

Abstract
Parkinson's disease (PD) is a debilitating motor and cognitive neurodegenerative disorder for which there is no cure. While aging is the major risk factor for developing PD, clear environmental risks have also been identified. Environmental exposure to the metal manganese (Mn) is a prominent risk factor for developing PD and occupational exposure to high levels of Mn can cause a syndrome known as manganism, which has symptoms that closely resemble PD. In this study, we developed a model of manganism in the environmentally tractable nematode, Caenorhabditis elegans. We find that, in addition to previously described modes of Mn toxicity, which primarily include mitochondrial dysfunction and oxidative stress, Mn exposure also significantly antagonizes protein homeostasis, another key pathological feature associated with PD and many age-related neurodegenerative diseases. Mn treatment activates the ER unfolded protein response, severely exacerbates toxicity in a disease model of protein misfolding, and alters aggregate solubility. Further, aged animals, which have previously been shown to exhibit decreased protein homeostasis, are particularly susceptible to Mn toxicity when compared to young animals, indicating the aging process sensitizes animals to metal toxicity. Mn exposure also significantly alters iron (Fe) and calcium (Ca) homeostasis, which are important for mitochondrial and ER health and which may further compound toxicity. These finding indicate that modeling manganism in C. elegans can provide a useful platform for identifying therapeutic interventions for ER stress, proteotoxicity, and age-dependent susceptibilities, key pathological features of PD and other related neurodegenerative diseases.
Introduction
Parkinson's disease (PD) is a multifactorial disease in which aging, genetic susceptibility, and environmental insults all contribute to its onset. It is characterized by dopaminergic degeneration, which leads to a host of characteristic motor dysfunctions, including tremor, rigidity, and bradykinesia. Environmental exposure to metals, especially manganese, is an established risk factor for the development of PD1. Manganese (Mn) can be found in water purifiers, drinking water, fungicides, gasoline additives, and airborne pollutants; thus, exposure to Mn poses a significant environmental health risk1. Further, Mn shares transporters with iron (Fe) and low levels of Fe (one of the most common nutritional deficiencies) can lead to elevated Mn uptake in the brain2-4. Occupational exposure to high levels of manganese, usually via mining or smelting, can lead to a PD-like syndrome called manganism5, 6. Manganism has motor symptoms that closely resemble PD, likely due to the preferential accumulation of Mn in the basal ganglia, the brain region most affected in PD6. However, manganism can be distinguished from PD as it preferentially affects the globus pallidus instead of the substantia nigra, leading to subtly distinct motor symptoms and poor response to L-DOPA, the dopamine precursor commonly used to treat PD6. Despite these differences, Mn toxicity can induce many of the same pathological cascades observed in PD, such as mitochondrial dysfunction, reactive oxygen species (ROS) production, and activation of apoptotic and necrotic signaling pathways7. Thus, identifying pathways that contribute to Mn toxicity may enhance our mechanistic understanding of PD.
In addition to mitochondrial dysfunction and oxidative stress, impaired protein homeostasis is another prominent feature of PD8, as well as many other age-dependent neurodegenerative diseases such as Alzheimer's disease (AD) and Huntington's disease (HD). PD patients form Lewy bodies, composed primarily of aggregated forms of the protein α-synuclein, AD patients form amyloid-β plaques and tau tangles, and HD patients form polyglutamine inclusions. Not surprisingly, many of these age-related neurodegenerative diseases show evidence of activation of the ER unfolded protein response (UPR), likely in an attempt to correct the improperly folded proteins that form these aggregates9, 10. Imbalances in metal homeostasis are another hallmark of age-dependent neurodegenerative diseases11. Elevated levels of Fe are found in affected brain regions of PD patients and pharmacological or genetic chelation of Fe has been demonstrated to halt progression of the disease in animal models of the disorder12. PD patients also exhibit aberrant sera levels of several metals including Mn, Fe, Cu, and Zn, further consistent with impaired metal homeostasis contributing to disease pathophysiology13. Several of these metals are capable of catalyzing the misfolding of aggregation/disease-prone proteins such as α-synuclein, amyloid-β, and prion proteins11, 14, 15. Thus, impaired metal homeostasis may help drive or exacerbate protein misfolding under disease contexts. In this study, we developed a model of manganism in the model organism C. elegans. We find that in addition to effects on mitochondrial function, Mn also results in activation of ER stress and impaired protein homeostasis and significantly disrupts Fe, Cu, and Ca homeostasis, key pathological features observed in many age-dependent neurodegenerative diseases.
Results
Manganese shortens lifespan and selectively disrupts iron, copper, and calcium homeostasis
To determine if Mn affects the lifespan of C. elegans, we exposed worms to increasing concentrations of manganese chloride (MnCl2). We found that lifetime exposure to 10 mM MnCl2 had no effect on lifespan and animals appear similar to controls. At 20 mM MnCl2, animals appear mostly healthy but display a significantly shortened lifespan. At 30 mM MnCl2, animals appear small and decrepit and die within days, reflecting extreme toxicity (Figure 1A, Suppl. Table 1). To determine how much Mn was taken up by worms and how it might affect other metals, we performed metallomic analysis by inductively-coupled plasma (ICP) spectroscopy on young adult whole animals that had been exposed to 10 mM, 20 mM, or 30 mM MnCl2 for 24 hours. As expected, we observed large increases of Mn in tissues in a dose-dependent manner (Fig. 1B). Furthermore, we observed that at 20 mM MnCl2, the abundance of Fe, Ca, and strontium (Sr), a metal that behaves physiologically similar to Ca, was lower in the worms. Interestingly, at 30 mM MnCl2, Fe and Cu become significantly more abundant. In contrast, we observed no significant changes in the levels of K, Mg, Na, P, S, or Zn (Fig. 1B). Based on these findings, we used 20 mM MnCl2 as an intermediate dose to study Mn toxicity in future studies.
