

Abstract
Plants produce structurally diverse secondary (specialized) metabolites to increase their fitness for survival under adverse environments. Several bioactive compounds for new drugs have been identified through screening of plant extracts. In this study, genome-wide association studies (GWAS) were conducted to investigate the genetic architecture behind the natural variation of rice secondary metabolites. GWAS using the metabolome data of 175 rice accessions successfully identified 323 associations among 143 single nucleotide polymorphisms (SNPs) and 89 metabolites. The data analysis highlighted that levels of many metabolites are tightly associated with a small number of strong quantitative trait loci (QTLs). The tight association may be a mechanism generating strains with distinct metabolic composition through the crossing of two different strains. The results indicate that one plant species produces more diverse phytochemicals than previously expected, and plants still contain many useful compounds for human applications.
Keywords: Oryza sativa, secondary metabolites, metabolome analysis, genome-wide association study, natural variation
Introduction
Plants have the ability to produce a wide range of structurally diverse secondary (specialized) metabolites to increase survival fitness in various adverse environments (Schwab, 2003; Pichersky and Lewinsohn, 2011; Saito, 2013). For instance, certain molecules play roles in plant–insect interactions, such as glucosinolates in Arabidopsis thaliana and flavone glycosides in cereals (Kliebenstein et al., 2001c; Simmonds, 2001; Beekwilder et al., 2008). Based on the structural diversity, several bioactive compounds for new drugs have been identified through screening of extracts of various plant species. Recently, metabolomics studies revealed that the composition of secondary metabolites in plants is an inherently variable phenotype, as genetic polymorphisms cause large qualitative and quantitative variations in metabolic phenotypes (metabolotypes) among cultivars and ecotypes (Chan et al., 2010a; Saito and Matsuda, 2010; Weigel, 2012; Carreno-Quintero et al., 2013). Although relatively tight genetic control of natural variations has been identified through metabolome quantitative trait loci (mQTL) analyses (Rowe et al., 2008; Schauer et al., 2008; Lisec et al., 2009; Matsuda et al., 2012; Gong et al., 2013), knowledge remains limited as to how diverse secondary metabolites are produced in a given plant species and the genetic architecture of qualitative and quantitative variations in the metabolic phenotype.
Genome-wide association study (GWAS) is a method for mapping the loci responsible for natural variations in a target phenotype by the identification of significantly associated genetic polymorphisms in a large population (Brachi et al., 2011; Weigel, 2012). GWAS has been widely used to identify loci that are related to various agronomically important traits, as well as to uncover the genetic architecture that controls these traits (Atwell et al., 2010; Huang et al., 2010; Chan et al., 2011; Zhao et al., 2011). The development of metabolomics tools in last decade has also facilitated the comprehensive phenotyping of metabolomic traits (Saito and Matsuda, 2010). Genome-wide association analyses of metabolomic traits of A. thaliana populations found that genotype–-metabolite associations form clusters of hotspots in regions under strong positive selection (Chan et al., 2010a). Metabolome-GWAS using maize also demonstrated that concentrations of multiple lignin precursors showed strong genetic associations with other agronomic traits (Riedelsheimer et al., 2012; Hill et al., 2013; Wen et al., 2014). Recently, metabolome-GWAS using rice showed that metabolic pathways could be reconstructed from genotype-metabolite associations (Chen et al., 2014; Dong et al., 2014). However, while complex modes of inheritance have been revealed by GWAS studies of metabolites, knowledge remains limited about the genetic architecture behind the structural diversity of secondary metabolism. Although GWAS of A. thaliana has confirmed the major polymorphic loci identified in biparental RIL populations controlled the large natural variation of glucosinolate levels, the applicability of these findings to other plant species requires more immense investigation (Kliebenstein et al., 2001a, b, c; Keurentjes et al., 2007; Chan et al., 2010b).
In this study, GWAS was conducted by analyzing the aerial part of 175 Japanese diverse rice (Oryza sativa) cultivar seedlings using liquid chromatography-tandem mass spectrometry (LC-MS/MS) for the non-targeted analysis of known and unknown metabolites (Bottcher et al., 2008; Matsuda et al., 2009). The analysis revealed that there are two types of genetic architectures responsible for the natural variations in the composition of secondary metabolites in the rice population. While the small number of mQTLs tightly associated with levels of one-third of analyzed metabolites, levels of other one-third of metabolites were under the smaller effect of multiple QTL.
Results and discussions
Large structural diversity of rice specialized metabolites
A metabolome dataset composed of 342 metabolite signals (peaks) in 668 samples was obtained using liquid chromatography-mass spectrometry (LC/MS) (Tables S1–S3) (Matsuda et al., 2009, 2010). Metabolite annotation successfully characterized the structures of 91 metabolites, demonstrating that phytochemicals produced in rice cultivars were more diverse than previously reported (Figure 1 and Table S4) (Besson et al., 1985; Mohanlal et al., 2011). Among the metabolite signals, 6 and 32 metabolite signals were ‘annotated’ and ‘identified’, respectively, on the basis of comparisons of MS/MS spectra, an exact mass number, and retention time with those of standards (Figure 2) (Yang et al., 2014). For further characterization of metabolite structure, a molecular MS/MS network was constructed by connecting two metabolite signals (nodes) that had similar MS/MS spectra (See Experimental procedures, blue edges in Figure 1). The molecular MS/MS network showed the presence of several clusters of metabolites. For instance, a cluster contains apigenin-6-C-α-l-arabinosyl-8-C-α-l-arabinoside 6 (referred to as apigenin-di-C-arabinoside, peak ID 33368), which has a MS/MS spectrum with a characteristic fragmentation pattern of flavone-C-glycoside (Figure 2a) (Cavaliere et al., 2005). The MS/MS spectrum of a neighborhood metabolite signal (ID 38198) in the network exhibits a similar fragmentation pattern, except for a larger mass number of the precursor ion (+CH2O, Figure 2b). Based on the similarity of MS/MS spectra, the metabolite signal was characterized to be apigenin-C-hexoside-C-pentoside 7. Using a similar procedure, 53 metabolite structures were partly ‘characterized’ in this study (Table S4).
Figure 1.
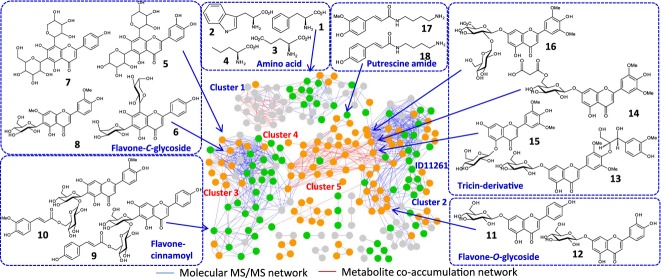
Combined metabolomics networks of rice.
Each node represents one metabolite signal. The molecular MS/MS network on MS/MS spectral similarity is shown as blue edges. Red edges represent the metabolite co-accumulation network on metabolites with similar accumulation patterns observed among the 175 rice cultivars. Interpretable networks were obtained by employing a threshold of similarity score above 0.7 for both networks. Clusters mentioned in the text are presented by circles. The structures of representative metabolites in each cluster are also shown. Metabolite names by the bold numbers are presented in Table S4. Nodes of metabolites with relatively large broad-sense heritability (H2 > 0.5) and significantly distorted from the normal distribution by Kolmogorov-Smirnov test (P < 0.01) are shown in orange color. Green nodes are metabolites with H2 > 0.5 and P > 0.01 (See legend of Figure 6b).
