

Abstract
In order to realize the goal of stratified and/or personalized medicine in the clinic, significant advances in the field of biomarker discovery are necessary. Adding to the abundance of nucleic acid biomarkers being characterized, additional protein biomarkers will be needed to satisfy diverse clinical needs. An appropriate source for finding these biomarkers is within blood, as it contains tissue leakage factors as well as additional proteins that reside in blood that can be linked to the presence of disease. Unfortunately, high abundant proteins and complexity of the blood proteome present significant challenges for the discovery of protein biomarkers from blood. Animal models often enable the discovery of biomarkers that can later be translated to humans. Therefore, determining appropriate sample preparation of proteomic samples in rodent models is an important research goal. Here, we examined both mouse and rat blood samples (including both serum and plasma), for appropriate high abundant protein removal techniques for subsequent gel-based proteomic experiments. We assessed four methods of albumin removal: antibody-based affinity chromatography (MARS), Cibacron® Blue-based affinity depletion (SwellGel® Blue Albumin Removal Kit), protein-based affinity depletion (ProteaPrep Albumin Depletion Kit) and TCA/acetone precipitation. Albumin removal was quantified for each method and SDS-PAGE and 2-DE gels were used to quantify the number of protein spots obtained following albumin removal. Our results suggest that while all four approaches can effectively remove high abundant proteins, antibody-based affinity chromatography is superior to the other three methods.
Introduction
Difficulties in sample preparation currently limit the discovery of protein biomarkers from biofluids, in particular blood plasma and serum. One of the biggest challenges in the study of blood plasma involves the broad concentration range of its protein constituents. In humans, there is approximately a 109 order of magnitude from most to least abundant proteins [1]. In addition, few high abundant proteins dominate the plasma, making biomarker discovery of lower abundance proteins even more difficult. For example, twenty-two proteins comprise over 90% of the total protein mass in human serum and albumin alone accounts for over 50%. These dominant species prevent the detection of lower-abundance proteins that may be of greater interest as putative biomarkers [2]. Therefore, a successful system of proteomic sample preparation to remove these high abundant proteins is needed to examine lower abundant proteins of interest and to reduce the complexity for improved biomarker discovery. Researchers have developed successful ways to remove these proteins, but these methods vary in the efficiency and mechanism for removing targeted highly abundant proteins [3,4,5,6,7].
Putative protein biomarkers discovered after the removal of high abundant proteins may serve to detect diseases earlier with higher accuracy, but may prove to be challenging for subsequent validation in humans. Therefore, animal models are necessary to validate these biomarkers and for the discovery of additional biomarkers. Initial 2-DE proteome maps of mouse and rat produced species specific patterns and showed serum proteins can vary substantially [8,9,10,11]. However, these samples have a similar wide dynamic range in protein concentrations as seen in human samples and therefore face some of the same technological challenges. Since the same high abundant proteins are found in blood of animals, their removal from these models is also necessary. There are many ways to accomplish high abundant protein removal for rodent blood including hydrophobic interactions [12], ammonium sulfate precipitation [13], ion exchange [10], antibody-based affinity chromatography [14,15], and TCA/acetone precipitation [16], and these approaches have been used to enable discovery of putative biomarkers [15,17,18,19,20]. In one of these studies, plasma protein biomarkers found in a mouse model of pancreatic cancer were used to translate to human protein orthologs, providing putative early detection markers applicable to human cancer [15]. These studies have focused on a single technique and have not directly compared removal methods to each other using the same samples. Moreover, each study has not compared these techniques for both serum and plasma obtained from both mice and rats.
In this study, four different methods for high abundant protein removal were compared using rat serum/plasma and mouse serum/plasma. SDS-PAGE was used to compare the extent of albumin removal between these methods. Further characterization using 2-D DIGE was done to assess the improvement in total protein spots after removal of high abundant proteins by each of the four different methods.