Figure 1. Manganese shortens lifespan and perturbs metal homeostasis.
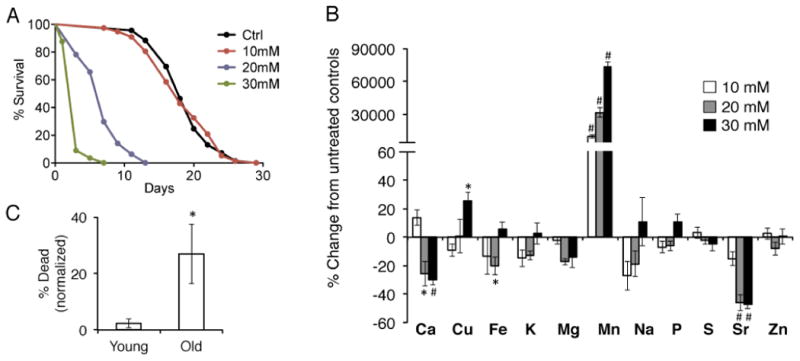
A, Lifespan of wild type N2 animals exposed to water (Ctrl), 10 mM, 20 mM, or 30mM MnCl2 beginning as young adults. 20 mM and 30 mM MnCl2 significantly shortens lifespan compared to controls. (p<0.0001, Log Rank Mantel-Cox) B, Metallomic ICP-AES analysis in day 1 wild type N2 young adult animals exposed to 10 mM, 20 mM, or 30 mM MnCl2 for 24 hours, compared to untreated water controls. (*=p<0.03, #=p<0.01, Student's t-test compared to untreated controls) C, Wild type N2 day 1 or day 10 adult worms were exposed to 20 mM MnCl2 and scored for lethality 48 hours later. Deaths were normalized the number of deaths in age-matched untreated controls. (*=p<0.05, Student's test)
Aged animals are susceptible to manganese toxicity
To determine how the aging process may interact with Mn toxicity, we exposed either young adult worms or worms that had been aged to day 10 of adulthood (approximately middle-aged) to 20 mM MnCl2 for 48-hours. Transient treatment of MnCl2 had virtually no effect on the viability of young adult animals, but significantly increased mortality in aged animals (adjusted to deaths observed in untreated age-matched controls)(Fig. 1C), indicating that the aging processes sensitizes animals to Mn toxicity.
Manganese activates the mitochondrial unfolded protein response and induces mitochondrial dysfunction
Previous studies have shown that acute exposure to manganese chloride (MnCl2) can cause increased formation of ROS and mitochondrial impairment when exposed during the larval stages of C. elegans16. In this study, we examined how Mn would affect adult hermaphrodite worms. We utilized a worm strain expressing a transcriptional GFP-reporter for the mitochondrial unfolded protein response (UPR) protein, phsp-6::GFP17, and exposed young adult worms to 10 mM or 20 mM MnCl2 for 24 hours. Consistent with our lifespan data (Fig 1A), we found that 10 mM MnCl2 was not toxic and did not elicit a mitochondrial UPR response (Fig. 2A-D). In contrast, exposure to 20 mM MnCl2 elicited a robust induction of the mitochondrial UPR (Fig. 2A-D). We also found that 20 mM MnCl2 severely depolarized the mitochondria in young adults as measured by tetramethylrhodamine methyl ester (TMRM) (Fig. 3), a cell-permeable fluorescent dye that is taken up by active mitochondria. Thus, our results with adult worms are consistent with previous reports that MnCl2 causes mitochondrial stress and dysfunction in C. elegans in larval forms16.
Figure 2. Acute Mn exposure activates the mitochondrial unfolded protein response.
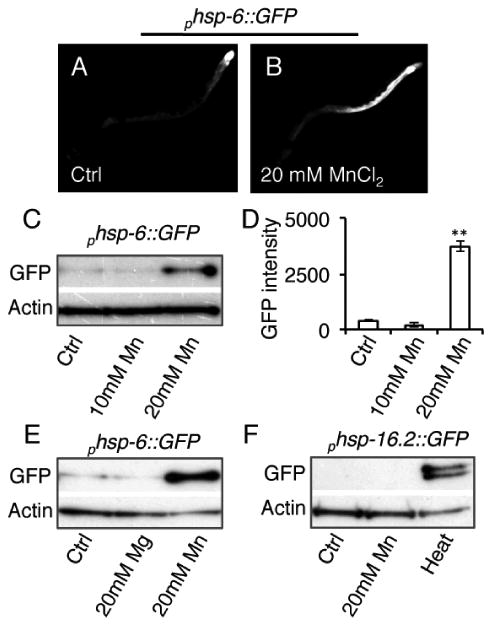
A, B, Fluorescent photomicrographs of day 1 adult hermaphrodite worms expressing phsp-6::GFP, a transcriptional reporter for the mitochondrial UPR, after 24 hour treatment with water (Ctrl) or 20 mM MnCl2. C, Immunoblots of protein extracts from day 1 adult animals expressing phsp-6::GFP after 24 hour treatment with water (Ctrl), 10 mM, or 20 mM MnCl2. D, Quantification of GFP intensity from phsp-6::GFP immunoblots in arbitrary units. (**=p<0.01, Student's t-test) E, Immunoblots of protein extracts from day 1 adult animals expressing phsp-6::GFP after 24 hour treatment with water (Ctrl), 20 mM MgCl2, or 20 mM MnCl2. F, Immunoblots of protein extracts from day 1 adult animals expressing phsp-16.2::GFP, a transcriptional reporter for heat stress, after 24 hour treatment with water (Ctrl), 20 mM MnCl2, or a 1 hour heat stress at 33°C.
