

Abstract
Mild cognitive impairment is a well-documented consequence of whole brain radiation therapy (WBRT) that affects 40-50% of long-term brain tumor survivors. The exact mechanisms for the decline in cognitive function post-WBRT remain elusive and no treatment or preventative measures are available for use in the clinic. Here, we review recent findings indicating how changes in the neurovascular unit may contribute to the impairments of learning and memory. In addition to affecting neuronal development, WBRT induces profound capillary rarefaction within the hippocampus-a region of the brain important for learning and memory. Therapeutic strategies such as hypoxia, which restore the capillary density, result in the rescue of cognitive function. In addition to decreasing vascular density, WBRT impairs vasculogenesis and/or angiogenesis, which may also contribute to radiation-induced cognitive decline. Further studies aimed at uncovering the specific mechanisms underlying these WBRT-induced changes in the cerebrovasculature are essential for developing therapies to mitigate the deleterious effects of WBRT on cognitive function.
Keywords: angiogenesis, cerebrovasculature, cognitive impairment, neurogenesis, vasculogenesis
Clinical Importance of WBRT
Close to 1.7 million new cases of cancer [1] and 69,720 primary brain tumors [2] are expected to be diagnosed in 2013 in the United States. Between 20 and 50% of patients with systemic cancer develop brain metastases [3]. Whole brain radiation therapy (WBRT) continues to be one of the most common forms of treatment for primary and metastatic brain tumors located in brain regions that are difficult to remove surgically, as well as for treatment of primary brain tumors following surgical intervention [4]. With progressive improvements in treatment regimens, the population of long-term cancer survivors continue to grow with 62% of adult brain cancer patients surviving beyond five years [5]. Although WBRT has proven to be effective in eliminating brain tumors, damage to normal brain tissue is inevitable and several complications are observed following treatment that decrease quality of life for survivors of the disease.
Radiation-Induced Brain Injury
Radiation-induced brain injury can be categorized as: acute, early delayed or late delayed reactions [6]. Acute injury is very rare and occurs hours to weeks after radiation therapy while early delayed injury occurs from 1-6 months post-irradiation and can involve transient demyelination and somnolence (drowsiness). Both acute and early-delayed reactions are normally reversible and resolve spontaneously. However, late delayed effects, characterized by demyelination, vascular abnormalities and ultimately white matter necrosis [7] are observed more than 6 months post-irradiation and are considered to be irreversible and progressive. The complications of WBRT greatly affect the quality of life of cancer survivors after treatment, and there is currently no accepted treatment to prevent or reverse these effects.
The consequences of radiation-induced injury can be highly variable and these differences are compounded by differences in treatment regimens (e.g. single vs. fractionated dosing - Table 1). Current treatments range from single, high doses to lower cumulative fractionated doses administered to the entire body or focused on specific organs These treatment regimens highlight the importance of selecting relevant experimental designs for studies aimed at understanding WBRT-induced tissue damage. [4]. Generally, higher doses of radiation are used for targeted therapy and may result in focal damage limited to the radiated area and the surrounding tissues. However, whole body radiation is generally done in smaller doses since doses of 10Gy or higher can be lethal if bone marrow cells are not replenished. Therefore, it is unlikely that a single dose of 10Gy radiation results in the identical biological response as the same cumulative dose of radiation delivered as 2Gy fractions over a several weeks period. It is imperative that experimental models appropriately mimic accepted treatment regimens in order to prevent erroneous conclusions regarding the actions of and treatment for radiation-induced damage.
Table 1. Summary of studies assessing radiation-induced brain injury.
| Reference | Dose | Effects/ Injury |
|---|---|---|
| [172] | 18 Gy | Increased blood-brain barrier permeability |
| [22] | 10 Gy | Transient increase in microglial proliferation, increased activated microglia |
| [173] | 8 Gy | Increased hypertrophy of astrocytes |
| [174] | 15 Gy | Increased histopathology in tissues |
| [175] | 2×0.75 Gy | Increased organelle damage (cytoplasmic vacuolation, dilation of endoplasmic reticulum, destruction of mitochondria and damage to plasma membrane). |
| [176] | 2×10 Gy | Time- and dose-dependent increase in hippocampal necrosis |
| 3×10 Gy 4×10 Gy | Increase in pial microvessel permeability (high at 40 Gy, modest at 30 Gy) | |
| [177, 178] | 3 Gy whole body | Acute decline in proliferation followed by increased proliferation in rostral migratory stream (RMS) |
| [179] | 2.5 Gy whole body | Decreased proliferation and neuron maturation in dentate gyrus of hippocampus |
| [33, 35] | 0–50 Gy to spinal cord | Decreased endothelial cell density (apoptosis) at 24h with 50 Gy; transient blood-brain barrier breakdown |
| [180] | 50, 75, 120 Gy to parietal cortex | Time- and dose-dependent gliosis, blood-brain barrier breakdown and necrosis |
| [181] | 4×5 Gy or 8×5 Gy | Transient cognitive impairments after 20 Gy and more severe cognitive impairments after 40 Gy. Blood-brain barrier disruption and astrogliosis after 40 Gy |
| [182] | 20×2 Gy | Increased astrocyte numbers in cortex and increased blood-brain barrier permeability |
WBRT Induces Cognitive Impairment
One of the most prevalent consequences of WBRT is the onset of cognitive decline, which occurs in 40-50% of long-term brain tumor survivors (more than 1 year post-irradiation) [8-10]. These patients exhibit significant impairments in tests of working memory [11], verbal memory [12] and general IQ [13]. Deterioration of cognitive function post-WBRT generally precedes the decline in quality of life, as measured by activities of daily living [14]. The clinical findings of WBRT-induced impairments in learning and memory have also been confirmed in animal models. Dose-and time-dependent deficits in cognitive function have been reported in rats [15] in response to single [16] and fractionated [17, 18] WBRT. Additionally, deficits in spatial learning have been reported in mouse models [19, 20]. Our laboratory recently demonstrated that fractionated WBRT induces time-dependent learning and memory deficits in both the Barnes maze and active avoidance tasks [21]. Importantly, spatial learning was progressively impaired after WBRT as mice exhibited increased latency to the escape box and made more errors in the Barnes maze in the months following treatment (Figure 1). Despite extensive studies demonstrating the effects of WBRT on cognition in multiple species, the etiology of WBRT-induced cognitive impairment remains poorly-understood.
Figure 1. Mice subjected to fractionated WBRT have progressive impairment in learning ability when assessed on the Barnes maze.
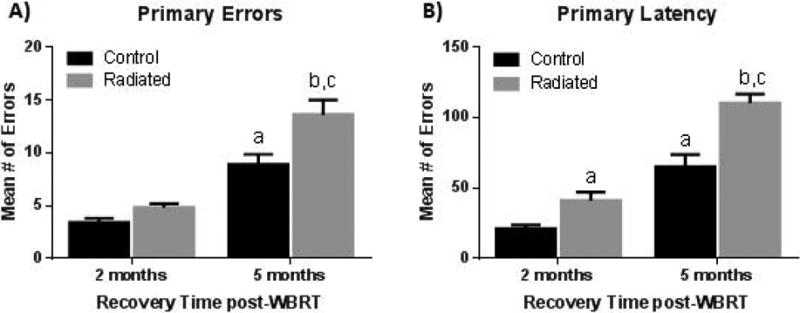
Compared to non-radiated controls, radiated mice make more errors (A) and exhibit increased latency (B) to locate the hidden escape box. This impairment is worse at 5 months post-WBRT. [“a” represents significance compared to control 2 months, “b” – compared to control 5 months, and “c” – compared to radiated 2 months; p<0.05]. Adapted from [21].
WBRT-induced Inflammatory Responses
It is well-established that radiation therapy induces chronic oxidative stress [5], neuro-inflammation (e.g. activated microglia and infiltrating peripheral monocytes [7, 20, 22]) and acute increases in the expression of pro-inflammatory cytokines [23]. There is strong evidence that some pro-inflammatory cytokines (e.g. TNF-α) induce endothelial cell damage and death [24-26]. Brain radiation, administered as single high doses, induces transient increases in tissue gene expression of various cytokines [23, 27-29]. Additionally, at the protein level, whole body radiation induces dose-and time-dependent increases in the production of IL-12 and IL-18 [30]. Moreover, chronic tissue inflammation in the form of increased gene expression of cytokines, chemokines and chemokine receptors has been reported in the lung [31] and thorax [32] in response to single doses of radiation. Unfortunately, the precise relationship between cytokine expression and cognitive impairment remains unclear. Previous reports indicate that tissue inflammation increases angiogenesis and blood flow but these responses are absent after WBRT. In addition, the time course for cytokine expression is more consistent with the early, transient disruption of some behavioral tests that occurs in the days or weeks immediately following radiation rather than the delayed, late effects of radiation that are the focus of this review.
Vasculature Effects of WBRT
Radiation has profound effects on the vasculature, more specifically, endothelial cells. Radiation therapy induces dose-dependent endothelial apoptosis [33], suppression of endothelial cell proliferation [34], disruption of the blood brain barrier [35], thickening and vacuolation of the vascular basement membrane [36], breakdown of the extracellular matrix [37] and microvascular rarefaction in rat brain as early as 10 weeks following fractionated WBRT [38]. Similarly, single doses of 5–20 Gy to the brain result in a 15% decrease in endothelial cell number within 1 day of irradiation that was maintained for at least one month [39]. Thus, both single, high doses of radiation as well as lower, cumulative fractionated doses have been shown to compromise the vasculature within the brain.
