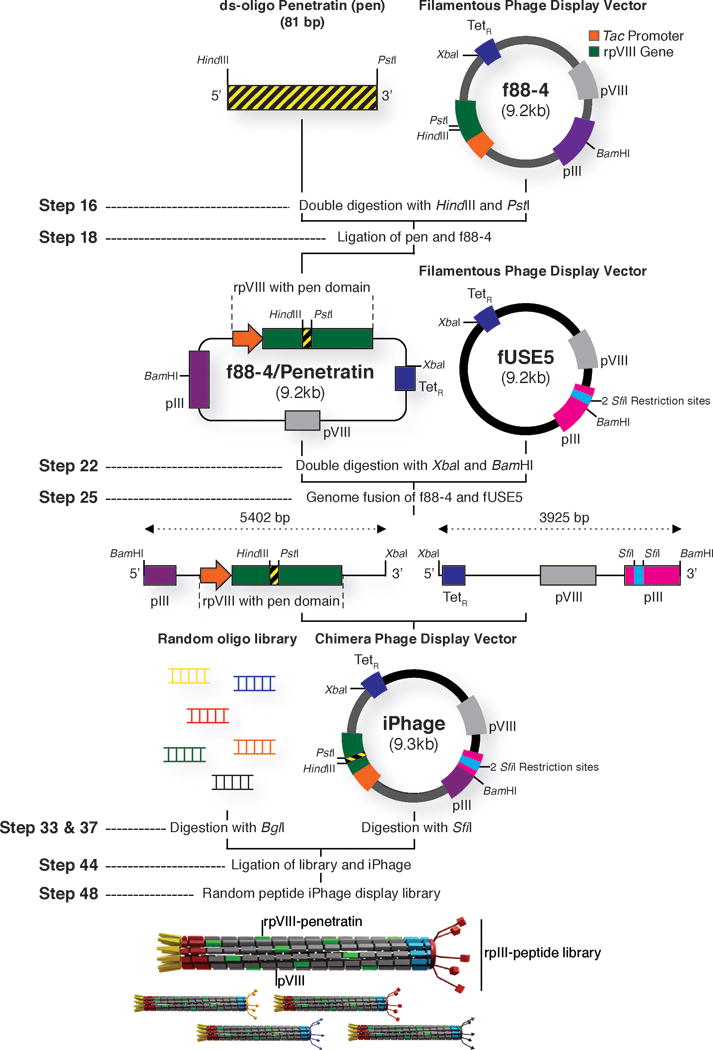
Generation of iPhage vector and library cloning strategy. The parental f88-4 phage vector contains two capsid genes encoding a wild-type protein VIII (pVIII, depicted in grey) and a recombinant protein VIII (rpVIII, depicted in green). The recombinant gene VIII contains a foreign DNA insert with a HindIII and a PstI cloning site, which allows the cloning of annealed oligonucleotides encoding the pen peptide in frame with the recombinant gene VIII. For generation of the iPhage vector, f88-4 displaying the pen peptide motif (RQIKIWFQNRRMKWKK) and fUSE5 phage plasmids are digested with BamHI and XbaI restriction enzymes, purified, and fused by standard ligation protocol. Next, an annealed random oligonucleotide library (e.g. X4YX4) is digested with the BglI restriction enzyme and cloned within the SfiI restriction site of the pIII coat protein gene (pIII, depicted in light blue) on the iPhage vector. MC1061 E. coli electro competent cells are transformed with the library cloned iPhage vector to produce a random peptide iPhage library. TetR – tetracycline resistance gene.
The fUSE5 vector used in our studies was engineered to be non-infective by disruption of the gene III reading frame with a 14-bp “stuffer”53. Infectivity is restored only when the stuffer sequence is replaced with an in-frame insertion. Removal of the fUSE5 stuffer sequence within gene III is achieved by digestion with the restriction enzyme SfiI (Figure 2). This process leaves two overhanging sites incompatible with each other, thereby allowing the unidirectional cloning of the DNA insert53. Also, it is important to note that the f88-4 display vector contains two VIII genes, encoding two different types of pVIII molecules. One pVIII is recombinant, fused to penetratin, and the other is wild type (WT). The recombinant gene (r)pVIII is synthetic and differs in its nucleotide sequence from the WT gene (Figure 2). Because the recombinant gene VIII is transcribed from a tac promoter, full expression in a lacIQ strain, such as K91Blu/kan E. coli, requires the inducer IPTG (1 mM) and absence of glucose. In a lacI strain such as MC1061 E. coli, the IPTG is not necessary.
For the purification of f88-4, fUSE5, and iPhage vectors, we recommend the use of a cesium chloride density gradient (Figure 3a). This method yields ultra-clean, high-quality phage plasmid preps appropriate for cloning of pen, construction of the chimera vector, and iPhage library construction (Figure 3b). However, if long spinning times and the use of EtBr are considered a major drawback, we recommend that the phage plasmid be purified twice on a Qiagen 500-tip to yield cleaner DNA preps.
Figure 3.
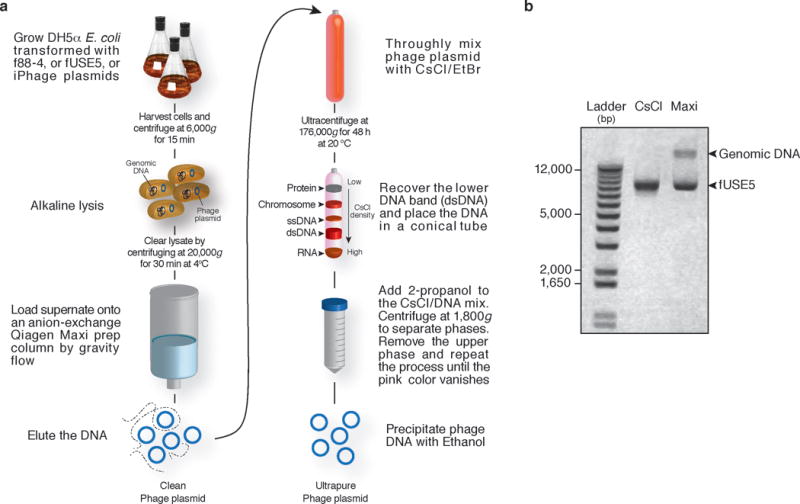
Overview of phage vector (f88-4, fUSE5, and iPhage) purification by CsCl. (a) Overnight bacterial culture is harvested and purified via a maxi-prep plasmid purification kit according to the manufacturer’s instructions ; a ‘clean phage plasmid’ prep is produced. For maximum phage plasmid purity (ultrapure), a CsCl/EtBr gradient is performed. After ultracentrifugation, the lower plasmid band (dsDNA band) is recovered and is precipitated by addition of 2-propanol into the DNA mix. 2-propanol will also remove the EtBr from the phage plasmid. Finally, addition of ethanol precipitates the DNA and removes any salt contaminants from the plasmid prep. (b) 0.8% E-gel analysis of fUSE5 purified with the Qiagen Maxi kit followed by CsCl/EtBr gradient purification (CsCl) or by Qiagen Maxi prep fUSE5 (Maxi) only. fUSE5 phage plasmid: 9206 Kb.
Because the library quality and diversity rely on the cloning protocol, we recommend that both phage vector and insert are added to the tubes, mixed with Milli-Q water, and warmed to 50°C for 3 min to melt any cohesive termini that have reannealed during DNA purification. Chill the DNA solution on ice before the rest of the ligation reagents are added. Purification of the ligated products and recovery in water allow high electroporation efficiency in the MC1061 E. coli strain.
Screening, selection, and receptor validation of candidate iPhage clones
The screening of a phage library consists of 3 main steps: (i) introduction of phage particles to an immobilized target, in this case the Kaposi Sarcoma (KS) 1767 cell line54; (ii) removal of unbound phage by washing; and (iii) elution of bound phage. Ideally, one cycle of selection should be sufficient, although in practice several rounds are necessary (typically two to four) to isolate target-specific binders. DNA sequencing is used to monitor enriched peptide frequency compared to the unselected library. Next, the peptide of interest is synthesized and used to identify and ultimately validate the receptor candidate (Figure 4). Enriched peptide sequences can also be further analyzed by comparison to a publically available peptide database (e.g. Basic Local Alignment Search Tool – BLAST) to identify proteins and/or ligands with the same sequence as the peptide.
Figure 4.
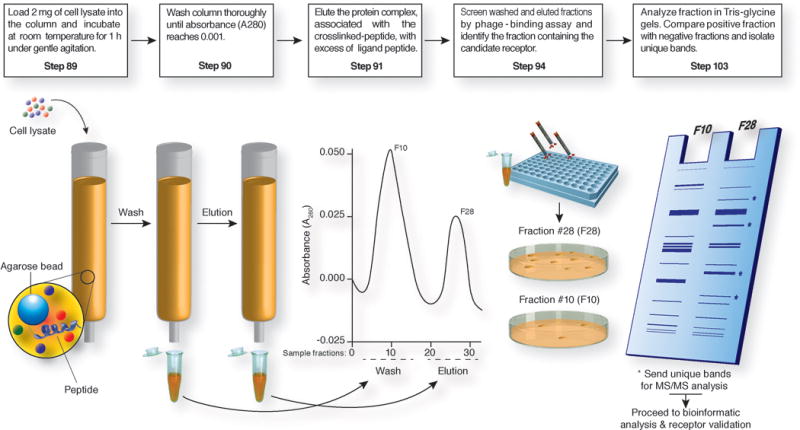
Systematic approach for affinity chromatography and receptor identification based on iPhage technology. Affinity columns are crafted by coupling of the selected synthetic peptide to agarose beads according to the manufacturer’s guidelines. Cell lysate is then loaded in the column and the receptor is eluted with the corresponding competitive peptide at a concentration of 5 mM. Eluted fractions are monitored by absorbance (optical density at 280 nm), desalted, and concentrated. Fraction screening is performed by the coating of equal amounts of protein on a 96-well plate and incubation with targeted iPhage or parental insertless iPhage (109 TU each). Each phage population is recovered by host bacterial infection. Positive fractions (high number of bacterial colonies) are further analyzed by gel electrophoresis. Finally, unique bands are identified and selected for MS/MS analysis.
For successful iPhage biopanning, we recommend titering the iPhage library or iPhage clone no longer than one week before each experiment to ensure that an accurate number of iPhage particles is used. When iPhage biopanning is done on a cell line, cells must be tested frequently for mycoplasma contamination. If possible, do not use a cell line that has been sub-cultured more than 5 times, because frequent passage could lead to genetic drift and other variations that would compromise the experimental results. Maintaining a fresh K91/kan E. coli stock for phage recovery is also important. Because there is always a contamination risk of K91/kan E. coli by phage present in the laboratory, use caution when handling bacteria and phage. Use only sterile single-use or autoclaved glassware and avoid spills. After handling phage purifications, glassware, and any other reusable equipment, materials must be soaked for 30 min in 10 % bleach and autoclaved.
Phage binding and immunocapture assays are commonly used to validate candidate receptors for peptides isolated from the combinatorial phage display screenings. Several factors can influence the results of the assays. For example, the molecular interaction between the peptide and receptor can be influenced by the amount of immobilized protein, the affinity of the peptide ligand for its receptor, ionic strength, the conformation of the protein, the existence of protein complexes, and temperature. During the phage binding assay, the cell lysates must be prepared with non-denaturing (i.e., non-ionic) detergents such as Triton X-100, NP-40, or Tween 20 to disrupt the cell membrane integrity, thereby facilitating lysis and extraction of soluble proteins, yet maintaining native structural conformations and protein complexes, and protease inhibitors to prevent protein degradation. Subcellular fractions, prepared by the differential centrifugation method55–57 (Figure 5a) or commercially available cell fractionation kits (e.g ProteoExtract Subcellular Proteome Extraction Kit, Millipore, Cat no. 539790), must be tested by Western blotting for specific markers such as cadherin (membrane marker), hydroxysteroid (17-beta) dehydrogenase 10 (ERAB; mitochondrial marker), high mobility group box 1 (HMGB1; nuclear marker) and actin (cytosol marker) to ensure proper organelle/compartment enrichment (Figure 5b). Further subcellular fractionation techniques57–61 to obtain highly pure subcellular fractions, such as sucrose density gradient centrifugation57 may be applied and are compatible with the iPhage technology. Use of either PBS or Tris-buffered saline for the phage binding assays is recommended. Note that other buffers might be used, but optimization would be required. The blocking solution (BSA) should always be filtered (0.22 μm) because this procedure minimizes non-specific control phage binding and bacterial colony background.
Figure 5.