Figure 2.

Tandem mass (MS/MS) spectra of rice metabolites.
(a) Apigenin-6-C-α-l-arabinosyl-8-C-α-l-arabinoside 6 (peak ID 33368), (b) apigenin-C-hexoside-C-pentoside 7 (ID 38198), and (c) unknown metabolite (ID 11261). MS2T ID indicates the code of the representative MS/MS spectral tag of each metabolite in the RIKEN PRIME MS2T library.
Clustering of metabolites by MS/MS spectral similarities revealed that, in addition to several amino acids and putrescine amides (compounds 1–4, 17, and 18 in Figure 1), a series of flavonoids was produced in rice, including flavone-C-glycosides, flavone-O-glycosides, and tricin derivatives (5–16, blue edges in Figure 1) (Dong et al., 2014; Yang et al., 2014). While no tricin-C-glycoside was found in the metabolome data, several tricin-specific derivatives were present, including flavonolignans and tricin-glycosides (for instance, compounds 13–16 in Figure 1). The flavonolignans with tricin aglycone such as tricin 4′-O-(erythro-β-guaiacylglycerol) ether 7-O-β-d-glucopyranoside 13 have been found from monocot plants (Bouaziz et al., 2002; Chang et al., 2010). Furthermore, tricin 7-O-(6′′-O-malonyl)-β-d-glucoside 14 was first reported in our previous study(Yang et al., 2014). The tricin derivatives may contribute to rice physiology; their unique biological activities have been previously reported (Mohanlal et al., 2011). Furthermore, the presence of two uncharacterized clusters (clusters 1 and 2 in Figure 1) indicated that rice contains unknown metabolic functions that produced unknown metabolites such as peak ID 11261 (Figure 2c).
To investigate the coordinated regulation of metabolite levels, the metabolite co-accumulation network was constructed (red edges in Figure 1) and superimposed on the molecular MS/MS network. The metabolite co-accumulation network revealed the presence of several clusters of co-accumulated metabolites overlapping with the clusters of structurally similar metabolites. This trend was observed for amino acid and flavone-O-hexoside, indicating coordinated regulation of these metabolite contents. In comparison, the cluster of flavone-C-glycoside in the molecular MS/MS network was separated into two clusters of the metabolite co-accumulation network, indicating complex control of flavone-C-glycoside biosynthesis (clusters 3 and 4 in Figure 1).
Genome-wide association studies
GWAS were conducted for 342 metabolites using the genotype data of 3168 SNPs (Table S5) to indentify the mQTL responsible for metabolic phenotype variations (Yonemaru et al., 2012). The distribution of −log10 (P values) determined by the naïve analysis was far from the expected distribution, probably because of the high level of false positive signals that were derived from the genetic model without considering a population structure (Figure S1). The inflation of P-values by the naïve analysis has also been reported in previous studies (Chan et al., 2010a; Riedelsheimer et al., 2012). Thus, in this study, the efficient mixed-model association (EMMA) was employed to correct the confounding effects of population and genetic relatedness in the association mapping (Kang et al., 2008). The P-value distribution was close to the expected distribution when using the mixed-model.
As shown in Figure 3(a), 323 significant associations among 143 SNPs and 89 metabolites were observed when employing a relatively strict threshold (α = 1.0 × 10−5, false discovery ratio: 3.4%, Table S6). Red lines in Figure 3(b) show the associations among the SNPs and the metabolites (aligned on the upper and lower boundaries in the figure, respectively). We found that one polymorphism tends to affect the levels of multiple metabolites, as 113 of 143 SNPs were significantly associated with more than two metabolites (Figure 3b). Furthermore, gene ontology (GO) enrichment analysis suggested that polymorphisms in genes related to glycosylation and protein–protein interaction might play important roles in metabolotype variations. It is because genes categorized in transferring glycosyl groups and protein binding are frequently observed among 2244 genes encoded near the SNPs (Table S7).
Figure 3.

Genetic architecture of rice secondary metabolism.
(a) Manhattan plot for genome-wide association mapping of rice metabolic phenotypes. SNPs significantly associated with some metabolite levels were plotted on the rice genome (α = 1.0 × 10−3).
(b) Associations between 3168 SNPs aligned on the upper boundary and 342 metabolites aligned on the lower boundary. Positions of SNPs correspond to the above panel. Red, blue, and gray lines represent significant associations between SNPs and metabolites with threshold levels of α = 1.0 × 10−5, 5.0 × 10−5, and 1.0 × 10−3, respectively. Positions of metabolite clusters and representative metabolites are also represented (Table S4 for metabolite names).
It is also assumed that metabolite levels are controlled by the interaction of mQTLs (Rowe et al., 2008; Kliebenstein, 2009; Lisec et al., 2009; Chen et al., 2014). If epistasis is a major mode-of-inheritance with large effect on rice metabolic phenotypes, the metabolite levels of a cultivar would be significantly lesser or greater compared to the two parental cultivars. Since the rice population used in this study includes 38 sets of the cultivar and their parental cultivars (Table S1), the occurrence of the epistatic effect was investigated by comparing the levels of a metabolite among the cultivar and its parents. Among the 12 312 tests in total (38 sets of 342 metabolites), a higher and lower metabolite levels was observed in 166 and 173 cases (one-sided t-test at α = 0.01), respectively. Since the probabilities were close to false positive levels, the epistatic effect is unlikely to be a major mode-of-inheritance in rice metabolic phenotypes.
mQTLs responsible for the natural variation of metabolic phenotypes
The GWAS clearly showed that there are several hotspots of significantly associated SNPs. Among these genetic hubs, one of the most prominent hotspot regions is located around the short arm of chromosome 6, where the SNP genotype NIAS_Os_ac06000458 with G/A alleles was tightly associated with the levels of various flavone-C-glycosides (Figure 4a). For instance, the SNP genotype explained 68.6% of the total variation of the levels of apigenin-di-C-arabinoside 6 for 175 cultivars. Near the SNP marker, there were OsCGT gene encoding flavone C-glucosyltransferase that functions in the selective formation of 6C-glucosylflavone (Brazier-Hicks et al., 2009) and its two homologous UGT genes (Os06g0289200 and Os06g0289900, Figure 4b). The protein sequences of the two genes were similar to that of OsCGT (E-values determined by blastx using RAPDB were 1e-142 and 2e-112, respectively). While 6C-glucosylation is a basal metabolic function in the rice population, the capability to produce flavone-6C-arabinosides such as apigenin-di-C-arabinoside 6 is strictly associated with G genotype of this SNP (Figure 4c). These results indicate that 6C-arabinosylation should be associated with polymorphism related to UGT genes as has been reported in the previous study (Chen et al., 2014). The detailed structural characterization of metabolite signals performed in this study highlighted that the polymorphism is responsible for the 6-C-α-l-arabinosylation of flavones. A similar tight association was observed between the SNP marker on chromosome 4 (NIAS_Os_ac04000042), and the unknown metabolites in cluster 2 (Figure 1) including ID 11155, ID11269, and ID11261 (Figure 5a,b). The gene annotation data indicated that, among the six genes positioned in the mQTL candidate region, arginine decarboxylase gene (Os04g0107600) play a role in the polyamine biosynthesis and suggests a possible precursor for the metabolite biosynthesis (Table S8).