In order to relate vascular changes to specific cognitive deficits, it is critical to demonstrate that radiation modifies vascular structure and/or function in brain regions important for learning and memory. Initial studies demonstrated that there is a rarefaction of vessels within the CA1 region of the hippocampus (an area important for spatial memory) 12 months after single doses as low as 0.5 Gy and 2 Gy high-linear energy transfer (LET) radiation [40]. More detailed analyses have shown that fractionated WBRT induces a significant capillary rarefaction in the CA1, CA3and dentate gyrus of the hippocampus (approximately a 40% reduction); a reduction that is also associated with a significant loss of endothelial cells and pericytes and is closely associated with deficits in spatial learning and memory [41].
The density of capillaries within any tissue is correlated with regional blood flow [42, 43]. Because the brain is a highly-metabolic organ, it requires a consistent and efficient supply of oxygen, nutrients and trophic factors to ensure normal function. This is accomplished through highly-organized capillary networks that minimize the diffusion distance between the blood vessels and neurons/glia. Indeed, our laboratory demonstrated that capillary rarefaction within the hippocampus occurs alongside the appearance of learning deficits [21], illustrating the close association between capillary density and neuronal function. Based on these studies and the increased expression of HIF-1a and VEGFR that occur in response to WBRT, we conclude that capillary rarefaction not only results in loss of nutrient delivery to tissues, but also results in localized tissue hypoxia and a reduced ability to remove products of cellular metabolism (including CO2) from brain tissues. Capillary rarefaction is therefore a key mechanism by which WBRT can induce neuronal dysfunction and as a result, contribute to cognitive impairment.
Several studies have assessed the changes in blood flow and metabolism within tumors or normal tissue following radiation treatment. Acute increases in blood flow are observed in both gliomas and normal brain regions following single, high doses of WBRT (20 Gy) [44]. This increase appears to be an acute phenomenon, as more recent studies have shown dose-dependent decreases in cerebral blood volume in the months following fractionated stereotactic radiotherapy [45]. Decreased glucose metabolism in the radiation-injured or peri-radiation-injured brain tissue [46] and in brain regions receiving over 40 Gy radiation [47] has also been reported. Similar decreases in cerebral blood flow were observed in the targeted and surrounding healthy tissue weeks following stereotaxic radiosurgery [48]. These studies illustrate that radiation therapy induces a long-term reduction in vascular density that is followed by decreased cerebral blood flow and metabolism.
The loss of blood vessels following WBRT should induce vessel repair mechanisms to minimize long-term damage to the tissue. The processes of angiogenesis and vasculogenesis are recognized as two primary mechanisms responsible for the development, maintenance and/or restoration of vascular integrity following disruption of vascular networks. Angiogenesis is a multi-step process that results in the formation of new blood vessels from pre-existing vascular networks [49]. Alternatively, vasculogenesis, a process of de novo vessel formation, involves the mobilization of populations of cells, generally referred to as endothelial progenitor cells (EPCs), from the bone marrow to sites of injury to stimulate repair [50-54]. Studies demonstrating that prevention of glioblastoma recurrence can only be accomplished when vasculogenesis, rather than angiogenesis, is inhibited provide evidence that these processes are activated by unique stimuli and utilize independent pathways [55].
Following vascular damage, EPCs, known to express surface markers such as VEGFR2 (KDR/ Flk-1), CD34 and CD133, are mobilized from the bone marrow and circulate in peripheral blood [56-58]. Several studies have reported changes in specific populations of EPCs after vascular trauma/injury (Table 2). Circulating EPCs are elevated in patients who experience burns, coronary artery bypass [59], congestive heart failure [60], musculoskeletal trauma [61], head and neck cancer [62], hind limb ischemia [63], radiation therapy [64], coronary angioplasty [65], or surgical injury (laparotomy) [66]. However, the surface markers used to identify EPCs in each study vary widely, emphasizing the controversy that exists in the field in terms of the characteristics of EPCs (Table 2). CD34+ cells were the first population of cells described as putative EPCs based on their ability to stimulate angiogenesis in vivo [56]. More recently, bone marrow transplantation of VEGFR2+ (Flk-1+) cells was shown to stimulate both angiogenesis and neurogenesis in a model of cerebral ischemia [67]. Despite these differences in cellular markers, it is evident that EPC recruitment has an important role in some types of vessel repair.
Table 2.
Summary of EPC characteristics used to detect changes after various types of vascular injury.
| Reference | Type of Vascular Trauma | Source | Markers for Putative EPCs | Response Characteristics |
|---|---|---|---|---|
| [59] | Burn, Coronary Artery Bypass Grafting | Blood | VEGFR2+ | Transient increase |
| [60] | Congestive Heart Failure | Blood | CD34+ CD34+/CD133+/VEGFR2+ | Transient increase |
| [61] | Musculoskeletal Trauma | Blood | CD34+ and CD133+ | Gradual increase. Decreased at day 7 |
| [62] | Head and Neck Cancer | Blood | CD133+/VEGFR2+ | Increased levels of cells. |
| [63] | Hind limb ischemia + nicotine treatment | Bone Marrow | CD34+/VEGFR2+ | Increased levels of cells |
| [64] | Radiation-treated cancer patients | Blood | CD34/CD133 and CD34/VEGFR2 | Increased after treatment |
| [183] | Percutaneous Coronary Intervention | Blood | In Vitro Colony formation of mononuclear cells | Early mobilization |
| [65] | Coronary Angioplasty | Blood | In Vitro colony formation | Increased colonies |
| [66] | Surgery – Laparotomy | Bone Marrow Blood Spleen |
Sca-1/ckit VEGF/MAC-1- Lectin & low-density |
Increased Increased Increased |
| [184] | Burn (thermal injury) | Blood | lipoprotein uptake CD45-(dim)/CD133/CD144/VEGFR2 | Rapid increase |
| [185] | Traumatic brain injury | Blood | CD34 CD133 CD34/CD133 |
Decrease Gradual increase Gradual increase |
In addition to EPCs, circulating mature endothelial cells have been described as promising indicators of endothelial cell injury since they are increased in peripheral blood of patients with vascular disorders [68]. Normally, mature endothelial cells are found lining blood and lymphatic vessels; therefore, detection of these cells in the circulation is hypothesized to indicate damage to the endothelium. For example, increased numbers of mature endothelial cells have been reported in patients with pulmonary hypertension [69], percutaneous coronary angioplasty [70]and atherosclerosis [71]. Exploring the effects of WBRT on circulating EPCs and mature endothelial cells would provide important information regarding the status of both vessel damage and repair in response to radiation treatment; providing data that are currently limited.
WBRT Impairs Angiogenesis
Angiogenesis (see [49, 72, 73]) occurs through a multistep process involving several key intra- and intercellular signaling processes. Generally, quiescent vessels are stabilized by pericytes that act to suppress endothelial cell proliferation. When an angiogenic signal is detected, pericytes detach from the endothelium mainly through the action of matrix metalloproteinases (MMPs). The blood vessel then dilates and endothelial cell permeability increases, allowing the extracellular matrix to be degraded. Endothelial cells then proliferate and begin to migrate (sprout). A lumen develops and the new vessel fuses with other vessel branches through myeloid bridge cells (leukocytes). Newly-formed vessels are stabilized by pericytes and the basement membrane is deposited.
As previously mentioned, radiation induces tissue hypoxia, characterized by increased hypoxia inducible factor 1 alpha (HIF-1α) expression and oxidative stress as early as 4 weeks post-treatment [74]. It is well-established that hypoxia is a potent angiogenic stimulus [75-77] that acts through the stabilization of HIF-1α and the activation of downstream angiogenic factors including, but not limited to, vascular endothelial growth factor (VEGF) and erythropoietin [77-79]. In response to WBRT, angiogenesis does not occur despite the presence of local tissue hypoxia and normal levels of hematopoietic cells in the circulation [80] suggesting that WBRT damages angiogenic mechanisms that would normally rebuild the cerebrovasculature. The ability of WBRT to impair angiogenesis may be key to understanding its effects on cognitive function.
Abrogating WBRT-induced Vascular Deficits with Hypoxia
Since hypoxia is a physiological stimulus for angiogenesis and radiation has been reported to inhibit angiogenesis, systemic hypoxia presents a potentially useful tool for dissecting the mechanisms for radiation-induced vascular rarefaction. Systemic hypoxia has been utilized in various studies to assess physiological changes that occur at high altitudes or to elucidate mechanisms of vessel growth. Following brief exposures to hypoxic conditions (acute hypoxia), cerebral arteries dilate [81] and cerebral autoregulation (the ability to maintain a steady blood perfusion) is suppressed [82] to ensure that oxygen supply to the brain is not reduced extensively and damage to the brain is minimized. Prolonged exposure to hypoxic conditions (chronic hypoxia) induces angiogenesis [41], mobilizes EPCs [83], recruits bone marrow-derived cells to the pulmonary vasculature [84], prevents hypertension in pre-hypertensive rats and reverses hypertension in hypertensive rats via activation of vascular endothelial growth factor A (VEGF-A) and increases capillary density [85]. Thus, systemic hypoxia is an excellent tool to assess the potential mechanisms for vascular growth or vascular rarefaction post-WBRT. In our own studies, we have utilized a 30-day systemic hypoxia challenge to assess the capacity for cerebrovascular angiogenesis. Importantly, systemic hypoxia was found to completely reverse the WBRT-induced deficits in cerebrovascular density (Figure 2) and the corresponding reduction in learning and memory. The recovery of cognitive function persisted for at least 2 months after the animals were returned to a normoxic environment [41][21]. Although systemic hypoxia is not a translationally relevant model, this is an important finding that provides insight into the mechanisms of vascular rarefaction after radiation and perhaps will provide clues to potential therapeutic interventions. Additionally, other tools such as gene transfer (developmental endothelial locus -1; Del-1) [86], cell therapy (CD34+cell delivery to the brain) [87], and exercise [88] have been used to stimulate angiogenesis and can be utilized as potential tools for studying angiogenic mechanisms post-WBRT.