Materials and Methods
Sample collection
Rodent blood was collected under IACUC protocols for (DH) and (PL). For mice, whole blood was collected by ocular bleed. For rats, blood was collected from the saphenous vein on the inside of the thigh using a 21 gauge needle. Serum was allowed to clot at room temperature for 2–5 hours followed by centrifugation at 5000 × g for 10 minutes. The supernatant was collected and stored at −80°C in fresh tube. For plasma, blood was collected into BD 0.5ml microtainer tubes containing Potassium EDTA (Becton Dickinson, Franklin Lakes, NJ). Blood was centrifuged at 15,000 × g for 10 minutes to separate the plasma from the red blood cells. Plasma was collected, aliquoted and stored at −80°C until analysis.
High abundant protein removal
Depletion of high abundant proteins was carried out according to the manufacturer’s instructions with minor modifications as detailed below. TCA/acetone was carried out similarly as previously published [16]. TCA was dissolved in water to make a 20% solution and this solution was diluted 1:1 with the protein sample on ice for 30 minutes. Following incubation the proteins were centrifuged and the protein pellet was washed 2 x with ice-cold acetone. The ProteaPrep procedure was carried out as described in the manufacturer’s protocol (Protea). Protein samples were diluted in sample buffer 1:4 and then loaded into pre-packed columns containing a proprietary dry powder that facilitated non-antibody, affinity-based serum albumin removal. The capture ligand is a recombinant protein that claims to be more specific than an antibody-based system with stronger binding constants. For SwellGell® Blue Albumin (Pierce), 40 μl samples of plasma or serum were diluted into 160 μl of bind/wash buffer. Albumin binding incubations were done for 2 minutes (twice). Incubations were washed 3 times with 200 μl. The flow through and washes were pooled as the albumin removed sample. For antibody-affinity chromatography using the MARS MS-3 (Agilent Technologies), rodent plasma or serum was diluted five times in Buffer A (40 μl sample and 160 μl of buffer, 200 μl total volume) and centrifuged through a 0.22 micron spin filter tube (Millipore) at 16,000 × g for 5 minutes to remove particulates. Then, plasma or serum was processed using 4.6 × 50 mm Multiple Affinity Removal Column Mouse-3 (Agilent Technologies), which specifically removes albumin, IgG, and transferrin. A low abundant protein fraction was collected for each sample. Fractions were concentrated by precipitating with an equal volume of 20% TCA solution and incubated at 4°C for 30 minutes. Precipitate was spun down and washed twice with cold 100% acetone, allowed to air dry and then resuspended in DIGE labeling buffer (7 M urea, 2 M thiourea, 4% CHAPS, 30 mM Tris, pH 8.5). Protein quantification was performed using Precision Red Advanced Protein Assay Reagent (Cytoskeleton).
SDS-PAGE
Crude and high abundant protein depleted plasma or serum samples (5 μg) were mixed with 5X sample loading buffer (0.2 M Tris pH 6.8, 20% glycerol, 10% SDS, 5% BME), boiled for 10 minutes at 100°C and resolved on a 4–20% Tris-Glycine gel (Invitrogen). Gels were stained for total protein using Sypro Ruby Protein Gel Stain (Invitrogen, S-12000) and visualized using the BioChemi system (UVP BioImaging Systems).
2-D DIGE
Crude and high abundant protein depleted plasma and serum samples were separated in two dimensions using the GE Life Sciences Ettan DIGE system protocol. Briefly, each sample (50 μg) was minimally labeled with 1 μl of 200 pM Cy2, Cy3 or Cy5 for 30 minutes. Labeling reactions were stopped by the addition of 1 μl of 1 mM lysine. The samples were pooled together and added to rehydration buffer (7 M urea, 2 M thiourea, 4% CHAPS, 1.2% DeStreak, 1% pharmalytes). A final volume of 450 μl sample was loaded onto 24 cm pH 3–10NL Immobiline DryStrips (GE Life Sciences) and focused by active overnight rehydration, followed by isoelectric focusing for a total of 62,500 Vhrs. Strips were equilibrated in SDS equilibration buffer (6 M urea, 30% glycerol, 2% SDS) for 15 min with 10 mg/ml DTT, then 15 min in fresh buffer with 25 mg/ml 15 min with IAA, then applied to DIGE gels (GE Life Sciences) for 2nd dimension separation. The resulting CyDye labeled protein gels were scanned using 100 micron resolution on Typhoon 9410 (GE Life Sciences).
