

Abstract
We combined functional information such as protein–protein interactions or metabolic networks with genome information in Saccaromyces cerevisiae to predict cis-regulatory motifs in the upstream region of genes. We developed a new scoring metric combining these two information sources and used this metric in motif discovery. To estimate the statistical significance of this metric, we used brute-force randomization, which shows a consistent well-behaved trend. In contrast, real data showed complex nonrandom behavior. With conservative parameters we were able to find 42 degenerate motifs (that touch 40% of yeast genes) based on 647 original patterns, five of which are well known. Some of these motifs also show limited spatial position in the promoter, indicative of a true motif. We also tested the metric on other known motifs and show that this metric is a good discriminator of real motifs. As well as a pragmatic motif discovery method, with many applications beyond this work, these results also show that interacting proteins are often coordinated at the level of transcription, even in the absence of obvious coregulation in gene expression data sets.
[Supplemental material is available online at http://www.ebi.ac.uk/∼ettwille/genome_research_paper_2003/result_overlap.html. Program available upon request.]
Regulation of transcription has been recognized as one of the major steps in a cascade of control points for gene expression and regulation. The simplest model of transcriptional regulation in both eukaryotic and prokaryotic cells is the activation or repression of the transcriptional apparatus by proteins that bind to a limited stretch of DNA sequence located (usually) upstream of a given transcript. This model leads to a straightforward computational statement that finding the regions for transcriptional control is about finding short “motifs” or sub-sequences of letters near genes to which transcription factors bind and subsequently influence transcription. Although potentially one could consider this problem starting de novo with just the genome sequence, most researchers have also drawn on other data sets. Such data sets have included experimental results of the DNA binding of a number of proteins (Lee et al. 2002), large-scale transcription experiments such as microarrays (Brazma et al. 1998; Holmes and Bruno 2000; Hughes et al. 2000; van Helden et al. 2000; Ohler and Niemann 2001; Blanchette and Tompa 2002), and genome comparisons (Wasserman et al. 2000).
In this work we bring a new class of information, functional information about genes, alongside genome information for discovering motifs in Saccaromyces cerevisiae. The rationale behind this is that proteins rarely act alone. Indeed, to be functionally active, proteins need to associate with others and form a functional group that can perform a given task. All of the essential proteins that form the functional group must be present at a same given time, and it therefore makes sense for the cell to have the proteins be regulated in a synergic fashion by some common transcription factor(s). This concept is often true in prokaryotes, where genes involved in the same metabolic or cellular function (and therefore more prone to interactions) are also part of the same multigenic transcript and consequently are regulated by the same set of transcription factors. This concept has also been found to be true in specific cases in eukaryotic organisms (Niehrs and Pollet 1999). For example, yeast genes coding for proteins involved in the metabolism of galactose are regulated by the same transcription factors, GAL4/GAL80 (Klar and Halvorson 1974).
We seek to identify motifs that are limited to a specific set of genes, and this set of genes should have a significantly nonrandom concordance with the input functional information (in our case, protein interaction networks and metabolic networks). Methods to assess the significance of concordances with such complex data are difficult to derive from first principles, so we have used brute-force randomization techniques to derive the statistical significance. We show that, despite the complex nature of the data, the randomized samples behave in a consistent manner, whereas with the real data, a far more complex picture is returned with many statistically interesting motifs. Some of these motifs are known, whereas the others represent potential novel motifs.
We also show that these significant motifs have other intriguing nonrandom properties, such as in some cases a consistent distance from the putative translation start. Conversely we show that some known motifs that we did not discover directly still show significant nonrandom behavior in our tests. The patterns we discovered are consistent when we use different functional information and are robust to different pattern discovery algorithms and parameters. These results show that, by using only information about protein interactions, it is possible to find known and novel DNA patterns that are used by S. cerevisiae in the regulation of gene transcription.