Figure 4.
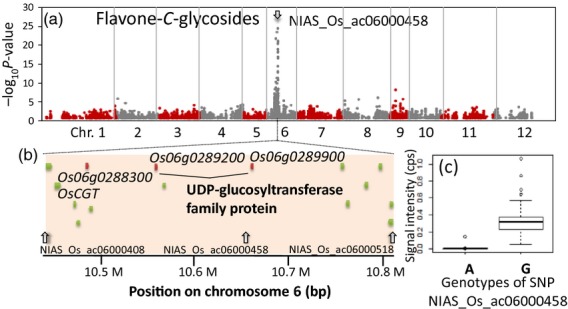
GWAS of 31 metabolites in flavone-C-glycoside cluster.
(a) Manhattan plot for 31 metabolites in flavone-C-glycoside cluster (α = 1.0 × 10−3). Position of SNPs associated with 6C-arabinosylation of flavone (NIAS_Os_ac06000458) is indicated by grey arrow.
(b) Rice genome region around the SNP marker, NIAS_Os_ac06000458 on chromosome 6.
(c) Associations between genotypes of NIAS_Os_ac06000458 and apigenin-di-C-arabinoside levels.
Figure 5.

Genome-wide association study of rice metabolites.
(a) Manhattan plot for unknown metabolite ID11621 and (b) its association with SNP genotypes NIAS_Os_ab040000042. (c) Manhattan plot for 9 flavone-O-glycosides including luteolin-7-O-glucoside and (d) association between luteolin-7-O-glucoside and NIAS_Os_aa01010133. (e) Manhattan plot for 7 amino acids and (f) association between phenylalaine and NIAS_Os_ab02000283.
The GWAS demonstrated a genetic background for the coordinated regulations of flavone-O-glycoside and amino acid contents, which were observed in the metabolite co-accumulation network (Figure 1). For instance, there was a SNP on chromosome 1 (NIAS_Os_aa01010133) that was significantly associated with luteolin-7-O-glucoside 12 and other flavone-O-glycoside contents (Figure 5c). The SNP genotype explained 21.5% of the total variance of luteolin-7-O-glucoside 12 levels in 175 rice accessions (Figure 5d). Since the position of NIAS_Os_aa01010133 was far from (0.31–0.38 Mb) the cluster of seven UDP-glucuronosyl/UDP-glucosyltransferase (UGT) genes such as UGT706D1 (Os01g0736300) responsible for the glucosylation of flavones (Ko et al., 2008), unknown molecular mechanism should be responsible for the coordinated regulation of the flavone-O-glycoside levels (Table S8).
GWAS data also showed that the genotype of SNP NIAS_Os_ab02000283 on chromosome 2 is significantly associated with phenylalanine 1 levels (−log10 P-value = 3.39 (Figure 5e). However, only 9% of the total variance could be explained by the SNP genotype (Figure 5f) indicating that amino acids levels are controlled by a relatively large number of weak mQTLs. This SNP was also associated with other amino acids in the cluster, such as leucine 4 and tryptophan 2. Among the four genes in the mQTL candidate region, Os02g0278700 showed homology with GA1 in Arabidopsis (At4g02780, E-value 6e-07 by TAIR) that encodes ent-copatyl diphosphate synthetase responsible for gibberellins biosynthesis (Table S8). However, since almost equal distribution was observed for the SNP genotypes (91 and 83 strains have ‘C’ and ‘G’ genotypes, respectively), the polymorphisms is unlikely to be associated with agronomically important traits such as plant height. It suggests that there should be other causal gene for the natural variation of amino acid levels. Many loci that were qualitatively associated with various metabolites were also found in this study. The detailed genotyping or genome sequencing of various rice cultivars will uncover the genetic polymorphisms controlling the observed natural variation (Gong et al., 2013; Wen et al., 2014).
Genetic architecture for house-keeping metabolites
The metabolite levels were controlled by both genetic and environmental factors. The relative standard deviations of each metabolite level largely varied across the 175 cultivars (Figure 6a). The variations were mainly derived from the genetic polymorphisms, because relatively large broad-sense heritabilities (H2 > 0.5) were observed for 234 (68.4%) metabolites (Figure 6b). For the metabolites that were dominantly controlled by genetic factors, a Kolmogorov–Smirnov test indicated that the quantitative variations of one-third (115/342) of the metabolites followed a normal distribution (P > 0.01) (Figure 6b). Metabolites in this group are represented as green nodes in Figure 1. This group included amino acids, some flavone glycosides, and flavonolignans such as phenylalanine 1 (Figure 6c), isovitexin 2″-O-(6‴-(E)-p-coumaroyl)-glucoside 9, and tricin 4′-O-(erythro-β-guaiacylglycerol) ether 7-O-β- d-glucopyranoside 13. Our GWAS demonstrated that the metabolites in this group are under the control of multiple mQTLs that are weakly associated with metabolic phenotypes. For instance, only 9% of the total variance in phenylalanine 1 levels can be explained by weakly associated SNP NIAS_Os_ab02000283 on chromosome 2 (Figure 5f). Similar GWAS results with marginal associations have been observed for several agronomically important traits, such as flowering time and grain weight (Huang et al., 2010, 2013; Zhao et al., 2011). Because a level of metabolite under the control of multiple QTLs hardly shows extreme phenotypes, this characteristic represents the genetic architecture for the consistent biosynthesis of essential amino acids and house-keeping flavonoids as defense compounds, such as a radical scavenging and UV absorbance activities (Simmonds, 2003).
Figure 6.
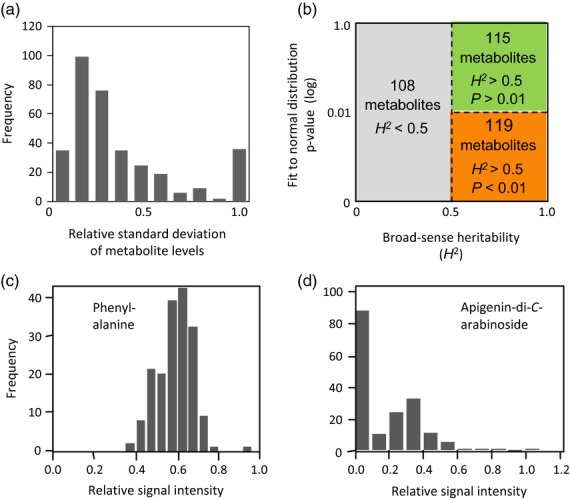
Natural variations in metabolite compositions.
(a) Relative standard deviation among the 175 rice cultivars.
(b) Grouping of metabolites by broad-sense heritability (H2) and Kolmogorov–Smirnov test for fitting a normal distribution (P-value). Numbers of metabolites classified in each group are shown in the figure. Group colors (green, orange, and gray) correspond to node colors in Figure 1.
(c, d) Variation in phenylalanine (c) and apigenin-di-C-arabinoside (d) levels among the 175 rice cultivars.