Image analysis
Data analysis was carried out using DeCyder 2-D 7.0 software (GE Life Sciences). Spot detection and abundance quantification was performed using the differential in-gel analysis (DIA) module of DeCyder. Densitometry, using ImageJ processing program (available free online at rsb.info.nih.gov/ij/), was performed on selected albumin bands to determine the percent removed.
Results
Four different methods were tested for their ability to remove albumin from both rodent blood samples. Both rat and mouse samples of plasma and serum were used. SDS-PAGE and 2-DE were used to evaluate the overall improvements in proteomic sample preparation following high abundant protein removal. Table 1 shows the recovery of the total protein following these different methods. Most of the protein remains in the high abundant fraction, but this table shows that the total protein obtained from these different methods does not vary substantially. Therefore, none of these methods reduce total protein recovery more than another.
Table 1. Protein recovered after high abundant protein removal is similar among four different methods.
For all methods used for high abundant protein removal, most of the protein was removed (about 90%). The remaining lower abundant protein samples were similar in total amount of protein obtained, ranging from 109 to 177 μg. Some differences between samples were found but further experimentation needs to be done to ensure these differences are statistically valid.
| Sample | Start Total (ug) | Method | |||
|---|---|---|---|---|---|
| SG | Protea | TCA/Ac | MARS | ||
| Mouse serum | 1960 | 174 | 160 | 152 | 150 |
| Mouse plasma | 1943 | 172 | 168 | 153 | 109 |
| Rat serum | 2082 | 171 | 148 | 177 | 174 |
| Rat plasma | 1942 | 151 | 174 | 137 | 155 |
After determining that the total amount of protein does not differ substantially due to sample preparation, we then examined the protein pattern using SDS-PAGE. We compared samples following each technique to each other and to raw serum and plasma. Figure 1 shows SDS-PAGE images for both serum and plasma from mice. All methods were able to reduce the amount of albumin and increase the overall number of protein bands that could be detected. Albumin depletion strategies for rat serum and plasma showed similar results with decreased albumin and increased total protein spots (Figure 2). The albumin removal results suggest that all methods chosen for study here can improve the proteomic spot pattern. However, important differences between techniques were found. For example, the method that removed the most albumin for mouse plasma was the antibody-based affinity chromatography, as it removed about twice as much albumin in some cases (Table 2).
Figure 1. Mouse Blood SDS-PAGE.
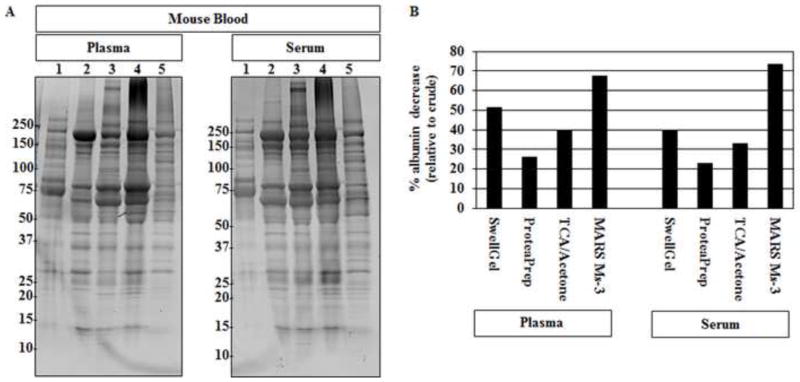
(A) Mouse plasma and serum, post-albumin removal by each method, was evaluated by SDS-PAGE. Lane 1 crude plasma (left); serum (right). 2. SwellGel® Blue Albumin 3. ProteaPrep Lane 4: TCA/acetone Lane 5: MARS Ms-3. Crude samples show a few protein bands and the presence of a dark band at roughly 70 kDa representing the main albumin band. All lanes show increased numbers of protein bands and a lower main albumin band following albumin removal. (B) Densitometry analysis of percent albumin decrease showed different depletion levels among the four tested methods. The antibody-based affinity chromatography method removed the most albumin for both plasma and serum samples. Percent albumin decrease was calculated using the main albumin band density, relative to each lane, divided by the percent albumin found in each crude sample.
