

Abstract
Objective:
To assess the relationship between CD56bright natural killer (NK) cells and multiple sclerosis (MS) disease activity in patients with relapsing-remitting MS treated with daclizumab high-yield process (DAC HYP).
Methods:
Data were from patients enrolled in a 52-week randomized, double-blind, placebo-controlled study of DAC HYP and its extension study. Assessments included relationships of CD56bright NK cell numbers (identified using fluorescence-activated cell sorting) at weeks 4 and 8 with the numbers of new or newly enlarging T2-hyperintense lesions between weeks 24 and 52 and the annualized relapse rate.
Results:
In DAC HYP–treated patients but not placebo-treated patients, the numbers of CD56bright NK cells increased over 52 weeks of treatment, and their numbers at weeks 4 and 8 predicted the number of new or newly enlarging T2-hyperintense lesions between weeks 24 and 52 of treatment (p ≤ 0.005 for each comparison). Similar but nonsignificant trends were observed between CD56bright NK cell counts and the annualized relapse rate in DAC HYP–treated patients. DAC HYP–treated patients who showed lower levels of expansion of CD56bright NK cells still developed fewer new or newly enlarging T2-hyperintense lesions than placebo-treated patients during the first year of treatment.
Conclusions:
CD56bright NK cells appear to mediate some of the treatment-related effects of DAC HYP, but their numbers do not account for the full effect of DAC HYP on MS-related outcomes.
Daclizumab (DAC) is a humanized monoclonal antibody that binds to CD25, the α-subunit of the interleukin 2 (IL-2) high-affinity receptor (IL-2R), and selectively inhibits high-affinity IL-2 receptor signaling.1 The blockade of IL-2 interaction with CD25 increases the availability of IL-2 to signal via the intermediate-affinity IL-2 receptor (CD122/CD132).2,3 DAC was initially evaluated as a treatment for relapsing-remitting multiple sclerosis (RRMS) based on the presumed direct inhibitory effects on CD25-expressing, activated T cells. However, robust expansion of intermediate-affinity IL-2R expressing CD3−CD16−/dimCD56bright natural killer (CD56bright NK) cells, an immune regulatory population capable of selectively killing autologous, activated T cells, was observed early during clinical evaluation.4
In a small open-label study of DAC-treated patients with MS, higher numbers of CD56bright NK cells after treatment were correlated with fewer new or newly enlarging MS lesions, as defined by cranial MRI during treatment.4 This observation was subsequently confirmed in a blinded analysis of a subset of patients who elected to participate in a substudy of a phase 2 trial that compared DAC vs placebo when coadministered with interferon β treatment.5 In the current analysis, we report results of a prospectively defined analysis of the relationship between CD56bright NK cell counts and treatment response to DAC high-yield process (HYP) when given as a monotherapy for up to 2 years during the randomized, placebo-controlled, multinational SELECT6 and SELECTION7 trials. Specifically, we sought to determine whether the numbers of CD56bright NK cells could be used to predict treatment response early in the course of therapy.
METHODS
Details of the design and methods for the SELECT study6 and the SELECTION extension study7 have been described previously. Briefly, SELECT was a double-blind parallel-group trial in which 621 male and female patients with RRMS were randomized 1:1:1 to treatment with once-monthly subcutaneous injections of DAC HYP 150 mg, DAC HYP 300 mg, or placebo for 52 weeks. Patients who were treated with DAC HYP in SELECT were randomized to continue on DAC HYP in the SELECTION extension study or to a 24-week treatment interruption followed by reinitiation. Cranial MRI was performed in all patients at baseline and at weeks 24, 52, 72, and 104. All study center personnel except the pharmacist were blinded to treatment assignment for each study (SELECT and SELECTION). All MRIs were read and interpreted at a central facility (Basel, Switzerland). CD56bright NK cells were measured in all study patients at baseline (screening visit and study entry visit at week 0) as well as before dosing on weeks 4, 8, 16, 24, 32, 48, 52, 56, 60, 64, 68, 72, and 104.
Sample analysis.