RESULTS
The basic workflow of our method is presented in Figure 1. The input data are the upstream regions of the yeast genes (see Methods) and functional information from either metabolic interactions or direct protein–protein interactions. Our starting points are sets of at least three genes linked to either the compounds or the complexes (depending on the network considered) in the bipartite graph. We then used a pattern discovery tool such as Teiresias to propose a set of “motifs” (see Methods). This generates many patterns, specifically 197,922 patterns from the entire KEGG network (http://www.genome.ad.jp/kegg/) and 197,111 and 320,405 patterns from the Cellzome (http://www.cellzome.com/) and MDS (http://www.mdsproteomics.com/yeast/) networks, respectively. Although these patterns are technically nonrandom, their number and distribution across genes is not a convincing signal for believable motifs. However, we noticed that certain patterns discovered in one area of the network had a surprising concordance with other areas of the input network. We developed a metric called “overlap score” (see Methods) which provides the degree of overlap between a proposed fully connected network of genes linked by a pattern and a set of genes linked by the functional information, discounting the original seed that built the pattern and normalizing for the connectivity of the nodes and size of the network. To test the significance of such concordance, we used a brute-force randomization technique generating 100,000 fully connected pattern networks of different sizes with the gene identifiers being random but all of the other aspects of the network remaining the same. We then calculated the “overlap score” of these random networks with the interaction network. This process is illustrated in Figure 2.
Figure 1.

Workflow diagram of the motif discovery method.
Figure 2.

Overlap score explanation. Only nodes that are connected by an edge in the functional network and the pattern network were used for the overlap score calculation, discounting the initial set used for the pattern discovery. The left panel shows a sample real network, with three edges forming an initial seed of nodes (A,B,C). For one of the patterns discovered using this seed, it also found genes D, E, F, and G, many of which share edges with the functional network. The overlap score in this case is 0.86. In contrast, the right panel shows a sample random network, which was chosen to have the same number of nodes (four) as the proposed pattern network. In this case however, only one edge is shared, and the overlap score is 0.44.
Figure 3A shows the overlap score of random pattern networks as a function of the size of the pattern network. The randomized networks show a consistent, well behaved trend of linear increase score with increasing number of nodes. For each network size, we assessed normality of distribution for the overlap score and found that, for small network sizes, many networks have an overlap score of zero, which makes the distribution skewed. This skewness becomes less significant as the network size increases. For network sizes of more than 150, we assumed normal distributions, because a high percentage of Shapiro-Wilk hypothesis tests (Shapiro and Wilk 1965) have a P-value greater than 0.02 (see Methods). We performed the same tests using the χ2 method, resulting in similar conclusions. A regression line corresponding to four standard deviations from the mean of overlap scores for each network size was constructed, and real networks having a score above this line were considered significant. Figure 3B shows the overlap score for real pattern networks. Most of the patterns found produce a network of genes that have little or no concordance with the functional network; however, 647 motifs have a network of genes that shows a much higher overlap score. In other words, these specific motifs are found upstream of genes that have a significantly higher probability of being interaction partners.
Figure 3.


Overlap score with (A) MDS random network and (B) MDS real network. The lines correspond to the average four standard deviations to the mean for the random pattern overlap with regression equation Y = 0.004336X + 0.2613.
These patterns required some further processing to be useful. Firstly, the pattern discovery systems output discrete patterns, so that, for example, GANTATG and GNATATG would be treated as two distinct patterns despite their obvious overlap. We cluster the patterns using their genomic location (see Methods). This procedure is then followed by a single linkage clustering, reducing the set of interesting patterns down to a total number of 42 motifs for the three functional networks considered in this study. Essentially equivalent results were achieved when varying the Teiresias parameters or when using another pattern discovery method, SPEXS (Vilo 2002). In the present study, we deliberately chose conservative parameters to be sure of finding interesting motifs.