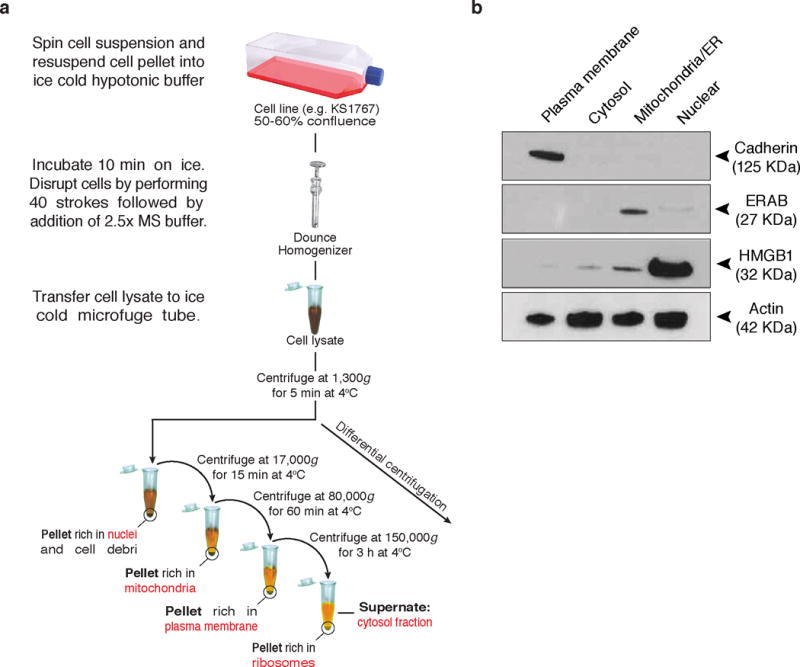
Overview of differential centrifugation, and subcellular fractionation quality analysis. (a) After an overnight incubation with the iPhage library, cells are recovered and transferred into a 15 ml conical tube. Cell pellet is washed and mixed with ice-cold hypotonic buffer. Cells are mechanically lysed with a Dounce homogenizer. Mitochondrial stabilization (MS) buffer is added to the cell lysate and after centrifuging the samples the supernate is recovered and transferred into clean microfuge tubes. The first pellet sample contains the nuclei, intact cells, and cell debri. Further fractionation may be performed to purify the nuclear fraction. Repeated centrifugation at progressively high speeds will separate cell components on the basis of size and density. (b) Equal protein concentrations were resolved in a 4-20% Tris-glycine gel and transferred onto a nitrocellulose membrane. Plasma membrane, cytosol, mitochondria/endoplasmic reticulum (ER), and nuclear fractions were tested with Cadherin (membrane marker), ERAB (mitochondrial marker), HMGB1 (nuclear marker), and actin (cytosol marker) antibodies.
Box 1. Rapid phage quantification (qPhage) TIMMING 2 h.
In this procedure phage content is determined by qPCR. Dilute the iPhage clone or library sample to 1:10,000 in Milli-Q water. Add 1 μl of diluted iPhage, Fast SYBR Green PCR Master Mix and 3.75 pmol of each oligonucleotide qPhage primer (Table 1) directed to the amplification of a fragment of the TetR gene, in a final reaction volume of 15 μl/well. The PCR thermal profile consists of:
|
| |||
| Cycle number | Segment 1 | Segment 2 | Segment 3 |
|
| |||
| 1 | 50 °C, 2 min | ||
| 2 | 95 °C, 10 min | ||
| 3–43 | 95 °C, 15 s 60 °C, 60 s |
||
|
| |||
Standard curves are generated with serial phage dilutions (from 10 to 108 plasmids) for each run. Each sample is amplified in triplicate. The standard curve is calculated by a linear regression analysis and serial dilutions. Amplification efficiency (AE) of each PCR cycle is calculated from the slope (s) of the standard curve by the equation: AE = 101/(−s). The value obtained is then multiplied by the dilution factor (i.e. 104) resulting in ‘number of phage particles/μl’.
In the immunocapture assay, a critical factor is the determination of candidate receptor expression levels. Lysates obtained from the cell line screened should always be tested by Western blotting to define the source of the protein extract. In addition, different concentrations of immobilized antibodies as well as cell extracts should be tested to determine optimal conditions prior to performing the immunocapture assay. It is important to ascertain the amount of immobilized protein on the plate. The best concentration for the assay can be estimated by a protein assay (Bradford or BCA) or by serial protein dilutions.
During affinity chromatography, multiple protein bands are often co-purified, indicating that the candidate receptor might be found in association with other molecular components (Figure 4). Tandem mass spectrometry and bioinformatics analysis (i.e., DAVID, String, or ingenuity pathway analysis) should indicate whether the proteins purified were previously reported to form protein complexes.
Rapid phage quantification
In this protocol we used the traditional phage display method of phage quantification by counting transduced units and individual sequencing of phage clones. However, to eliminate the reliance on host bacteria, one can utilize real-time PCR for rapid phage quantification (qPhage)62. The qPhage protocol is described in Box 1 and can be used to replace the traditional colony count method in STEPS 57, 72, 77, and 94. Note that with the qPhage method, template optimization may be required.
MATERIALS
REAGENTS
Plasmids: fUSE5 (GenBank: AF218364) and f88-4 (GenBank: AF218363.1) phage plasmids53, 63. For instructions of how to request and obtain phage plasmids, access: http://www.biosci.missouri.edu/smithgp/PhageDisplayWebsite/PhageDisplayWebsiteIndex.html
Electrocompetent DH5α cells (Life Technologies, cat no. 18258-012)
Escherichia coli (MC1061 and K91/kan)40. For instructions of how to request and obtain bacteria strains, access: http://www.biosci.missouri.edu/smithgp/PhageDisplayWebsite/PhageDisplayWebsiteIndex.html
BamHI endonuclease (Thermo Fisher Scientific, cat. no. ER0052)
BglI endonuclease (Thermo Fisher Scientific, cat. no. ER0071)
HindIII endonuclease (Thermo Fisher Scientific, cat. no. ER0505)
PstI endonuclease (Thermo Fisher Scientific, cat. no. ER0612)
SfiI endonuclease (Thermo Fisher Scientific, cat. no. ER1821)
XbaI endonuclease (Thermo Fisher Scientific, cat. no. ER0682)
Oligonucleotides (Sigma-Genosys) (see Table 1)
Kaposi Sarcoma (KS) 1767 cell line54 or cell line of interest. The KS1767 cell line can be obtained by contacting Dr. Renata Pasqualini (rpasqual@mdanderson.org) or Dr. Wadih Arap (warap@mdanderson.org).
0.05% Trypsin-EDTA (Life Technologies, cat. no. 25300054)
FBS (Life Technologies, cat. no. 10437-028)
L-Glutamine, 100x (Life Technologies, cat. no. 25030081)
MEM (Cellgro, cat. no. 15-010)
MEM Non-essential amino acids solution (100x, Life Technologies, cat. no. 11140050)
MEM-vitamin solution (100x, Life Technologies, cat. no. 11120052)
Penicillin-Streptomycin solution (100x, Life Technologies, cat. no. 15240-062)
PBS, pH 7.4 (Thermo Fisher Scientific, cat. no. BP2438-20)
Alexa Fluor 488 Goat Anti-Rabbit IgG (Life Technologies, cat. no. A-11008)
beta Actin antibody (Abcam, cat no. ab8226)
ERAB antibody (Abcam, cat no. ab137455)
fd Bacteriophage antibody (SIGMA-ALDRICH, cat. no. B7786)
HMGB1 antibody – ChIP Grade (Abcam, cat no. ab18256)
Normal rabbit IgG (Millipore, cat. no. Nl01-100UG)
pan Cadherin antibody (Abcam, cat no. ab22744)
10 mM dNTPs (Life Technologies, cat. no. 18427-013)
2-Mercaptoethanol (SIGMA-ALDRICH, cat. no. M3148) CAUTION Harmful if swallowed. Toxic in contact with skin. Causes burns. Work in a fume hood and wear proper protective equipment.
2-Propanol (SIGMA-ALDRICH, cat no. I9516)
BSA (SIGMA-ALDRICH, cat. no. A9418-100G)
CaCl2 (SIGMA-ALDRICH, cat. no. 449709)
CarboxyLink Immobilization Kit (Thermo Fisher Scientific, cat. no. 44899)
Cell proliferation kit (Roche; MTT (cat. no. 11465007001) or WST-1 (cat. no. 11644807001)]
Cesium chloride (CsCl) (Thermo Fisher Scientific, cat. no. BP1595-500)
Coomassie blue stain (SimplyBlue SafeStain, Life Technologies, cat. no. LC6060)
DMSO (SIGMA-ALDRICH, cat. no. D8418-50ML) CAUTION DMSO is readily absorbed through the skin and has been linked to aplastic anemia. When handling DMSO, wear appropriate gloves, safety glasses and use a pipetting aid under a safety chemical hood.
EtBr (Bio-Rad, cat. no. 161-0433) CAUTION EtBr is a potent mutagen. May be fatal if inhaled and is harmful if swallowed or absorbed through skin. Causes irritation to eyes, respiratory tract, and skin. May cause heritable genetic damage. Wear gloves and safety glasses. It must be handled with extreme caution and decontaminated on activated charcoal or amberlite ion exchange resins prior to disposal.
Ethanol (Thermo Fisher Scientific, cat. no. BP2818-100)
Fast SYBR Green (Life Technologies, cat. no. 4385612) CAUTION May be carcinogenic when ingested or absorbed by the skin.
Glucose (SIGMA-ALDRICH, cat. no. D9434)
Glycerol (SIGMA-ALDRICH, cat. no. G5516)
Glycine (SIGMA-ALDRICH, cat. no. G8898)
GoTaq DNA polymerase (Promega, cat. no. M8295)
IPTG (Thermo Fisher Scientific, cat. no. BP175510)
K2HPO4 (SIGMA-ALDRICH, cat. no. P3786)
Kanamycin monosulfate (SIGMA-ALDRICH, cat no. K4000)
KCl (SIGMA-ALDRICH, cat. no. P9541-500)
KH2PO4 (SIGMA-ALDRICH, cat. no. P9791)
LB Agar (Thermo Fisher Scientific, cat. no. BP9735)
Mannitol (SIGMA-ALDRICH, cat. no. M4125)
MgCl2 (SIGMA-ALDRICH, cat. no. M8266)
MgSO4 (SIGMA-ALDRICH, cat. no. M7506)
n-Octyl glucoside (SIGMA-ALDRICH, cat. no. O8001-100G)
NaCl (Thermo Fisher Scientific, cat. no. BP3581)
NaOH (SIGMA-ALDRICH, cat. no. 221465)
Paraformaldehyde (PFA) Aqueous Solution (Electron Microscopy Sciences, cat. no. 15710) CAUTION PFA is highly toxic. Avoid contact with eyes, skin, and mucous membranes. Use skin and eye protection during PFA manipulation.
Phenylmethylsulfonyl fluoride (PMSF) (SIGMA-ALDRICH, cat. no. P7626) CAUTION PMSF powder is hazardous. Use skin and eye protection when preparing solutions of PMSF.
Plasmid Maxi Kit (QIAGEN, cat. no. 12963)
Polyethylene glycol-8000 (SIGMA-ALDRICH, cat. no. P5413)
Protease inhibitor cocktail (Roche, cat. no. 11836170001)
QIAprep Spin Miniprep Kit (QIAGEN, cat. no. 27106)
QIAquick Gel Extraction Kit (QIAGEN, cat. no. 28704)
QIAquick nucleotide removal Kit (QIAGEN, cat. no. 28304)
Quanti-Marker 100 bp (Bioexpress, cat. no. C-5088-200)
Quanti-Marker 1Kb (Bioexpress, cat. no. C-5087-200)
Sodium azide (SIGMA-ALDRICH, cat. no. S2002-5G) CAUTION Sodium azide powder is hazardous. Use skin and eye protection when preparing solutions containing sodium azide.
Streptomycin sulfate (SIGMA-ALDRICH, cat. no. S6501)
Sucrose (SIGMA-ALDRICH, cat. no. 84097)
Super Optimal Broth with Catabolite repression (SOC) (Life Technologies, cat. no. 15544-034).
Synthetic peptides (PolyPeptide Laboratories or CPC Scientific)
T4 DNA ligase (Life Technologies, cat. no. 15224-041)
TE Buffer (Promega, cat. no. V6232)
Tetracycline (SIGMA-ALDRICH, cat. no. T8032)
Triton X-100 (SIGMA-ALDRICH, cat. no. T8787)
Tryptone (Thermo Fisher Scientific, cat. no. BP1421500)
UltraPure 0.5M EDTA (Life Technologies, cat. no. 15575-020)
UltraPure 1M Tris-HCl, pH 7.5 (Life Technologies, cat. no. 15567-027)
Yeast extract (Thermo Fisher Scientific, cat. no. BP1422-500)
TABLE 1.
Primer set sequences
| Primer set | Sequence | Purification |
|---|---|---|
| Penetratin | Fwd: 5′-CACAAGCTTTGCCAACGTCCCTCGACAGATAAAGATTTGGTTCCAAAACCGGCGCATGAAGTGGAAGAAGCCTGCAGCACA-3′ Rev: 5′-TGTGCTGCAGGCTTCTTCCACTTCATGCGCCGGTTTTGGAACCAAATCTTTATCTGTCGAGGGACGTTGGCAAAGCTTGTG-3′ |
PAGE or HPLC |
|
| ||
| Random library |
*TPL: 5′-CACTCGGCCGACGGGGCTNNKNNKNNKNNKTATNNKNNKNNKNNKGGGGCCGCTGGGGCCGAA-3′ Fwd: 5′-CACTCGGCCGACG-3′ Rev: 5′-TTCGGCCCCAGCGGC-3′ |
Desalted |
|
| ||
| f88-4seq | Fwd: 5′-GCTCCTTTCGCTTTCTTCCCTTCC-3′ Rev: 5′-TCAGGGGAGTAAACAGGAGACAAG-3′ |
Desalted |
|
| ||
| qPhage | Fwd: 5′-TGAGGTGGTATCGGCAATGA-3′ Rev: 5′-GGATGCTGTATTTAGGCCGTTT-3′ |
Desalted |
|
| ||
| pIIIseq | Fwd: 5′-AGCAAGCTGATAAACCGATACAATT-3′ Rev: 5′-CCCTCATAGTTAGCGTAACGATCT-3′ |
Desalted |
|
| ||
| NLS oligo sequence | Fwd: 5′-GGGCTCCGAAAAAAAAACGCAAAGTGGGGGCCGCTG-3′ Rev: 5′-CGGCCCCCACTTTGCGTTTTTTTTTCGGAGCCCCGT-3′ |
Desalted |
|
| ||
| YKWYYRGAA oligo sequence | Fwd: 5′- GGGCTTATAAATGGTATTATCGCGGCGCGGCGGGGGCCGCTG -3′ Rev: 5′- CGGCCCCCGCCGCGCCGCGATAATACCATTTATAAGCCCCGT -3′ |
Desalted |
Template: N – indicates all four nucleotides; K – indicates an equimolar mixture of G and T to prevent the introduction of stop codons into the sequence.