Anticoagulated whole blood samples for lymphocyte quantification were collected in acid citrate dextrose vacutainer tubes (BD Biosciences, San Jose, CA) and shipped at ambient temperature for next-day analysis. Fluorescence-activated cell sorting was performed using validated assays at Esoterix Clinical Trials Services (Mechlen, Belgium). Absolute lymphocyte, T, B, and NK cell counts were determined using whole blood samples, Multitest antibody cocktail reagents, and Trucount technology (BD Biosciences). Percentages of CD56bright NK cells were determined using the following antibodies (BD Biosciences): anti-CD3-PerCP (clone SK7), anti-CD16-FITC (clone NKP15), and anti-CD56-APC (clone NCAM16.2). The CD56bright NK cell assay included staining of 100 µL of whole blood for 20 minutes with the antibody conjugates described above, followed by the addition of 4 mL whole blood lysing solution (BD Biosciences), centrifugation, washing with phosphate-buffered saline containing 2% calf serum, and resuspension in 500 µL 1% paraformaldehyde solution. A total of 100,000 lymphocyte events were collected for each analysis. Percentages of CD122 (clone TU27)–positive cells were determined by setting gates for positive staining when compared with levels observed for an isotype control antibody (clone MOPC21) using antibodies from BD Biosciences. Intensity levels of CD122 were determined using mean fluorescence intensity (MFI) and converted to mean equivalents of soluble fluorescence using QuantiBRITE phycoerythrin beads (BD Biosciences). A representative CD122 MFI determination for CD3−CD16+CD56dim and CD56bright NK cells is shown in figure e-1 at Neurology.org/nn. Analyses were performed using WinList analysis software (Verity Software House, Topsham, ME).
Analysis plan.
To assess the relationship between CD56bright NK cells and DAC HYP efficacy, a prespecified analysis plan was developed to determine whether CD56bright NK cells could identify DAC HYP-treated patients who would have the largest reductions in MS disease activity. The following MS-related endpoints were used to measure MS activity: new or newly enlarging T2-hyperintense lesions between weeks 0 and 52, new or newly enlarging T2-hyperintense lesions between weeks 24 and 52 (to assess predictive value for lesions known to be forming after measurement of the CD56bright NK cells), new or newly enlarging T2-hyperintense lesions between weeks 52 and 104, and the annualized relapse rate (ARR). Based on analyses of a previous phase 2 trial,5 the primary predictor for analyses of efficacy response was the absolute number of CD56bright NK cells measured at the week 4 and week 8 time points. In sensitivity analyses, change from baseline and percent change from baseline in the absolute number of CD56bright NK cells were also considered predictors of efficacy. Relationships between treatment response and CD56bright NK cell measurements made at later time points were also estimated.
For the primary prespecified analysis, negative binomial regression was used to test whether the absolute number of CD56bright NK cells at week 4 (and in a separate model at week 8) was a significant predictor of new or newly enlarging T2-hyperintense lesions in DAC HYP–treated patients after adjustment for the baseline number of new T2-hyperintense lesions and gadolinium-enhancing lesions. Because 2 hypotheses were tested (week 4 and week 8 counts), the predetermined significance value to declare a positive prediction was 0.025. To describe the relationships and account for potential nonlinearity between CD56bright NK cells and new or newly enlarging T2-hyperintense lesions, effects by quartile of CD56bright NK cells defined at the specified time points were also estimated. Unless otherwise stated, p values shown are the likelihood ratio statistic.
Analyses of data from year 1 were performed in the subset of the intention-to-treat (ITT) population of the SELECT trial (all patients who had been randomized except for 21 patients from one study center who were excluded due to systematic misdosing at that center) with available data for CD56bright NK cells. Analyses of data from year 2 were performed on the subset of the ITT population of SELECTION with available data for CD56bright NK cells, excluding patients who either were randomized to treatment interruption or had a delay of more than 55 days between the final dose of DAC HYP in SELECT and the first dose in SELECTION.
RESULTS
Among the demographic factors and baseline markers of MS disease activity analyzed at study entry in relationship to CD56bright NK cell counts, there was no association at baseline between CD56bright NK cell numbers and either markers of MS activity (e.g., baseline T2-hyperintense lesions, p = 0.652; baseline gadolinium-enhancing lesions, p = 0.649; relapses in the 12 months before randomization, p = 0.315) or weight (p = 0.226). There was a trend for lower baseline CD56bright NK cell counts with older age, but it did not reach statistical significance (p = 0.073).
CD56bright NK cells in placebo-treated patients.