Genetic architecture to generate intra-species diversity of phytochemicals
The metabolome data also revealed that quantitative variations of the other 119 metabolites are predominantly controlled by heritable factors (H2 > 0.5) and significantly deviated from the normal distribution (P < 0.01, Figure 6b, orange nodes in Figure 1). The group included apigenin-di-C-arabinoside 6 (Figure 6d) and an unknown metabolite ID11261. These distortions were associated with low numbers of QTLs that strongly control the content of multiple metabolites (Figure 3b). Indeed, pedigree data showed that the haplotype around the NIAS_Os_ac06000458 was tightly linked to the high apigenin-di-C-arabinoside phenotype (Figure 7, highlighted in red). A similar tight association was observed between the unknown metabolite (ID11261) and the haplotype around SNP marker NIAS_Os_ab04000042 (Figure 7, highlighted in blue). This is another type of genetic architecture that produces progenies with various phytochemical compositions. As shown in Figure 7, Norin 22 exhibited low level of apigenin-di-C-arabinoside 6 (signal intensity was <0.01) and high accumulation of ID11261 (signal intensity was 0.11). In contrast, Norin 1 exhibited a high apigenin-di-C-arabinoside level (0.43) and low ID11261 level (<0.01), respectively. By crossing two cultivars, new patterns of apigenin-di-C-arabinoside and ID11261 accumulation level were generated, such as the high apigenin-di-C-arabinoside and high ID11261 in Hatsunishiki and the low apigenin-di-C-arabinoside and low ID11261 in Koshihikari. The strong effects of the small number of mQTLs on metabolic phenotypes were also supported by comparing genotype and metabolic phenotype similarities among cultivars, as dissimilar metabolic phenotypes were observed between several pairs of genetically similar cultivars (Figure S2). The genetic architecture is a mechanism generating strains with distinct metabolic composition through the crossing of two different strains. The recombined metabolite composition may have some advantages in interactions with herbivores and microbes because some insects recognize the composition of plant metabolites before feeding or spawning (Nguyen et al., 2013).
Figure 7.
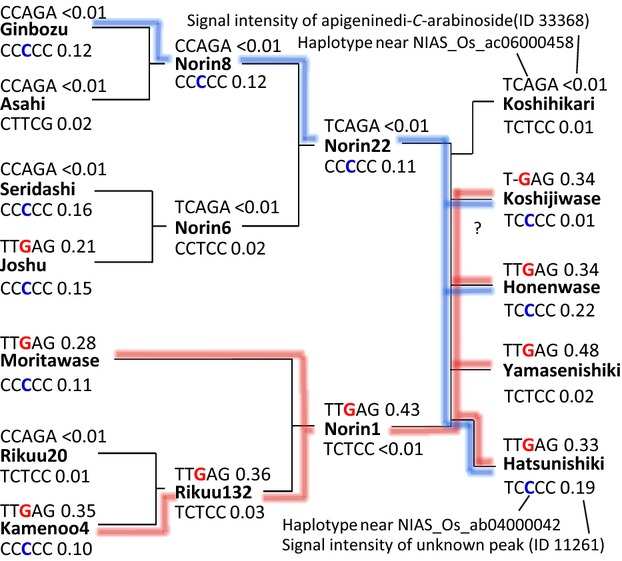
Phylogenetic tree of the five rice cultivars developed by crossing Norin 1 and Norin 22.
Cultivar names are shown in bold. The five nucleotide sequences above the cultivar names represent the window of haplotypes around the SNP marker, NIAS_Os_ac06000458, that was significantly associated with the levels of apigenin-di-C-arabinoside (ID 33368). The signal intensities of the unknown metabolite in each cultivar are also shown. The data below the cultivar names indicate the haplotypes around SNP marker, NIAS_Os_ab040000042, that was significantly associated with an unknown metabolite (ID 11261). Red and blue lines represent the phylogenic origins of high apigenin-di-C-arabinoside genotypes and unknown metabolite, respectively. ‘?’ indicates an inconsistent between genotype and metabolotype.
Greater phytochemical variation in the world rice population
It has been demonstrated that Japanese cultivated rice population have rather limited genotypic diversity (Yonemaru et al., 2012) and that there is a greater phytochemical variation in the world rice population (Chen et al., 2014). For instance, in the co-accumulation network, there is a large cluster of structurally unrelated metabolites (cluster five in Figure 1) including isoorientin 7,3′-dimethyl ether 8 (ID 23522), tricin 7-O-(2″-O-β-d-glucosyl)-β-d-glucuronoside 16 (ID 54700), and tricin 7-O-(6″-O-malonyl)-β-d-glucoside 14. The detailed analysis indicated that a small number of landrace accessions actively synthesize these metabolites, whereas these metabolites were rarely observed in the improved cultivars (Figure 8a). This finding indicates that the genetic variation of this trait is due to rare alleles in the population while the linkage disequilibrium decay in japonica is generally larger than that in indica (McNally et al., 2009; Huang et al., 2010).
Figure 8.
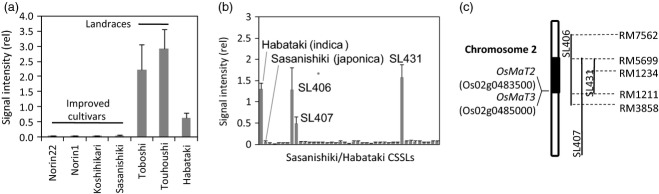
Mapping of the locus controlling tricin 7-O-(6′′-O-malonyl)-β-d-glucoside 14 content.
(a) Levels of 14 in rice cultivars. Relative levels in leaf blades of 2-week-old seedlings from four improved cultivars, 2 landraces, and Habataki are represented. Each data point expresses the mean of three experiments ± standard deviation (SD).
(b) Relative levels of 14 in the shoots of the Sasanishiki/Habataki chromosomal segment substitution lines (CSSLs) and their parental varieties. Levels in the shoot are expressed as relative values. Each data point presents the mean of three experiments ± SD.
(c) Schematic representation of chromosomal substitutions on chromosome 2, showing the genomic region controlling the level of 14 as black. Lines of each CSSL each indicate the genome regions derived from Habataki. Dashed lines represent molecular markers, and lines represent malonyl transferase genes on this chromosome.
To explore additional genetic polymorphism associated with metabolic phenotype variation, the levels of these metabolites were determined for the seedlings of Sasanishiki (japonica rice)/Habataki (indica rice) chromosome segment substitution lines (CSSLs). Habataki (indica rice) is able to produce 8, 16, and 14 (Figure 8a), whereas this variation is not present in this GWAS population. For instance, production of 14 was associated with the Habataki genotype of SSR marker RM1234 on chromosome 2 (Figure 8b,c). Gene annotation data indicated that two genes, OsMaT2 and OsMaT3, are encoded near the marker. Previous in vitro and genetic analyses suggested that these genes encode flavonoid malonyltransferase, and a probable position of malonylation is the 6″-hydroxyl group of the flavone glycosides (Kim et al., 2009; Gong et al., 2013). Since the metabolite structure of tricin 7-O-(6″-O-malonyl)-β-d-glucoside was unequivocally identified, the result indicated that an in vivo function of these genes are malonylation of 6″ position of tricin 7-O-β-d-glucoside. Similar genotype–metabolotype associations were observed for 8 and 16, whose candidate loci were mapped on chromosomes 4 and 12, respectively (Figure S3).