Figure 2. Rat Blood SDS-PAGE.
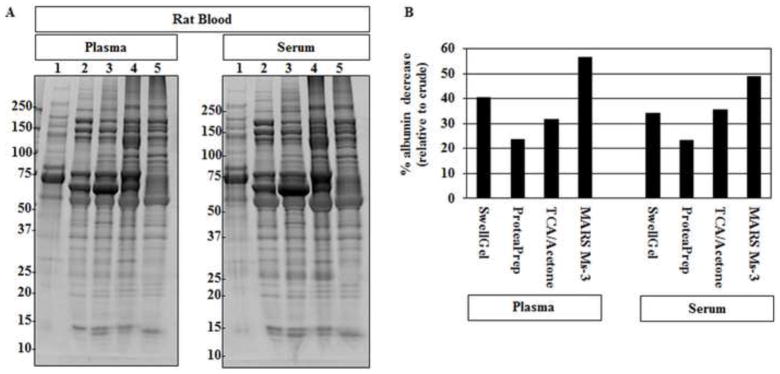
(A) Rat plasma and serum, post-albumin removal by each method, was evaluated by SDS-PAGE. Lane 1 crude plasma (left); serum (right). 2. SwellGel® Blue Albumin 3. ProteaPrep Lane 4: TCA/acetone Lane 5: MARS Ms-3. Crude samples show a few protein bands and the presence of a dark band at roughly 70 kDa representing the main albumin band. All lanes show increased numbers of protein bands and a lower main albumin band following albumin removal. (B) Densitometry analysis of percent albumin decrease showed different depletion levels among the four tested methods. The antibody-based affinity chromatography method removed the most albumin for both plasma and serum samples. Percent albumin decrease was calculated using the main albumin band density, relative to each lane, divided by the percent albumin found in each crude sample.
Table 2. Different sample preparation techniques yield significant albumin removal with different total protein spots.
For all four sample types, antibody-based affinity chromatography proved to be the best technique for albumin removal and increased protein spots found in a 2D gel. For (A) mouse plasma/serum and (B) rat serum, TCA/acetone method provides the second most albumin removal and yields the second most 2-D gel spots.
| A | ||||||||
|---|---|---|---|---|---|---|---|---|
| Species | Mouse | |||||||
| Sample Type | Plasma | Serum | ||||||
| Albumin Removal Method | SwellGel | Protea Prep | TCA acetone | MARS Ms-3 | SwellGel | Protea Prep | TCA acetone | MARS Ms-3 |
| Albumin Removed (SDS-PAGE) | 51% | 26% | 39% | 68% | 39% | 23% | 33% | 73% |
| Total 2-D Spots Detected | 876 | 851 | 965 | 1352 | 974 | 1025 | 1083 | 1311 |
| B | ||||||||
|---|---|---|---|---|---|---|---|---|
| Species | Rat | |||||||
| Sample Type | Plasma | Serum | ||||||
| Albumin Removal Method | SwellGel | Protea Prep | TCA acetone | MARS Ms-3 | SwellGel | Protea Prep | TCA acetone | MARS Ms-3 |
| Albumin Removed (SDS-PAGE) | 40% | 24% | 32% | 56% | 34% | 23% | 36% | 49% |
| Total 2-D Spots Detected | 1008 | 943 | 1157 | 1285 | 1069 | 1069 | 1155 | 1263 |
Another issue for improved biomarker discovery using proteomics involves the concentration of the protein sample and the use of buffer exchange to ensure proper buffer conditions for subsequent proteomic analysis following high abundant protein removal. Specifically, after high abundant protein removal using SwellGel® Blue Albumin, ProteaPrep, and MARS, we examined differences between TCA precipitation and molecular weight cutoff in this subsequent concentration and buffer exchange step. SDS-PAGE was used to compare the two procedures (Supplemental Figure 1). SDS-PAGE showed very similar banding patterns for both procedures, following the 3 different types of high abundant removal protocols. Since TCA/acetone and molecular weight cutoff removal were found to be similar, TCA/acetone was chosen for subsequent proteomic analysis.