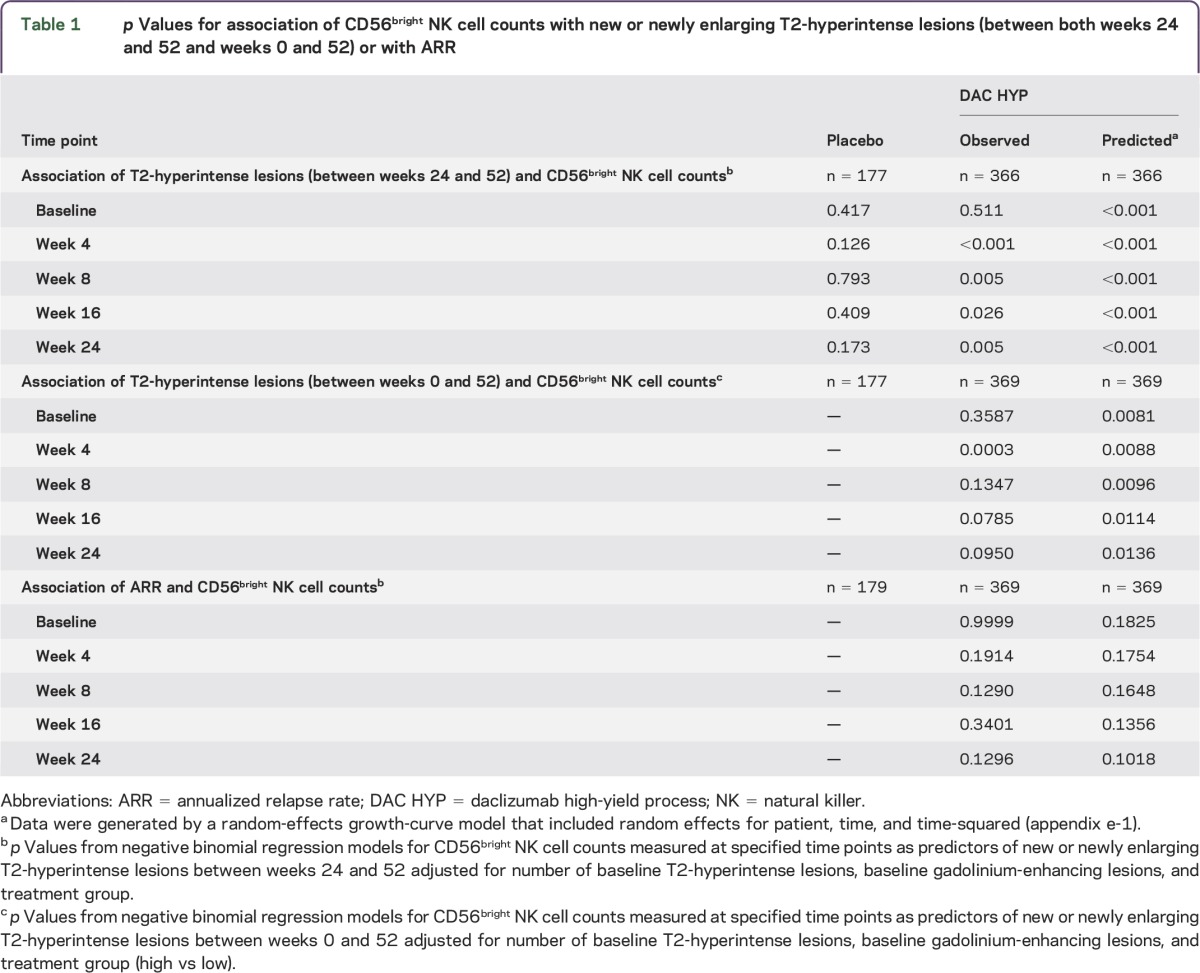
To assess longitudinal stability of CD56bright NK cell counts in untreated patients with RRMS, we analyzed visit-to-visit correlations in placebo-treated patients at weeks 0 and 52. Although large variability in CD56bright NK cell counts was observed between patients, CD56bright NK cell counts for individual patients demonstrated a high degree of longitudinal stability over a 1-year period (Spearman r = 0.758 between baseline and week 52; figure 1, table e-1). In placebo-treated patients, CD56bright NK cell counts were not associated with the number of new or newly enlarging T2-hyperintense lesions at baseline or at any postbaseline time point (table 1).
Figure 1. Relationship between CD56bright natural killer (NK) cell counts at baseline and study end in placebo-treated patients.