Figure 3. Acute Mn exposure depolarizes the mitochondria.
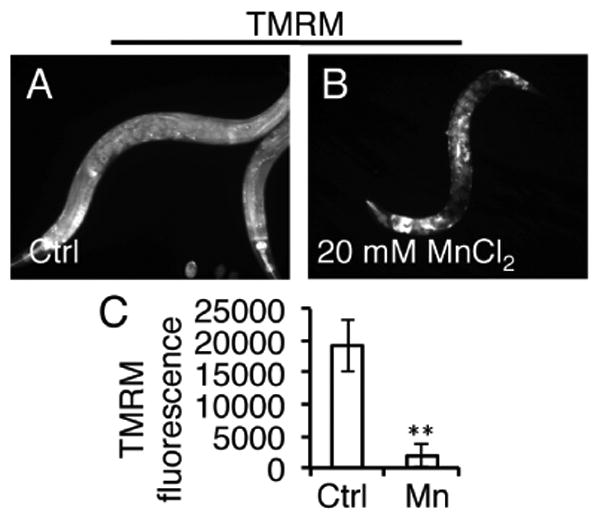
A, B, Fluorescent photomicrographs of day 1 adult worms collected after 24 hour treatment with 20 mM MnCl2 in the presence of TMRM. C, Quantification of TMRM levels in day 1 adult worms after 24 hour treatment with water (Ctrl) or 20 mM MnCl2. (**=p<0.001, Student's t-test.)
To test for specificity of MnCl2, we exposed young adult worms to 20 mM of another divalent metal, magnesium chloride (MgCl2), and found that it did not induce the mitochondrial UPR (Fig. 2E). In addition, MnCl2 did not activate the transcriptional GFP-reporter for the cytoplasmic heat-shock protein, phsp-16.2::GFP18, while heat stress did so as expected (Fig. 2F), indicating that MnCl2 does not induce a cytoplasmic or global stress response. Thus, we observe some specificity of MnCl2 to mitochondrial dysfunction in C. elegans.
Manganese activates the ER unfolded protein response
To determine if Mn was capable of activating UPR pathways other than the mitochondria, we next analyzed the ER UPR. We utilized the ER UPR transcriptional GFP-reporter, phsp-4::GFP, a homologue of the ER UPR sensing chaperone, BiP19. Acute exposure to 20 mM MnCl2 in young adults was also found to activate the ER UPR while20 mM MgCl2 did not (Fig. 4). This suggests that in addition to effects on mitochondrial stress, Mn also elicits effects on the ER.
Figure 4. Acute Mn exposure activates the ER unfolded protein response.
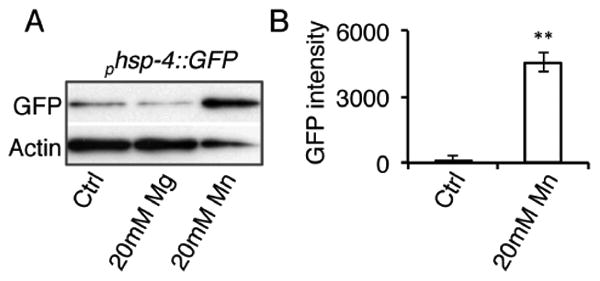
A, Immunoblots of protein extracts from day 1 adult animals expressing phsp-4::GFP, a transcriptional reporter for the ER UPR, after 24 hour treatment with water (Ctrl), 20 mM MgCl2, or 20 mM MnCl2. B, Quantification of GFP intensity from phsp-4::GFP immunoblots in arbitrary units. (**=p<0.001, Student's t-test)
Manganese antagonizes protein homeostasis in a model of protein misfolding
To determine if elevated mitochondrial and/or ER stress correlate with dopaminergic neurodegeneration akin to that seen in human disease, we next examined the effects of acute Mn exposure in adult wild type animals whose dopaminergic neurons were labeled with GFP (pdat-1::GFP) or in adult animals that also expressed α-synucleinin their dopaminergic neurons (pdat-1::GFP; pdat-1::α-synuclein) and that exhibit age-dependent neuronal degeneration20. We aged animals to day 5 of adulthood and then exposed aimals to 20 mM of MnCl2 for 48 hours. While MnCl2 was toxic as measured by organismal viability (data not shown), acute MnCl2 exposure did not induce dopaminergic degeneration in control animals nor did we see additional degeneration in α-synuclein-expressing animals (Fig. 5). Conflicting reports exist as to whether MnCl2 exposure results in frank dopaminergic cell loss in C. elegans, alone or in the context of α-synuclein expression16, 21, 22. Settivari et. al. and Benedetto et. al. previously reported that MnCl2 causes dopaminergic degeneration in C. elegans when exposed as larva, whereas a more recent report by Borhorst et. al. reported that MnCl2 did not induce degeneration under similar conditions16, 21, 22. We tested whether 50 mM MnCl2 could induce dopaminergic degeneration in larval worms as originally reported16 and observed miniscule changes in dopaminergic integrity with MnCl2 treatment (100% preservation of neurons in control versus 98% preservation in MnCl2 treated, data not shown), supporting the latter findings that MnCl2 does not effectively induce dopaminergic degeneration.