Figure 2. WBRT reduces hippocampal capillary density while systemic hypoxia reverses capillary rarefaction.
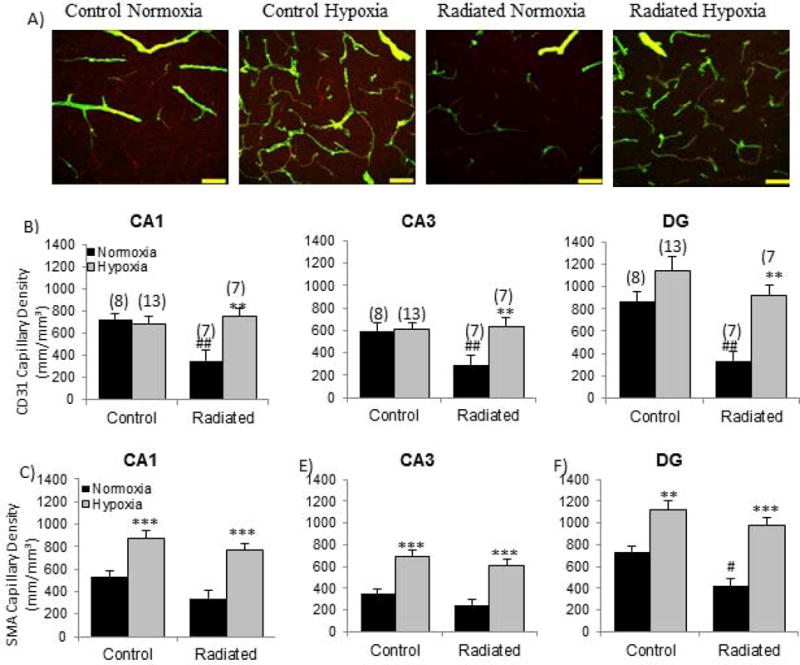
(A) CD31 (red) and smooth muscle actin (green) capillaries are reduced following WBRT and restored with systemic hypoxia. Quantification of capillary density measured by (B) endothelial cell and (C) smooth muscle cell staining in CA1, CA3 and dentate gyrus (DG) of the hippocampus. **p<0.01, ***p<0.001 compared to Normoxia; #p<0.05, ##p<0.01 compared to Control Normoxia. Adapted from [41].
Radiation-induced Changes in the Neurovascular Unit
Compromising the vasculature of the brain, as WBRT has been shown to do, can be detrimental to neuronal function and survival. Neurons are highly-metabolic cells that require a constant supply of nutrients, oxygen and growth factors not only for proper development, but also for survival. Moreover, neurons are dependent on the efficient removal of carbon dioxide and metabolic waste products. In order for these functions to occur successfully, an intact vascular network surrounding the neurons is required. Thus, neurons are found located within a specialized niche comprised of astrocytes, pericytes, smooth muscle cells and endothelial cells that work in coordination to orchestrate proper neuro-vascular communication [89]. The loss of cells within this neurovascular niche could have profound consequences. As mentioned, WBRT leads to both a reduction in endothelial cells as well as pericytes. Because of this, other cells within the neurovascular unit suffer from a loss of nutrient delivery, metabolic waste removal and localized hypoxia, all of which may ultimately compromise cellular survival.
In addition to disturbing the neurovascular unit as a whole, WBRT can negatively impact neurons directly. It is well-established that brain radiation inhibits neurogenesis [90-96]. Because neurogenesis within the hippocampus is required during development and throughout life for proper learning and memory [97-99], it is likely that the decreased neurogenesis following radiation contributes directly to impairments in cognition [100]. Interestingly, the decrease in neurogenesis after WBRT can be reversed by the introduction of human embryonic stem cells into the mouse hippocampus, resulting in improved cognitive function [101, 102]. These findings provide prima facie evidence that radiation-induced decreases in neuronal stem cells and neurogenesis are important contributing factors to the cognitive deficits following WBRT. Unfortunately, these studies did not analyze whether the treatment regimen increased angiogenesis in the radiated animals. Based on the neurovascular interactions that are necessary for neurogenesis, this is an important question that needs to be explored. A recent study designed to identify the major contributions of angiogenesis and neurogenesis to learning (through targeted inhibition of each process) determined that angiogenesis is the critical component for learning acquisition while inhibiting neurogenesis paradoxically improves performance on the water maze task [103]. Additionally, there is strong evidence that microvascular angiogenesis is disrupted if neurogenesis is inhibited [104]. These studies and others provide important support for the interaction between these two processes and the importance of vascular plasticity in cognitive performance. Furthermore, it has been recognized for some time that trophic factors, supplied by the microvasculature, regulate neurogenesis [105]. Factors such as VEGF [106, 107], VEGF-C [108], hepatocyte growth factor (HGF) [109] and granulocyte colony-stimulating factor (G-CSF) [110] have strong neurogenic effects further supporting the hypothesis that capillary density within the tissues must be maintained in order for learning and memory to occur.
There is compelling evidence that radiation leads to significant impairments in both neurogenesis and angiogenesis [19, 90-93, 95, 96]. Interestingly, both chronic [111, 112] and intermittent [113] systemic hypoxia stimulate these processes. Additional studies will be required to assess the specific molecular mechanisms for the effects of radiation on neurogenesis and angiogenesis but, based on our current understanding of the field, radiation-induced impairments that are initiated within the neurovascular niche are likely a primary factor in the decline in cognitive function.
Vascular Recovery after WBRT – The Role of Bone Marrow Derived Cells
As previously noted, there is strong evidence that bone marrow derived cells contribute to recovery of the vasculature in different organs/tissues. For example, in response to hypoxia bone marrow-derived cells were found to be closely associated with blood vessels in the mouse spinotrapezius muscle [114]. In a rat model of stroke, bone marrow stromal cell transplantation resulted in the recovery of motor behavior, along with new blood vessel formation in the infarct region [115]. Although these and other, transplantation studies provide evidence for functional recovery of the vasculature and improvements in cognitive function, there is still no consensus as to whether these cells differentiate into vascular cells and incorporate into the vessel wall. There is strong evidence to support the role of EPCs in re-endothelialization, neovascularization and endothelial repair [50, 116-126]. However, other studies have reported a supportive role for transplanted bone marrow cells in cytokine secretion [53, 127], monocyte development [128] and/or the recruitment of monocytes and macrophages to sites of injury [129]. These conflicting reports necessitate further studies addressing the roles of EPCs and transplanted bone marrow-derived cells in vascular recovery.
If EPCs and bone marrow-derived transplanted cells do not become part of the vascular wall, one can hypothesize that they differentiate into cell types that comprise the neurovascular unit, including pericytes. Pericytes are a heterogeneous population of mural cells associated with the microvasculature [130, 131] and have an important role in endothelial proliferation [132], blood brain barrier integrity [133], contraction of capillaries and regulation of capillary blood flow [134]. Pericytes have been shown to guide and precede proliferating endothelial cells during embryonic angiogenesis [135], stabilize newly formed blood vessels and maintain endothelial cells in a quiescent state [77]. Importantly, pericytes derived from the bone marrow have been detected at sites of angiogenesis and vasculogenesis [136, 137]. Because of the vital roles that pericytes play in maintaining blood brain barrier integrity and regulating cerebral blood flow [138], it is likely that the loss of pericytes we observe following WBRT [41] may contribute to cognitive dysfunction. Thus, the potential recruitment of pericytes from the bone marrow to the brain following chronic systemic hypoxia should be explored further.
It is also possible that hypoxia induces the recruitment of bone marrow derived glial cells to the brain. Glia, such as astrocytes and microglia, are known to influence not only neuronal physiology but also cerebrovasculature physiology. In addition to serving as a key component of the neurovascular unit, astrocytes regulate neurovascular coupling [139, 140] as well as cerebral blood flow [141]. Furthermore, these cells release angiogenic signaling molecules such as VEGF [142, 143] and modulate cerebral blood flow via release of adenosine and prostaglandin E2 [139, 144]. Interestingly, astrocytes are recruited to the brain from the bone marrow following ischemic stroke [145, 146]. Following transplantation, bone marrow derived astrocyte incorporation is associated with improved functional recovery following stroke and traumatic brain injury [147-149]. Astrocytes are not the only bone marrow derived glial cell within the brain, as several studies indicate that microglial-like cells are released into the circulation from the bone marrow and home into the brain [115, 150-152]. While these monocytic, microglial-like macrophages are not of the same lineage of resident microglia [153, 154], both cell types respond to sites of neuronal damage and actively phagocytize debris [154-158]. Similar to astrocytes, microglia can release angiogenic factors such as VEGF2, TGF-β and FGF [159-162]. Moreover, microglia have a critical role in maintaining neurovascular integrity [163] and participate in synaptic pruning [164, 165]. Thus, the recruitment and/or activation of glial cells, both astrocytes and microglia (microglial-like cells), could greatly influence the neurovascular unit and, ultimately cognitive function, following WBRT and/or hypoxia.