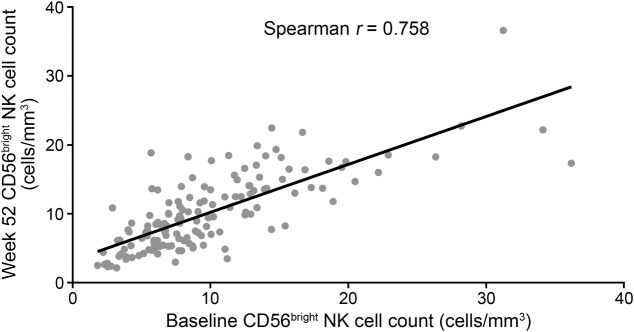
Table 1.
p Values for association of CD56bright NK cell counts with new or newly enlarging T2-hyperintense lesions (between both weeks 24 and 52 and weeks 0 and 52) or with ARR
CD56bright NK cell expansion in DAC HYP–treated patients.
In DAC HYP–treated patients, the median CD56bright NK cell count was higher than the median count in the placebo group at all postbaseline time points during the first year of treatment. By week 52, the numbers of CD56bright NK cells in DAC HYP–treated patients had stabilized and remained relatively constant thereafter (figure 2, table e-1). At the dose levels evaluated in the SELECT trial, there was no evidence of a dose-dependent effect, as the median CD56bright NK cell count was similar between the DAC HYP 150-mg and 300-mg groups at all time points. By week 52, CD56bright NK cell counts in DAC HYP–treated patients were approximately 5-fold higher than at baseline (median 4.9x increase; interquartile range [IQR] 2.6–8.8x). As a percentage of total lymphocytes, the number of CD56bright NK cells increased from a median of 0.6% (IQR 0.4%–0.9%) at baseline to a median of 3.6% (IQR 2.2%–5.7%) at week 52. In patients switched to DAC HYP at week 52, the initial expansion of CD56bright NK cells resembled that observed in the continuous treatment group, and numbers at week 104 were similar between those groups (figure 2).
Figure 2. CD56bright natural killer (NK) cell counts in daclizumab high-yield process (DAC HYP)-treated patients.

Data are medians with 25th and 75th percentiles in samples from DAC HYP–treated patients over 2 years (pooled 150-mg and 300-mg groups) or patients switched from placebo to DAC HYP (pooled 150-mg and 300-mg groups).
At the patient level, relative differences in CD56bright NK cell counts between DAC HYP–treated patients were relatively stable longitudinally even while the absolute numbers of these cells were expanding (baseline–week 52 CD56bright NK cell counts: Spearman r = 0.506, p < 0.001; week 4–week 52 CD56bright NK cell counts: Spearman r = 0.511, p < 0.001). When expansion of CD56bright NK cells was defined by the upper 95% confidence interval (CI) for the maximum increase from baseline over 1 year in the placebo-treated patients (an increase of 19 CD56bright NK cells/μL), 90% of DAC HYP–treated patients could be classified as expanders.
CD56bright NK cell numbers and treatment response.
In contrast to placebo-treated patients, higher numbers of CD56bright NK cells measured at week 4 predicted fewer new or newly enlarging T2-hyperintense lesions between weeks 0 and 52 in DAC HYP–treated patients (p < 0.001). When considering all postbaseline time points between weeks 4 and 24, CD56bright NK cell counts at week 4 had the strongest predictive association with new or newly enlarging T2-hyperintense lesions occurring between weeks 24 and 52 (table 1). In sensitivity analyses using the predicted number of CD56bright NK cells (from a random-effects model), CD56bright NK cell counts predicted new or newly enlarging T2-hyperintense lesions (weeks 0–52 and weeks 24–52) at all time points. A similar directional relationship was present for prediction of the ARR, but it was not statistically significant (table 1).
Quartile analysis was used to describe the magnitude of the statistical associations. Among patients treated with DAC HYP during the first year, those patients in the highest quartile of CD56bright NK cell counts at week 8 had 62% fewer new or newly enlarging T2-hyperintense lesions between weeks 24 and 52 than DAC HYP–treated patients who were in the lowest quartile of CD56bright NK cell counts at week 8 (p < 0.001; figure 3A). Analyses of CD56bright NK cell numbers at other time points between weeks 4 and 24 yielded similar results as at week 8 (table e-2 and data not shown). Similar trends were seen across quartiles of DAC HYP–treated patients using the ARR as the outcome measure, but the results were not statistically significant (figure 3B).