EQUIPMENT
0.22 μm Syringe filter (Millipore, cat. no. SLGVR25NB)
0.45 μm Syringe filter (Millipore, cat. no. SLHA025NB)
0.5 ml microfuge tubes (Thermo Fisher Scientific, cat. no. 05-408-120)
1 ml syringe (BD Biosciences, cat. no. 309628)
1.5 ml microfuge tubes (Thermo Fisher Scientific, cat. no, 05-408-129)
15-ml screw-cap polypropylene tubes (BD Biosciences, cat. no. 352196)
175-cm2 tissue culture flasks (BD Biosciences, cat. no. 353028)
18g needles (BD Biosciences, cat. no. 305196)
2 L flask (Thermo Fisher Scientific, cat. no. 10-041D)
2.0 ml microfuge tubes (Thermo Fisher Scientific, cat. no, 05-408-138)
500 ml Centrifuge bottles (Thermo Fisher Scientific, cat. no. 3141-0500)
75-cm2 tissue culture flasks (BD Biosciences, cat. no. 353136)
96-Well Disposable Electroporation Plate, 2 mm gap (BTX Harvard Apparatus, cat. no. 45-0450)
96-well microplates, flat bottom (BD Biosciences, cat. no. 353077)
96-well PCR plates (SIGMA-ALDRICH, cat. no. Z605298)
96-well U-bottom plates (BD Biosciences, cat. no. 351177)
Dounce tissue grinder with glass pestle (Thomas Scientific, cat. no. 3432N79)
E-Gel 0.8%, 2.0% and 4.0% agarose (Life Technologies, cat. no. G5018-08, G5018-02, G5018-04)
ECM 630 High Throughput Electroporation System (BTX Harvard Apparatus, cat. no. 45-0051)
Gene pulser/Micropulser Cuvettes 0.1 cm gap (Bio-Rad, cat. no. 165-2083)
Glass chamber slide system; 16-well (Thermo Fisher Scientific, cat. no. 178599)
Glass Pasteur pipettes (Thermo Fisher Scientific, cat. no. 13-678-20A)
HT-100 plate handler (BTX Harvard Apparatus, cat. no. 45-0400)
Novex 4-20% Tris-glycine SDS-PAGE gels (Life Technologies, cat. no. EC6025BX5)
Parafilm (Fisher Scientific, cat. no. 13-374-10)
Phase-contrast microscope (e.g. Zeiis Axioscope)
Protein A-coated 96-well plates (Thermo Fisher Scientific, cat. no. 15130)
Quick-seal ultracentrifuge tubes (Beckman cat. no. 342412)
Spin column concentrators (Thermo Fisher Scientific, cat no. UFC501096)
Stainless steel blade (Thermo Fisher Scientific, cat. no. 08-916-5B)
Ultracentrifuge Optima XPN (Beckman Coulter, cat. no. A94469) or equivalent
Ultracentrifuge rotor VTi65.2 (Beckman Coulter, cat. no. 362754) or equivalent
Ultraviolet transilluminator (Thermo Fisher Scientific, cat. no. UVP95-0283-04)
REAGENTS SETUP
2.5x Mitochondrial stabilization (MS) buffer
Mix 525 mM mannitol, 175 mM sucrose, 2.5 mM EDTA (pH 7.5), and 12.5 mM Tris-HCl (pH 7.5). Store at 4°C for up to 2 weeks.
Column buffer
Prepare the following buffer in PBS: 0.01 mM CaCl2, 0.01 mM MgCl2, and 50 mM n-Octyl glucoside. Add 0.2 mM PMSF and protease inhibitor cocktail (1 tablet per 50 ml of buffer) CRITICAL Because protease inhibitors degrade quickly in solution always use freshly prepared column buffer. The column buffer without PMSF and protease inhibitors can be stored at 4°C for up to 1 week.
Glycine buffer (pH 2.3)
Mix 20 mM glycine in Milli-Q water. Adjust the pH of the solution with 4N HCl to pH 2.3. Store at room temperature for up to 1 month.
Glycine buffer (pH 3.0)
To prepare glycine buffer mix 100 mM glycine in Milli-Q water. Adjust the pH of the solution with 4N HCl to pH 3.0. Glycine buffer can be stored at room temperature for up to 1 month.
Hypotonic buffer
Mix 10 mM NaCl, 1.5 mM MgCl2, and 10 mM Tris-HCl (pH 7.5). Hypotonic buffer is stable for up to 1 month at 4°C.
IPTG stock
Dissolve 2.38 g of IPTG in 10 ml sterile distilled water to make 1M IPTG solution. Filter-sterilize and store in aliquots at −20°C. IPTG is stable for several months at −20°C.
Kanamycin stock
Dissolve kanamycin at 50 mg ml−1 in distilled water and filter sterilize. Divide into 1 ml aliquots and store in the dark at −20°C. Kanamycin is stable for several months if properly stored. Working concentration is usually 50 μg ml−1.
Luria-Bertani (LB) medium and agar plates
For 1 L, dissolve 10 g tryptone, 5 g yeast extract, 15 g agar, and 10 g NaCl in 950 ml deionized water. Adjust the pH of the solution to 7.0 with NaOH and bring the volume up to 1 L. Autoclave in liquid cycle for 20 min at 15 psi. Allow the solution to cool to 55°C, add antibiotic (e.g., kanamycin, streptomycin, and/or tetracycline) and pour into 10 cm plates. Allow to solidify and invert plates. Agar plates can be stored for up to 6 months at 4°C in the dark.
PEG (16.7%, wt/vol)-NaCl (13.3M) stock (PEG/NaCl)
Dissolve 100 g of polyethylene glycol-8000 and 116.9 g of NaCl in 475 ml water. Adjust volume to 600 ml. Brief heating to 65°C will be necessary to dissolve solids. Alternatively, the solution can be autoclaved and subsequently agitated while hot until the liquid reaches room temperature. PEG-NaCl solution can be stored for up to 6 months at 4°C.
Protein extraction buffer
Prepare the following buffer in PBS: 1 mM CaCl2, 1 mM MgCl2, 50 mM n-Octyl glucoside, 1% (vol/vol) Triton X-100, 0.2 mM PMSF, and protease inhibitor cocktail (1 tablet per 50 ml of buffer) CRITICAL Because protease inhibitors degrade quickly in solution, always use freshly-prepared protein extraction buffer. The protein extraction buffer without PMSF and protease inhibitors can be stored at −20°C for up to 6 months.
Streptomycin stock
Dissolve streptomycin sulfate in water for a 50 mg ml−1 stock, and filter sterilize. Streptomycin can be stored at −20°C for several months. Working concentration is usually 50 μg ml−1.
Super optimal Broth (SOB) Media
For 1 L, dissolve 20 g bacto tryptone, 5 g bacto yeast extract, 2 ml of 5M NaCl, 2.5 ml 1M KCl, 10 ml of 1M MgCl2, 10 ml of 1M MgSO4 in 900 ml of distilled water and adjust to 1 L with distilled water. Sterilize by autoclaving. Media can be stored at room temperature for several months.
Super Optimal Broth with Catabolite repression (SOC)
Prepare as the SOB media and add 20 ml of 1M glucose. Sterilize by autoclaving. Media can be stored at room temperature for several months.
Terrific broth (TB)
Dissolve 12 g tryptone, 24 g yeast extract, and 4 ml glycerol in 900 ml of distilled water. Sterilize by autoclaving and allow cooling to room temperature. Adjust volume to 1 L with 100 ml of a filter-sterilized solution of 0.17M K gH2PO4 and 0.72M K2HPO4. Media can be stored at room temperature for several months.
Tetracycline stock
Dissolve 20 mg ml−1 of tetracycline in ethanol. Tetracycline can be stored at −20°C in the dark for several months. Working concentration is usually 40 μg ml−1.
PROCEDURE
Preparation of the f88-4 and fUSE5 phage vectors TIMING 4 d
1| Chill 0.1 cm electroporation cuvettes and 0.5 ml micro centrifuge tubes on ice 15 min before electroporation. Thaw the electrocompetent DH5α E. coli on ice and place in chilled 0.5 ml microfuge tubes. Mix the plasmid and bacteria and let stand on ice for 15 min. Transfer the mixture into a 0.1 cm electroporation cuvette. Electroporate 50 ng of phage vector (i.e. f88-4, fUSE5) in 20 μl of DH5α per cuvette in the following conditions: 2.0 kV, 200 ohms, 25 μF (Bio-Rad). CRITICAL STEP Avoid the introduction of air bubbles into the cell/DNA mixture to prevent arcing during electroporation.
2| Immediately remove the cuvette and add 1 ml of SOC medium. With a Pasteur pipette transfer the cell suspension to a 1.5 ml tube and incubate at 37°C for 1 h.
3| Plate 100 μl of serial dilutions (1:10, 1:100, 1:1,000) on LB-tetracycline (tet) (40 μg ml−1) plates and incubate overnight at 37°C. Note that E. coli transformed with phage vectors grow slower and might require longer incubation times to obtain visible colonies. PAUSE POINT Bacterial plates are stable for at least a week when wrapped with parafilm and stored at 4°C.
4| Prepare a seed culture from a single colony in 5 ml of LB-tet (40 μg ml−1) media and grow in agitation (225–250 rpm) for 8 h at 37°C.
5| Add the starter culture to 500 ml of LB-tet media (40 μg ml−1) and agitate at 250 rpm for 12–18 h at 37°C. Use a 2 L flask to provide sufficient air for the overnight culture. The culture should reach a cell density of approximately 3 – 4 × 109 cells per ml, which typically corresponds to a pellet wet weight of approximately 3 g/L medium.
6| Centrifuge the culture at 6,000g for 15 min at 4°C, and purify with the maxi-prep plasmid purification kit, according to the manufacturer’s instructions. PAUSE POINT If desired, cell pellets can be stored at −20°C for 12 h. CRITICAL STEP To increase the plasmid yield, warm the elution buffer to 50°C. TROUBLESHOOTING
7| Prepare a 1:100 dilution of the DNA, mix well, and measure the absorbance at 260 nm (A260) in a spectrophotometer blanked against 10 mM Tris-HCl, pH 7.5. Calculate the concentration of DNA using the following formula:
8| Measure the plasmid DNA volume, and for each ml of plasmid DNA add 1.1 g of CsCl and 100 μl of EtBr, 10 mg ml−1 (the volume will increase by 25–30%). Dissolve the CsCl by mixing gently. Cover with aluminum foil and incubate at room temperature for 30 min. CRITICAL STEP Perform the following steps under low light intensity to avoid DNA mutations due to EtBr exposure. CAUTION EtBr is a potent mutagen. May be fatal if inhaled and is harmful if swallowed or absorbed through skin. Causes irritation to eyes, respiratory tract and skin. May cause heritable genetic damage. Wear gloves and safety glasses. It must be handled with extreme caution and decontaminated on activated charcoal or amberlite ion exchange resins prior to disposal.
9| To remove any precipitate present in the CsCl/DNA solution, spin tubes at 6,000g for 5 min at room temperature (debris may either float or sink). Carefully transfer only the clear reddish supernate to a quick-seal ultracentrifuge test tube. Using a 20-gauge syringe, completely fill the tube by adding the equivalent TE/CsCl/EtBr solution (i.e., without DNA) as prepared in STEP 8. Dry the neck of each tube, place metal caps on the quick-seal tubes, heat-seal them and re-check the balance. Place each tube and its counterbalance tube in an ultracentrifuge rotor (e.g., VTi65.2 rotor). Spin at 176,000g for 48 h at 20°C. CRITICAL STEP It is advisable to first practice sealing on tubes filled only with water. Check each seal by pointing the top of the tube into the sink and applying pressure. Failure to seal tubes appropriately may cause the tubes to collapse during ultracentrifugation.
10| Carefully remove tubes from rotor so as to not disturb the gradient. Follow the methods detailed in Sambrook & Russell64 to assemble materials used to extract the plasmid DNA. In summary, with an 18g needle make a vent in the tube by puncturing it at the top; leave the needle hanging in the tube to prevent leakage. Pull out the lower plasmid band (the lower band contains the double-stranded plasmid; the upper band contains the single-stranded DNA) with an 18g needle attached to a 1 ml syringe (Figure 3A). Place the DNA in a 15 ml Falcon tube. CRITICAL STEP To avoid DNA shearing during sample collection, remove the needle from the syringe and transfer the plasmid DNA to a clean 50-ml collection tube. Repeat this as many times as necessary. TROUBLESHOOTING
11| Measure the sample volume and mark its level on the surface of the collection tube. Remove the EtBr by addition of 15 ml of 2-propanol per 1 ml CsCl/DNA solution. Vortex for 10s (a fluffy white precipitate will appear) and centrifuge at 1,800g for 1 min at room temperature to separate phases. Remove the upper phase (pink; isopropanol) and add TE buffer up to the mark indicating the original volume. Mix until the precipitate is dissolved. Repeat the process until the pink color disappears (3–4 times), each time adding TE as necessary.
12| In a 50 ml Falcon tube, measure the final plasmid DNA volume and add 2.5 volumes of fresh TE to the sample. Mix thoroughly. Add 2 volumes of ethanol (e.g., 3.5 ml of original DNA solution + 7.0 ml ethanol), vortex, and incubate at −20°C for at least 2 h. PAUSE POINT DNA can be precipitated overnight at −20°C.
13| Centrifuge sample at 15,000g for 30 min at 4°C. Decant all the ethanol, air-dry the pellet, and resuspend with 500 μl of 10 mM Tris-HCl (pH 8.0). Measure the DNA concentration as previously described in step 7. Figure 3B depicts a fUSE5 prep before and after CsCl purification. PAUSE POINT DNA may be stored at −20°C for up to 6 months.