The final set of 42 motifs connects to a total of 2457 genes, about 40% of the yeast genome. These motifs are tabulated in Table 1 and its Web site complement. Some of these are well known motifs that bind to known transcription factors in yeast, and the regulated genes predicted by our analysis match the experimental evidence previously published. For example, the motif GGTGGCAAA identified in cluster 6 is known to bind to Rpn4p, a transcription factor that controls expression of the proteasome (Mannhaupt et al. 1999). Interestingly, we also found the reverse-complementary motif TTTGCCACC of the Rpn4p binding site identified in cluster 23. This motif is also found upstream of proteasomal genes, but not the same set of genes as cluster 6 (see Web site complement). Another well known example is the GCN4 binding site TGACTC represented in cluster 33 (Arndt and Fink 1986). The motif AAAATTTT is an interesting motif that scores very highly in all three of the functional networks we used. This low-complexity motif has been extensively studied for its strong DNA bending properties (Koo et al. 1986). In a promoter context, the adenine-thymine track increases the accessibility of transcription factors bound to nearby sequences (Iyer and Struhl 1995) and therefore has an effect on transcription rates. In the present study, we found that this apparently wildly occurring pattern is found very often upstream of genes that are involved in transcription and translation processes. The ubiquity of this motif for such basic processes suggests that it could be a “global state” switch for yeast. For example, one hypothesis is that it could be involved in a cell response to constantly changing conditions. Re-adaptation often involves production of proteins and enzymes for the cell to be able to use the new resources of that environment. Having a common and simple regulatory element such as the adenine-thymine track that controls the rate of production of most of the genes that are involved in the transcription/translation machinery could enable the cell to rapidly boost the production of new proteins and therefore quickly adapt to new situations.
Table 1.
Summary of All of the Motifs
 |
 |
 |
 |
 |
“Occurrence” is the number of genes in the overlap network derived from the relevant functional network(s) (see network column). The column network shows where the motif was initially found having a significant overlap score. K, KEGG; C, Cellzome; M, MDS. The standard deviation columns, SD KEGG, SD cell, and SD MDS are the cluster standard deviations from the mean of random overlap scores applied to the functional network KEGG, Cellzome, and MDS, respectively. The Function column is a functional annotation based on the overlap genes annotation.
Certain motifs with significant overlap scores also display other nonrandom behavior such as a tight positional distribution relative to the start codon. This is reflected by the standard deviation (SD) score (see Methods), which calculates the significance of the positional distribution for a certain motif in the overlap genes versus random genes that also have the motif. Figure 4 shows the location of pattern 4 (SD score = 0.00 against Cellzome network) for the overlap genes. A total of 15 motifs showed a significantly narrow spatial distribution.
Figure 4.

Motif locations relative to start codon of overlap genes from cluster 4.
The process of finding new potential motifs depends on Teiresias parameters. Nevertheless, known motifs that do not satisfy this initial step of generating patterns can still display significant overlapping scores. From a list of putative transcription factor binding sites, about 20% appear to have a significant overlap score for at least one of the networks. Table 2 shows the known sites that have significant overlap score(s).
Table 2.