Figure 5. Mn does not cause dopaminergic degeneration in aged animals.
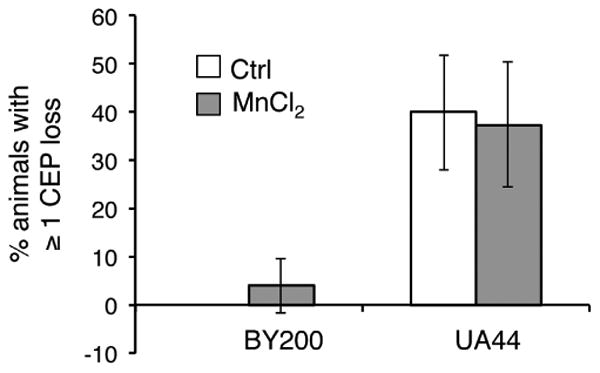
Quantification of animals displaying dendritic loss in one or more of the their dopaminergic neurons located in the head region (CEP). Worms expressing pdat-1::GFP (BY200) or pdat-1::GFP; pdat-1::a-synuclein (UA44) were aged until day 5 of adulthood and then exposed to water (Ctrl) or 20 mM MnCl2 for 48 hours. No significant differences were observed between Ctrl and Mn-treated animals, Student's t-test.
Given that ER stress is an indication of impaired protein homeostasis, we next examined the effects of Mn in a robust model of protein misfolding. We examined a polyglutamine model in which animals express a stretch of 40 polyglutamines in their body wall muscle (AM141 [rmIs133(P(unc-54) Q40::YFP)])23. We found that exposure to 20 mM MnCl2 as young adults caused severe toxicity that resulted in ∼ 75% lethality rate after just 24 hours (Fig. 6A). Animals that remained alive were unhealthy in appearance and smaller in size (Figs. 6B-E). The effect was specific to Mn and not the chloride ion as the metabolite choline chloride (C5H14ClNO abbreviated to ChCl) had no effect on polyglutamine-expressing animals at the same concentration (Fig. 6). Mn-treated animals also displayed fewer inclusions that were smaller in size (Fig. 7A,B). Further, Mn treatment altered the SDS-solubility of polyglutamine aggregates, resulting in significantly less SDS-soluble aggregates with a concomitant trend of increased SDS-insolubility, suggesting that Mn can alter protein conformation in vivo (Fig. 7C, D). Thus, Mn appears to significantly antagonize proteostasis in a non-neuronal model of protein misfolding.
Figure 6. Acute Mn exposure is toxic in a polyglutamine model of protein misfolding.
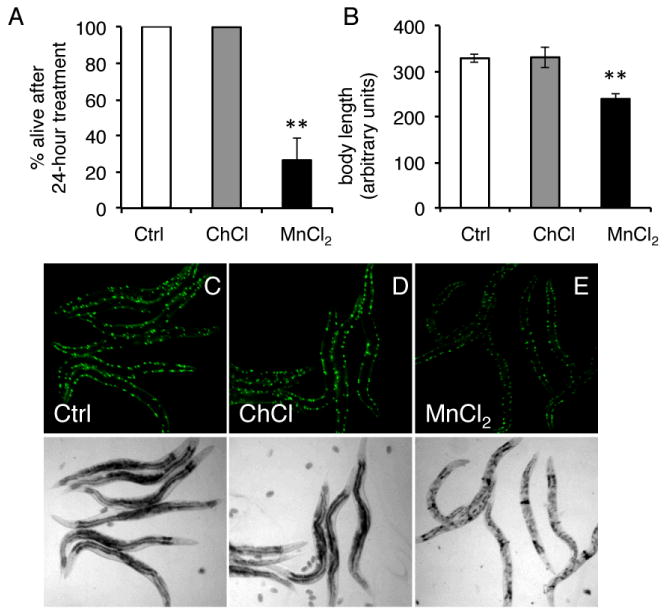
A, Quantification of number of day 1 animals that remained alive after a 24 hour treatment with water (Ctrl), 20 mM ChCl, or 20 mM MnCl2. All animals remained alive in Ctrl and ChCl-treated animals, while only ∼26% survive MnCl2 treatment (**=p<0.005, Student's Test). B, Quantification of body length of day 1 animals after 24 hour treatment with water (Ctrl), 20 mM ChCl, or 20 mM MnCl2. (**p=<0.0001, Student's t-test) C-E, Top panel, fluorescent photomicrographs of day 1 adults worms expressing 40 polyglutamines tagged to YFP in their body wall muscle (AM141 [rmIs133(P(unc-54) Q40::YFP)]) after 24 hour exposure to water (Ctrl), 20 mM ChCl, or 20 mM MnCl2. Bottom panel, bright field photomicrographs from top panels.
Figure 7. Acute Mn exposure alters the SDS-solubility of aggregates in a polyglutamine model.