Insertion of the penetratin peptide into the recombinant pVIII coat protein of the f88-4 phage TIMING 7–8 d
14| Generate a phage clone displaying the penetratin peptide by cloning of the Penetratin oligonucleotides containing flanking HindIII and PstI restriction sites (Table 1) into the rpVIII gene of the f88-4 phage plasmid. Resuspend the oligonucleotides in 10 mM Tris-HCl (pH 8.0) at a concentration of 1 μg μl−1. Mix equimolar amounts of oligonucleotides (1 to 5 μg in total is recommended) in a 0.5-mL microfuge tube. Bring the final volume to 50 μl with 10 mM Tris-HCl (pH 8.0).
15| Anneal the oligonucleotide mix in a thermo cycler according to the program shown below:
|
| ||
| Step | Temperature | Time |
|
| ||
| 1 | 93°C | 3 min |
| 2 | 80°C | 20 min |
| 3 | 75°C | 20 min |
| 4 | 70°C | 20 min |
| 5 | 65°C | 20 min |
| 6 | 40°C | 60 min |
| 7 | 04°C | Hold |
|
| ||
PAUSE POINT Annealed oligonucleotides can be stored at −20°C for several months.
16| Double-digest the annealed oligonucleotides (1–5 μg) and f88-4 plasmid (50 μg) with HindIII and PstI restriction endonucleases as shown below and incubate overnight at 37°C.
|
| ||
| Component | Amount per reaction | Final |
|
| ||
| Penetratin oligos | Depends on concentration | 1–5 μg |
| OR: f88-4 vector | Depends on concentration | 50 μg |
| HindIII endonuclease | 0.5 μl | 5U |
| PstI endonuclease | 0.5 μl | 5U |
| 10x endonuclease buffer R | 5.0 μl | 1x |
| Milli-Q water | Up to 50 μl | |
|
| ||
17| Purify the oligonucleotides and plasmid with the QIAquick gel extraction kit according to the manufacturer’s instructions. Analyze the purity of the oligonucleotides and of the plasmid samples in 2% and 0.8% E-gels, respectively. To quantify the DNA, use UV-band intensity (100 bp Quanti-Markers) and check the absorbance at 260 nm.
18| In a 0.5 ml microfuge tube, prepare ligations with 50 ng of vector and different insert (i.e. oligonucleotides) molar ratios (e.g., 1:1, 1:3, 1:5, and 1:10) with 1 unit of T4 DNA ligase in a final volume of 20 μl as depicted below and incubate the reaction at 16°C for 12 h. Include a control sample without insert to verify that the vector was completely digested. The insert-to-vector molar ratio can have a significant effect on the outcome of a ligation and subsequent transformation step. The formula to calculate the insert amount is:
Example for 1:3 molar ratio:
|
| ||
| Component | Amount per reaction | Final |
|
| ||
| Vector | Depends on concentration | 50 ng |
| Insert | Depends on concentration | e.g. 1.02 ng |
| 5x Ligation buffer | 4.0 μl | 1x |
| T4 DNA ligase (5 U μl−1) | 1.0 μl | 5 U |
| Milli-Q water | Up to 20.0 μl | |
|
| ||
CRITICAL STEP To improve the ligation efficiency, mix the vector, insert, and water in a microfuge tube. Next, incubate the mix at 50°C for 3 min and immediately chill the reaction on ice. Proceed with the ligation reaction by adding the ligation buffer and T4 DNA ligase as depicted above.
19| Heat inactivate the T4 ligase at 65°C for 20 min. Add 1 μl of ligation product into 20 μl of electrocompetent DH5α E. coli. Follow the transformation steps as described in STEPS 1–3. TROUBLESHOOTING
20| Pick single bacterial colonies and inoculate in 3 ml of LB-tet (40 μg ml−1) media. Incubate overnight at 37°C under agitation (225 rpm).
21| Process the bacterial culture for plasmid purification with the mini-prep plasmid purification kit according to the manufacturer’s instructions. Use 1 μg of plasmid and 1 pmol of each f88-4 sequencing primer (Table 1) for SANGER-based DNA sequencing (http://www.mdanderson.org/education-and-research/resources-for-professionals/scientific-resources/core-facilities-and-services/dna-analysis-facility/services-and-fees/index.html). The f88-4 primer set flanks the TAC promoter and the rpVIII gene to confirm the correct peptide-protein fusion in the f88-4/penetratin vector.
22| In two separate 0.5-mL microfuge tubes, double-digest 50 μg of f88-4/penetratin and of fUSE5 phage plasmid with XbaI and BamHI restriction enzymes at 37°C as follows:
|
| ||
| Component | Amount per reaction | Final |
|
| ||
| f88-4 vector | Depends on concentration | 50 μg |
| OR: fUSE5 vector | Depends on concentration | 50 μg |
| XbaI endonuclease | 0.5 μl | 5U |
| BamHI endonuclease | 0.5 μl | 5U |
| 10x endonuclease buffer Y | 5.0 μl | 1x |
| Milli-Q water | Up to 50 μl | |
|
| ||
23| After a 4 h incubation, load the DNA fragments onto an agarose gel (0.8%) and run at 100 volts for 45 min. Under an ultraviolet transilluminator, excise the DNA fragment of 3,925 bp of fUSE5 (this contains the rpIII for peptide library cloning) and the 5,402 bp fragment of the f88-4/penetratin vector (this contains the rpVIII-penetratin) (Figure 2).
24| Place the agarose-DNA fragments in 1.5 ml microfuge tubes and purify with the QIAquick gel-extraction kit according to the manufacturer’s protocol. Measure DNA concentration as described in STEP 7.
25| Ligate 50 ng of fUSE5 DNA fragment to 68.8 ng of f88-4 DNA fragment with 1 unit of T4 DNA ligase in a final volume of 20 μl as shown in STEP 18. Incubate the reaction overnight at 16°C. TROUBLESHOOTING
26| Purify the ligation product with the QIAquick Nucleotide Removal kit according to the manufacturer’s instructions and perform the transformation procedure (STEPS 1–3).
27| Pick 10 single colonies and inoculate 10 cultures of 3 ml LB tet medium. Incubate for 16 h at 37°C under agitation (250 rpm). Isolate the phage plasmid with the plasmid mini-prep kit according to the manufacturer’s instructions. To verify accurate assembly of the bifunctional iPhage vector, sequence each sample with f88-4seq (1pmol of each Fwd and Rev) and pIIIseq (1pmol of each Fwd and Rev) primers sets separately (Table 1). After sequencing confirmation of the iPhage chimera, make a 30% glycerol-LB bacteria stock65 and store samples at −80°C. Use proper aseptic technique when handling bacteria glycerol stocks. PAUSE POINT Glycerol stocks are stable for up to one year.
iPhage peptide library construction TIMING 12–14 d
28| Streak a tetracycline plate with DH5α E. coli containing the iPhage plasmid from the glycerol stock prepared in STEP 27 and incubate the plate overnight at 37°C. Pick a single colony from the freshly-streaked plate, inoculate the bacteria into 5 ml of LB-tet (40 μg ml−1) medium, and follow the procedure for phage plasmid isolation and purification (STEPs 4–13). TROUBLESHOOTING
29| Inoculate a seed culture of electrocompetent MC1061 E. coli for overnight growth in 10 ml of SOB containing 50 μg ml−1 of streptomycin sulfate.
30| Dilute 2 ml of the overnight culture into 2 L of SOB (w/o magnesium) supplemented with 50 μg ml−1 of streptomycin. Distribute the 2 L culture into four 2 L flasks (500 ml/flask) and grow bacteria at 37ºC under agitation (250 rpm). Monitor the bacterial growth by optical density until absorbance reaches 0.8 at 600 nm (around 3–4 h culture). Immediately centrifuge the bacteria cells at 6,000g for 10 min at 4°C.
31| Wash the bacterial pellet twice with 150 ml ice-cold 10% (vol/vol) glycerol diluted in double-distilled water, centrifuge at 6,000g for 15 min at 4°C. CRITICAL STEP Keep all solutions cold and maintain the bacteria on ice at all times.
32| To the pellet (STEP 31) add 2 ml of ice-cold 10% glycerol. With a 2 ml serological pipet, gently loosen the bacteria pellet; 5 to 6 ml of bacterial suspension should be expected. Prepare aliquots of 200 μl or 1 ml in microfuge tubes and snap-freeze in liquid nitrogen. Store the aliquots containing the electrocompetent MC1061 E. coli at −80°C. Test cell competency by electroporation of 20 μl of MC1061 with 10 pg of high-copy-plasmid DNA (e.g. pUC19). Plate 100 μl of several dilutions of the transformation on selective plates and incubate at 37°C overnight. Count the number of colonies and calculate the number of CFU (colony-forming units) per μg of plasmid DNA. Suitable electrocompetent MC1061 cells should make well above 1×109 colonies/μg plasmid DNA. CRITICAL STEP The total volume of electrocompetent MC1061 cells needed to generate the iPhage library (STEP 48) is 20 ml. To obtain large amounts of MC1061 cells, one may increase proportionally the bacteria culture volume or repeat STEPs 29 – 32 as necessary.
33| With the appropriate endonuclease buffer, digest 100 μg of the iPhage plasmid from STEP 28 with 200 units of SfiI restriction endonuclease, as shown in STEP 16. Incubate the reaction at 50°C for 4 h.
34| After digestion, the SfiI restriction enzyme will generate non-identical, non-complementary three base 3′-overhangs. Run the linearized iPhage plasmid on an E-Gel (0.8%) for 30 min at 200 V to confirm digestion. Digestion with SfiI will linearize the 9300 bp-long iPhage plasmid into one single 9300bp fragment. In the agarose gel, the single linear iPhage plasmid will appear to migrate slower than the same plasmid when circular, resulting in a higher DNA band (linear plasmid) and a lower DNA band (circular plasmid). Purify the digested iPhage vector with the gel-extraction kit according to the manufacturer’s instructions. This step allows the complete removal of the 14 bp DNA stuffer (5′-TGGCCTGGCCTCTG-3′) located in the pIII gene. Measure the DNA concentration as described in STEP 7. Store linearized iPhage plasmid at −20°C. TROUBLESHOOTING
35| Resuspend the oligonucleotide template (TPL) and the random library primer (Fwd and Rev) set (Table 1) with 10 mM Tris-HCl (pH 8.0) for a stock concentration of 1 μg μl−1 each. Convert the synthetic oligonucleotide template X4YX4 (X, any residue; Y, tyrosine), flanked by BglI restriction sites, to double-stranded DNA by PCR amplification as shown:
|
| ||
| Component | Amount per reaction | Final |
|
| ||
| X4YX4 Template | 0.1 μl | 100 ng |
| Library Forward primer | 3.0 μl | 3 μg |
| Library Reverse primer | 3.0 μl | 3 μg |
| DMSO | 1.0 μl | 2% |
| 10 mM dNTPs | 2.0 μl | 0.4 mM |
| 25 mM MgCl2 | 2.4 μl | 1.2 mM |
| 5x GoTaq buffer | 10.0 μl | 1x |
| GoTaq polymerase (5U μl−1) | 1.0 μl | 5U |
| Milli-Q water | Up to 50 μl | |
|
| ||
Use the following PCR conditions:
|
| ||||
| Cycle number | Denature | Anneal | Extend | Hold |
|
| ||||
| 1 | 94 °C, 2 min | |||
| 2–36 | 94 °C, 30 s | 60 °C, 30 s | 72 °C, 30 s | |
| 37 | 72 °C, 5 min | |||
| 38 | 4 °C | |||
|
| ||||
CRITICAL STEP Perform at least 16 PCR reactions to generate enough double-stranded insert for the ligation reactions. For effective PCR, addition of DMSO (2% final) is recommended to weaken hydrogen bonding and prevent formation of hairpin structures.
36| Purify and elute the PCR products (the library PCR-insert) containing BglI restriction sites using the QIAquick nucleotide removal kit according to the manufacturer’s instructions, and measure the DNA concentration at A260.
37| Digest 1 μg of library PCR-insert with 100 units of BglI restriction enzyme in a final volume of 50 μl, as described in STEPs 16 or 22. Incubate overnight at 37°C.
38| Purify, elute, and measure the concentration of the BglI digested PCR-inserts as described in STEP 36.
39| To determine the optimal vector:insert molar ratio, perform test ligations with 50 ng of linearized iPhage vector and different BglI digested PCR-insert ratios (1:1, 1:3, 1:5, and 1:10), as described in STEP 18. Also, set up a reaction with the linearized iPhage plasmid alone to estimate the level of bacterial transformation due to non-digested iPhage plasmid.
40| Heat inactivate the T4 ligase at 65°C for 20 min. Thaw an aliquot of MC1061 E. coli on ice and add 1 μl of the ligation products to 20 μl of bacteria. Follow the transformation steps as described in STEPs 1–3.
41| Immediately remove the cuvette and add 200 μl of SOC medium. With a Pasteur pipette transfer the cell suspension to a 1.5 ml microfuge tube and incubate at 37°C for 1 h under agitation (250 – 300 rpm).
42| Plate 1, 10, and 50 μl of transformed bacteria on LB-tet agar plates and incubate overnight at 37°C.