Known Transcription Factor Binding Sites That Have a Significant Overlap Score
| Transcription factors | References | Observed genes | Consensus | KEGG | Cellzome | MDS |
| MET 31/32 | (Blaiseau et al. 1997) | Methionine biosynthesis | AAACTGTG | 5.25 | 1.46 | 0.96 |
| HAP2 | (Mantovani 1998) | Oxidative phosphorylation | ACCAAT.A | 6.51 | 0.11 | 1.36 |
| GRF2/REB1 | (Chasman et al. 1990) | Unknown | [TC]..[TC][TC]ACCCG | 1.88 | 4.62 | 3.45 |
| [TC]..[TC][TC]ACCCT | 1.27 | 2.82 | 4.38 | |||
| PHO4 | (Hayashi and Oshima 1991) | Met, Thr, Asn synthesis | CACGTG | 6.36 | 1.95 | 1.94 |
| MBP1/MBF1 | (Lowndes et al. 1991) | DNA replication | ACGCGT.A | 4.41 | 4.74 | 3.81 |
| RPN4p | (Mannhaupt et al. 1999) | Proteasome | GGTGGCAAA | 0.33 | 11.62 | 13.46 |
| GCN4 | (Hope and Struhl 1985) | Amino acids synthesis | TGACTCA | 8.66 | 3.39 | 2.87 |
| CBF1 | (Dowell et al. 1992) | Unknown | TCAC.TGA | 5.21 | 0.5 | 1.23 |
| TFIID-TBP | (Struhl 1995) | Unknown | TATATAAA | 4.91 | 1.39 | 3.61 |
The values are the standard deviations from the mean of random overlap score. Values in bold are significant. The “Observed gene” column is a functional annotation based on the overlap gene annotations.
The motif TATATAAA (TATA box) shows a surprisingly high overlap score with the metabolic and MDS networks, even though the TATA box is present in most of the yeast genes. The consensus TATATAAA is only present in 463 genes in the yeast genome. The 71 overlap genes do not belong to any well defined functional group, but most of them are genes that code for enzymes used in basal metabolism.
DISCUSSION
Our approach identifies 42 potential sites that we strongly suspect are involved in gene expression, most likely via transcriptional regulation. In our set we see some well known motifs and other novel cases. Some of these novel motifs are shared by genes that have a convincing biological reason to be coregulated. These 42 motifs constitute a conservatively selected subset of those that could be identified using this approach. Firstly, we generated in the course of this analysis over 700,000 different patterns, most of which were rejected on the basis of our functional concordance criteria. As more functional data sets are produced, such as other protein–protein interactions or the results of gene deletion experiments, we feel confident that we can expand our list. Second, this analysis is limited by the power of the motif discovery tool, which in this case is Teiresias with parameters of eight conserved nucleotides with two wild cards. Because known motifs which did not meet these criteria still provided significant overlap scores, there is clearly considerable potential to improve motif discovery using this highly specific discriminator. We are currently experimenting with brute-force scan methods across the entire genome, with positive results. Thirdly, more sophisticated scoring functions could be combined, for example, using explicit background models of the genome (Stormo and Tan 2002) or combining archetypal information content profiles (M. Eisen, pers. comm.).
Of particular interest is the idea of combining our analysis with evolutionary approaches. Sequence differences between closely related organisms can, for most genes, be considered due mainly to the action of random mutations and negative selection. We are currently working on using comparative information both as an additional signal in motif discovery and as information in the final scoring function.
Our method is certainly not the first motif discovery method applied to yeast. For example, methods which use gene expression clusters for motif discovery have had particular success (Eisen et al. 1998; Holmes and Bruno 2000; Vilo et al. 2000). Some of the motifs found in these works are similar to those we found. However, because motifs are described using information content based on different types of input data, a one-to-one comparison is not possible. We believe our method is complementary to gene expression clustering, because even when a gene has a number of different regulation pressures placed on it, and thus it is difficult to assign to any common cluster on the basis of co-expression, the overlap score of the motifs involved in regulation and the functional network can still be significant.
The final set of motifs for yeast is likely to be defined by the analysis of large-scale deletion sets, in particular deletions of transcription factors followed by gene expression experiments. In addition, data from chromatin immunoprecipitation experiments (Ren et al. 2000; Lee et al. 2002) applied to transcription factors binding to DNA regulatory regions will be very useful. Even though we might imagine that such techniques will be trivial to interpret, we believe that a scoring scheme similar to that presented here will provide the best interpretation of these data. More generally, in other organisms, in particular metazoans, it is likely that we need to combine many threads of evidence to provide a decoding of transcriptional regulation. We hope that the basic method presented here will provide a framework for this decoding and eventually result in a more complete understanding of transcriptional regulation for many organisms.