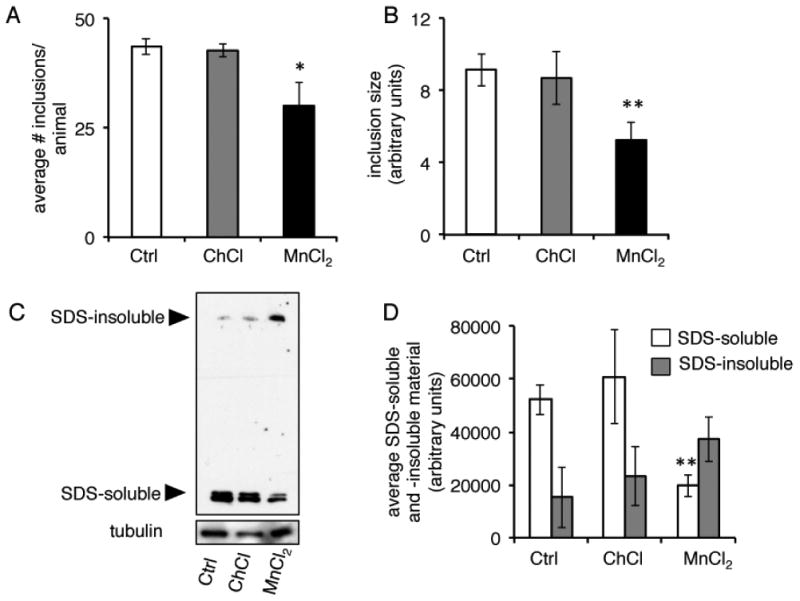
A, Quantification of number of YFP inclusions in day 1 adult worms expressing a 40 polyglutamine repeat tagged to YFP in their body wall muscle (AM141 [rmIs133(P(unc-54) Q40::YFP)]) after 24 hour treatment with water (Ctrl), 20 mM ChCl, or 20 mM MnCl2 (*=p<0.03, Student's t-test). B, Quantification of relative size of YFP inclusions in day 1 animals after 24 hour treatment with water (Ctrl), 20 mM ChCl, or 20 mM MnCl2 (**p=<0.01, Student's t-test). C, Immunoblots of protein extracts from day 1 animals after 24 hour treatment with water (Ctrl), 20 mM ChCl, or 20 mM MnCl2. Aggregates were separated into SDS-soluble or SDS-insoluble fractions (higher molecular weights). D, Quantification of SDS-soluble and -insoluble material from immunoblots (n=3, **p=<0.01, Student's t-test).
Discussion
In this study, we describe a model of manganism in C. elegans that encompasses several modes of toxicity: metallomic imbalances, age-dependent susceptibility, mitochondrial and ER stress, and protein misfolding (see model, Fig. 8). Mn can directly disrupt mitochondria function and affect ER-mediated protein folding7, 15. In addition, we find that at intermediate levels of toxicity (20 mM), Mn selectively lowers Fe and Ca levels, which may further exacerbate losses in mitochondria and protein homeostasis (Fig. 8). In C. elegans, Mn shares transport with Fe via the divalent metal transporter (DMT-1) orthologs2. Thus, it is not surprising that Fe is lowered in response to excess Mn, likely as a consequence of transport competition, which has been observed in mammalian models following Mn-treatment24, 25. Lowered Fe in response to Mn-treatment is associated with a shift in Fe2+ to Fe3+, which would allow Fe to more readily participate in the Fenton reaction leading to generation of free radicals and enhanced oxidative stress24 (Fig. 8). Moreover, some evidence exists suggesting that Mn and Ca also share transporters in the mitochondria and Golgi apparatus14, 26, 27, supporting a model in which excess Mn also outcompetes Ca for transport. In C. elegans, Mn and Ca appear to have similar subcellular localizations and the Golgi ATPase, PMR-1, has been identified as a Mn2+/Ca2+ transporter27, 28. Lowered Ca levels are associated with impaired ER-Golgi trafficking in addition to impaired mitochondrial function9, 29. Crosstalk between mitochondria and ER stress may further drive pathology (Fig. 8).
Figure 8. Modes of Mn toxicity.
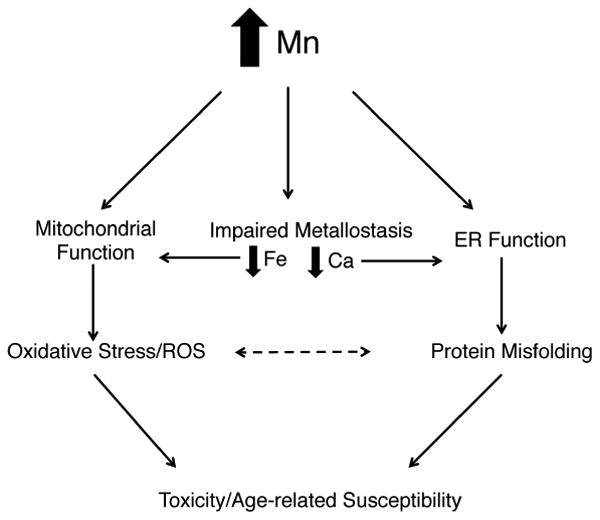
Schematic of potential avenues of Mn-toxicity. Mn directly affects mitochondrial function by disrupting mitochondria complex I and depleting mitochondrial ATP stores, which activates the mitochondrial stress response and stimulates ROS production, leading to toxicity and cell death. Mn can also directly perturb protein misfolding, which activates the ER stress response and leads to toxicity and cell death. Mn alters Fe homeostasis, which can lead to mitochondrial dysfunction and increase its participation in the Fenton reaction and ROS production. Mn alters Ca homeostasis, which can lead to ER dysfunction and exacerbates protein misfolding. Oxidative stress may exacerbate protein misfolding and vice versa. All pathways can eventually contribute to toxicity and cell death.