Additional Effects of Radiation that Suppress Angiogenesis
The prevailing view in the field is that one of the primary consequences of radiation is the suppression of cellular proliferation. Cellular senescence, associated with aging and cancer suppression, is recognized as one of the processes by which cellular proliferation is regulated [166, 167]. Senescent cells express markers such as p16ink4a also known as CDKN2A [167-169], p38MAPK [170] and have high senescence associated beta galactosidase (SA β-gal) activity [171, 172]. Recent studies demonstrate that radiation induces premature senescence in vitro [173] and in vivo, senescent cells can persist at least to six months after radiation in mice [174]. Even though cellular senescence occurs following radiation, it remains unclear whether senescence per se is a contributing factor in impaired angiogenesis and it is not known whether specific cells are more susceptible to senescence than others. Determining whether endothelial cells, pericytes and/or microglia in the brain adapt a senescent phenotype will be important in understanding why capillary density is not restored following WBRT.
Summary & Conclusions
We and others have demonstrated that WBRT induces significant cerebral microvascular rarefaction in brain regions important for learning and memory. The relationship between the reduction in capillary density and impaired cognitive function is based on the close temporal association between these events, the well-recognized role of the vasculature in maintenance of neuronal function, and studies indicating that interventions that restore vascular density are able to recover cognitive function. Importantly, these studies indicate that the WBRT-induced decline in cognitive function does not result from an intractable change in brain structure.
The profound decrease in vascular density that occurs after WBRT results in a localized tissue hypoxia which would normally be repaired by stimulation of local angiogenic mechanisms. However, these processes appear to be damaged by WBRT whereas vasculogenesis, which depends on cells derived from the bone marrow, remains intact. The mechanisms for the deficits in local angiogenesis are poorly understood but may be related to the development of an accelerated ‘senescent cell phenotype’ in the endothelial cells that remain after WBRT and fail to undergo apoptosis. Cellular senescence and the development of a ‘secretory associated senescent phenotype’ may, in part, explain the chronic inflammation and oxidative stress that persist in the brain after WBRT. Nevertheless if properly stimulated, bone marrow derived cells can home to sites of vascular injury where they participate in new vessel formation or replacement of damaged endothelial cells. An important question remains, however, about the specific cell types that are transported to the brain and the mechanisms that contribute to vascular repair and ultimately cognitive recovery. Theoretically EPCs may home to brain and participate in rebuilding the vasculature. More likely however, circulating myeloid progenitor cells may be transported to brain and develop into glial-like cells that stimulate proliferation of existing vascular networks. This is a complex area of investigation, and the cell types and their secretory products will need to be identified before any therapeutic interventions can be designed. Thus, the specific mechanisms that contribute to vascular recovery after WBRT will only be addressed through additional research.
Acknowledgments
Sources of Support: This work was supported by grants from the American Heart Association (Pre-doctoral Fellowship to JPW), American Federation for Aging Research (to AC), the Oklahoma Center for the Advancement of Science and Technology (to AC, ZU, WES), the American Heart Association (to AC and ZU), the NIH (AG031085 to AC; AT006526 to ZU; AG038747, NS056218 and P01 AG11370 to WES) and the Ellison Medical Foundation (to WES). The authors would like to express their gratitude for the support of the Donald W. Reynolds Foundation, which funds aging research at the University of Oklahoma Health Sciences Center under its Aging and Quality of Life Program.
Footnotes
Disclosure/Conflict of Interest: None.
Literature Cited
- 1.American Cancer Society. Cancer Facts & Figures 2013. American Cancer Society; 2013. [Google Scholar]
- 2.American Brain Tumor Association. Facts & Statistics, 2013 2013 [Google Scholar]
- 3.Zhang X, et al. A Review of Current Management of Brain Metastases. Annals of surgical oncology. 2011 doi: 10.1245/s10434-011-2019-2. [DOI] [PubMed] [Google Scholar]
- 4.Khuntia D, et al. Whole-brain radiotherapy in the management of brain metastasis. J Clin Oncol. 2006;24:1295–1304. doi: 10.1200/JCO.2005.04.6185. [DOI] [PubMed] [Google Scholar]
- 5.Robbins M, et al. Radiation-induced kidney injury: a role for chronic oxidative stress? Micron. 2002;33:133–141. doi: 10.1016/s0968-4328(01)00006-3. [DOI] [PubMed] [Google Scholar]
- 6.Tofilon P, Fike J. The radioresponse of the central nervous system: a dynamic process. Radiat Res. 2000;153:357–370. doi: 10.1667/0033-7587(2000)153[0357:trotcn]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 7.Schultheiss T, Stephens L. Invited review: permanent radiation myelopathy. Br J Radiol. 1992;65:737–753. doi: 10.1259/0007-1285-65-777-737. [DOI] [PubMed] [Google Scholar]
- 8.Crossen J, et al. Neurobehavioral sequelae of cranial irradiation in adults: a review of radiation-induced encephalopathy. J Clin Oncol. 1994;12:627–642. doi: 10.1200/JCO.1994.12.3.627. [DOI] [PubMed] [Google Scholar]
- 9.Imperato J, et al. Effects of treatment on long-term survivors with malignant astrocytomas. Ann Neurol. 1990;28:818–822. doi: 10.1002/ana.410280614. [DOI] [PubMed] [Google Scholar]
- 10.Johannesen T, et al. Radiological and clinical assessment of long-term brain tumour survivors after radiotherapy. Radiother Oncol. 2003;69:169–176. doi: 10.1016/s0167-8140(03)00192-0. [DOI] [PubMed] [Google Scholar]
- 11.Welzel G, et al. Acute neurocognitive impairment during cranial radiation therapy in patients with intracranial tumors. Strahlenther Onkol. 2008;184:647–654. doi: 10.1007/s00066-008-1830-6. [DOI] [PubMed] [Google Scholar]
- 12.Welzel G, et al. Memory function before and after whole brain radiotherapy in patients with and without brain metastases. Int J Radiat Oncol Biol Phys. 2008;72:1311–1318. doi: 10.1016/j.ijrobp.2008.03.009. [DOI] [PubMed] [Google Scholar]
- 13.Silber J, et al. Whole-brain irradiation and decline in intelligence: the influence of dose and age on IQ score. J Clin Oncol. 1992;10:1390–1396. doi: 10.1200/JCO.1992.10.9.1390. [DOI] [PubMed] [Google Scholar]
- 14.Li J, et al. Relationship between neurocognitive function and quality of life after whole-brain radiotherapy in patients with brain metastasis. Int J Radiat Oncol Biol Phys. 2008;71:64–70. doi: 10.1016/j.ijrobp.2007.09.059. [DOI] [PubMed] [Google Scholar]
- 15.Liu Y, et al. An experimental study of acute radiation-induced cognitive dysfunction in a young rat model. AJNR Am J Neuroradiol. 2010;31:383–387. doi: 10.3174/ajnr.A1801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Akiyama K, et al. Cognitive dysfunction and histological findings in adult rats one year after whole brain irradiation. Neurol Med Chir (Tokyo) 2001;41:590–598. doi: 10.2176/nmc.41.590. [DOI] [PubMed] [Google Scholar]
- 17.Shi L, et al. Spatial learning and memory deficits after whole-brain irradiation are associated with changes in NMDA receptor subunits in the hippocampus. Radiat Res. 2006;166:892–899. doi: 10.1667/RR0588.1. [DOI] [PubMed] [Google Scholar]
- 18.Brown W, et al. Capillary loss precedes the cognitive impairment induced by fractionated whole-brain irradiation: a potential rat model of vascular dementia. J Neurol Sci. 2007;257:67–71. doi: 10.1016/j.jns.2007.01.014. [DOI] [PubMed] [Google Scholar]
- 19.Rola R, et al. Radiation-induced impairment of hippocampal neurogenesis is associated with cognitive deficits in young mice. Experimental neurology. 2004;188:316–330. doi: 10.1016/j.expneurol.2004.05.005. [DOI] [PubMed] [Google Scholar]
- 20.Raber J, et al. Radiation-induced cognitive impairments are associated with changes in indicators of hippocampal neurogenesis. Radiat Res. 2004;162:39–47. doi: 10.1667/rr3206. [DOI] [PubMed] [Google Scholar]