METHODS
The basic workflow of our method is presented in Figure 1.
Network Generation
First we represented the interactions in bipartite networks having two types of entities, compounds (for the metabolic network) or complexes (for the interaction networks) and proteins that are linked to either the compounds or the complexes. The resulting bipartite representations from both the metabolic and protein–protein interactions provided our starting point for finding the initial patterns. A monopartite representation (Fig. 5) with only one type of node (protein) can be derived and was used to assess putative motifs found in the upstream region of the corresponding gene (see overlap score).
Figure 5.

Monopartite (or unipartite) representation derived from a bipartite representation of a graph. Labels “compounds” and “enzymes” are for the KEGG network; labels “complexes” and “proteins” are for the protein interactions networks.
Metabolic Network
The KEGG database (Kanehisa 1997) was used for this study. Only compounds linked to enzymes of the yeast S. cerevisiae were used. If the number of yeast enzymes that act on a given compound exceeded 60, this compound was removed from the data set (a total of 17 compounds were removed including H2O, O2…). All reactions were considered reversible, resulting in an undirected graph. Interactions are only presented once to avoid signal amplification. A BLAST all-versus-all (ftp://ftp.mcbi.mih.gov/blast) was performed for the upstream sequences (600 bp) of all yeast genes, and interactions that involved genes with homologous upstream sequences were removed from the network (BLASTN on plus/plus strands with all default parameters except for expectation value “e” set at 0.000001). A total of 24 interactions were removed for the monopartite metabolic network. This was done in order to avoid false-positive patterns.
Protein Interaction Network
Protein–protein interaction data were derived from two data sets of experimental results, identified as Cellzome (Gavin et al. 2002) and MDS (Ho et al. 2002) data sets. Both are based on a large-scale approach to systematically identify protein complexes in S. cerevisiae. As for the metabolic network, the same BLAST all-versus-all was performed for the upstream sequences of all yeast genes, and protein interactions that involved corresponding genes with homologous upstream sequences were removed from the networks. A total of two and 30 interactions were removed from the monopartite networks derived from Cellzome and MDS, respectively.
Pattern Search
The DNA promoter regions considered are of a fixed length of 600 bp upstream of S. cerevisiae genes (we also tried 400 and 300 bp, with essentially equivalent results). The genome data used are the S. cerevisiae strain S288C complete genome (The yeast genome directory 1997). The pattern searching program used for this study is Teiresias (Rigoutsos and Floratos 1998) with the following parameters: L = 8, W = 10, C = 7, K = 3 with −v for complexes or compounds with less than 10 genes. L = 8, W = 10, C = 7, K = 4 with −v otherwise. The patterns obtained are at least eight defined nucleotides long with a maximum of two wild cards allowed. For each pattern obtained, a pattern network was created by linking all of the genes that possess the pattern in their upstream regions, and the resulting fully connected pattern graph was used for calculating the overlap score.
Overlap Score
The overlap score represents the number of common edges between the initial monopartite representation of the functional network and the proposed pattern network, normalized by the number of edges connected to the considered nodes. Each common edge is counted once but divided by the total number of functional edges from the two nodes; in addition the total number is raised to the power 0.5, as this corrects for the tendency of larger networks producing large scores (see equation 1).
We do not include the initial seed edges, because these generated the initial set of patterns evaluated in the scoring function.
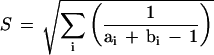 |
1 |
Summation is over all common edges (i) present in both networks connecting node Ai to node Bi. The denominator ai + bi − 1 is the total number of edges in the initial functional network from both nodes Ai and Bi, discounting the edge being counted.
In order to model the overlap score, random networks of the same size as the proposed pattern network were created by choosing genes at random, including the seed nodes (200 random pattern networks for each size ranging from 2 to 500, and thus a total of 100,000 networks). The significance of the real overlap scores was assessed using this model. We also experimented with other randomization procedures (randomization of the functional network) with essentially identical results. There is an observed linear relationship between the number of nodes in the pattern network and the score.