Mn induces ER stress in conjunction with alterations in Ca homeostasis
While transient activation of the ER UPR is a protective mechanism that ensures the proper folding of proteins, constitutive activation of the ER UPR is a hallmark of age-dependent neurodegenerative diseases that may further drive or exacerbate pathology30, 31. Previous studies have shown that Mn can upregulate the ER UPR sensor protein, BiP, in a cell model32 and that Mn exposure in the presence of Fe deficiency can also induce ER stress and cell death33, findings that are consistent with our results in C. elegans. We also observed decreased Ca levels in Mn-treated animals. Ca homeostasis has repeatedly been shown to be vital for the proper functioning of the ER. High Ca levels in the ER are important for proper function of the ER and of BiP, while depletion of Ca stores disrupts ER function and activates the ER UPR29. The impact of Mn on Ca homeostasis may therefore represent a novel avenue of exploration towards identifying additional mechanisms contributing to Mn toxicity.
Role of Mn in protein aggregation and aging
Aged animals are characterized by the accumulation of insoluble proteins34, 35, indicating that aging may be due, in part, to impaired protein homeostasis. We also observed that treatment with Mn late in life (day 10 worms) leads to a ∼30% increase in mortality rate after 48 hours (normalized to untreated age-matched controls), while virtually no young animals died after a 48 hour treatment. Thus, aged animals may be particularly susceptible to the mitochondrial and proteotoxic stresses of Mn due to their already impaired proteome.
Mn has been shown to stimulate the aggregation of α-synuclein in vitro and potentially exacerbate other neurodegenerative diseases such as amyotrophic lateral sclerosis (ALS) and prion disease14, 15. Here, we show that Mn also has the ability to alter aggregation states in vivo by shifting the unstable polyglutamine aggregates from an SDS-soluble state to a more SDS-insoluble state. Though a change in solubility of aggregates is not necessarily an indication of toxicity on its own, we concomitantly observed that Mn treatment resulted in nearly a 75% mortality rate in the polyglutamine model after just 24 hours of exposure. Animals that remained alive were decrepit and died within another 24 hours (data not shown). Thus, Mn-induced impairments in protein homeostasis combined with mitochondria dysfunction may result in widespread collapse of cellular functions and lead to rapid death in the polyglutamine model.
Mn and dopaminergic degeneration
Notably absent from our model is the ability of Mn exposure to induce dopaminergic degeneration in C. elegans, for which conflicting reports exist16, 21, 22. Previous studies have reported dopaminergic degeneration with MnCl2 when exposed during larval stages. In this study, we found that MnCl2 did not induce dopaminergic degeneration in either larva or aged worms, consistent with recent reports that also failed to find Mn-induced neurodegeneration22. Authors from this study cited the purity of the MnCl2 batches used as a possible discrepancy between their results and those reported previously. Taken together, our results suggest that MnCl2 does not directly target dopaminergic neurons. Close analysis of patient cases of manganism show that the region of the basal ganglia most affected by Mn is the globuspallidus, which receives inputs downstream of the dopaminergic-rich substania nigra6. Thus, Mn may only indirectly affect dopaminergic outputs rather than having direct affects on dopaminergic neurons.
Conclusions
Given the multifactorial causes of PD, C. elegans provides a tractable model to study the combined impact of environment, age, and genetic susceptibilities. Our findings suggest the modeling manganism in C. elegans could provide a platform for drug discovery that could lead to the identification of therapeutic avenues that allow a better understand of these factors as they relate to the interplay between mitochondrial dysfunction, protein misfolding, and metallomic imbalances.
Methods
C. elegans strains
The following strains were cultured using standard conditions and can be obtained via the Caenorhabditis Genetics Center (CGC, University of Minnesota)36: N2 Bristol (wild type), SJ4100 [zcIs13(hsp-6p::GFP)], SJ6 [zcIs4(hsp-4p::GFP)], CL2070 [dvIs70(phsp16.2p::GFP; rol-6(su1006)], and BA671 [spe-9(hc88)]. The following were kind gifts from the following laboratories: BY200 [vtIs1(pdat-1::GFP)] (provided by Aschner laboratory, Vanderbilt University)22, UA44 [baIn11(pdat-1::α-synuclein, pdat-1::GFP)] (provided by Caldwell laboratory, University of Alabama)20, and AM141 [rmIs133(P(unc-54) Q40::YFP)] (provided by Morimoto laboratory, Northwestern University)23.
Preparation on manganese plates
Stock solutions of 3M manganese (II) chloride tetra-hydrate (Sigma-Aldrich) were made in deionized/distilled water immediately before use. MnCl2 solution was added directly to NGM plates at the indicated final concentrations. E. coli OP50 was spotted on NGM plates and spread by gently swirling and allowed to grow for 24 hours at room temperature. Plates were stored at 4°C for no longer than 2 weeks.
Microscopy and quantification
Day 1 adult worms were collected and paralyzed in 1mM levamisole, mounted on agar pads with glass coverslips, and analyzed using an Olympus BX51 upright microscope. Worms expressing GFP were analyzed using 470/40 nm excitation and 525/50 nm emission wavelengths. Worms exposed to TMRM were analyzed using 535/50 nm excitation and 610/75 nm emission wavelengths.
Approximately 20 animals per condition were used and fluorescent intensity was normalized to the body-length (mid-line length of the animal). Quantification of pixel densities and polyglutamine inclusions were analyzed with Image J™. All experiments were repeated at least three times with similar results.