To determine if a more sensitive proteomic technique could differentiate among these high abundant protein removal procedures, we used 2-D DIGE. Figure 3 shows 2-D DIGE images of the mouse serum and plasma samples following high abundant protein removal. Successful high abundant protein removal is demonstrated by the increased number of total spots and diverse spot pattern as compared to crude serum and plasma. The antibody-based affinity chromatography method showed significantly better albumin removal and more total protein spots for both mouse sample types. The three other methods removed substantial albumin and had similar numbers of total protein spots, but each had roughly half of the albumin removal and 200–300 fewer protein spots than the affinity chromatography.
Figure 3. Mouse Blood 2-D DIGE.

(A) Albumin depleted mouse plasma and (B) serum. 2-D DIGE gel images are shown with corresponding number of detected protein spots. The antibody-based affinity chromatography method removed the most albumin for both plasma and serum samples.
Figure 4 shows similar results of improved proteomic separation for rat plasma. Again, antibody-based affinity chromatography removed significantly more albumin than the other three techniques, which were similar with respect to the amount of albumin removed. Total protein spots were also highest for the affinity chromatography by at least 100 spots. Over 1200 protein spots were found following the removal of the top three serum/plasma proteins using affinity chromatography. Even though this antibody column was optimized for mouse, the results shown here clearly demonstrate that the column can be useful for removal of high abundant rat serum/plasma proteins.
Figure 4. Rat Blood 2-D DIGE.

(A) Albumin depleted rat plasma and (B) serum. 2-D DIGE gel images are shown with corresponding number of detected protein spots. Similar to the mouse blood data, antibody-based affinity chromatography shows the highest number of protein spots, specifically in the lower half of the gel.
Table 2 shows the results of the albumin removal and the total protein spots found for all four sample types for all four albumin depletion methods, showing percent albumin decrease for each technique used. To validate which techniques removed substantial albumin, the amount of albumin that was removed was quantified using densitometry of the SDS-PAGE. These data show that antibody-based affinity chromatography removes the most albumin and shows the greatest number of total protein spots by a significant amount.
Discussion
Successful proteomic sample preparation from blood often requires high abundant protein removal. High abundant proteins have been shown to be responsible for concealing putative markers. For example, albumin was found to obscure sex differences in blood plasma of rats and humans [2]. A failure to effectively remove high abundant proteins can also result in failed or incomplete biomarker studies. To determine the optimal protocol for subsequent biomarker discovery, we completed four methods of high abundant protein removal for both rat and mouse serum and plasma. Our results clearly show that antibody-based affinity chromatography is the superior method for this approach, similar to results we previously showed for human serum [3].
Several reports in the literature have addressed the issue of serum complexity and proteomic analyses. Pieper and co-workers used immunoaffinity subtraction chromatography to remove 10 proteins from human blood plasma. Following protein depletion, Coomassie blue stained 2-dimensional electrophoresis (2-DE) gels revealed approximately 650 protein spots compared with only 220 spots visible in a sample of crude serum. Silver staining of the protein-depleted sample revealed an even larger number, 950 spots [9]. Chan and coworkers used an affinity spin tube filter method to remove albumin and IgG to enrich for low-abundant cancer biomarkers in serum. Over 250 potential biomarkers for breast cancer were identified in this study. TCA-acetone has been used to remove albumin from serum [5]. Finally, Steel and co-workers also used an immunoaffinity resin to remove albumin and IgG from human serum samples in order to simplify the serum proteome [21]. Our data demonstrate that antibody-based affinity chromatography removes the greatest percentage of albumin and results in the highest total number of lower abundant protein spots relative to any published work to date. We used 2-D DIGE [3], which can detect protein spots as low as 150–500 pg of a single protein, with a linear response in protein concentration over five orders of magnitude [22]. This method allowed us to detect over 1200 protein spots in each of these four rodent samples, by far the most protein spots detected for these sample types.