Normality assessment for each random network size was done using the Shapiro-Wilk test (Shapiro and Wilk 1965). This test calculates a W-statistic that tests whether a random sample of continuous values, x1, x2, …, xn comes from (specifically) a normal distribution. For random networks with a size greater than 150 nodes, the percentage values of the Shapiro-Wilk hypothesis test with a P-value greater than 0.02 are 84%, 92.5%, and 95% for the Cellzome, KEGG, and MDS networks, respectively.
Standard Deviation Score to Assess Upstream Positional Distribution of Patterns
Given a set of upstream regions containing a pattern A, the standard deviation of the different locations of this pattern with respect to the start codon of the genes is calculated as:
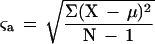 |
2 |
with ςa being the standard deviation of the pattern a, N the number of sequences in the set, μ the average location in respect to the start codon, and X the location. The standard deviation score is based on comparing the standard deviation for the set of X genes that comprise an overlap network with the standard deviation for a set of X random genes that have the same pattern. This comparison is done 100 times per pattern, and a P-value called standard deviation score can be calculated from these comparisons. This score reflects a better conservation of the upstream location of the pattern within the overlap network. We assumed here that a real pattern should conserve its position relative to the transcription starting site and that the UTR regions in yeast are about the same length for all of the genes within a set.
Pattern Clustering and Sequence Logo Generation
Clustering was based on the genomic location of the patterns. For each pattern we derived all of the exact locations of its occurrence in the upstream regions of all of the genes in the yeast genome. Two patterns were linked together if they shared at least 40% of genomic locations (exact location +/-−5 bp) for at least one of the two pattern location profiles. A final cluster contains all the patterns that are linked together (single linkage clustering). For each cluster of more than one motif, a sequence logo was then derived by retrieving all sequences in the upstream region of overlap genes that match at least one of the motifs in the cluster. The sequences obtained were then aligned (Eddy 1998), and a profile logo was built based on the information content of each position in the alignment using SEQLOGO (Vilo et al. 2003; http://ep.ebi.ac.uk/EP/).
WEB SITE REFERENCES
http://www.genome.ad.jp/kegg/; KEGG Kyoto Encyclopedia of Genes and Genomes.
http://www.mdsproteomics.com/yeast/; MDS proteomics.
http://www.cellzome.com/; Cellzome.
http://ep.ebi.ac.uk/EP/; Expression Profiler.
http://www.ebi.ac.uk/∼ettwille/genome_research_paper_2003/result_overlap.html; Web complement.
Acknowledgments
L.E., J.R., and E.B. are supported by EMBL funding. J.R also acknowledges funding from the TEMBLOR project of the European Union. Some of this work used the ENSEMBL computer facility, provided by the Wellcome Trust. We thank James Cuff for systems support, Guy Slater and Martin Hammond for reading the manuscript, and Jaak Vilo and Alvis Brazma for helpful discussion.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
E-MAIL birney@ebi.ac.uk; FAX 44-1223-494919.
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.866403.