The analysis of Sasanishiki/Habataki CSSL also showed that much greater levels of apigenin-di-C-arabinoside 6 in Habataki compared to Sasanishiki (Figure S4). Interestingly, Sasanishiki has high levels of apigenin-di-C-arabinoside phenotypes, and has an identical genotype to Norin 1 (Figure 7). This result indicates that Habataki has other genetic polymorphisms related to apigenin-di-C-arabinoside biosynthesis that did not exist in the population used for GWAS. As shown in our previous metabolome-QTL analysis using Sasanisiki/Habataki CSSLs, the candidate region of QTL overlapped with the position of NIAS_Os_ac06000458 (Matsuda et al., 2012), indicating the important role of the hotspot on chromosome 6 for the natural variation in composition of flavone-C-glycosides in japonica and indica rice varieties.
Conclusion
LC-MS-based-metabolomics revealed the structural diversity of flavone glycosides produced in rice cultivars, that is mainly derived from various modifications of apigenin, luteolin, and chrysoeriol aglycones (Figure 1). GWAS highlighted that approximately one-third of metabolites are mainly regulated by mQTLs that have a large effect ( Figures 3b and 6b), and the genotypes of a small number of loci affect an intra-species diversity of metabolic compositions (Figures 7 and 8). Metabolome-GWAS of other crops would further uncover the genetic architectures generating the diversity of secondary metabolites to adapt various environments that will be useful information for future crop improvement. The findings also indicate that when screening for biologically active compounds, intra-species variation of secondary metabolite compositions must be taken into consideration. A further understanding of the genetic architecture for generating phytochemical diversity will guide the discovery of novel pharmaceuticals from plants (Wang et al., 2012).
Experimental procedures
Plant materials
A Japanese rice collection of 175 accessions were used in this study (Table S1) (Yonemaru et al., 2012). The Sasanishiki/Habataki chromosome segment substitution lines (CSSLs, 39 accessions) were also used (Ando et al., 2008). Seeds were sterilized in 10% sodium hypochloric acid solution by vacuum infiltration for 1 h, and then immersed in aqueous 2% PPM™ solution (Nacalai Tesque, Kyoto, Japan, http://www.nacalai.co.jp/) at 28°C for 1 day in darkness. Seeds were sown in wet commercial fertilized soil (Bonsol II; Sumitomo Chemical, Tokyo, Japan, http://www.sumitomochem.co.jp/), and maintained under a 12-h light (28°C)/12-h dark (20°C) cycle for germination. Plants were kept under constant subirrigation conditions with tap water. After 2 weeks of growth, the entire aboveground (or aerial) part of one seedling was collected, weighed, and frozen in liquid nitrogen for analysis. Samples were stored at −80°C until analysis.
Metabolome analysis using liquid chromatography quadrupole time-of-flight mass spectrometry (LC-QToF-MS)
Analysis was performed using samples with three or four biological replicates per cultivar. Frozen rice tissue was homogenized in five volumes of cold 80% aqueous methanol containing an internal standard (0.5 mg L−1 lidocaine, Tokyo Kasei, Tokyo, Japan, http://www.tcichemicals.com/), using a mixer mill (MM 300, Retsch, Haan, Germany, http://www.retsch.com/) and a zirconia bead for 6 min at 20 Hz. Samples were centrifuged at 15 000 g for 10 min. The supernatant (3 μl) were subsequently subjected to metabolome analysis using liquid chromatography coupled with electrospray quadrupole time-of-flight tandem mass spectrometry with an Acquity BEH ODS column (LC-ESI-QToF/MS, HPLC: Waters Acquity UPLC system; MS: Waters QToF Premier, http://www.waters.com/). Metabolome analysis and data processing were conducted according to a previously described method (Matsuda et al., 2009, 2010). Briefly, metabolome data were obtained in positive ion mode (m/z 100–2000; dwell time: 0.5 sec), from which a data matrix was generated by MetAlign2 (Lommen and Kools, 2012). Signal intensities were normalized by dividing them by the intensities of the internal standard (lidocaine). A data matrix containing the 342 metabolite intensities from 668 runs was produced for the Japanese rice population (Tables S2 and S3).
Metabolite annotation
For structural elucidation of metabolite signals, MS/MS spectral tag (MS2T) libraries were constructed (Matsuda et al., 2011). The extracts of 14–15 cultivars were mixed and utilized for MS/MS spectra data acquisition. Analyses were repeated for 12 mixtures using automated data acquisition methods as previously described (Matsuda et al., 2009). Each MS2T entry was assigned a unique code, OSAXXpXXXXX, indicating the library name (OSAXXp) and entry number. MS2T libraries containing 164 051 entries were constructed. MS2Ts were added to metabolite signals, from which the structure of each metabolite signal was elucidated by searching the ReSpect (RIKEN MS/MS spectra database for phytochemicals) (Sawada et al., 2012), MassBank (Horai et al., 2010), KNApSAcK (Afendi et al., 2012), and PRIMe standard compound database (Sakurai et al., 2013). The two spectra were considered to be similar when the similarity score of the ReSpect search was greater than 0.6. Thresholds were set at m/z Δ0.05 and 0.15 min, respectively, for the molecular formula search on the KNApSAcK database and comparison of retention times. Based on the criteria proposed by the metabolome standard initiative (MSI) (Sumner et al., 2007), metabolite signals were ‘characterized’ when parts of a structure were deduced from mass data. Metabolite signals were ‘annotated’ when a common metabolite was observed in the outputs from both the ReSpect and KNApSAcK searches. Metabolite signals were considered to be ‘identified’ when three distinct pieces of information, including the MS/MS spectra, exact mass number, and chromatographic behavior, were matched to identical metabolites (Table S4). Data obtained in this study are available on the PRIMe website (http://prime.psc.riken.jp/) (Sakurai et al., 2013). Isolation and structural determination of rice flavones has been reported previously (Yang et al., 2014).
Broad-sense heritability
In this study, the total variance of metabolite level was calculated as the sum of genetic and environmental factors, expressed as: Var(P) = Var(G) + Var(E) (Visscher et al., 2008). Here, Var(G) and Var(E) represent the variance derived from genetic and environmental effects, respectively. Broad-sense heritability (H2) was estimated to be H2 = Var(G)/Var(P) using one-way analysis of variance by treating 175 cultivars as a random effect and biological replicates as the replication effect.
Network analyses
A molecular MS/MS network was constructed using previously reported methods with some modifications (Watrous et al., 2012). For each metabolite signal, an MS/MS spectrum of MS2T with the most intense base peak was used as the representative spectrum. Pairwise similarities with cosine ≥0.7 were used to define molecular MS/MS networks. To construct the metabolite co-accumulation network, Pearson product-moment correlation coefficients were determined using mean values of signal intensities (Table S3). A pairwise similarity with a score of ≥0.7 was used to construct the metabolite co-accumulation network. Networks were visualized using Cytoscape 2.8.3 (Assenov et al., 2008).