43| Count the number of colonies and determine the optimal vector-insert molar ratio for ligation based on transformation efficiency and background from the negative control ligation. TROUBLESHOOTING
44| Set up the large-scale library ligation reaction as follows:
|
| ||
| Component | Amount per reaction | Final |
|
| ||
| Linearized iPhage vector (STEP 34) | Depends on concentration | 10 μg |
| Library insert (STEP 38) | Optimized quantity from test ligation | |
| 5x Ligation buffer | 400 μl | 1x |
| T4 DNA ligase (5 U μl−1) | 100 μl | 500 U |
| Milli-Q water | Up to 2,000 μl | |
|
| ||
CRITICAL STEP To improve the ligation efficiency, mix the vector, insert, and water in a microfuge tube. Next, incubate the mix at 50°C for 3 min and immediately chill the reaction on ice. Proceed with the ligation reaction by adding the ligation buffer and T4 DNA ligase as depicted above.
45| Aliquot the ligation reaction in twenty 0.5 ml microfuge tubes (100 μl/tube) and incubate at 22°C for 2 h. Transfer tubes to 16°C and incubate overnight.
46| Mix the ligation products with 5 volumes of binding buffer (PB buffer) and load the samples into 20 mini-prep QIAprep columns. Let the column stand 3 min at room temperature.
47| Centrifuge at 10,000g for 30s at 4°C. Wash columns with 500 μl of washing buffer (PE buffer) and centrifuge for 30–60s. Discard the flow-through and centrifuge for an additional 1 min to remove residual wash buffer. To elute DNA, add 50 μl of water and centrifuge columns for 2 min. CRITICAL STEP To increase X4YX4-iPhage plasmid yield, warm the water to 50°C prior to adding it to the columns.
48| Measure the DNA concentration as shown in STEP 7 and perform one thousand electroporations with MC1061 E. coli as described in STEP 1. Electroporate 10 ng of DNA in 20 μl of MC1061 per cuvette. Alternatively, a high-throughput electroporation system can be used (ECM 630 + HT-100 BTX Harvard Apparatus). If this method is desired, mix 1 ml of X4YX4-iPhage (STEP 47) with 22 ml of MC1061 E. coli, and let stand on ice for 15 min. Transfer 50 μl of the mixture into each well of a 96-well high throughput electroporation plate (Multi-well electroporation plate cat. no. 450450, Harvard Apparatus). Electroporate five 96-well plates under the following conditions: 2.4 kV, 750 ohms, 25 μF. CRITICAL STEP Avoid introduction of air bubbles into the cell/DNA mixture to prevent arcing during electroporation.
49| Combine the electroporations into 380 ml of prewarmed SOC media. Gently agitate (150 rpm) the bacteria at 37°C for 1 h.
50| Add the 400 ml culture (STEP 49) to 3.6 L of LB-tetracycline (40 μg ml−1) /streptomycin (50 μg ml−1) and divide the culture into eight 2 L baffled Fernbach flasks (500 ml/flasks) to provide sufficient air for the overnight culture. Agitate culture (250–300 rpm) at 37°C for 12–18 h.
51| Transfer the culture to eight 500 ml centrifuge bottles and centrifuge at 6,000 g for 15 min at 4°C. Transfer supernate to clean centrifuge bottles and keep at 4°C. Recover and wash the bacterial pellet with 500 ml ice-cold 10% glycerol, centrifuge at 6,000 g for 15 min at 4°C, and repeat. After the second centrifugation, resuspend pellet in 2 ml of 50% glycerol and aliquot cells into ten chilled 0.5 ml centrifuge tubes (0.2 ml/tube). Snap-freeze pellet in liquid nitrogen and store samples at −80°C. CRITICAL STEP The bacterial pellet is used for library amplification. A comprehensive library amplification protocol is described in Box 2.
Box 2. Amplifying a Library TIMMING 3 d.
Follow the steps below to amplify an iPhage library with minimal risk of reduction in its diversity.
Grow an overnight culture of K91/kan in LB medium containing 100 μg ml−1 kanamycin.
Inoculate two 1 L culture flasks containing 100 ml Terrific broth with 1 ml of the overnight K91/kan culture. Agitate culture (250-300 rpm) at 37ºC until the bacteria reach an optical density ranging between 1.6 and 2.0 at a wavelength of 600 nm. Reduce shaking to 100 rpm for 5 min.
Into each flask add 5 × 1010 TU of the original iPhage library to be amplified. Continue slow shaking for 15 min.
Split each culture into four 2 L baffled Fernbach flasks containing pre-warmed 500 ml LB supplemented with 0.22 μg ml−1 tetracycline. Agitate culture (250-300 rpm) for 30 min at 37ºC.
Bring the tetracycline concentration to 18 μg ml−1 by addition of 20 mg ml−1 of tetracycline to each flask. Continue shaking the flasks overnight at 37°C.
Purify and titer iPhage particles as described in STEPs 52-57.
52| To remove residual bacterial debris, centrifuge the supernate at maximum speed at 4°C for 20 min. Collect and transfer supernate into 4 clean 2 L flasks (1 L/flask). Add 150 ml of PEG/NaCl solution into 1 L supernate (0.15 volume of PEG/NaCl per volume of supernate) to precipitate iPhage particles. Stir supernate overnight at 4°C.
53| Transfer the precipitation solution equally into ten clean 500 ml centrifuge bottles and centrifuge at 10,000g for 30 min at 4°C. A white pellet should be obtained. Discard the supernate and centrifuge samples again at 10,000g for 10 min. Carefully decant the supernate.
54| Resuspend each iPhage pellet in 2 ml of sterile PBS and combine into a 50 ml conical tube. Centrifuge tube at 10,000g at 4°C for 10 min, discard pellet, and transfer iPhage solution into a new 50 ml conical tube.
55| Repeat the precipitation by adding 3 ml of PEG/NaCl solution (0.15 volume) into 20 ml iPhage suspension for 1 h on ice. Centrifuge at 10,000g for 30 min at 4°C and resuspend pellet in 0.5 ml up to 1 ml of PBS depending on the size of the pellet.
56| Transfer the solution to a 1.5 microfuge tube, centrifuge at 14,000g for 10 min at 4°C. Discard pellet and transfer supernate to a new 1.5 microfuge tube. Recentrifuge to remove residual bacteria and debris. Discard pellet. Filter the resulting supernate containing the iPhage library particles through a 0.45μm filter.
57| Titrate iPhage by preparing serial dilutions of 10−6, 10−7, and 10−8 of the phage library in PBS (10 μl/dilution). Prepare dilutions in triplicate. For each 10 μl of dilution add 100 μl of log phase K91/kan E. coli. Allow iPhage infection for 30 min at room temperature. Plate 100 μl of each dilution in triplicates on LB plates containing tetracycline (40 μg ml−1) and kanamycin (50 μg ml−1), and incubate at 37°C overnight. The iPhage titers are expressed as bacterial transducing units (TUs)/μl. Calculate iPhage titer using the following formula:
For example, if 80 colonies are counted in the 10−7 dilution plate:
Alternatively, the qPhage protocol may be used for iPhage titration (Box 1). CRITICAL STEP K91/kan E. coli viability plays an important role in iPhage titration. Always infect a log-phase growing bacteria with an optical density ranging between 1.6 and 2.0 at a wavelength of 600 nm (OD600). PAUSE POINT iPhage titers are relatively stable; the preparations can be stored at 4°C for long periods of time (several months) without significant decrease in the titers. For longer storage times, one should check the titer of the preparation before use. TROUBLESHOOTING
iPhage library screening in live cells TIMING 2 d
58| Grow the KS1767 cell line to 50–60% confluence in 75-cm2 culture flasks. The cells are cultured in complete MEM containing 10% (vol/vol) FBS, MEM-vitamin solution, MEM Non-essential amino acids, penicillin-streptomycin, and L-glutamine at 37°C in a 5% CO2-humidified incubator.
59| Prepare 5×1011 TU of the iPhage library in 8 ml of MEM containing 10% (vol/vol) FBS. Filter the iPhage library solution (8 ml) through a 0.22 μm filter. Remove the media from the overnight culture and add the iPhage library (8 ml). Gently agitate flask to cover the cell monolayer evenly. Incubate overnight at 37°C in a 5% CO2.
60| On the following day, warm 0.05% trypsin-EDTA, PBS, and complete growth media in a 37ºC water bath. Remove and discard iPhage/growth media from the culture flask with a sterile pipette. Wash cells with 10 ml of warm PBS to remove any remaining media. Replace PBS with 3 ml of trypsin-EDTA. Incubate for 10 min at room temperature. Check culture with an inverted optical microscope to ensure that the cells are detached from the surface.
61| Neutralize trypsin/EDTA by adding 10 ml of warm growth media. Draw the cell suspension with a sterile pipette. Rinse the inside surface of the flask several times with the cell suspension to ensure that the most of the cells are in suspension. Check under an inverted optical microscope if necessary. Transfer the cell suspension into a 15 ml conical tube and centrifuge at 150g for 5 min at room temperature. Wash the cell pellet three times with ice-cold PBS to remove traces of trypsin/EDTA and growth media. Resuspend pellet in 1 ml of PBS.
62| Mix the pellet suspension (1 ml) with 11 ml of ice-cold hypotonic buffer. Transfer cell suspension into a Dounce homogenizer and incubate for 10 min on ice.
63| Perform 15 up-and-down strokes with a loose-fitting pestle and 25 up-and-down strokes with a tight-fitting pestle on the KS1767 cells, to disrupt the membranes, and add 8 ml of 2.5× MS buffer. Seal the tube with parafilm and mix thoroughly by inversion.
64| Aliquot the 20 ml cell lysate suspension into ten 2 ml ice-cold microfuge tubes, and centrifuge at 1,300g for 5 min at 4°C. Recover and transfer supernate into clean microfuge tubes. Centrifuge samples twice more. The pellet contains the nuclei, intact cells, and large membrane fragments.
65| Transfer supernate into clean 2 ml microfuge tubes and pellet the mitochondria at 17,000g for 15 min at 4°C. Wash the mitochondria by resuspension of the pellet in 500 μl MS buffer. Centrifuge samples once more at 17,000g for 15 min at 4°C. Discard the supernate and keep the mitochondrial pellet on ice. PAUSE POINT The mitochondrial pellet can be stored at −80°C for further assays. CRITICAL STEP If the iPhage recovery (STEP 66) is performed on the same day, we recommend starting a K91/kan E. coli culture (STEP 57) 2–3 h before the subcellular fractionation (STEP 62).
66| Add 200 μl of the K91/kan E. coli onto each mitochondrial pellet aliquot (10 aliquots in total – STEP 65). With a micropipette gently resuspend and mix the pellet. Incubate sample for 1 h at room temperature.
67| Transfer the 200 μl of bacteria + pellet to 10 ml of pre-warmed LB medium supplemented with 40 μg ml−1 tetracycline and 100 μg ml−1 kanamycin and incubate for 30 min at room temperature.
68| Dilute sample to 1:10, 1:100, and 1:1,000, plate 100 μl of each dilution in triplicate onto LB-tet/kan plates, and incubate plates overnight at 37°C. Transfer the remaining culture (~10 ml) into 300 ml of pre-warmed LB-tet/kan containing 1 mM IPTG and incubate overnight at 37°C.
69| Pick single colonies and proceed to iPhage PCR and insert sequencing (STEP 73). Alternatively, colony plates can be stored at 4°C and processed together after the last iPhage screening round is complete (STEP 72).
Phage recovery by salt precipitation TIMING 1 d
70| Centrifuge the bacterial culture (STEP 68) at 6,000g for 20 min at 4°C, and transfer the supernate into new bottles containing 0.15 volumes of PEG/NaCl solution (e.g. 45 ml PEG/NaCl : 300 ml culture). Incubate for 2 h on ice. After incubation, follow STEPS 53–55.
71| After the second phage precipitation (STEP 55), centrifuge samples at 10,000g for 30 min at 4°C. Carefully decant supernate and resuspend the iPhage pellet in 100 μl of PBS. Recover and transfer resuspended iPhage particles to a clean 1.5 ml microfuge tube. To remove insoluble material, centrifuge samples at 14,000g for 10 min at 4°C and transfer supernate to a sterile 1.5 ml microfuge tube.
72| Filter the iPhage particles through a 0.22 μm filter and titrate the iPhage particles as described in STEP 57. Alternatively, the qPhage protocol may be used to replace colony count method (Box 1). Store the iPhage particles at 4°C for further rounds of selection on KS167 cells. Based on our studies, a total of three rounds of selection is necessary to enrich specific peptide sequences and reduce false-positive ligands. For each round of selection follow STEPs 58 – 71. PAUSE POINT The recovered iPhage particles are stable for at least two weeks without reduction in titer.
PCR-sequencing of iPhage inserts TIMING 1 d
73| The peptide identity is determined by sequencing the DNA corresponding to the insert in the recombinant pIII. Thus, to determine the insert sequence of recovered iPhage clones, pick 96 well-separated bacterial colonies from each round of selection (STEP 68). For each round prepare a 96-well U-bottom plate and inoculate each colony in 50 μl of 30% (vol/vol) glycerol-LB. Store plates at −80°C.
74| Prepare a PCR reaction as described below. For high throughput screening use a 96-well PCR plate. Include a negative control of an iPhage without insert.
|
| ||
| Component | Amount per reaction | Final |
|
| ||
| Bacterial suspension | 2.0 μl | |
| pIIIseq forward primer* | 1.0 μl | 8 pmol |
| pIIIseq reverse primer* | 1.0 μl | 8 pmol |
| 10 mM DNTP | 0.5 μl | 0.25 mM |
| 5x GoTaq buffer | 4.0 μl | 1x |
| DMSO | 0.4 μl | 2% |
| GoTaq polymerase (5U μl−1) | 0.4 μl | 2U |
| Milli-Q water | Up to 20 μl | |
|
| ||
| * pIIIseq primer set (Table 1) | ||
Use the following PCR conditions:
|
| ||||
| Cycle number | Denature | Anneal | Extend | Hold |
|
| ||||
| 1 | 94 °C, 3 min | |||
| 2–36 | 94 °C, 10 s | 60 °C, 30 s | 72 °C, 60 s | |
| 37 | 72 °C, 3 min | |||
| 38 | 4 °C | |||
|
| ||||
75| Run 2 μl of ten PCR reactions randomly selected from each PCR plate on a 2% E-Gel. Positive iPhage colonies will have PCR products of around 300 bp-long, whereas the negative control will generate a PCR product of around 250 bp-long. Prepare a 1:10 dilution with water from each PCR reaction and submit samples for nucleotide sequencing with the pIIIseq Rev primer (Table 1). The peptide sequences and sequence enrichment are analyzed based on bioinformatics tools and are described elsewhere62.