Antibody-based affinity chromatography often is the best choice for high abundant protein removal as we showed here for rodent samples. Importantly, since this column was designed using mouse antibodies, it was not clearly evident that it would work well with rat samples. However, our work shows that both rat serum and plasma can be improved for biomarker discovery using this approach. The specificity and efficiency of the microbead, IgY-based anti-rat immunoaffinity LC column has been previously examined and has improved protein detection using several different techniques including SELDI-TOF MS, 2-dimensional SDS-PAGE, and 2-dimensional liquid chromatography [14]. Although results here show more protein spots, we do not know if this is because of the relative improvement of the column used here or the highly sensitive visualization technique of 2-D DIGE, which is also highlighted as integral for improved protein biomarker identification [14]. Future work should be done to see if species specific antibodies improve upon the results found here. Species-specific differences may also explain why the ProteaPrep depletion kit was not better than antibody-affinity despite claims to that the recombinant protein capture ligand is more efficient than antibody-based methods. The ProteaPrep capture ligand is claimed to be more specific than antibodies and has a stronger binding constant to human serum albumin. The results here do not support this, which might be due to species-specific differences. Further investigation into human albumin removal is necessary to compare these removal methods.
In addition, we showed that TCA precipitation enables the complete removal of buffer for both concentration and exchange purposes for these sample types. TCA precipitation has been widely used to concentrate protein samples and exchange existing buffers in proteomic sample preparation [23]. However, protein is denatured following TCA precipitation, so protein activity cannot be assessed. In addition, proteins cannot be resuspended easily in any non-denaturing buffer (such as PBS). Molecular weight cutoff membranes have also been widely used for concentration and buffer exchange. Molecular weight membranes enable the removal of buffer by centrifugation through filtration, while retaining proteins of at least 3 kDa. This procedure offers a single step sample concentration in a single tube for minimal sample handling and reduced sample loss. When comparing a single TCA/acetone step with molecular weight cutoff membranes following other types of high abundant protein removal, we did not notice substantial differences (Supplementary Figure 1).
Supplementary Material
SDS-PAGE showing no substantial differences between TCA precipitation and molecular weight cutoff membranes. Following three different high abundant protein removal methods, differences in protein samples were compared using TCA precipitation (T) and centrifugation using molecular weight cutoff membranes (M). The three different methods are SwellGel® Blue Albumin (S), ProteaPrep (P), and affinity chromatography (MARS). No substantial differences can be seen between these two methods for different rodent blood samples, mouse serum (Left) and rat plasma (Right).
Acknowledgments
The authors would like to thank Sasha Silvestrini for technical assistance. Part of this work is based upon work supported by the S.D. Bechtel, Jr. Foundation and by the National Science Foundation under Grant No. 0952013 and Grant No. 0733758. Any opinions, findings, and conclusions or recommendations expressed in this material are those of the authors and do not necessarily reflect the views of the S.D. Bechtel, Jr. Foundation. The authors would like to acknowledge funding from NIH R21-AR063348 to DRH, NIH R01-ES016308 to PJL and Dept. Pathology proteomics initiative funds to BAC.