- 21.Warrington JP, et al. Whole brain radiation-induced impairments in learning and memory are time-sensitive and reversible by systemic hypoxia. PloS one. 2012;7:e30444. doi: 10.1371/journal.pone.0030444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schindler M, et al. Aging-dependent changes in the radiation response of the adult rat brain. Int J Radiat Oncol Biol Phys. 2008;70:826–834. doi: 10.1016/j.ijrobp.2007.10.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee WH, et al. Irradiation induces regionally specific alterations in pro-inflammatory environments in rat brain. Int J Radiat Biol. 2010;86:132–144. doi: 10.3109/09553000903419346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rastogi S, et al. TNF-α response of vascular endothelial and vascular smooth muscle cells involve differential utilization of ASK1 kinase and p73. Cell Death Differ. 2011 doi: 10.1038/cdd.2011.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Desouza CV, et al. Role of inflammation and insulin resistance in endothelial progenitor cell dysfunction. Diabetes. 2011;60:1286–1294. doi: 10.2337/db10-0875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Quillard T, et al. Inflammation dysregulates Notch signaling in endothelial cells: implication of Notch2 and Notch4 to endothelial dysfunction. Biochem Pharmacol. 2010;80:2032–2041. doi: 10.1016/j.bcp.2010.07.010. [DOI] [PubMed] [Google Scholar]
- 27.Kalm M, et al. Transient inflammation in neurogenic regions after irradiation of the developing brain. Radiat Res. 2009;171:66–76. doi: 10.1667/RR1269.1. [DOI] [PubMed] [Google Scholar]
- 28.Lee WH, et al. Aging attenuates radiation-induced expression of pro-inflammatory mediators in rat brain. Neuroscience letters. 2010;476:89–93. doi: 10.1016/j.neulet.2010.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Linard C, et al. Abdominal irradiation increases inflammatory cytokine expression and activates NF-kappaB in rat ileal muscularis layer. Am J Physiol Gastrointest Liver Physiol. 2003;285:G556–565. doi: 10.1152/ajpgi.00094.2003. [DOI] [PubMed] [Google Scholar]
- 30.Shan Y, et al. Ionizing radiation stimulates secretion of pro-inflammatory cytokines: dose-response relationship, mechanisms and implications. Radiat Environ Biophys. 2007;46:21–29. doi: 10.1007/s00411-006-0076-x. [DOI] [PubMed] [Google Scholar]
- 31.Haase MG, et al. Sustained elevation of NF-kappaB DNA binding activity in radiation-induced lung damage in rats. Int J Radiat Biol. 2003;79:863–877. doi: 10.1080/09553000310001632903. [DOI] [PubMed] [Google Scholar]
- 32.Johnston CJ, et al. Radiation-induced pulmonary fibrosis: examination of chemokine and chemokine receptor families. Radiat Res. 2002;157:256–265. doi: 10.1667/0033-7587(2002)157[0256:ripfeo]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 33.Li Y, et al. Endothelial apoptosis initiates acute blood-brain barrier disruption after ionizing radiation. Cancer Res. 2003;63:5950–5956. [PubMed] [Google Scholar]
- 34.Lee WH, et al. Radiation attenuates physiological angiogenesis by differential expression of VEGF, Ang-1, tie-2 and Ang-2 in rat brain. Radiat Res. 2011;176:753–760. doi: 10.1667/rr2647.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li Y, et al. Early radiation-induced endothelial cell loss and blood-spinal cord barrier breakdown in the rat spinal cord. Radiat Res. 2004;161:143–152. doi: 10.1667/rr3117. [DOI] [PubMed] [Google Scholar]
- 36.Kamiryo T, et al. Radiosurgery-induced microvascular alterations precede necrosis of the brain neuropil. Neurosurgery. 2001;49:409–414. doi: 10.1097/00006123-200108000-00026. discussion 414-405. [DOI] [PubMed] [Google Scholar]
- 37.Lee WH, et al. Irradiation alters MMP-2/TIMP- 2 system and collagen type IV degradation in brain. International Journal of Radiation Oncology Biology Physics. 2011 doi: 10.1016/j.ijrobp.2010.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Brown W, et al. Vascular damage after fractionated whole-brain irradiation in rats. Radiat Res. 2005;164:662–668. doi: 10.1667/rr3453.1. [DOI] [PubMed] [Google Scholar]
- 39.Ljubimova N, et al. Endothelial cell population dynamics in rat brain after local irradiation. Br J Radiol. 1991;64:934–940. doi: 10.1259/0007-1285-64-766-934. [DOI] [PubMed] [Google Scholar]
- 40.Mao X, et al. High-LET radiation-induced response of microvessels in the Hippocampus. Radiat Res. 2010;173:486–493. doi: 10.1667/RR1728.1. [DOI] [PubMed] [Google Scholar]
- 41.Warrington JP, et al. Cerebral microvascular rarefaction induced by whole brain radiation is reversible by systemic hypoxia in mice. Am J Physiol Heart Circ Physiol. 2011;300:H736–744. doi: 10.1152/ajpheart.01024.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gjedde A, Diemer N. Double-tracer study of the fine regional blood-brain glucose transfer in the rat by computer-assisted autoradiography. Journal of cerebral blood flow and metabolism : official journal of the International Society of Cerebral Blood Flow and Metabolism. 1985;5:282–289. doi: 10.1038/jcbfm.1985.36. [DOI] [PubMed] [Google Scholar]
- 43.Zhao R, Pollack G. Regional differences in capillary density, perfusion rate, and P-glycoprotein activity: a quantitative analysis of regional drug exposure in the brain. Biochem Pharmacol. 2009;78:1052–1059. doi: 10.1016/j.bcp.2009.06.001. [DOI] [PubMed] [Google Scholar]
- 44.Spence AM, et al. Blood flow changes following 137Cs irradiation in a rat glioma model. Radiat Res. 1988;115:586–594. [PubMed] [Google Scholar]
- 45.Fuss M, et al. Radiation-induced regional cerebral blood volume (rCBV) changes in normal brain and low-grade astrocytomas: quantification and time and dose-dependent occurrence. Int J Radiat Oncol Biol Phys. 2000;48:53–58. doi: 10.1016/s0360-3016(00)00590-3. [DOI] [PubMed] [Google Scholar]
- 46.Clavo B, et al. Modification of glucose metabolism in radiation-induced brain injury areas using cervical spinal cord stimulation. Acta Neurochir (Wien) 2009;151:1419–1425. doi: 10.1007/s00701-009-0400-8. [DOI] [PubMed] [Google Scholar]
- 47.Hahn C, et al. Dose-dependent effects of radiation therapy on cerebral blood flow, metabolism, and neurocognitive dysfunction. Int J Radiat Oncol Biol Phys. 2009;73:1082–1087. doi: 10.1016/j.ijrobp.2008.05.061. [DOI] [PubMed] [Google Scholar]
- 48.Taki S, et al. Changes in regional cerebral blood flow in irradiated regions and normal brain after stereotactic radiosurgery. Ann Nucl Med. 2002;16:273–277. doi: 10.1007/BF03000106. [DOI] [PubMed] [Google Scholar]
- 49.Conway E, et al. Molecular mechanisms of blood vessel growth. Cardiovasc Res. 2001;49:507–521. doi: 10.1016/s0008-6363(00)00281-9. [DOI] [PubMed] [Google Scholar]
- 50.Caballero S, et al. Ischemic vascular damage can be repaired by healthy, but not diabetic, endothelial progenitor cells. Diabetes. 2007;56:960–967. doi: 10.2337/db06-1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hunting C, et al. Circulating endothelial (progenitor) cells reflect the state of the endothelium: vascular injury, repair and neovascularization. Vox Sang. 2005;88:1–9. doi: 10.1111/j.1423-0410.2005.00589.x. [DOI] [PubMed] [Google Scholar]
- 52.Yoder M. Defining human endothelial progenitor cells. J Thromb Haemost. 2009;7(Suppl 1):49–52. doi: 10.1111/j.1538-7836.2009.03407.x. [DOI] [PubMed] [Google Scholar]
- 53.Ahn G, Brown J. Role of endothelial progenitors and other bone marrow-derived cells in the development of the tumor vasculature. Angiogenesis. 2009;12:159–164. doi: 10.1007/s10456-009-9135-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Leone A, et al. From bone marrow to the arterial wall: the ongoing tale of endothelial progenitor cells. Eur Heart J. 2009;30:890–899. doi: 10.1093/eurheartj/ehp078. [DOI] [PubMed] [Google Scholar]
- 55.Kioi M, et al. Inhibition of vasculogenesis, but not angiogenesis, prevents the recurrence of glioblastoma after irradiation in mice. J Clin Invest. 2010;120:694–705. doi: 10.1172/JCI40283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Asahara T, et al. Isolation of putative progenitor endothelial cells for angiogenesis. Science. 1997;275:964–967. doi: 10.1126/science.275.5302.964. [DOI] [PubMed] [Google Scholar]
- 57.Peichev M, et al. Expression of VEGFR-2 and AC133 by circulating human CD34(+) cells identifies a population of functional endothelial precursors. Blood. 2000;95:952–958. [PubMed] [Google Scholar]
- 58.Urbich C, Dimmeler S. Endothelial progenitor cells: characterization and role in vascular biology. Circ Res. 2004;95:343–353. doi: 10.1161/01.RES.0000137877.89448.78. [DOI] [PubMed] [Google Scholar]
- 59.Gill M, et al. Vascular trauma induces rapid but transient mobilization of VEGFR2(+)AC133(+) endothelial precursor cells. Circ Res. 2001;88:167–174. doi: 10.1161/01.res.88.2.167. [DOI] [PubMed] [Google Scholar]
- 60.Valgimigli M, et al. CD34+ and endothelial progenitor cells in patients with various degrees of congestive heart failure. Circulation. 2004;110:1209–1212. doi: 10.1161/01.CIR.0000136813.89036.21. [DOI] [PubMed] [Google Scholar]
- 61.Laing A, et al. Mobilization of endothelial precursor cells: systemic vascular response to musculoskeletal trauma. J Orthop Res. 2007;25:44–50. doi: 10.1002/jor.20228. [DOI] [PubMed] [Google Scholar]