Amplification of iPhage clones TIMING 3 d
76| iPhage clones of choice can be amplified by inoculation of 3 ml of LB tet-strep containing 1 mM IPTG with 1 μl of the bacteria glycerol stock (STEP 73) and incubation of the seed culture at 37°C for 8 h. Transfer the seed culture to 100 ml of LB tet-strep containing 1 mM IPTG and follow the iPhage-precipitation protocol as described in STEPS 52–56.
77| Titrate each iPhage clone (STEP 57) no longer than a week prior to performing the cell viability assay. Alternatively, iPhage titration may be achieved by using the qPhage protocol as described in Box 1.
Screening of bioactive iPhage clones by cell viability assay TIMING 4 d
78| Seed 2.5×104 KS1767 cells in each well of a 96-well flat-bottom tissue culture plate in a final volume of 100 μl of complete growth media and incubate cell culture overnight at 37°C in 5% CO2.
79| Remove growth media and avoid disturbing the cell monolayer. Carefully add 100 μl of complete growth media containing 1×109 TU of iPhage in triplicate and incubate plate overnight at 37°C in 5% CO2. Include insertless-iPhage and YKWYYRGAA-iPhage32 as negative and positive controls, respectively. The oligonucleotide sequence that encodes the YKWYYRGAA peptide is depicted in the TPL primer in Table 1.
80| After 24 h, take 10 random phase-contrast microscopy images from each triplicate and determine cell viability by addition of 10 μl of MTT (12 mM) according to the manufacturer’s instructions. Alternatively, cell viability may be determined by the WST-1 assay according to the manufacture’s protocol.
81| Mix each sample thoroughly with a micropipette and read absorbance at 570 nm. Plot data and correlate with the cell density/morphology images obtained with the phase-contrast microscopy. Bioactive iPhage can be selected based on reduced cell growth rate and cell viability measured by the MTT or WST-1 assays as well as cell shrinkage or pyknosis (chromatin condensation) visible in light microscopy.
82| Once the bioactive iPhage clone is identified, synthesize in house or order its corresponding peptide ligand for further receptor identification and validation. We recommend peptide purity of 95% and the use of acetate as a counter-ion.
Ligand receptor purification by affinity chromatography TIMING 2 d
83| Conjugate 10 mg of a bioactive peptide to a 2 ml agarose column matrix (Carboxylink immobilization kit), according to the manufacturer’s instructions.
84| Culture KS1767 cells to 90% confluence in six 175-cm2 culture flasks. One day prior to use, replace media with complete growth media and incubate overnight at 37°C in a 5% CO2.
85| Wash and detach cells by standard tissue culture procedures.
86| Wash the cell pellet once with 10 ml of plain MEM and resuspend the pellet in a 1:1 volume of ice-cold protein extraction buffer (i.e., 500 μl MEM : 500 μl extraction buffer). Incubate the cell suspension at 4°C for 1 h on a rocking plate.
87| Centrifuge cell lysate (suspension) at 20,000g for 15 min at 4°C and keep the supernate on ice. Measure the protein concentration using the BCA protein assay kit according to the manufacturer’s protocol. PAUSE POINT Cell protein extracts can be aliquoted and stored at −80°C for at least one week.
88| Equilibrate the affinity column prepared in STEP 83 and column buffer (see REAGENT SETUP) to room temperature. Equilibrate column by addition of 10 ml of column buffer passed through the column. CRITICAL STEP Throughout the procedure, do not allow the resin to become dry. Replace bottom cap as soon as buffer drains down to the top of the resin bed.
89| Add 1–2 mg of total cell extract to the column (STEP 87). Cap and seal the column at both ends with parafilm, and rock for 1 h at room temperature.
90| Place the column in a base support stand, remove top and bottom caps from column, and wash the column with 20 ml of column buffer. Monitor the absorbance at 280 nm until it reaches 0.001. To remove any residual protein, wash the column with 10 ml of column buffer.
91| Elute the bound protein by adding 2 ml of the bioactive peptide (5 mM) diluted in column buffer and collect 0.5 ml fractions. Continually add 20 ml of column buffer to the column and monitor elution by absorbance at 280 nm until it reaches 0.001 (approx. 20 samples). The excess of bioactive peptide will compete with the protein complex associated with the peptide crosslinked to the column and will thereby release the protein receptor. TROUBLESHOOTING
92| Measure the protein concentration of each fraction and use a 2D gel clean-up kit according to the manufacturer’s instructions to desalt and concentrate fractions of interest. Store samples at −80°C. PAUSE POINT Samples can be stored at −80°C for no longer than one week.
93| To remove any residual protein and reactivate the resin, wash the column with 10 ml of glycine buffer. Wash the column with 10 ml of column buffer and equilibrate with 8 ml of PBS containing 0.05% sodium azide. Store the column at 4°C. CRITICAL STEP The columns are stable for 12 months if properly washed and stored. CAUTION Sodium azide is a common preservative of samples and stock solutions. Because it is a hazardous chemical, wear gloves and facemask during the preparation of solutions.
Screening of the eluted receptor fraction by phage-binding assay TIMING 3 d
94| This part enables the determination of the receptor candidate distribution among fractions of interest. Before starting, titer the bioactive iPhage clone and insertless iPhage as described in STEP 57. Alternatively, iPhage titration may be achieved by using the qPhage protocol as depicted in Box 1.
95| Immobilize 5 μg of protein diluted in PBS (pH 7.2) from each fraction of interest in triplicate on a 96-well flat-bottom plate. As a negative control, immobilize 50 μl of 1% (wt/vol) BSA in PBS. Incubate plate overnight at 4°C.
96| The next day, wash the wells once with 200 μl of PBS.
97| Block each well with 200 μl of PBS containing 1% (wt/vol) BSA and incubate at room temperature for 2 h. CRITICAL STEP To reduce non-specific phage binding with BSA, filter the blocking solution through a 0.22 μm filter before use.
98| Carefully wash each well twice with 200 μl of PBS.
99| Add 1×109 iPhage particles/well of bioactive iPhage clone or insertless iPhage diluted in 50 μl of PBS containing 0.1% (wt/vol) BSA into wells containing the fraction of interest and negative control wells (STEP 95). Incubate plate at room temperature for 2 h.
100| Wash the 96-well plate 12 times with 200 μl of PBS.
101| Remove the PBS and add 200 μl of K91/kan E. coli in each well and incubate the plate for 1 h at room temperature. CRITICAL STEP K91/kan E. coli must be grown on the day of panning. For infection use log-phase growing bacteria with an optical density ranging between 1.6 and 2.0 at a wavelength of 600 nm (OD600).
102| Transfer bacteria from each replicate well into a 2 ml microfuge tube and dilute to 1:5, 1:20, and 1:200. Plate 100 μl on kan/tet plates in triplicate from each dilution and incubate overnight at 37°C. Count bacterial colonies on the following day. The elution fraction containing the most colonies represents the fraction containing the receptor for the ligand-peptide. TROUBLESHOOTING
103| After identifying the fraction containing the candidate receptor, prepare 5 – 20 μg of protein with 4x NuPAGE LDS sample buffer containing 2-mercaptoethanol from a negative control fraction (low or no colonies) and from the fraction of interest. Load protein samples onto a precast Novex 4–20% Tris-glycine gel and run at 200 volts for 50 min. CAUTION 2-mercaptoethanol is harmful if swallowed. It is toxic in contact with skin and causes burns. Work in a fume hood and wear proper protective equipment.
104| Wash the gel twice with 200 ml of Milli-Q water for 5 min and then, stain with SimplyBlue according to the manufacturer’s instructions. Photograph and scan the gel to detect the unique protein bands in the fraction of interest.
105| Using a clean scalpel, cut and transfer the gel fragments to a clean 1.5 ml microfuge tube. Perform mass spectrometry (MS) on the samples. If the purified fractions contain multiple bands, it is necessary to perform 2D gel electrophoresis. TROUBLESHOOTING
106| Contact your MS facility or the protein core facility, prior to sending the samples for tandem mass spectrometry (MS/MS), for further instructions on how to submit samples. The MS/MS analysis provides a list of candidate receptors that will need experimental validation. Receptor candidates should be prioritized based on ions score significance thresholds, protein scores, protein inference, and the targeting context. More information can be found at http://www.matrixscience.com. Use commercially available recombinant protein in a phage binding or an immunocapture assay to confirm receptor-ligand interactions. If recombinant proteins are not available, use antibodies to immunocapture the protein of interest from cell lysates. TROUBLESHOOTING
Phage binding assay of recombinant proteins TIMING 3 d
107| Immobilize 0.1–0.5 μg of recombinant protein in 50 μl of PBS in triplicate on a 96-well flat-bottom plate. If recombinant proteins are tagged (e.g. with GST), use the tag alone as a negative control. Otherwise, immobilize 50 μl of 1% (wt/vol) BSA in triplicate. Incubate plate overnight at 4°C.
108| Repeat STEPs 96–102.
Immunocapture assay TIMING 4 d
109| Use protein A-coated 96-well plates and add 10 μg ml−1 of desired antibody (polyconal or monoclonal) in triplicate. Alternatively, dilute antibody 1:100 in 50 μl of PBS. As a negative control, add 10 μg ml−1 or equal dilution of immunoglobulin isotype control in triplicate. Leave at least 3 empty wells (i.e. without antibodies) to be used as ‘blank’ (e.g. BSA-blocked protein A coated wells). Incubate plate overnight at 4°C.
110| Add 200 μl of PBS containing 2% (wt/vol) BSA into each well and incubate for 2 h at room temperature.
111| Add the cell extract (STEPs 84 – 87) containing 30–60 μg of protein in 50 μl of PBS into each well and incubate overnight at 4°C.
112| Remove the plate from 4°C and leave it at room temperature for 1 h.
113| Repeat STEPs 99–102
TROUBLESHOOTING
Troubleshooting advice can be found in Table 2.
TABLE 2.
Troubleshooting table.
| Step | Problem | Possible reason | Solution |
|---|---|---|---|
| 6, 28 | Little DNA recovered or poor quality DNA prep | Slow growth of bacteria culture or poor DNA purification (multiple bands observed in agarose gel) | Grow large culture volumes: fUSE5, f88-4 and iPhage plasmids are considered low-copy number plasmids. Perform CsCl purification as described in STEP 8. |
| 10, 28 | Difficult to visualize the plasmid DNA band | Poor CsCl gradient separation | Make sure the proper amount of CsCl was used; changing the ultracentrifuge rotor could increase band definition/separation (e.g. try 70i.Ti). |
| 19, 25, 43 | Low ligation efficiency | Non-digested plasmids and/or insert; overexposure of DNA fragments to UV during the gel extraction procedure. | Ensure the complete digestion of the vector in an agarose gel by analyzing the DNA running pattern (see Step 34), the linear plasmid (cut) will appear to migrate slower (higher band) in the agarose gel than the same plasmid when circular (uncut – lower band); although the restriction enzymes used to create the iPhage vector and library generate incompatible ends, dephosphorylating the vector may help increase the ligation efficiency; increase restriction enzyme incubation time; use as little as possible of the vector plasmid (e.g 10 ng); purify ligation before electroporation as described in STEP 46; perform gel extraction by blind excision or use a 365 nm UV transilluminator instead of the usual 312 nm. |
| Insufficient insert ligated to the vector | Optimize the vector:insert molar ratio as described in STEP 18. | ||
| 34 | Strong star activity in the iPhage DNA | Excess of SfiI endonuclease or prolonged incubation | Use fewer units (e.g. 50U) of SfiI or decrease incubation time (e.g. 2 h) to achieve complete digestion and avoid overdigestion. |
| 57 | Low iPhage titers | Old K91/kan E. coli | Check bacterial viability. Repeat infection of 1 ml of K91/kan E. coli bacteria with the new iPhage preparation for 1 h (10 ml at least), and grow in 500 ml for up to 16 h. |
| Incorrect phage assembly | Sequence iPhage plasmid with the f88-4seq and pIIIseq primers sets (Table 1) to verify accurate assembly of the bifunctional iPhage vector. | ||
| 91 | Low or no protein recovered | Insufficient protein loaded in the column | Measure protein concentration by BCA or Bradford methods; optimize the amount of protein to be loaded on the column. |
| Sample loading or elution conditions not favorable for protein purification | Optimize buffer pH; elution buffer is not optimal, increase concentration of bioactive peptide (e.g. 10 mM); alternatively, perform the elution step with 20 ml of Glycine buffer (pH 3.0) and recover 0.5 ml aliquots always monitoring the absorbance at 280 nm. | ||
| Excess of nonspecific proteins | Improper column wash | Wash column more thoroughly before elution; wash columns with a more stringent second wash buffer (for example, with higher salt). The stringency may be increased by increasing the sodium chloride (NaCl) concentration to 0.5 M or even 1 M to reduce ionic and electrostatic attractions. Low levels of reducing agents (such as 1-2 mM DTT or BME) can help disrupt non-specific interactions mediated by disulfide bridges. | |
| 102 | Too many colonies to count; no difference between fractions | Inaccurate phage titer | Check bacterial viability. Repeat infection of 1 ml of K91/kan E. coli bacteria with the new iPhage preparation for 1 h (10 ml at least), and grow in 500 ml for up to 16 h. |
| Insufficient blocking or poor plate washing | Prepare fresh 1% BSA blocking solution and incubate plates at 4°C overnight. Add 0.05% Tween-20 (T) into PBS and wash the 96-well plate 20 times with 200 μl T-PBS. | ||
| Excess of protein coated on the plate | Remeasure protein concentration by BCA or Bradford methods; optimize the amount of protein to be coated on the plate. | ||
| 105 | Difficulty identifying/extracting unique protein bands | Excess of receptor candidates in the fraction of interest | Perform a 2D gel analysis and remove protein spots of interest for mass spec analysis. |
| Low protein concentration in the fraction of interest | Stain the gel with SYPRO or silver stain. | ||
| 106 | Keratin is the major protein identified by MS/MS | Sample contamination through environment rather than natural abundance | Use a biological safety cabinet or laminar flow hood; wear disposable lab coats; wear powder-free nitrile gloves; clean all surfaces with water and ethanol. |
TIMMING
Steps 1 – 13, Preparation of the f88-4 and fUSE5 phage vectors: 4 d
Steps 14 – 27, Insertion of the penetratin peptide into the recombinant pVIII coat protein of the f88-4 phage: 7–8 d
Steps 28 – 57, iPhage peptide library construction: 12–14 d
Steps 58 – 69, iPhage library screening in live cells: 2 d
Steps 70 – 72, Phage recovery by salt precipitation: 1 d
Steps 73 – 75, PCR-sequencing of iPhage inserts: 1 d
Steps 76 and 77, Amplification of iPhage clones: 3 d
Steps 78 – 82, Screening of bioactive iPhage clones by cell viability assay: 4 d
Steps 83 – 93, Ligand receptor purification by affinity chromatography: 2 d
Steps 94 – 106, Screening of the eluted receptor fraction by phage-binding assay: 3 d
Steps 107 and 108, Phage binding assay of recombinant proteins: 3 d
Steps 109 – 113, Immunocapture assay: 4 d
Box 1, Rapid phage quantification (qPhage): 2 h
Box 2, Amplifying a Library: 3 d
Box 3, Subcellular localization of iPhage particles: 2 d
Box 3. Subcellular localization of iPhage particles TIMMING 2 d.