Western blot assays
For western blots, approximately 20-40 worms were collected at day 1 of adulthood. Worms were flash frozen and resuspended in a final concentration of 2% SDS sample buffer with 2.5% β-mercaptoethanol. Immunoblots were probed with anti-mouse GFP (B2) (Santa Cruz Biotechnology, 1:1000) or anti-rabbit tubulin (LL-17) (Sigma Aldrich, 1:1000) for loading controls.
TMRM assays
To assess mitochondrial membrane potential, N2 wild type worms were reared from synchronized eggs at 20°C until the 4th larval stage and then transferred to control plates or plates containing MnCl2 in the presence of tetramethylrhodamine methyl ester (TMRM). TMRM was prepared as previously described to a final concentration of 0.1uM in NGM agar plates17. Worms were collected for microscopic analysis and quantified 24 hours later.
Scoring of dopaminergic degeneration
Synchronous populations of worms were aged until day 5 of adulthood at 20°C and then transferred to control or MnCl2 plates. After 48 hours, animals were collected for microscopic analysis. Animals were scored as having 0, 1, 2, 3, or 4 cephalic dendrites as measured by GFP fluorescence. Approximately 30 animals per condition were used and experiments were repeated at least three times with similar results.
Polyglutamine analysis
AM141 [rmIs133(P(unc-54) Q40::YFP)] worms were grown at 20°C until the 4th larval stage and then transferred to control or MnCl2 plates and simultaneously shifted to 25°C. After 24 hours, worms were collected for analysis for western blot, for microscopy, or for survival (scored by gentle prodding).
Lifespan analysis
Synchronized populations of worms were cultured at 20°C and transferred to control or MnCl2 plates beginning as young adults that had been supplemented with 10μg/μl 5-fluoro 2-deoxyuridine (FUdR) to prevent progeny production. Animals that crawled off the plates or died due to internal gut or vulval extrusions were censored from the population. We found that in the absence of FUdR, concentrations higher than 10 mM MnCl2 elicited high levels of matricide (larval explosions), thus making lifespan analysis difficult to interpret (data not shown). Log rank (Mantel-Cox) statistics were used for analysis of lifespans using Prism™.
Inductively-coupled plasma atomic emission spectroscopy analysis
ICP analysis was conducted as previously described37. Briefly, synchronized populations of arrested L1s were grown en masse (25,000-40,000 worms) at 25°C to induce sterility in the temperature-sensitive mutant, spe-9(hc88). Worms were shifted to control or MnCl2 plates at the 4th larval stage worms and collected in M9 buffer 24 hours later. Worms were washed three times in M9 buffer and a final wash in 150 mM choline chloride, 1mM HEPES. Worms were dried at 60°C for 48 hours. Dried pellets were acid digested with Omnitrace 70% HNO3 at 60°C overnight. Samples were diluted with Omnitrace water for a final concentration of 5% HNO3.
Supplementary Material
Acknowledgments
We thank members of the Lithgow and Andersen laboratories for helpful discussion. We thank Tai C. Holland for technical support for ICP analysis. This work was supported by a Larry L. Hillblom Foundation grant as well as NIH grants (R21 AGGSS2620). S.A. was supported by NIH Training Grants AG000266 and 1F32ES022370-01A1.
References
- 1.Aschner M, Erikson KM, Herrero Hernandez E, Tjalkens R. Neuromolecular medicine. 2009;11:252–266. doi: 10.1007/s12017-009-8083-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Au C, Benedetto A, Anderson J, Labrousse A, Erikson K, Ewbank JJ, Aschner M. PloS one. 2009;4:e7792. doi: 10.1371/journal.pone.0007792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Erikson KM, Shihabi ZK, Aschner JL, Aschner M. Biological trace element research. 2002;87:143–156. doi: 10.1385/BTER:87:1-3:143. [DOI] [PubMed] [Google Scholar]
- 4.Au C, Benedetto A, Aschner M. Neurotoxicology. 2008;29:569–576. doi: 10.1016/j.neuro.2008.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Racette BA. Neurotoxicology. 2013 doi: 10.1016/j.neuro.2013.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Olanow CW. Annals of the New York Academy of Sciences. 2004;1012:209–223. doi: 10.1196/annals.1306.018. [DOI] [PubMed] [Google Scholar]
- 7.Benedetto A, Au C, Aschner M. Chemical reviews. 2009;109:4862–4884. doi: 10.1021/cr800536y. [DOI] [PubMed] [Google Scholar]
- 8.Moore DJ, West AB, Dawson VL, Dawson TM. Annu Rev Neurosci. 2005;28:57–87. doi: 10.1146/annurev.neuro.28.061604.135718. [DOI] [PubMed] [Google Scholar]
- 9.Cali T, Ottolini D, Brini M. BioFactors (Oxford, England) 2011;37:228–240. doi: 10.1002/biof.159. [DOI] [PubMed] [Google Scholar]
- 10.Lindholm D, Wootz H, Korhonen L. Cell Death Differ. 2006;13:385–392. doi: 10.1038/sj.cdd.4401778. [DOI] [PubMed] [Google Scholar]