- 62.Brunner M, et al. Elevated levels of circulating endothelial progenitor cells in head and neck cancer patients. J Surg Oncol. 2008;98:545–550. doi: 10.1002/jso.21139. [DOI] [PubMed] [Google Scholar]
- 63.Heeschen C, et al. Endothelial progenitor cells participate in nicotine-mediated angiogenesis. J Am Coll Cardiol. 2006;48:2553–2560. doi: 10.1016/j.jacc.2006.07.066. [DOI] [PubMed] [Google Scholar]
- 64.Allan D, et al. Mobilization of circulating vascular progenitors in cancer patients receiving external beam radiation in response to tissue injury. Int J Radiat Oncol Biol Phys. 2009;75:220–224. doi: 10.1016/j.ijrobp.2009.04.033. [DOI] [PubMed] [Google Scholar]
- 65.Bonello L, et al. Endothelial injury induced by coronary angioplasty triggers mobilization of endothelial progenitor cells in patients with stable coronary artery disease. J Thromb Haemost. 2006;4:979–981. doi: 10.1111/j.1538-7836.2006.01858.x. [DOI] [PubMed] [Google Scholar]
- 66.Condon E, et al. Surgical injury induces the mobilization of endothelial progenitor cells. Surgery. 2004;135:657–661. doi: 10.1016/j.surg.2003.10.012. [DOI] [PubMed] [Google Scholar]
- 67.Bao X, et al. Transplantation of Flk-1+ human bone marrow-derived mesenchymal stem cells promotes angiogenesis and neurogenesis after cerebral ischemia in rats. Eur J Neurosci. 2011;34:87–98. doi: 10.1111/j.1460-9568.2011.07733.x. [DOI] [PubMed] [Google Scholar]
- 68.Woywodt A, et al. Circulating endothelial cells: life, death, detachment and repair of the endothelial cell layer. Nephrol Dial Transplant. 2002;17:1728–1730. doi: 10.1093/ndt/17.10.1728. [DOI] [PubMed] [Google Scholar]
- 69.Amabile N, et al. Circulating endothelial microparticle levels predict hemodynamic severity of pulmonary hypertension. Am J Respir Crit Care Med. 2008;177:1268–1275. doi: 10.1164/rccm.200710-1458OC. [DOI] [PubMed] [Google Scholar]
- 70.Bonello L, et al. Level of adenosine diphosphate receptor P2Y12 blockade during percutaneous coronary intervention predicts the extent of endothelial injury, assessed by circulating endothelial cell measurement. J Am Coll Cardiol. 2010;56:1024–1031. doi: 10.1016/j.jacc.2010.01.072. [DOI] [PubMed] [Google Scholar]
- 71.Gao Y, et al. Circulating endothelial cells as potential markers of atherosclerosis. Can J Neurol Sci. 2008;35:638–642. doi: 10.1017/s0317167100009446. [DOI] [PubMed] [Google Scholar]
- 72.Carmeliet P. Mechanisms of angiogenesis and arteriogenesis. Nat Med. 2000;6:389–395. doi: 10.1038/74651. [DOI] [PubMed] [Google Scholar]
- 73.Carmeliet P, Jain RK. Molecular mechanisms and clinical applications of angiogenesis. Nature. 2011;473:298–307. doi: 10.1038/nature10144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Rabbani Z, et al. Hypoxia inducible factor 1alpha signaling in fractionated radiation-induced lung injury: role of oxidative stress and tissue hypoxia. Radiat Res. 2010;173:165–174. doi: 10.1667/RR1816.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Cao R, et al. Hypoxia-induced retinal angiogenesis in zebrafish as a model to study retinopathy. PloS one. 2008;3:e2748. doi: 10.1371/journal.pone.0002748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ingraham JP, et al. Aging reduces hypoxia-induced microvascular growth in the rodent hippocampus. J Gerontol A Biol Sci Med Sci. 2008;63:12–20. doi: 10.1093/gerona/63.1.12. [DOI] [PubMed] [Google Scholar]
- 77.Liao D, Johnson R. Hypoxia: a key regulator of angiogenesis in cancer. Cancer Metastasis Rev. 2007;26:281–290. doi: 10.1007/s10555-007-9066-y. [DOI] [PubMed] [Google Scholar]
- 78.Denko N. Hypoxia, HIF1 and glucose metabolism in the solid tumour. Nat Rev Cancer. 2008 doi: 10.1038/nrc2468. [DOI] [PubMed] [Google Scholar]
- 79.Pugh C, Ratcliffe P. Regulation of angiogenesis by hypoxia: role of the HIF system. Nat Med. 2003;9:677–684. doi: 10.1038/nm0603-677. [DOI] [PubMed] [Google Scholar]
- 80.Udagawa T, et al. Chronic suppression of angiogenesis following radiation exposure is independent of hematopoietic reconstitution. Cancer Res. 2007;67:2040–2045. doi: 10.1158/0008-5472.CAN-06-2877. [DOI] [PubMed] [Google Scholar]
- 81.Wilson MH, et al. Cerebral artery dilatation maintains cerebral oxygenation at extreme altitude and in acute hypoxia-an ultrasound and MRI study. Journal of cerebral blood flow and metabolism : official journal of the International Society of Cerebral Blood Flow and Metabolism. 2011 doi: 10.1038/jcbfm.2011.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Subudhi AW, et al. Acute hypoxia impairs dynamic cerebral autoregulation: results from two independent techniques. J Appl Physiol. 2009;107:1165–1171. doi: 10.1152/japplphysiol.00498.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Schröder K, et al. NADPH oxidase Nox2 is required for hypoxia-induced mobilization of endothelial progenitor cells. Circ Res. 2009;105:537–544. doi: 10.1161/CIRCRESAHA.109.205138. [DOI] [PubMed] [Google Scholar]
- 84.Angelini DJ, et al. Hypoxia-induced mitogenic factor (HIMF/FIZZ1/RELM alpha) recruits bone marrow-derived cells to the murine pulmonary vasculature. PloS one. 2010;5:e11251. doi: 10.1371/journal.pone.0011251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Vilar J, et al. Chronic hypoxia-induced angiogenesis normalizes blood pressure in spontaneously hypertensive rats. Circ Res. 2008;103:761–769. doi: 10.1161/CIRCRESAHA.108.182758. [DOI] [PubMed] [Google Scholar]
- 86.Fan Y, et al. Del-1 gene transfer induces cerebral angiogenesis in mice. Brain Res. 2008;1219:1–7. doi: 10.1016/j.brainres.2008.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Chen SH, et al. Umbilical cord blood-derived CD34+ cells improve outcomes of traumatic brain injury in rats by stimulating angiogenesis and neurogenesis. Cell Transplant. 2013 doi: 10.3727/096368913X667006. [DOI] [PubMed] [Google Scholar]
- 88.Zhang P, et al. Early exercise improves cerebral blood flow through increased angiogenesis in experimental stroke rat model. J Neuroeng Rehabil. 2013;10:43. doi: 10.1186/1743-0003-10-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Fisher M. Pericyte signaling in the neurovascular unit. Stroke. 2009;40:S13–15. doi: 10.1161/STROKEAHA.108.533117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Zhu C, et al. Irradiation to the immature brain attenuates neurogenesis and exacerbates subsequent hypoxic-ischemic brain injury in the adult. Journal of neurochemistry. 2009;111:1447–1456. doi: 10.1111/j.1471-4159.2009.06413.x. [DOI] [PubMed] [Google Scholar]
- 91.Jahanshahi M, et al. Radiation-induced lowered neurogenesis associated with shortened latency of inhibitory avoidance memory response. Folia Neuropathol. 2011;49:103–108. [PubMed] [Google Scholar]
- 92.Kim S, et al. Gamma-irradiation is more efficient at depleting hippocampal neurogenesis than D-galactose/NaNO2. Neuroscience letters. 2011;498:47–51. doi: 10.1016/j.neulet.2011.04.059. [DOI] [PubMed] [Google Scholar]
- 93.Achanta P, et al. Ionizing radiation impairs the formation of trace fear memories and reduces hippocampal neurogenesis. Behav Neurosci. 2009;123:1036–1045. doi: 10.1037/a0016870. [DOI] [PubMed] [Google Scholar]
- 94.Fishman K, et al. Radiation-induced reductions in neurogenesis are ameliorated in mice deficient in CuZnSOD or MnSOD. Free Radic Biol Med. 2009;47:1459–1467. doi: 10.1016/j.freeradbiomed.2009.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Monje M. Cranial radiation therapy and damage to hippocampal neurogenesis. Dev Disabil Res Rev. 2008;14:238–242. doi: 10.1002/ddrr.26. [DOI] [PubMed] [Google Scholar]
- 96.Manda K, et al. Cranial irradiation-induced inhibition of neurogenesis in hippocampal dentate gyrus of adult mice: attenuation by melatonin pretreatment. J Pineal Res. 2009;46:71–78. doi: 10.1111/j.1600-079X.2008.00632.x. [DOI] [PubMed] [Google Scholar]
- 97.Noguès X, et al. Functions for adult neurogenesis in memory: An introduction to the neurocomputational approach and to its contribution. Behav Brain Res. 2011 doi: 10.1016/j.bbr.2011.08.009. [DOI] [PubMed] [Google Scholar]
- 98.Marín-Burgin A, Schinder AF. Requirement of adult-born neurons for hippocampus-dependent learning. Behav Brain Res. 2011 doi: 10.1016/j.bbr.2011.07.001. [DOI] [PubMed] [Google Scholar]