Seed 5×103 KS1767 cells in a 16-well glass chamber plate and allow cells to grow overnight at 37°C. The next day prepare per well 1×109 TU of iPhage particles in 100 μl of MEM containing 2% (vol/vol) FBS (filtered through a 0.22 μm filter). Remove the media from the overnight-grown culture and add the iPhage particles. Incubate the chamber overnight at 37°C in a 5% CO2-humidified incubator. After incubation, add the desired organelle dye (e.g., Mitotracker dye solution for mitochondria labeling) and incubate chamber at 37°C for 30 min. The population of cell membrane-bound phage is removed by washing of the cells 3 times with 200 μl per well of glycine buffer (20 mM glycine; pH 2.3). For each wash, incubate chamber at room temperature for 3 min under gentle agitation. Glycine is neutralized by washing the chamber three times with 200 μl of PBS and fixed with 4% (vol/vol) paraformaldehyde (PFA) in PBS at room temperature for 15 min. CAUTION PFA is highly toxic. Avoid contact with eyes, skin, and mucous membranes. Use skin and eye protection during PFA manipulation and application. Note that different fixatives may be used such as methanol or acetone but conditions will require optimization depending on the organelle dye used. After three washes with PBS, fixed cells are rendered permeable with 200 μl of PBS containing 0.2% (vol/vol) Triton X-100, blocked with 200 μl of PBS containing 1% (wt/vol) BSA, and incubated with 100 μl of the primary fd bacteriophage antibody (1:200 dilution) or normal rabbit IgG in PBS containing 1% (wt/vol) BSA for 2 h at room temperature. Next, 100 μl of secondary Alexa Fluor 488 anti-rabbit IgG (1:300) in 1% BSA-PBS is added to each well, and incubation is continued for 1 h at room temperature in the dark. Cells are washed with PBS, fixed with PBS containing 4% (vol/vol) PFA, mounted in the presence of DAPI, and visualized under a fluorescence microscope or by confocal microscopy (Supplementary Figure 1).
ANTICIPATED RESULTS
The iPhage technology is a unique platform for ligand-directed targeting of organelles and molecular pathways within live cells with applications ranging from fundamental cell biology to drug development. For creation of the chimeric iPhage vector, the backbone of phage vector f88-4 displaying the peptide RQIKIWFQNRRMKWKK (penetratin) on the rpVIII is ligated into the fUSE5 phage plasmid genome (Figure 2). If the iPhage constructs are properly assembled, generation of iPhage particles is often abundant and non-toxic to the host bacteria K91/kan E. coli. iPhage library titers of 1–5×1010 TU ml−1 and clone diversity up to 109 are routinely obtained and are consistent with the titers generated with the parental phage. Functional assays to evaluate either correct display of penetratin and/or the capacity of targeted iPhage to home to different intracellular compartments, such as mitochondria (exemplified in this protocol), can be performed by either infection of host E. coli or by the qPhage method (Box 1) and complemented by the iPhage internalization assay (Supplementary Figure 1 – Box 3) in virtually any cell line32. For instance, iPhage particles displaying the SV40 large T-antigen nuclear localization signal (NLS – PKKKRKV)66 can be enriched specifically in the nuclear fraction of KS1767, whereas parental phage and non-targeted (insertless) iPhage show no significant enrichment towards any subcellular fraction (Figure 6a). (Note that the oligonucleotide sequence encoding the NLS peptide is shown in Table 1.) Similarly, fluorescence microscopy reveals that NLS-iPhage is specifically directed to the nuclei of live KS1767 cells (Figure 6b). Although not required, designing and producing iPhage particles displaying a NLS targeting sequence will allow the investigator to become acquainted with the iPhage technology and related methods before the production and screening of an iPhage library. Additionally, NLS-iPhage particles can serve as positive controls for functional assays during receptor identification and validation.
Figure 6.
iPhage organelle-targeting properties. (a) KS1767 cells were incubated with insertless Phage, insertless iPhage, and iPhage-NLS for 24 h at 37°C. Phage particles were recovered from mitochondria/ER and nuclear fractions, and phage homing was determined by bacterial infection and colony count. Experiments were performed in triplicates and bars represent mean values for phage TU recovered from each fraction ± SEM. (b) KS1767 cells were incubated with iPhage-NLS for 24 h at 37°C. iPhage-NLS particles were detected in the nucleus by phage antibody. Arrowheads indicate merged red-blue fluorescence. Scale Bar, 100 μm.
Three rounds of synchronous selection (STEPs 58 – 69) are performed to isolate organelle-enriched iPhage clones (Figure 7a). The recovered iPhage pool can be evaluated by either infection of host E. coli or by the qPhage method (Box 1). After round 3, a significant increase in iPhage particles should be observed in each of the subcellular fractions (Figure 7b). Appropriate bioinformatic analyses must be applied to identify enriched sequences. The selected peptide candidate is synthesized by solid-phase method and used in functional assays as well as for receptor identification. Thus far, several peptides with intracellular homing and biological properties have been identified by this technology (not shown). Figure 7c depicts the intracellular distribution of a biotinylated intracellular ligand peptide (iLP – GKKRYDHEL) fused to penetratin (pen). The iLP candidate exhibits a unique, reproducible, and specific nuclear membrane distribution in KS1767 cells. After the peptide candidate is characterized in vitro, the peptide is coupled to agarose beads and utilized to affinity-purify its binding partner or intracellular receptor. Fractions obtained from the affinity chromatography are individually tested against the iPhage candidate (Figure 7d). Positive fractions are further analyzed by SDS-PAGE (Figure 7e), or alternatively by two-dimensional gel electrophoresis, to identify a distinct set of proteins present in the peptide eluate. Unique bands should be methodically and carefully isolated and analyzed by tandem mass spectrometry (MS/MS) to reveal a list of reasonable receptor candidates. The use of bioinformatic tools (e.g., DAVID, String, or IPA) to mine the MS/MS data is encouraged and should be performed at an early stage of the investigation. In summary, our method allows an unbiased combinatorial selection, isolation, identification, and validation of intracellular receptors directing iPhage particles or ligand peptides to distinct and specific cell compartments.
Figure 7.
Combinatorial screening of peptides targeting subcellular compartments and identification of candidate receptors by affinity chromatography and MS/MS analysis. (a) Diagram of combinatorial screening and peptide validation steps. (b) Selection of an iPhage display peptide library from nuclei, cytosol, and mitochondrial/ER fractions produces serial sequence enrichment. (c) Cellular distribution of internalizing candidate peptide fused to pen depicts peptide homing and binding to the nuclei of KS1767 cells. Intracellular peptide location was revealed by avidin-FITC and DAPI counterstaining. Arrows indicate peptide localization. Scale Bar, 100 μm. (d) Wash, peptide-elution, and glycine elution fractions were immobilized on 96-well plates and tested for binding with iPhage harboring the iLP peptide. Insertless iPhage and BSA were used as negative controls. Phage binding assays were run in triplicates; bars represent mean values for phage TU recovered from immobilized proteins ± SEM. (e) Wash (F22) and elution (F46) fractions were analyzed on a Tris-glycine gel (4% – 20%), and proteins were visualized by Coomassie blue staining. Arrows indicate proteins extracted for mass spectrometric analysis.
Supplementary Material
Supplementary Figure 1: Subcellular iPhage localization. Confocal fluorescence microscopy analysis of KS1767 cells exposed to parental phage, iPhage, and MLS-iPhage. Intracellular localization was revealed by anti-rabbit Alexa Fluor 488, orange-Mitotracker, and DAPI counterstaining. Here we show individual slice acquisitions and 3D multiplanar reformations, with the appearance of true co-localization both on individual 0.5 μm optical slices. Scale Bar, 10 μm. Yellow fluorescence indicates co-localization.
Acknowledgments
This work was supported by grants from the NIH, the DOD, and by awards from The University of Texas M. D. Anderson Cancer Center Trust, the Marcus Foundation, AngelWorks, and from the Gillson-Longenbaugh Foundation (all to W.A. and R.P.). R.R. received support from the Odyssey Scholar Program at the University of Texas M.D. Anderson Cancer Center.
Footnotes
AUTHOR CONTRIBUTIONS
R.R., A.S.D., L.G-R., J.G.G., R.L.S., R.P., and W.A. designed experiments. R.R., A.S.D., L.G-R., and C.C.S., performed the experiments. R.R., A.S.D., L.G-R., C.C.S., J.G.G., R.L.S., R.P., and W.A. analyzed data. R.R., A.S.D., L.G-R., R.L.S., R.P., and W.A. wrote the manuscript. R.P. and W.A. supervised the project.
COMPETING FINANCIAL INTERESTS
The authors declare competing financial interests.