Figure 3. Clinical and MRI outcomes by quartiles of increasing CD56bright natural killer (NK) cell counts.
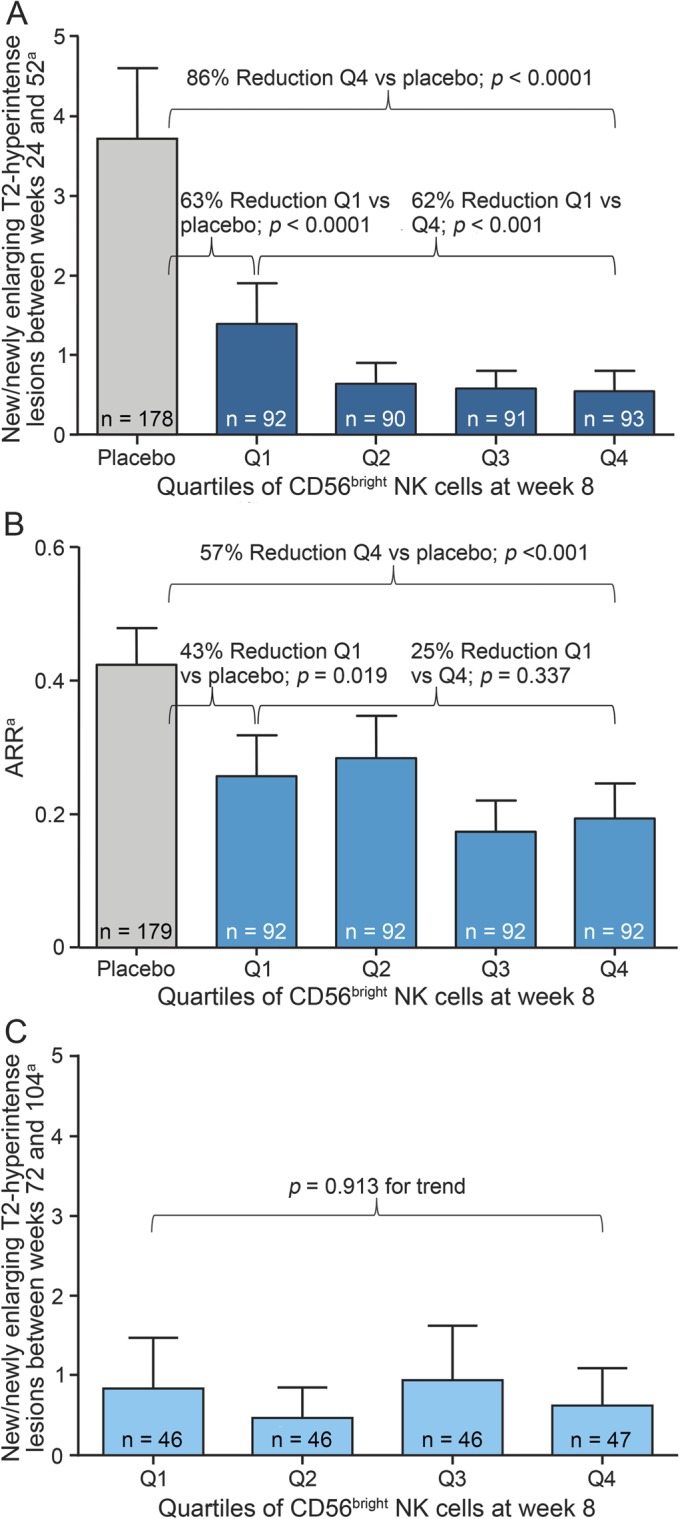
All data are mean and upper 95% confidence interval. (A) Numbers of new or newly enlarging T2-hyperintense lesions between weeks 24 and 52 in daclizumab high-yield process (DAC HYP)-treated patients and placebo-treated patients. (B) Annualized relapse rates (ARRs) between weeks 0 and 52 in DAC HYP–treated patients and placebo-treated patients. (C) Numbers of new or newly enlarging T2-hyperintense lesions between weeks 52 and 104 in DAC HYP–treated patients. aData were estimated from a negative binomial regression model adjusted for baseline T2-hyperintense lesions, baseline gadolinium-enhancing lesions, and week 8 CD56bright quartile.