- 99.Palmer T, et al. Vascular niche for adult hippocampal neurogenesis. J Comp Neurol. 2000;425:479–494. doi: 10.1002/1096-9861(20001002)425:4<479::aid-cne2>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- 100.Coremans V, et al. Impaired neurogenesis, learning and memory and low seizure threshold associated with loss of neural precursor cell survivin. BMC Neurosci. 2010;11:2. doi: 10.1186/1471-2202-11-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Acharya M, et al. Rescue of radiation-induced cognitive impairment through cranial transplantation of human embryonic stem cells. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:19150–19155. doi: 10.1073/pnas.0909293106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Acharya MM, et al. Human neural stem cell transplantation ameliorates radiation-induced cognitive dysfunction. Cancer Res. 2011;71:4834–4845. doi: 10.1158/0008-5472.CAN-11-0027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Kerr AL, et al. Angiogenesis but not neurogenesis is critical for normal learning and memory acquisition. Neuroscience. 2010;171:214–226. doi: 10.1016/j.neuroscience.2010.08.008. [DOI] [PubMed] [Google Scholar]
- 104.Monje ML, et al. Irradiation induces neural precursor-cell dysfunction. Nat Med. 2002;8:955–962. doi: 10.1038/nm749. [DOI] [PubMed] [Google Scholar]
- 105.Plane JM, et al. Intact and injured endothelial cells differentially modulate postnatal murine forebrain neural stem cells. Neurobiology of disease. 2010;37:218–227. doi: 10.1016/j.nbd.2009.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Wang YQ, et al. VEGF overexpression enhances striatal neurogenesis in brain of adult rat after a transient middle cerebral artery occlusion. Journal of neuroscience research. 2007;85:73–82. doi: 10.1002/jnr.21091. [DOI] [PubMed] [Google Scholar]
- 107.Zhang H, et al. VEGF is a chemoattractant for FGF-2-stimulated neural progenitors. J Cell Biol. 2003;163:1375–1384. doi: 10.1083/jcb.200308040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Calvo CF, et al. Vascular endothelial growth factor receptor 3 directly regulates murine neurogenesis. Genes Dev. 2011;25:831–844. doi: 10.1101/gad.615311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Shang J, et al. Strong neurogenesis, angiogenesis, synaptogenesis, and antifibrosis of hepatocyte growth factor in rats brain after transient middle cerebral artery occlusion. Journal of neuroscience research. 2011;89:86–95. doi: 10.1002/jnr.22524. [DOI] [PubMed] [Google Scholar]
- 110.Sehara Y, et al. Potentiation of neurogenesis and angiogenesis by G-CSF after focal cerebral ischemia in rats. Brain research. 2007;1151:142–149. doi: 10.1016/j.brainres.2007.01.149. [DOI] [PubMed] [Google Scholar]
- 111.Fagel DM, et al. Cortical neurogenesis enhanced by chronic perinatal hypoxia. Experimental neurology. 2006;199:77–91. doi: 10.1016/j.expneurol.2005.04.006. [DOI] [PubMed] [Google Scholar]
- 112.Jin K, et al. Stem cell factor stimulates neurogenesis in vitro and in vivo. J Clin Invest. 2002;110:311–319. doi: 10.1172/JCI15251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Zhu X, et al. Intermittent hypoxia promotes hippocampal neurogenesis and produces antidepressant-like effects in adult rats. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2010;30:12653–12663. doi: 10.1523/JNEUROSCI.6414-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.O'Neill TJ, et al. Mobilization of bone marrow-derived cells enhances the angiogenic response to hypoxia without transdifferentiation into endothelial cells. Circ Res. 2005;97:1027–1035. doi: 10.1161/01.RES.0000189259.69645.25. [DOI] [PubMed] [Google Scholar]
- 115.Yang M, et al. Changes in host blood factors and brain glia accompanying the functional recovery after systemic administration of bone marrow stem cells in ischemic stroke rats. Cell Transplant. 2010;19:1073–1084. doi: 10.3727/096368910X503415. [DOI] [PubMed] [Google Scholar]
- 116.Cho H, et al. Mobilized endothelial progenitor cells by granulocyte-macrophage colony-stimulating factor accelerate reendothelialization and reduce vascular inflammation after intravascular radiation. Circulation. 2003;108:2918–2925. doi: 10.1161/01.CIR.0000097001.79750.78. [DOI] [PubMed] [Google Scholar]
- 117.Ii M, et al. Endothelial progenitor thrombospondin-1 mediates diabetes-induced delay in reendothelialization following arterial injury. Circ Res. 2006;98:697–704. doi: 10.1161/01.RES.0000209948.50943.ea. [DOI] [PubMed] [Google Scholar]
- 118.Iwakura A, et al. Estrogen-mediated, endothelial nitric oxide synthase-dependent mobilization of bone marrow-derived endothelial progenitor cells contributes to reendothelialization after arterial injury. Circulation. 2003;108:3115–3121. doi: 10.1161/01.CIR.0000106906.56972.83. [DOI] [PubMed] [Google Scholar]
- 119.Lin H, et al. After vascular injury, heme oxygenase-1/carbon monoxide enhances re-endothelialization via promoting mobilization of circulating endothelial progenitor cells. J Thromb Haemost. 2009;7:1401–1408. doi: 10.1111/j.1538-7836.2009.03478.x. [DOI] [PubMed] [Google Scholar]
- 120.Walter D, et al. Statin therapy accelerates reendothelialization: a novel effect involving mobilization and incorporation of bone marrow-derived endothelial progenitor cells. Circulation. 2002;105:3017–3024. doi: 10.1161/01.cir.0000018166.84319.55. [DOI] [PubMed] [Google Scholar]
- 121.Hristov M, et al. Importance of CXC chemokine receptor 2 in the homing of human peripheral blood endothelial progenitor cells to sites of arterial injury. Circ Res. 2007;100:590–597. doi: 10.1161/01.RES.0000259043.42571.68. [DOI] [PubMed] [Google Scholar]
- 122.Kähler C, et al. Peripheral infusion of rat bone marrow derived endothelial progenitor cells leads to homing in acute lung injury. Respir Res. 2007;8:50. doi: 10.1186/1465-9921-8-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Kwon O, et al. Bone marrow-derived endothelial progenitor cells and endothelial cells may contribute to endothelial repair in the kidney immediately after ischemia-reperfusion. J Histochem Cytochem. 2010;58:687–694. doi: 10.1369/jhc.2010.956011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Murayama T, et al. Determination of bone marrow-derived endothelial progenitor cell significance in angiogenic growth factor-induced neovascularization in vivo. Exp Hematol. 2002;30:967–972. doi: 10.1016/s0301-472x(02)00867-6. [DOI] [PubMed] [Google Scholar]
- 125.Shao H, et al. Statin and stromal cell-derived factor-1 additively promote angiogenesis by enhancement of progenitor cells incorporation into new vessels. Stem Cells. 2008;26:1376–1384. doi: 10.1634/stemcells.2007-0785. [DOI] [PubMed] [Google Scholar]
- 126.Au P, et al. Differential in vivo potential of endothelial progenitor cells from human umbilical cord blood and adult peripheral blood to form functional long-lasting vessels. Blood. 2008;111:1302–1305. doi: 10.1182/blood-2007-06-094318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Zhang Y, et al. Release of proinflammatory mediators and expression of proinflammatory adhesion molecules by endothelial progenitor cells. Am J Physiol Heart Circ Physiol. 2009;296:H1675–1682. doi: 10.1152/ajpheart.00665.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Capoccia B, et al. G-CSF and AMD3100 mobilize monocytes into the blood that stimulate angiogenesis in vivo through a paracrine mechanism. Blood. 2006;108:2438–2445. doi: 10.1182/blood-2006-04-013755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Suh W, et al. Transplantation of endothelial progenitor cells accelerates dermal wound healing with increased recruitment of monocytes/macrophages and neovascularization. Stem Cells. 23:1571–1578. doi: 10.1634/stemcells.2004-0340. [DOI] [PubMed] [Google Scholar]
- 130.Armulik A, et al. Endothelial/pericyte interactions. Circ Res. 2005;97:512–523. doi: 10.1161/01.RES.0000182903.16652.d7. [DOI] [PubMed] [Google Scholar]
- 131.Lu C, Sood AK. Role of Pericytes in Angiogenesis. In: Teicher BA, editor. Cancer Drug Discovery and Development Angiogenic Agents in Cancer Therapy. Humana Press; 1999. [Google Scholar]
- 132.Shepro D, Morel N. Pericyte physiology. FASEB J. 1993;7:1031–1038. doi: 10.1096/fasebj.7.11.8370472. [DOI] [PubMed] [Google Scholar]
- 133.Hayashi K, et al. Effects of hypoxia on endothelial/pericytic co-culture model of the blood-brain barrier. Regul Pept. 2004;123:77–83. doi: 10.1016/j.regpep.2004.05.023. [DOI] [PubMed] [Google Scholar]
- 134.Rucker H, et al. Cellular mechanisms of CNS pericytes. Brain Res Bull. 2000;51:363–369. doi: 10.1016/s0361-9230(99)00260-9. [DOI] [PubMed] [Google Scholar]
- 135.Virgintino D, et al. An intimate interplay between precocious, migrating pericytes and endothelial cells governs human fetal brain angiogenesis. Angiogenesis. 2007;10:35–45. doi: 10.1007/s10456-006-9061-x. [DOI] [PubMed] [Google Scholar]