References
- 1.Pasqualini R, Ruoslahti E. Organ targeting in vivo using phage display peptide libraries. Nature. 1996;380:364–366. doi: 10.1038/380364a0. [DOI] [PubMed] [Google Scholar]
- 2.Arap W, Pasqualini R, Ruoslahti E. Cancer treatment by targeted drug delivery to tumor vasculature in a mouse model. Science. 1998;279:377–380. doi: 10.1126/science.279.5349.377. [DOI] [PubMed] [Google Scholar]
- 3.Pasqualini R, Arap W, Rajotte D, Ruoslahti E. In: Phage Display: A Laboratory Manual. Barbas CF, Burton DR, Scott JK, Silverman GJ, editors. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, New York: 2001. pp. 1–24. [Google Scholar]
- 4.Pasqualini R, et al. Aminopeptidase N is a receptor for tumor-homing peptides and a target for inhibiting angiogenesis. Cancer Res. 2000;60:722–727. [PMC free article] [PubMed] [Google Scholar]
- 5.Arap W, et al. Steps toward mapping the human vasculature by phage display. Nat Med. 2002;8:121–127. doi: 10.1038/nm0202-121. [DOI] [PubMed] [Google Scholar]
- 6.Laakkonen P, Porkka K, Hoffman JA, Ruoslahti E. A tumor-homing peptide with a targeting specificity related to lymphatic vessels. Nat Med. 2002;8:751–755. doi: 10.1038/nm720. [DOI] [PubMed] [Google Scholar]
- 7.Mintz PJ, et al. Fingerprinting the circulating repertoire of antibodies from cancer patients. Nat Biotechnol. 2003;21:57–63. doi: 10.1038/nbt774. [DOI] [PubMed] [Google Scholar]
- 8.Higgins LM, et al. In vivo phage display to identify M cell-targeting ligands. Pharm Res. 2004;21:695–705. doi: 10.1023/b:pham.0000022418.80506.9a. [DOI] [PubMed] [Google Scholar]
- 9.Kolonin MG, Saha PK, Chan L, Pasqualini R, Arap W. Reversal of obesity by targeted ablation of adipose tissue. Nat Med. 2004;10:625–632. doi: 10.1038/nm1048. [DOI] [PubMed] [Google Scholar]
- 10.Ballard VL, Holm JM, Edelberg JM. Quantitative PCR-based approach for rapid phage display analysis: a foundation for high throughput vascular proteomic profiling. Physiol Genomics. 2006;26:202–208. doi: 10.1152/physiolgenomics.00025.2006. [DOI] [PubMed] [Google Scholar]
- 11.Kolonin MG, et al. Synchronous selection of homing peptides for multiple tissues by in vivo phage display. FASEB J. 2006;20:979–981. doi: 10.1096/fj.05-5186fje. [DOI] [PubMed] [Google Scholar]
- 12.Zhang L, Giraudo E, Hoffman JA, Hanahan D, Ruoslahti E. Lymphatic zip codes in premalignant lesions and tumors. Cancer Res. 2006;66:5696–5706. doi: 10.1158/0008-5472.CAN-05-3876. [DOI] [PubMed] [Google Scholar]
- 13.Hardy B, Battler A, Weiss C, Kudasi O, Raiter A. Therapeutic angiogenesis of mouse hind limb ischemia by novel peptide activating GRP78 receptor on endothelial cells. Biochem Pharmacol. 2008;75:891–899. doi: 10.1016/j.bcp.2007.10.008. [DOI] [PubMed] [Google Scholar]
- 14.Mintz PJ, et al. An unrecognized extracellular function for an intracellular adapter protein released from the cytoplasm into the tumor microenvironment. Proc Natl Acad Sci U S A. 2009;106:2182–2187. doi: 10.1073/pnas.0807543105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Staquicini FI, et al. Vascular ligand-receptor mapping by direct combinatorial selection in cancer patients. Proc Natl Acad Sci U S A. 2011;108:18637–18642. doi: 10.1073/pnas.1114503108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Joliot A, Pernelle C, Deagostini-Bazin H, Prochiantz A. Antennapedia homeobox peptide regulates neural morphogenesis. Proc Natl Acad Sci U S A. 1991;88:1864–1868. doi: 10.1073/pnas.88.5.1864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Derossi D, Joliot AH, Chassaing G, Prochiantz A. The third helix of the Antennapedia homeodomain translocates through biological membranes. J Biol Chem. 1994;269:10444–10450. [PubMed] [Google Scholar]
- 18.Derossi D, et al. Cell internalization of the third helix of the Antennapedia homeodomain is receptor-independent. J Biol Chem. 1996;271:18188–18193. doi: 10.1074/jbc.271.30.18188. [DOI] [PubMed] [Google Scholar]
- 19.Théodore L, et al. Intraneuronal delivery of protein kinase C pseudosubstrate leads to growth cone collapse. J Neurosci. 1995;15:7158–7167. doi: 10.1523/JNEUROSCI.15-11-07158.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen YN, et al. Selective killing of transformed cells by cyclin/cyclin-dependent kinase 2 antagonists. Proc Natl Acad Sci U S A. 1999;96:4325–4329. doi: 10.1073/pnas.96.8.4325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mi Z, Mai J, Lu X, Robbins PD. Characterization of a class of cationic peptides able to facilitate efficient protein transduction in vitro and in vivo. Mol Ther. 2000;2:339–347. doi: 10.1006/mthe.2000.0137. [DOI] [PubMed] [Google Scholar]
- 22.Cardó-Vila M, Arap W, Pasqualini R. Alpha v beta 5 integrin-dependent programmed cell death triggered by a peptide mimic of annexin V. Mol Cell. 2003;11:1151–1162. doi: 10.1016/s1097-2765(03)00138-2. [DOI] [PubMed] [Google Scholar]
- 23.Davidson TJ, et al. Highly efficient small interfering RNA delivery to primary mammalian neurons induces MicroRNA-like effects before mRNA degradation. J Neurosci. 2004;24:10040–10046. doi: 10.1523/JNEUROSCI.3643-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Muratovska A, Eccles MR. Conjugate for efficient delivery of short interfering RNA (siRNA) into mammalian cells. FEBS Lett. 2004;558:63–68. doi: 10.1016/S0014-5793(03)01505-9. [DOI] [PubMed] [Google Scholar]
- 25.Apostolopoulos V, et al. Delivery of tumor associated antigens to antigen presenting cells using penetratin induces potent immune responses. Vaccine. 2006;24:3191–3202. doi: 10.1016/j.vaccine.2006.01.032. [DOI] [PubMed] [Google Scholar]
- 26.Fabani MM, Gait MJ. miR-122 targeting with LNA/2’-O-methyl oligonucleotide mixmers, peptide nucleic acids (PNA), and PNA-peptide conjugates. RNA. 2008;14:336–346. doi: 10.1261/rna.844108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Blobel G, Sabatini DD. Controlled proteolysis of nascent polypeptides in rat liver cell fractions. I. Location of the polypeptides within ribosomes. J Cell Biol. 1970;45:130–145. doi: 10.1083/jcb.45.1.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Blobel G, Dobberstein B. Transfer of proteins across membranes. I. Presence of proteolytically processed and unprocessed nascent immunoglobulin light chains on membrane-bound ribosomes of murine myeloma. J Cell Biol. 1975;67:835–851. doi: 10.1083/jcb.67.3.835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Blobel G. Intracellular protein topogenesis. Proc Natl Acad Sci U S A. 1980;77:1496–1500. doi: 10.1073/pnas.77.3.1496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Walter P, Blobel G. Purification of a membrane-associated protein complex required for protein translocation across the endoplasmic reticulum. Proc Natl Acad Sci U S A. 1980;77:7112–7116. doi: 10.1073/pnas.77.12.7112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pain D, Murakami H, Blobel G. Identification of a receptor for protein import into mitochondria. Nature. 1990;347:444–449. doi: 10.1038/347444a0. [DOI] [PubMed] [Google Scholar]
- 32.Rangel R, et al. Combinatorial targeting and discovery of ligand-receptors in organelles of mammalian cells. Nat Commun. 2012;3:788. doi: 10.1038/ncomms1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Barnhart KF, et al. A peptidomimetic targeting white fat causes weight loss and improved insulin resistance in obese monkeys. Sci Transl Med. 2011;3:108ra112. doi: 10.1126/scitranslmed.3002621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zurita AJ, et al. Combinatorial screenings in patients: the interleukin-11 receptor alpha as a candidate target in the progression of human prostate cancer. Cancer Res. 2004;64:435–439. doi: 10.1158/0008-5472.can-03-2675. [DOI] [PubMed] [Google Scholar]
- 35.Lewis VO, et al. The interleukin-11 receptor alpha as a candidate ligand-directed target in osteosarcoma: consistent data from cell lines, orthotopic models, and human tumor samples. Cancer Res. 2009;69:1995–1999mass. doi: 10.1158/0008-5472.CAN-08-4845. [DOI] [PubMed] [Google Scholar]
- 36.Cardó-Vila M, et al. From combinatorial peptide selection to drug prototype (II): targeting the epidermal growth factor receptor pathway. Proc Natl Acad Sci U S A. 2010;107:5118–5123. doi: 10.1073/pnas.0915146107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Giordano RJ, et al. From combinatorial peptide selection to drug prototype (I): targeting the vascular endothelial growth factor receptor pathway. Proc Natl Acad Sci U S A. 2010;107:5112–5117. doi: 10.1073/pnas.0915141107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Marchiò S, et al. Aminopeptidase A is a functional target in angiogenic blood vessels. Cancer Cell. 2004;5:151–162. doi: 10.1016/s1535-6108(04)00025-x. [DOI] [PubMed] [Google Scholar]
- 39.Hajitou A, et al. A hybrid vector for ligand-directed tumor targeting and molecular imaging. Cell. 2006;125:385–398. doi: 10.1016/j.cell.2006.02.042. [DOI] [PubMed] [Google Scholar]
- 40.Hajitou A, et al. Design and construction of targeted AAVP vectors for mammalian cell transduction. Nat Protoc. 2007;2:523–531. doi: 10.1038/nprot.2007.51. [DOI] [PubMed] [Google Scholar]
- 41.Hajitou A, et al. A preclinical model for predicting drug response in soft-tissue sarcoma with targeted AAVP molecular imaging. Proc Natl Acad Sci U S A. 2008;105:4471–4476. doi: 10.1073/pnas.0712184105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Soghomonyan S, et al. Molecular PET imaging of HSV1-tk reporter gene expression using [18F]FEAU. Nat Protoc. 2007;2:416–423. doi: 10.1038/nprot.2007.49. [DOI] [PubMed] [Google Scholar]
- 43.Paoloni MC, et al. Launching a novel preclinical infrastructure: comparative oncology trials consortium directed therapeutic targeting of TNFalpha to cancer vasculature. PLoS One. 2009;4:e4972. doi: 10.1371/journal.pone.0004972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tandle A, et al. Tumor vasculature-targeted delivery of tumor necrosis factor-alpha. Cancer. 2009;115:128–139. doi: 10.1002/cncr.24001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Staquicini FI, et al. Systemic combinatorial peptide selection yields a non-canonical iron-mimicry mechanism for targeting tumors in a mouse model of human glioblastoma. J Clin Invest. 2011;121:161–173. doi: 10.1172/JCI44798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Xu X, et al. Dominant effector genetics in mammalian cells. Nat Genet. 2001;27:23–29. doi: 10.1038/83717. [DOI] [PubMed] [Google Scholar]
- 47.Schlabach MR, et al. Cancer proliferation gene discovery through functional genomics. Science. 2008;319:620–624. doi: 10.1126/science.1149200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Silva JM, et al. Profiling essential genes in human mammary cells by multiplex RNAi screening. Science. 2008;319:617–620. doi: 10.1126/science.1149185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Peelle B, et al. Intracellular protein scaffold-mediated display of random peptide libraries for phenotypic screens in mammalian cells. Chem Biol. 2001;8:521–534. doi: 10.1016/s1074-5521(01)00031-x. [DOI] [PubMed] [Google Scholar]
- 50.Norman TC, et al. Genetic selection of peptide inhibitors of biological pathways. Science. 1999;285:591–595. doi: 10.1126/science.285.5427.591. [DOI] [PubMed] [Google Scholar]
- 51.Zhong J, Zhang H, Stanyon CA, Tromp G, Finley RL. A strategy for constructing large protein interaction maps using the yeast two-hybrid system: regulated expression arrays and two-phase mating. Genome Res. 2003;13:2691–2699. doi: 10.1101/gr.1134603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Staquicini FI, Sidman RL, Arap W, Pasqualini R. Phage display technology for stem cell delivery and systemic therapy. Adv Drug Deliv Rev. 2010;62:1213–1216. doi: 10.1016/j.addr.2010.09.014. [DOI] [PubMed] [Google Scholar]
- 53.Smith GP, Scott JK. Libraries of peptides and proteins displayed on filamentous phage. Methods Enzymol. 1993;217:228–257. doi: 10.1016/0076-6879(93)17065-d. [DOI] [PubMed] [Google Scholar]
- 54.Herndier BG, et al. Characterization of a human Kaposi’s sarcoma cell line that induces angiogenic tumors in animals. AIDS. 1994;8:575–581. doi: 10.1097/00002030-199405000-00002. [DOI] [PubMed] [Google Scholar]
- 55.Claude A. FRACTIONATION OF MAMMALIAN LIVER CELLS BY DIFFERENTIAL CENTRIFUGATION : I. PROBLEMS, METHODS, AND PREPARATION OF EXTRACT. J Exp Med. 1946;84:51–59. [PubMed] [Google Scholar]
- 56.Castle JD. Purification of organelles from mammalian cells. Curr Protoc Immunol. 2003;Unit 8:1B. doi: 10.1002/0471142735.im0801bs56. Chapter 8. [DOI] [PubMed] [Google Scholar]
- 57.de Araùjo ME, Huber LA, Stasyk T. Isolation of endocitic organelles by density gradient centrifugation. Methods Mol Biol. 2008;424:317–331. doi: 10.1007/978-1-60327-064-9_25. [DOI] [PubMed] [Google Scholar]
- 58.Huber LA, Pfaller K, Vietor I. Organelle proteomics: implications for subcellular fractionation in proteomics. Circ Res. 2003;92:962–968. doi: 10.1161/01.RES.0000071748.48338.25. [DOI] [PubMed] [Google Scholar]
- 59.Cox B, Emili A. Tissue subcellular fractionation and protein extraction for use in mass-spectrometry-based proteomics. Nat Protoc. 2006;1:1872–1878. doi: 10.1038/nprot.2006.273. [DOI] [PubMed] [Google Scholar]
- 60.Hoffmann K, et al. New application of a subcellular fractionation method to kidney and testis for the determination of conjugated linoleic acid in selected cell organelles of healthy and cancerous human tissues. Anal Bioanal Chem. 2005;381:1138–1144. doi: 10.1007/s00216-004-3009-z. [DOI] [PubMed] [Google Scholar]
- 61.Spann TP. In: Cells: A laboratory manual, Vol. 1: Culture and Biochemical Analysis of Cells. Spector DL, Goldman RD, Leinwand LA, editors. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, NY: 1998. [Google Scholar]
- 62.Dias-Neto E, et al. Next-generation phage display: integrating and comparing available molecular tools to enable cost-effective high-throughput analysis. PLoS One. 2009;4:e8338. doi: 10.1371/journal.pone.0008338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Parmley SF, Smith GP. Antibody-selectable filamentous fd phage vectors: affinity purification of target genes. Gene. 1988;73:305–318. doi: 10.1016/0378-1119(88)90495-7. [DOI] [PubMed] [Google Scholar]
- 64.Sambrook JJ, Russell DW. Molecular Cloning: A Laboratory Manual, Vol. 1. 3. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, New York: 2001. [Google Scholar]
- 65.Sambrook J, Fritsch EF, Maniatis T. Molecular Cloning: A Laboratory Manual, Vol. 3. 2. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, New York: 1989. [Google Scholar]
- 66.Kalderon D, Roberts BL, Richardson WD, Smith AE. A short amino acid sequence able to specify nuclear location. Cell. 1984;39:499–509. doi: 10.1016/0092-8674(84)90457-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1: Subcellular iPhage localization. Confocal fluorescence microscopy analysis of KS1767 cells exposed to parental phage, iPhage, and MLS-iPhage. Intracellular localization was revealed by anti-rabbit Alexa Fluor 488, orange-Mitotracker, and DAPI counterstaining. Here we show individual slice acquisitions and 3D multiplanar reformations, with the appearance of true co-localization both on individual 0.5 μm optical slices. Scale Bar, 10 μm. Yellow fluorescence indicates co-localization.