Genome-wide association studies
The SNP dataset and population structure of the Japanese rice population were obtained from the published literature (Yonemaru et al., 2012). Genotype data for 3168 SNPs with polymorphisms sharing at least 5% of 175 cultivars were used for the GWAS (Table S5). For the naïve model, a simple linear model, without correcting for population structure, was used with the following equation:
A mixed-model approach implemented in R package EMMA was employed to correct for confounding effects of population structure using the equation (Kang et al., 2008):
where Y, X, P, and β represent the phenotype vector, the SNP genotype vector, the population structure vector (K = 4), and the SNP effect, respectively. The association of each SNP was tested using a null hypothesis (H0), in which metabolite levels were assumed not to be associated with the SNP genotype. All statistical analyses were performed in r 2.15.1 (http://www.r-project.org/). For 1 SNP marker associated with a metabolic phenotype, a genome region between two neighborhood SNPs was considered to be the candidate region of QTL. This is because the mean size of the candidate region (0.24 Mb) is similar to that of linkage disequilibrium in rice (Yonemaru et al., 2012). A list of genes encoded in the candidate region was obtained based on SNP and open reading frame (ORF) positions in the rice genome (RAP builds 4 and 5) (Itoh et al., 2007). The list was used for gene enrichment analysis with agriGO to investigate the gene ontology frequently observed in the candidate region (GO type: Completed GO, Background/Reference: Rice MSU6.1 non-TE transcript ID) (Du et al., 2010).
Data availability
Raw metabolome data obtained in this study are available on the PRIMe website (http://prime.psc.riken.jp/).
Acknowledgments
We thank K. Akiyama, T. Sakurai, M. Suzuki, Dr. K Yonekura-Sakakibara (RIKEN Center for Sustainable Resource, Japan), and Dr. M. Yamasaki (Kobe University) for technical support and helpful comments. This work was partly supported by a grant from the Ministry of Agriculture, Forestry, and Fisheries of Japan (Genomics for Agricultural Innovation, NVR-0005) and JST, Strategic International Collaborative Research Program, SICORP for JP-US Metabolomics.
Conflict of interest
The authors declare no conflict of interest.
SUPPORTING INFORMATION
Additional Supporting Information may be found in the online version of this article.
Figure S1. Comparison of QQ plots between 2 GWAS using the naïve approach and mixed model approach (EMMA) for correcting population structure.
Figure S2. Similarities between two rice cultivars determined by genotype and metabolic phenotype similarities.
Figure S3. Mapping of locus controlling isoorientin 7,3′-dimethyl ether 8 and tricin 7-O-(2″-O-β-d-glucosyl)-β-d-glucuronoside 16 contents.
Figure S4. Levels of apigenin-6-C-α-l-arabinosyl-8-C-α-l-arabinoside 6 in the rice cultivars.
Table S1. List of rice accessions used in this study.
Table S2. Metabolome Dataset OryzaMetExpress 175 accessions.
Table S3. Dataset of mean intensity data.
Table S4. List of metabolites identified, annotated, characterized in this study.
Table S5. Genotypes of 3168 SNPs used in this study.
Table S6. Associations between SNPs and metabolite levels (α = 1.0 × 10−3).
Table S7. Gene ontology enrichment analysis of genes in the candidate regions of the detected metabolite-SNP associations when employing a relatively strict threshold (α = 1.0 × 10−5).
Table S8. Genes near SNPs associated with metabolic phenotypes.
References
- Afendi FM, Okada T, Yamazaki M, et al. KNApSAcK family databases: integrated metabolite-plant species databases for multifaceted plant research. Plant Cell Physiol. 2012;53:e1. doi: 10.1093/pcp/pcr165. [DOI] [PubMed] [Google Scholar]
- Ando T, Yamamoto T, Shimizu T, Ma XF, Shomura A, Takeuchi Y, Lin SY, Yano M. Genetic dissection and pyramiding of quantitative traits for panicle architecture by using chromosomal segment substitution lines in rice. Theor. Appl. Genet. 2008;116:881–890. doi: 10.1007/s00122-008-0722-6. [DOI] [PubMed] [Google Scholar]
- Assenov Y, Ramirez F, Schelhorn SE, Lengauer T, Albrecht M. Computing topological parameters of biological networks. Bioinformatics. 2008;24:282–284. doi: 10.1093/bioinformatics/btm554. [DOI] [PubMed] [Google Scholar]
- Atwell S, Huang YS, Vilhjalmsson BJ, et al. Genome-wide association study of 107 phenotypes in Arabidopsis thaliana inbred lines. Nature. 2010;465:627–631. doi: 10.1038/nature08800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beekwilder J, van Leeuwen W, van Dam NM, et al. The impact of the absence of aliphatic glucosinolates on insect herbivory in Arabidopsis. PLoS One. 2008;3:e2068. doi: 10.1371/journal.pone.0002068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Besson E, Dellamonica G, Chopin J, Markham KR, Kim M, Koh H-S, Fukami H. C-Glycosylflavonoids from Oryza sativa. Phytochemistry. 1985;24:1061–1064. [Google Scholar]
- Bottcher C, Roepenack-Lahaye EV, Schmidt J, Schmotz C, Neumann S, Scheel D, Clemens S. Metabolome analysis of biosynthetic mutants reveals diversity of metabolic changes and allows identification of a large number of new compounds in Arabidopsis thaliana. Plant Physiol. 2008;147:2107–2120. doi: 10.1104/pp.108.117754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouaziz M, Veitch NC, Grayer RJ, Simmonds MS, Damak M. Flavonolignans from Hyparrhenia hirta. Phytochemistry. 2002;60:515–520. doi: 10.1016/s0031-9422(02)00145-0. [DOI] [PubMed] [Google Scholar]
- Brachi B, Morris GP, Borevitz JO. Genome-wide association studies in plants: the missing heritability is in the field. Genome Biol. 2011;12:232. doi: 10.1186/gb-2011-12-10-232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brazier-Hicks M, Evans KM, Gershater MC, Puschmann H, Steel PG, Edwards R. The C-glycosylation of flavonoids in cereals. J. Biol. Chem. 2009;284:17926–17934. doi: 10.1074/jbc.M109.009258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carreno-Quintero N, Bouwmeester HJ, Keurentjes JJ. Genetic analysis of metabolome-phenotype interactions: from model to crop species. Trends Genet. 2013;29:41–50. doi: 10.1016/j.tig.2012.09.006. [DOI] [PubMed] [Google Scholar]
- Cavaliere C, Foglia P, Pastorini E, Samperi R, Lagana A. Identification and mass spectrometric characterization of glycosylated flavonoids in Triticum durum plants by high-performance liquid chromatography with tandem mass spectrometry. Rapid Commun. Mass Spectrom. 2005;19:3143–3158. doi: 10.1002/rcm.2185. [DOI] [PubMed] [Google Scholar]