- 136.Rajantie I, et al. Adult bone marrow-derived cells recruited during angiogenesis comprise precursors for periendothelial vascular mural cells. Blood. 2004;104:2084–2086. doi: 10.1182/blood-2004-01-0336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Ozerdem U, et al. Contribution of bone marrow-derived pericyte precursor cells to corneal vasculogenesis. Investigative ophthalmology & visual science. 2005;46:3502–3506. doi: 10.1167/iovs.05-0309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Bonkowski D, et al. The CNS microvascular pericyte: pericyte-astrocyte crosstalk in the regulation of tissue survival. Fluids Barriers CNS. 2011;8:8. doi: 10.1186/2045-8118-8-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Koehler RC, et al. Astrocytes and the regulation of cerebral blood flow. Trends Neurosci. 2009;32:160–169. doi: 10.1016/j.tins.2008.11.005. [DOI] [PubMed] [Google Scholar]
- 140.Petzold GC, Murthy VN. Role of astrocytes in neurovascular coupling. Neuron. 2011;71:782–797. doi: 10.1016/j.neuron.2011.08.009. [DOI] [PubMed] [Google Scholar]
- 141.Allan S. The neurovascular unit and the key role of astrocytes in the regulation of cerebral blood flow. Cerebrovasc Dis. 2006;21:137–138. doi: 10.1159/000090447. [DOI] [PubMed] [Google Scholar]
- 142.Scott A, et al. Astrocyte-derived vascular endothelial growth factor stabilizes vessels in the developing retinal vasculature. PloS one. 2010;5:e11863. doi: 10.1371/journal.pone.0011863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Bozoyan L, et al. Astrocytes control the development of the migration-promoting vasculature scaffold in the postnatal brain via VEGF signaling. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2012;32:1687–1704. doi: 10.1523/JNEUROSCI.5531-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Koehler RC, et al. Role of astrocytes in cerebrovascular regulation. J Appl Physiol. 2006;100:307–317. doi: 10.1152/japplphysiol.00938.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Borlongan CV, et al. The great migration of bone marrow-derived stem cells toward the ischemic brain: therapeutic implications for stroke and other neurological disorders. Progress in neurobiology. 2011;95:213–228. doi: 10.1016/j.pneurobio.2011.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Eglitis MA, et al. Targeting of marrow-derived astrocytes to the ischemic brain. Neuroreport. 1999;10:1289–1292. doi: 10.1097/00001756-199904260-00025. [DOI] [PubMed] [Google Scholar]
- 147.Lu D, et al. Adult bone marrow stromal cells administered intravenously to rats after traumatic brain injury migrate into brain and improve neurological outcome. Neuroreport. 2001;12:559–563. doi: 10.1097/00001756-200103050-00025. [DOI] [PubMed] [Google Scholar]
- 148.Mahmood A, et al. Long-term recovery after bone marrow stromal cell treatment of traumatic brain injury in rats. Journal of neurosurgery. 2006;104:272–277. doi: 10.3171/jns.2006.104.2.272. [DOI] [PubMed] [Google Scholar]
- 149.Lu J, et al. Adult bone marrow cells differentiate into neural phenotypes and improve functional recovery in rats following traumatic brain injury. Neuroscience letters. 2006;398:12–17. doi: 10.1016/j.neulet.2005.12.053. [DOI] [PubMed] [Google Scholar]
- 150.Recio JS, et al. Bone marrow contributes simultaneously to different neural types in the central nervous system through different mechanisms of plasticity. Cell Transplant. 2011 doi: 10.3727/096368910X552826. [DOI] [PubMed] [Google Scholar]
- 151.Opydo-Chanek M, Dąbrowski Z. Response of astrocytes and microglia/macrophages to brain injury after bone marrow stromal cell transplantation: a quantitative study. Neuroscience letters. 2011;487:163–168. doi: 10.1016/j.neulet.2010.10.014. [DOI] [PubMed] [Google Scholar]
- 152.Brevet M, et al. Chronic foot-shock stress potentiates the influx of bone marrow-derived microglia into hippocampus. Journal of neuroscience research. 2010;88:1890–1897. doi: 10.1002/jnr.22362. [DOI] [PubMed] [Google Scholar]
- 153.Ginhoux F, et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science. 2010;330:841–845. doi: 10.1126/science.1194637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Ajami B, et al. Infiltrating monocytes trigger EAE progression, but do not contribute to the resident microglia pool. Nature neuroscience. 2011;14:1142–1149. doi: 10.1038/nn.2887. [DOI] [PubMed] [Google Scholar]
- 155.Hill WD, et al. SDF-1 (CXCL12) is upregulated in the ischemic penumbra following stroke: association with bone marrow cell homing to injury. Journal of neuropathology and experimental neurology. 2004;63:84–96. doi: 10.1093/jnen/63.1.84. [DOI] [PubMed] [Google Scholar]
- 156.Beck H, et al. Participation of bone marrow-derived cells in long-term repair processes after experimental stroke. Journal of cerebral blood flow and metabolism : official journal of the International Society of Cerebral Blood Flow and Metabolism. 2003;23:709–717. doi: 10.1097/01.WCB.0000065940.18332.8D. [DOI] [PubMed] [Google Scholar]
- 157.Tanaka R, et al. Migration of enhanced green fluorescent protein expressing bone marrow-derived microglia/macrophage into the mouse brain following permanent focal ischemia. Neuroscience. 2003;117:531–539. doi: 10.1016/s0306-4522(02)00954-5. [DOI] [PubMed] [Google Scholar]
- 158.Simard AR, et al. Bone marrow-derived microglia play a critical role in restricting senile plaque formation in Alzheimer's disease. Neuron. 2006;49:489–502. doi: 10.1016/j.neuron.2006.01.022. [DOI] [PubMed] [Google Scholar]
- 159.Lu YZ, et al. Molecular mechanisms responsible for microglia-derived protection of Sprague-Dawley rat brain cells during in vitro ischemia. Neuroscience letters. 2005;373:159–164. doi: 10.1016/j.neulet.2004.10.004. [DOI] [PubMed] [Google Scholar]
- 160.Shimojo M, et al. Production of basic fibroblast growth factor in cultured rat brain microglia. Neuroscience letters. 1991;123:229–231. doi: 10.1016/0304-3940(91)90937-o. [DOI] [PubMed] [Google Scholar]
- 161.Liu X, et al. Basic FGF and FGF receptor 1 are expressed in microglia during experimental autoimmune encephalomyelitis: temporally distinct expression of midkine and pleiotrophin. Glia. 1998;24:390–397. doi: 10.1002/(sici)1098-1136(199812)24:4<390::aid-glia4>3.0.co;2-1. [DOI] [PubMed] [Google Scholar]
- 162.Rymo SF, et al. A two-way communication between microglial cells and angiogenic sprouts regulates angiogenesis in aortic ring cultures. PloS one. 2011;6:e15846. doi: 10.1371/journal.pone.0015846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Han HS, Suk K. The function and integrity of the neurovascular unit rests upon the integration of the vascular and inflammatory cell systems. Curr Neurovasc Res. 2005;2:409–423. doi: 10.2174/156720205774962647. [DOI] [PubMed] [Google Scholar]
- 164.Tremblay M, et al. Microglial interactions with synapses are modulated by visual experience. PLoS Biol. 2010;8:e1000527. doi: 10.1371/journal.pbio.1000527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Paolicelli RC, et al. Synaptic pruning by microglia is necessary for normal brain development. Science. 2011;333:1456–1458. doi: 10.1126/science.1202529. [DOI] [PubMed] [Google Scholar]
- 166.Rodier F, Campisi J. Four faces of cellular senescence. J Cell Biol. 2011;192:547–556. doi: 10.1083/jcb.201009094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Rayess H, et al. Cellular senescence and tumor suppressor gene p16. Int J Cancer. 2011 doi: 10.1002/ijc.27316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Coppé JP, et al. Tumor Suppressor and Aging Biomarker p16INK4a Induces Cellular Senescence without the Associated Inflammatory Secretory Phenotype. The Journal of biological chemistry. 2011;286:36396–36403. doi: 10.1074/jbc.M111.257071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Bazarov AV, et al. p16(INK4a) -mediated suppression of telomerase in normal and malignant human breast cells. Aging Cell. 2010;9:736–746. doi: 10.1111/j.1474-9726.2010.00599.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Freund A, et al. p38MAPK is a novel DNA damage response-independent regulator of the senescence-associated secretory phenotype. EMBO J. 2011;30:1536–1548. doi: 10.1038/emboj.2011.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Geng YQ, et al. Senescence-associated beta-galactosidase activity expression in aging hippocampal neurons. Biochem Biophys Res Commun. 2010;396:866–869. doi: 10.1016/j.bbrc.2010.05.011. [DOI] [PubMed] [Google Scholar]
- 172.Itahana K, et al. Methods to detect biomarkers of cellular senescence: the senescence-associated beta-galactosidase assay. Methods Mol Biol. 2007;371:21–31. doi: 10.1007/978-1-59745-361-5_3. [DOI] [PubMed] [Google Scholar]
- 173.Yentrapalli R, et al. Quantitative proteomic analysis reveals induction of premature senescence in human umbilical vein endothelial cells exposed to chronic low-dose rate gamma radiation. Proteomics. 2013 doi: 10.1002/pmic.201200463. [DOI] [PubMed] [Google Scholar]
- 174.Seol MA, et al. Prolonged expression of senescence markers in mice exposed to gamma-irradiation. J Vet Sci. 2012;13:331–338. doi: 10.4142/jvs.2012.13.4.331. [DOI] [PMC free article] [PubMed] [Google Scholar]