- 11.Barnham KJ, Bush AI. Current opinion in chemical biology. 2008;12:222–228. doi: 10.1016/j.cbpa.2008.02.019. [DOI] [PubMed] [Google Scholar]
- 12.Kaur D, Yantiri F, Rajagopalan S, Kumar J, Mo JQ, Boonplueang R, Viswanath V, Jacobs R, Yang L, Beal MF, DiMonte D, Volitaskis I, Ellerby L, Cherny RA, Bush AI, Andersen JK. Neuron. 2003;37:899–909. doi: 10.1016/s0896-6273(03)00126-0. [DOI] [PubMed] [Google Scholar]
- 13.Ahmed SS, Santosh W. PloS one. 2010;5:e11252. doi: 10.1371/journal.pone.0011252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bowman AB, Kwakye GF, Herrero Hernandez E, Aschner M. J Trace Elem Med Biol. 2011;25:191–203. doi: 10.1016/j.jtemb.2011.08.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Santner A, Uversky VN. Metallomics : integrated biometal science. 2010;2:378–392. doi: 10.1039/b926659c. [DOI] [PubMed] [Google Scholar]
- 16.Settivari R, Levora J, Nass R. The Journal of biological chemistry. 2009;284:35758–35768. doi: 10.1074/jbc.M109.051409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yoneda T, Benedetti C, Urano F, Clark SG, Harding HP, Ron D. Journal of cell science. 2004;117:4055–4066. doi: 10.1242/jcs.01275. [DOI] [PubMed] [Google Scholar]
- 18.Link CD, Cypser JR, Johnson CJ, Johnson TE. Cell stress & chaperones. 1999;4:235–242. doi: 10.1379/1466-1268(1999)004<0235:doosri>2.3.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Calfon M, Zeng H, Urano F, Till JH, Hubbard SR, Harding HP, Clark SG, Ron D. Nature. 2002;415:92–96. doi: 10.1038/415092a. [DOI] [PubMed] [Google Scholar]
- 20.Ruan Q, Harrington AJ, Caldwell KA, Caldwell GA, Standaert DG. Neurobiology of disease. 2010;37:330–338. doi: 10.1016/j.nbd.2009.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Benedetto A, Au C, Avila DS, Milatovic D, Aschner M. PLoS genetics. 2010;6 doi: 10.1371/journal.pgen.1001084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bornhorst J, Chakraborty S, Meyer S, Lohren H, Grosse Brinkhaus S, Knight AL, Caldwell KA, Caldwell GA, Karst U, Schwerdtle T, Bowman A, Aschner M. Metallomics : integrated biometal science. 2014;6:476–490. doi: 10.1039/c3mt00325f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Morley JF, Brignull HR, Weyers JJ, Morimoto RI. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:10417–10422. doi: 10.1073/pnas.152161099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fernsebner K, Zorn J, Kanawati B, Walker A, Michalke B. Metallomics : integrated biometal science. 2014;6:921–931. doi: 10.1039/c4mt00022f. [DOI] [PubMed] [Google Scholar]
- 25.Moldovan N, Al-Ebraheem A, Miksys NA, Farquharson MJ, Bock NA. Biometals. 2013;26:179–187. doi: 10.1007/s10534-012-9605-z. [DOI] [PubMed] [Google Scholar]
- 26.Gavin CE, Gunter KK, Gunter TE. Neurotoxicology. 1999;20:445–453. [PubMed] [Google Scholar]
- 27.Van Baelen K, Vanoevelen J, Missiaen L, Raeymaekers L, Wuytack F. The Journal of biological chemistry. 2001;276:10683–10691. doi: 10.1074/jbc.M010553200. [DOI] [PubMed] [Google Scholar]
- 28.McColl G, James SA, Mayo S, Howard DL, Ryan CG, Kirkham R, Moorhead GF, Paterson D, de Jonge MD, Bush AI. PloS one. 2012;7:e32685. doi: 10.1371/journal.pone.0032685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brostrom MA, Brostrom CO. Cell Calcium. 2003;34:345–363. doi: 10.1016/s0143-4160(03)00127-1. [DOI] [PubMed] [Google Scholar]
- 30.Moreno JA, Radford H, Peretti D, Steinert JR, Verity N, Martin MG, Halliday M, Morgan J, Dinsdale D, Ortori CA, Barrett DA, Tsaytler P, Bertolotti A, Willis AE, Bushell M, Mallucci GR. Nature. 2012;485:507–511. doi: 10.1038/nature11058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Halliday M, Mallucci GR. Neuropharmacology. 2014;76 Pt A:169–174. doi: 10.1016/j.neuropharm.2013.08.034. [DOI] [PubMed] [Google Scholar]
- 32.Chun HS, Lee H, Son JH. Neuroscience letters. 2001;316:5–8. doi: 10.1016/s0304-3940(01)02341-2. [DOI] [PubMed] [Google Scholar]
- 33.Seo YA, Li Y, Wessling-Resnick M. Neurotoxicology. 2013;38:67–73. doi: 10.1016/j.neuro.2013.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.David DC, Ollikainen N, Trinidad JC, Cary MP, Burlingame AL, Kenyon C. PLoS biology. 2010;8:e1000450. doi: 10.1371/journal.pbio.1000450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Reis-Rodrigues P, Czerwieniec G, Peters TW, Evani US, Alavez S, Gaman EA, Vantipalli M, Mooney SD, Gibson BW, Lithgow GJ, Hughes RE. Aging cell. 2011 doi: 10.1111/j.1474-9726.2011.00765.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sulston J HJ. In: The Nematode Caenorhabditis elegans. W BW, editor. Cold Spring Harbor Laboratory Press; Cold Spring Harbor: 1988. pp. 587–606. [Google Scholar]
- 37.Page KE, White KN, McCrohan CR, Killilea DW, Lithgow GJ. Metallomics : integrated biometal science. 2012;4:512–522. doi: 10.1039/c2mt00146b. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.