- Chan EK, Rowe HC, Hansen BG, Kliebenstein DJ. The complex genetic architecture of the metabolome. PLoS Genet. 2010a;6:e1001198. doi: 10.1371/journal.pgen.1001198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan EK, Rowe HC, Kliebenstein DJ. Understanding the evolution of defense metabolites in Arabidopsis thaliana using genome-wide association mapping. Genetics. 2010b;185:991–1007. doi: 10.1534/genetics.109.108522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan EK, Rowe HC, Corwin JA, Joseph B, Kliebenstein DJ. Combining genome-wide association mapping and transcriptional networks to identify novel genes controlling glucosinolates in Arabidopsis thaliana. PLoS Biol. 2011;9:e1001125. doi: 10.1371/journal.pbio.1001125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang CL, Wang GJ, Zhang LJ, Tsai WJ, Chen RY, Wu YC, Kuo YH. Cardiovascular protective flavonolignans and flavonoids from Calamus quiquesetinervius. Phytochemistry. 2010;71:271–279. doi: 10.1016/j.phytochem.2009.09.025. [DOI] [PubMed] [Google Scholar]
- Chen W, Gao Y, Xie W, et al. Genome-wide association analyses provide genetic and biochemical insights into natural variation in rice metabolism. Nat. Genet. 2014;46:714–721. doi: 10.1038/ng.3007. [DOI] [PubMed] [Google Scholar]
- Dong X, Chen W, Wang W, Zhang H, Liu X, Luo J. Comprehensive profiling and natural variation of flavonoids in rice. J. Integr. Plant Biol. 2014;56:876–886. doi: 10.1111/jipb.12204. [DOI] [PubMed] [Google Scholar]
- Du Z, Zhou X, Ling Y, Zhang Z, Su Z. agriGO: a GO analysis toolkit for the agricultural community. Nucleic Acids Res. 2010;38:W64–W70. doi: 10.1093/nar/gkq310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong L, Chen W, Gao Y, Liu X, Zhang H, Xu C, Yu S, Zhang Q, Luo J. Genetic analysis of the metabolome exemplified using a rice population. Proc. Natl Acad. Sci. USA. 2013;110:20320–20325. doi: 10.1073/pnas.1319681110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill CB, Taylor JD, Edwards J, Mather D, Bacic A, Langridge P, Roessner U. Whole-genome mapping of agronomic and metabolic traits to identify novel quantitative trait Loci in bread wheat grown in a water-limited environment. Plant Physiol. 2013;162:1266–1281. doi: 10.1104/pp.113.217851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horai H, Arita M, Kanaya S, et al. MassBank: a public repository for sharing mass spectral data for life sciences. J. Mass Spectrom. 2010;45:703–714. doi: 10.1002/jms.1777. [DOI] [PubMed] [Google Scholar]
- Huang X, Wei X, Sang T, et al. Genome-wide association studies of 14 agronomic traits in rice landraces. Nat. Genet. 2010;42:961–967. doi: 10.1038/ng.695. [DOI] [PubMed] [Google Scholar]
- Huang X, Zhao Y, Wei X, et al. Genome-wide association study of flowering time and grain yield traits in a worldwide collection of rice germplasm. Nat. Genet. 2013;44:32–39. doi: 10.1038/ng.1018. [DOI] [PubMed] [Google Scholar]
- Itoh T, Tanaka T, Barrero RA, et al. Curated genome annotation of Oryza sativa ssp. japonica and comparative genome analysis with Arabidopsis thaliana. Genome Res. 2007;17:175–183. doi: 10.1101/gr.5509507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang HM, Zaitlen NA, Wade CM, Kirby A, Heckerman D, Daly MJ, Eskin E. Efficient control of population structure in model organism association mapping. Genetics. 2008;178:1709–1723. doi: 10.1534/genetics.107.080101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keurentjes JJ, Fu J, Terpstra IR, Garcia JM, van den Ackerveken G, Snoek LB, Peeters AJ, Vreugdenhil D, Koornneef M, Jansen RC. Regulatory network construction in Arabidopsis by using genome-wide gene expression quantitative trait loci. Proc. Natl Acad. Sci. USA. 2007;104:1708–1713. doi: 10.1073/pnas.0610429104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim DH, Kim SK, Kim JH, Kim BG, Ahn JH. Molecular characterization of flavonoid malonyltransferase from Oryza sativa. Plant Physiol. Biochem. 2009;47:991–997. doi: 10.1016/j.plaphy.2009.08.004. [DOI] [PubMed] [Google Scholar]
- Kliebenstein D. Advancing genetic theory and application by metabolic quantitative trait loci analysis. Plant Cell. 2009;21:1637–1646. doi: 10.1105/tpc.109.067611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kliebenstein DJ, Gershenzon J, Mitchell-Olds T. Comparative quantitative trait loci mapping of aliphatic, indolic and benzylic glucosinolate production in Arabidopsis thaliana leaves and seeds. Genetics. 2001a;159:359–370. doi: 10.1093/genetics/159.1.359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kliebenstein DJ, Kroymann J, Brown P, Figuth A, Pedersen D, Gershenzon J, Mitchell-Olds T. Genetic control of natural variation in Arabidopsis glucosinolate accumulation. Plant Physiol. 2001b;126:811–825. doi: 10.1104/pp.126.2.811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kliebenstein DJ, Lambrix VM, Reichelt M, Gershenzon J, Mitchell-Olds T. Gene duplication in the diversification of secondary metabolism: tandem 2-oxoglutarate-dependent dioxygenases control glucosinolate biosynthesis in Arabidopsis. Plant Cell. 2001c;13:681–693. doi: 10.1105/tpc.13.3.681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko JH, Kim BG, Kim JH, Kim H, Lim CE, Lim J, Lee C, Lim Y, Ahn JH. Four glucosyltransferases from rice: cDNA cloning, expression, and characterization. J. Plant Physiol. 2008;165:435–444. doi: 10.1016/j.jplph.2007.01.006. [DOI] [PubMed] [Google Scholar]
- Lisec J, Steinfath M, Meyer RC, Selbig J, Melchinger AE, Willmitzer L, Altmann T. Identification of heterotic metabolite QTL in Arabidopsis thaliana RIL and IL populations. Plant J. 2009;59:777–788. doi: 10.1111/j.1365-313X.2009.03910.x. [DOI] [PubMed] [Google Scholar]
- Lommen A, Kools HJ. MetAlign 3.0: performance enhancement by efficient use of advances in computer hardware. Metabolomics. 2012;8:719–726. doi: 10.1007/s11306-011-0369-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuda F, Yonekura-Sakakibara K, Niida R, Kuromori T, Shinozaki K, Saito K. MS/MS spectral tag (MS2T)-based annotation of non-targeted profile of plant secondary metabolites. Plant J. 2009;57:555–577. doi: 10.1111/j.1365-313X.2008.03705.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuda F, Hirai MY, Sasaki E, Akiyama K, Yonekura-Sakakibara K, Provart NJ, Sakurai T, Shimada Y, Saito K. AtMetExpress development: a phytochemical atlas of Arabidopsis development. Plant Physiol. 2010;152:566–578. doi: 10.1104/pp.109.148031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuda F, Nakabayashi R, Sawada Y, Suzuki M, Hirai MY, Kanaya S, Saito K. Mass spectra-based framework for automated structural elucidation of metabolome data to explore phytochemical diversity. Front Plant Sci. 2011;2:40. doi: 10.3389/fpls.2011.00040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuda F, Okazaki Y, Oikawa A, Kusano M, Nakabayashi R, Kikuchi J, Yonemaru J, Ebana K, Yano M, Saito K. Dissection of genotype-phenotype associations in rice grains using metabolome quantitative trait loci analysis. Plant J. 2012;70:624–636. doi: 10.1111/j.1365-313X.2012.04903.x. [DOI] [PubMed] [Google Scholar]