REFERENCES
- 1.Arndt K. and Fink, G.R. 1986. GCN4 protein, a positive transcription factor in yeast, binds general control promoters at all 5′ TGACTC 3′ sequences. Proc. Natl. Acad. Sci. 83: 8516-8520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Blaiseau P.L., Isnard, A.D., Surdin-Kerjan, Y., and Thomas, D. 1997. Met31p and Met32p, two related zinc finger proteins, are involved in transcriptional regulation of yeast sulfur amino acid metabolism. Mol. Cell. Biol. 17: 3640-3648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Blanchette M. and Tompa, M. 2002. Discovery of regulatory elements by a computational method for phylogenetic footprinting. Genome Res. 12: 739-748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brazma A., Jonassen, I., Vilo, J., and Ukkonen, E. 1998. Predicting gene regulatory elements in silico on a genomic scale. Genome Res. 8: 1202-1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chasman D.I., Lue, N.F., Buchman, A.R., LaPointe, J.W., Lorch, Y., and Kornberg, R.D. 1990. A yeast protein that influences the chromatin structure of UASG and functions as a powerful auxiliary gene activator. Genes & Dev. 4: 503-514. [DOI] [PubMed] [Google Scholar]
- 6.Dowell S.J., Tsang, J.S., and Mellor, J. 1992. The centromere and promoter factor 1 of yeast contains a dimerisation domain located carboxy-terminal to the bHLH domain. Nucleic Acids Res. 20: 4229-4236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Eddy S.R. 1998. Profile hidden Markov models. Bioinformatics 14: 755-763. [DOI] [PubMed] [Google Scholar]
- 8.Eisen M.B., Spellman, P.T., Brown, P.O., and Botstein, D. 1998. Cluster analysis and display of genome-wide expression patterns. Proc. Natl. Acad. Sci. 95: 14863-14868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gavin A.C., Bosche, M., Krause, R., Grandi, P., Marzioch, M., Bauer, A., Schultz, J., Rick, J.M., Michon, A.M., Cruciat, C.M., et al. 2002. Functional organization of the yeast proteome by systematic analysis of protein complexes. Nature 415: 141-147. [DOI] [PubMed] [Google Scholar]
- 10.Hayashi N. and Oshima, Y. 1991. Specific cis-acting sequence for PHO8 expression interacts with PHO4 protein, a positive regulatory factor, in Saccharomyces cerevisiae. Mol. Cell. Biol. 11: 785-794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ho Y., Gruhler, A., Heilbut, A., Bader, G.D., Moore, L., Adams, S.L., Millar, A., Taylor, P., Bennett, K., Boutilier, K., et al. 2002. Systematic identification of protein complexes in Saccharomyces cerevisiae by mass spectrometry. Nature 415: 180-183. [DOI] [PubMed] [Google Scholar]
- 12.Holmes I. and Bruno, W.J. 2000. Finding regulatory elements using joint likelihoods for sequence and expression profile data. Proc. Int. Conf. Intell. Syst. Mol. Biol. 8: 202-210. [PubMed] [Google Scholar]
- 13.Hope I.A. and Struhl, K. 1985. GCN4 protein, synthesized in vitro, binds HIS3 regulatory sequences: Implications for general control of amino acid biosynthetic genes in yeast. Cell 43: 177-188. [DOI] [PubMed] [Google Scholar]
- 14.Hughes J.D., Estep, P.W., Tavazoie, S., and Church, G.M. 2000. Computational identification of cis-regulatory elements associated with groups of functionally related genes in Saccharomyces cerevisiae. J. Mol. Biol. 296: 1205-1214. [DOI] [PubMed] [Google Scholar]
- 15.Iyer V. and Struhl, K. 1995. Poly(dA:dT), a ubiquitous promoter element that stimulates transcription via its intrinsic DNA structure. EMBO J. 14: 2570-2579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kanehisa M. 1997. A database for postgenome analysis. Trends Genet. 13: 375-376. [DOI] [PubMed] [Google Scholar]