Abbreviations
- TCA
trichloroacetic acid
- SDS-PAGE
sodium dodecyl sulfate polyacrylamide gel electrophoresis
- 2-DE
two dimensional electrophoresis
- 2-D DIGE
two-dimensional difference gel electrophoresis
- MARS
Multiple Affinity Removal system
- PBS
phosphate buffered saline
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Anderson NL, Anderson NG. The human plasma proteome: history, character, and diagnostic prospects. Molecular & Cellular Proteomics. 2002;1:845–867. doi: 10.1074/mcp.r200007-mcp200. [DOI] [PubMed] [Google Scholar]
- 2.Gersten DM, Khirabadi BS, Kurian P, Ledley RS, Mahany T, Ramey ER, Ramwell PW. Albumin obscures sex differences in blood protein patterns of rats and humans. Biochem J. 1980;191:869–872. doi: 10.1042/bj1910869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chromy BA, Gonzales AD, Perkins J, Choi MW, Corzett MH, Chang BC, Corzett CH, McCutchen-Maloney SL. Proteomic analysis of human serum by two-dimensional differential gel electrophoresis after depletion of high-abundant proteins. J Proteome Res. 2004;3:1120–1127. doi: 10.1021/pr049921p. [DOI] [PubMed] [Google Scholar]
- 4.Adkins JN, Varnum SM, Auberry KJ, Moore RJ, Angell NH, Smith RD, Springer DL, Pounds JG. Toward a human blood serum proteome: analysis by multidimensional separation coupled with mass spectrometry. Molecular & Cellular Proteomics. 2002;1:947–955. doi: 10.1074/mcp.m200066-mcp200. [DOI] [PubMed] [Google Scholar]
- 5.Pieper R, Gatlin CL, Makusky AJ, Russo PS, Schatz CR, Miller SS, Su Q, McGrath AM, Estock MA, Parmar PP, Zhao M, Huang ST, Zhou J, Wang F, Esquer-Blasco R, Anderson NL, Taylor J, Steiner S. The human serum proteome: display of nearly 3700 chromatographically separated protein spots on two-dimensional electrophoresis gels and identification of 325 distinct proteins. Proteomics. 2003;3:1345–1364. doi: 10.1002/pmic.200300449. [DOI] [PubMed] [Google Scholar]
- 6.Polaskova V, Kapur A, Khan A, Molloy MP, Baker MS. High-abundance protein depletion: comparison of methods for human plasma biomarker discovery. Electrophoresis. 2010;31:471–482. doi: 10.1002/elps.200900286. [DOI] [PubMed] [Google Scholar]
- 7.Amhad Y, Sharma N. An Effective Method for the Analysis of Human Plasma Proteome using Two-dimensional Gel Electrophoresis. J Proteomics Bioinform. 2009;2:495–499. [Google Scholar]
- 8.Haynes P, Miller I, Aebersold R, Gemeiner M, Eberini I, Lovati MR, Manzoni C, Vignati M, Gianazza E. Proteins of rat serum: I. Establishing a reference two-dimensional electrophoresis map by immunodetection and microbore high performance liquid chromatography-electrospray mass spectrometry. Electrophoresis. 1998;19:1484–1492. doi: 10.1002/elps.1150190845. [DOI] [PubMed] [Google Scholar]
- 9.Duan X, Yarmush DM, Berthiaume F, Jayaraman A, Yarmush ML. A mouse serum two-dimensional gel map: application to profiling burn injury and infection. Electrophoresis. 2004;25:3055–3065. doi: 10.1002/elps.200406039. [DOI] [PubMed] [Google Scholar]
- 10.Hood BL, Zhou M, Chan KC, Lucas DA, Kim GJ, Issaq HJ, Veenstra TD, Conrads TP. Investigation of the mouse serum proteome. J Proteome Res. 2005;4:1561–1568. doi: 10.1021/pr050107r. [DOI] [PubMed] [Google Scholar]
- 11.Ritorto MS, Borlak J. A simple and reliable protocol for mouse serum proteome profiling studies by use of two-dimensional electrophoresis and MALDI TOF/TOF mass spectrometry. Proteome Sci. 2008;6:25. doi: 10.1186/1477-5956-6-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mahn A, Reyes A, Zamorano M, Cifuentes W, Ismail M. Depletion of highly abundant proteins in blood plasma by hydrophobic interaction chromatography for proteomic analysis. J Chromatogr B Analyt Technol Biomed Life Sci. 2010;878:1038–1044. doi: 10.1016/j.jchromb.2010.03.006. [DOI] [PubMed] [Google Scholar]