- McNally KL, Childs KL, Bohnert R, et al. Genomewide SNP variation reveals relationships among landraces and modern varieties of rice. Proc. Natl Acad. Sci. USA. 2009;106:12273–12278. doi: 10.1073/pnas.0900992106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohanlal S, Parvathy R, Shalini V, Helen A, Jayalekshmy A. Isolation, characterization and quantification of tricin and flavonolignans in the medicinal rice Njavara (Oryza sativa L.), as compared to staple varieties. Plant Foods Hum. Nutr. 2011;66:91–96. doi: 10.1007/s11130-011-0217-5. [DOI] [PubMed] [Google Scholar]
- Nguyen DD, Wu CH, Moree WJ, et al. MS/MS networking guided analysis of molecule and gene cluster families. Proc. Natl Acad. Sci. USA. 2013;110:E2611–E2620. doi: 10.1073/pnas.1303471110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pichersky E, Lewinsohn E. Convergent evolution in plant specialized metabolism. Annu. Rev. Plant Biol. 2011;62:549–566. doi: 10.1146/annurev-arplant-042110-103814. [DOI] [PubMed] [Google Scholar]
- Riedelsheimer C, Lisec J, Czedik-Eysenberg A, Sulpice R, Flis A, Grieder C, Altmann T, Stitt M, Willmitzer L, Melchinger AE. Genome-wide association mapping of leaf metabolic profiles for dissecting complex traits in maize. Proc. Natl Acad. Sci. USA. 2012;109:8872–8877. doi: 10.1073/pnas.1120813109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowe HC, Hansen BG, Halkier BA, Kliebenstein DJ. Biochemical networks and epistasis shape the Arabidopsis thaliana metabolome. Plant Cell. 2008;20:1199–1216. doi: 10.1105/tpc.108.058131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito K. Phytochemical genomics—a new trend. Curr. Opin. Plant Biol. 2013;16:373–380. doi: 10.1016/j.pbi.2013.04.001. [DOI] [PubMed] [Google Scholar]
- Saito K, Matsuda F. Metabolomics for functional genomics, systems biology, and biotechnology. Annu. Rev. Plant Biol. 2010;61:463–489. doi: 10.1146/annurev.arplant.043008.092035. [DOI] [PubMed] [Google Scholar]
- Sakurai T, Yamada Y, Sawada Y, Matsuda F, Akiyama K, Shinozaki K, Hirai MY, Saito K. PRIMe Update: innovative content for plant metabolomics and integration of gene expression and metabolite accumulation. Plant Cell Physiol. 2013;54:e5. doi: 10.1093/pcp/pcs184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawada Y, Nakabayashi R, Yamada Y, et al. RIKEN tandem mass spectral database (ReSpect) for phytochemicals: a plant-specific MS/MS-based data resource and database. Phytochemistry. 2012;82:38–45. doi: 10.1016/j.phytochem.2012.07.007. [DOI] [PubMed] [Google Scholar]
- Schauer N, Semel Y, Balbo I, Steinfath M, Repsilber D, Selbig J, Pleban T, Zamir D, Fernie AR. Mode of inheritance of primary metabolic traits in tomato. Plant Cell. 2008;20:509–523. doi: 10.1105/tpc.107.056523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwab W. Metabolome diversity: too few genes, too many metabolites? Phytochemistry. 2003;62:837–849. doi: 10.1016/s0031-9422(02)00723-9. [DOI] [PubMed] [Google Scholar]
- Simmonds MS. Importance of flavonoids in insect—plant interactions: feeding and oviposition. Phytochemistry. 2001;56:245–252. doi: 10.1016/s0031-9422(00)00453-2. [DOI] [PubMed] [Google Scholar]
- Simmonds MS. Flavonoid-insect interactions: recent advances in our knowledge. Phytochemistry. 2003;64:21–30. doi: 10.1016/s0031-9422(03)00293-0. [DOI] [PubMed] [Google Scholar]
- Sumner LW, Amberg A, Barrett D, et al. Proposed minimum reporting standards for chemical analysis. Metabolomics. 2007;3:211–221. doi: 10.1007/s11306-007-0082-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visscher PM, Hill WG, Wray NR. Heritability in the genomics era—concepts and misconceptions. Nat. Rev. Genet. 2008;9:255–266. doi: 10.1038/nrg2322. [DOI] [PubMed] [Google Scholar]
- Wang H, Khor TO, Shu L, Su ZY, Fuentes F, Lee JH, Kong AN. Plants vs. cancer: a review on natural phytochemicals in preventing and treating cancers and their druggability. Anticancer Agents Med. Chem. 2012;12:1281–1305. doi: 10.2174/187152012803833026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watrous J, Roach P, Alexandrov T, et al. Mass spectral molecular networking of living microbial colonies. Proc. Natl Acad. Sci. USA. 2012;109:E1743–E1752. doi: 10.1073/pnas.1203689109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weigel D. Natural variation in Arabidopsis: from molecular genetics to ecological genomics. Plant Physiol. 2012;158:2–22. doi: 10.1104/pp.111.189845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen W, Li D, Li X, et al. Metabolome-based genome-wide association study of maize kernel leads to novel biochemical insights. Nat. Commun. 2014;5:3438. doi: 10.1038/ncomms4438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z, Nakabayashi R, Okazaki Y, Mori T, Takamatsu S, Kitanaka S, Kikuchi J, Saito K. Toward better annotation in plant metabolomics: isolation and structure elucidation of 36 specialized metabolites from Oryza sativa (rice) by using MS/MS and NMR analyses. Metabolomics. 2014;10:543–555. doi: 10.1007/s11306-013-0619-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yonemaru J, Yamamoto T, Ebana K, Yamamoto E, Nagasaki H, Shibaya T, Yano M. Genome-wide haplotype changes produced by artificial selection during modern rice breeding in Japan. PLoS One. 2012;7:e32982. doi: 10.1371/journal.pone.0032982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao K, Tung CW, Eizenga GC, et al. Genome-wide association mapping reveals a rich genetic architecture of complex traits in Oryza sativa. Nat. Commun. 2011;2:467. doi: 10.1038/ncomms1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Comparison of QQ plots between 2 GWAS using the naïve approach and mixed model approach (EMMA) for correcting population structure.
Figure S2. Similarities between two rice cultivars determined by genotype and metabolic phenotype similarities.
Figure S3. Mapping of locus controlling isoorientin 7,3′-dimethyl ether 8 and tricin 7-O-(2″-O-β-d-glucosyl)-β-d-glucuronoside 16 contents.
Figure S4. Levels of apigenin-6-C-α-l-arabinosyl-8-C-α-l-arabinoside 6 in the rice cultivars.
Table S1. List of rice accessions used in this study.
Table S2. Metabolome Dataset OryzaMetExpress 175 accessions.
Table S3. Dataset of mean intensity data.
Table S4. List of metabolites identified, annotated, characterized in this study.
Table S5. Genotypes of 3168 SNPs used in this study.
Table S6. Associations between SNPs and metabolite levels (α = 1.0 × 10−3).
Table S7. Gene ontology enrichment analysis of genes in the candidate regions of the detected metabolite-SNP associations when employing a relatively strict threshold (α = 1.0 × 10−5).
Table S8. Genes near SNPs associated with metabolic phenotypes.
Data Availability Statement
Raw metabolome data obtained in this study are available on the PRIMe website (http://prime.psc.riken.jp/).