- 17.Klar A.J. and Halvorson, H.O. 1974. Studies on the positive regulatory gene, GAL4, in regulation of galactose catabolic enzymes in Saccharomyces cerevisiae. Mol. Gen. Genet. 135: 203-212. [DOI] [PubMed] [Google Scholar]
- 18.Koo H.S., Wu, H.M., and Crothers, D.M. 1986. DNA bending at adenine-thymine tracts. Nature 320: 501-506. [DOI] [PubMed] [Google Scholar]
- 19.Lee T.I., Rinaldi, N.J., Robert, F., Odom, D.T., Bar-Joseph, Z., Gerber, G.K., Hannett, N.M., Harbison, C.T., Thompson, C.M., Simon, I., et al. 2002. Transcriptional regulatory networks in Saccharomyces cerevisiae. Science 298: 799-804. [DOI] [PubMed] [Google Scholar]
- 20.Lowndes N.F., Johnson, A.L., and Johnston, L.H. 1991. Coordination of expression of DNA synthesis genes in budding yeast by a cell-cycle regulated trans factor. Nature 350: 247-250. [DOI] [PubMed] [Google Scholar]
- 21.Mannhaupt G., Schnall, R., Karpov, V., Vetter, I., and Feldmann, H. 1999. Rpn4p acts as a transcription factor by binding to PACE, a nonamer box found upstream of 26S proteasomal and other genes in yeast. FEBS Lett. 450: 27-34. [DOI] [PubMed] [Google Scholar]
- 22.Mantovani R. 1998. A survey of 178 NF-Y binding CCAAT boxes. Nucleic Acids Res. 26: 1135-1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Niehrs C. and Pollet, N. 1999. Synexpression groups in eukaryotes. Nature 402: 483-487. [DOI] [PubMed] [Google Scholar]
- 24.Ohler U. and Niemann, H. 2001. Identification and analysis of eukaryotic promoters: Recent computational approaches. Trends Genet. 17: 56-60. [DOI] [PubMed] [Google Scholar]
- 25.Ren B., Robert, F., Wyrick, J.J., Aparicio, O., Jennings, E.G., Simon, I., Zeitlinger, J., Schreiber, J., Hannett, N., Kanin, E., et al. 2000. Genome-wide location and function of DNA binding proteins. Science 290: 2306-2309. [DOI] [PubMed] [Google Scholar]
- 26.Rigoutsos I. and Floratos, A. 1998. Combinatorial pattern discovery in biological sequences: The TEIRESIAS algorithm. Bioinformatics 14: 55-67. [DOI] [PubMed] [Google Scholar]
- 27.Shapiro S.S. and Wilk, M.B. 1965. An analysis of variance test for normality (complete sample). Biometrika 52: 591-611. [Google Scholar]
- 28.Stormo G.D. and Tan, K. 2002. Mining genome databases to identify and understand new gene regulatory systems. Curr. Opin. Microbiol. 5: 149-153. [DOI] [PubMed] [Google Scholar]
- 29.Struhl K. 1995. Yeast transcriptional regulatory mechanisms. Annu. Rev. Genet. 29: 651-674. [DOI] [PubMed] [Google Scholar]
- 30.van Helden J., Rios, A.F., and Collado-Vides, J. 2000. Discovering regulatory elements in noncoding sequences by analysis of spaced dyads. Nucleic Acids Res. 28: 1808-1818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vilo J., 2002. “Pattern discovery from biosequences.” Thesis. University of Helsinki, Finland. ISBN 952-10-0819-9.
- 32.Vilo J., Brazma, A., Jonassen, I., Robinson, A., and Ukkonen, E. 2000. Mining for putative regulatory elements in the yeast genome using gene expression data. Proc. Int. Conf. Intell. Syst. Mol. Biol. 8: 384-394. [PubMed] [Google Scholar]
- 33.Vilo J., Kapushesky, M., Kemmeren, P., Sarkans, U., Brazma, A., et al. 2003. Expression profiler. In The analysis of gene expression data: Methods and software (ed. G. Parmigiani) Springer Verlag, New York, NY.
- 34.Wasserman W.W., Palumbo, M., Thompson, W., Fickett, J.W., and Lawrence, C.E. 2000. Human–mouse genome comparisons to locate regulatory sites. Nat. Genet. 26: 225-228. [DOI] [PubMed] [Google Scholar]
- 35.The yeast genome directory 1997. Nature 387: 5. [PubMed] [Google Scholar]