- 13.Mahn A, Ismail M. Depletion of highly abundant proteins in blood plasma by ammonium sulfate precipitation for 2D-PAGE analysis. J Chromatogr B Analyt Technol Biomed Life Sci. 2011;879:3645–3648. doi: 10.1016/j.jchromb.2011.09.024. [DOI] [PubMed] [Google Scholar]
- 14.Linke T, Doraiswamy S, Harrison EH. Rat plasma proteomics: effects of abundant protein depletion on proteomic analysis. J Chromatogr B Analyt Technol Biomed Life Sci. 2007;849:273–281. doi: 10.1016/j.jchromb.2006.11.051. [DOI] [PubMed] [Google Scholar]
- 15.Faca VM, Song KS, Wang H, Zhang Q, Krasnoselsky AL, Newcomb LF, Plentz RR, Gurumurthy S, Redston MS, Pitteri SJ, Pereira-Faca SR, Ireton RC, Katayama H, Glukhova V, Phanstiel D, Brenner DE, Anderson MA, Misek D, Scholler N, Urban ND, Barnett MJ, Edelstein C, Goodman GE, Thornquist MD, McIntosh MW, DePinho RA, Bardeesy N, Hanash SM. A mouse to human search for plasma proteome changes associated with pancreatic tumor development. PLoS Med. 2008;5:e123. doi: 10.1371/journal.pmed.0050123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen YY, Lin SY, Yeh YY, Hsiao HH, Wu CY, Chen ST, Wang AH. A modified protein precipitation procedure for efficient removal of albumin from serum. Electrophoresis. 2005;26:2117–2127. doi: 10.1002/elps.200410381. [DOI] [PubMed] [Google Scholar]
- 17.Amacher DE, Adler R, Herath A, Townsend RR. Use of proteomic methods to identify serum biomarkers associated with rat liver toxicity or hypertrophy. Clin Chem. 2005;51:1796–1803. doi: 10.1373/clinchem.2005.049908. [DOI] [PubMed] [Google Scholar]
- 18.Kruger AJ, Yang C, Tam SW, Hinerfeld D, Evans JE, Green KM, Leszyk J, Yang K, Guberski DL, Mordes JP, Greiner DL, Rossini AA, Bortell R. Haptoglobin as an early serum biomarker of virus-induced autoimmune type 1 diabetes in biobreeding diabetes resistant and LEW1.WR1 rats. Exp Biol Med (Maywood) 2010;235:1328–1337. doi: 10.1258/ebm.2010.010150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Karthik D, Ilavenil S, Kaleeswaran B, Sunil S, Ravikumar S. Proteomic analysis of plasma proteins in diabetic rats by 2D electrophoresis and MALDI-TOF-MS. Appl Biochem Biotechnol. 2012;166:1507–1519. doi: 10.1007/s12010-012-9544-8. [DOI] [PubMed] [Google Scholar]
- 20.Ivancic MM, Huttlin EL, Chen X, Pleiman JK, Irving AA, Hegeman AD, Dove WF, Sussman MR. Candidate serum biomarkers for early intestinal cancer using 15N metabolic labeling and quantitative proteomics in the ApcMin/+ mouse. J Proteome Res. 2013;12:4152–4166. doi: 10.1021/pr400467c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Steel LF, Trotter MG, Nakajima PB, Mattu TS, Gonye G, Block T. Efficient and specific removal of albumin from human serum samples. Molecular & Cellular Proteomics. 2003;2:262–270. doi: 10.1074/mcp.M300026-MCP200. [DOI] [PubMed] [Google Scholar]
- 22.Lilley KS, Friedman DB. All about DIGE: quantification technology for differential-display 2D-gel proteomics. Expert Rev Proteomics. 2004;1:401–409. doi: 10.1586/14789450.1.4.401. [DOI] [PubMed] [Google Scholar]
- 23.Koontz L. TCA Precipitation. Methods Enzymol. 2014;541:3–10. doi: 10.1016/B978-0-12-420119-4.00001-X. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
SDS-PAGE showing no substantial differences between TCA precipitation and molecular weight cutoff membranes. Following three different high abundant protein removal methods, differences in protein samples were compared using TCA precipitation (T) and centrifugation using molecular weight cutoff membranes (M). The three different methods are SwellGel® Blue Albumin (S), ProteaPrep (P), and affinity chromatography (MARS). No substantial differences can be seen between these two methods for different rodent blood samples, mouse serum (Left) and rat plasma (Right).