In contrast to the associations between CD56bright NK cell counts and T2-hyperintense lesions seen in the first year of DAC HYP therapy, there was no evidence that the differences in CD56bright NK cell counts early during DAC HYP treatment had longer-term predictive value. During the second year of DAC HYP treatment, quartiles of CD56bright NK cell counts at week 4 or week 8 showed no trend to indicate an association with the numbers of new or newly enlarging T2-hyperintense lesions between weeks 72 and 104 (week 8, p = 0.913 for trend; figure 3C). Similarly, no trend was apparent for the association of quartiles of CD56bright NK cell counts with the ARR (quartile 1: 0.06 vs quartile 4: 0.09; p = 0.938 for trend; data not shown).
Despite the stronger MRI-defined response during the first year of treatment among DAC HYP–treated patients in the highest quartile of CD56bright NK cell counts, patients in the lowest quartile of CD56bright NK cell counts still showed evidence of a robust treatment effect from DAC HYP when compared with the placebo group. DAC HYP–treated patients in the lowest quartile of CD56bright NK cell counts at week 8 showed a 63% reduction in new or newly enlarging T2-hyperintense lesions between weeks 24 and 52 compared with placebo-treated patients (p < 0.0001), as opposed to an 86% reduction in DAC HYP–treated patients in the highest quartile at week 8 (p < 0.0001; figure 3A). Even among the 10% (n = 37) of DAC HYP–treated patients who could not be classified as having more expanded CD56bright NK cells than the placebo group, there was still evidence of treatment efficacy, as there was a 54.2% reduction (95% CI 20.3%–73.7%) in the number of new T2-hyperintense lesions over 52 weeks compared with the placebo group (p = 0.010).
DISCUSSION
These analyses provide an independent prospective assessment of a potential treatment response biomarker for RRMS within the setting of a randomized placebo-controlled trial. Consistent with smaller published studies examining the relationship between CD56bright NK cell counts and DAC response,4,5 we observed a statistically significant inverse relationship between CD56bright NK cell counts measured after DAC HYP treatment and numbers of new or newly enlarging T2-hyperintense lesions developing post treatment. Although these differences in MRI lesion formation were potentially meaningful, DAC HYP–treated patients with the lowest CD56bright NK cell counts and those whose CD56bright NK cells did not expand more than placebo-treated patients still had substantially fewer new T2-hyperintense lesions than placebo-treated patients. Furthermore, CD56bright NK cell counts measured early during the DAC HYP treatment period were not predictive of MS activity after the first year, a finding that may be related to the dynamic process of expansion of these cells over time. Finally, the current study provides a large longitudinal dataset indicating that CD56bright NK cell counts are not related to the natural history of MS in untreated patients with RRMS.
The current findings support an important role of CD56bright NK cells in the therapeutic effects of DAC in MS. Recent evidence suggests a potential immunoregulatory role for these cells. For example, depletion of NK cells exacerbates symptoms of experimental autoimmune encephalitis in mice,8 whereas treatment with CD122-directing anti-IL-2 antibody (to stimulate NK cells in a manner similar to DAC in humans) attenuates the severity of symptoms in this murine disease model.9 This may reflect a direct cytotoxic effect of NK cells on autoantigen-specific encephalitic T cells.10 In humans, reduced NK cytolytic activity has been observed in patients with MS,11–13 and reductions in NK cell function have been reported to accompany the development of large asymptomatic lesions.14 The expansion of CD56bright NK cells during DAC HYP treatment is believed to be due to increased IL-2 bioavailability after CD25 blockade and the high concentration of intermediate-affinity IL-2 receptors expressed on these cells.2 Our results indicate that this expansion occurs most rapidly during the first 6 months of treatment and then reaches a new equilibrium approximately 12 months after initiation of treatment. The temporal dynamics of the CD56bright NK cell expansion, establishment of new equilibrium of the expanded NK cells, and their contraction after DAC HYP withdrawal are consistent with in vitro modeling indicating that CD56bright NK cells compete with T cells for limited IL-2.2 Although a correlation between CD56bright NK cell numbers and MS activity was seen in DAC-treated and not placebo-treated patients, this difference may have been due to changes in CD56bright NK cell effector function induced by DAC treatment that affect the ability of these cells to influence MS-related inflammation.15 However, given the treatment effects observed in patients without expansion of CD56bright NK cells, the current study results are consistent with the hypothesis that other immunologic effects observed with DAC, such as the reduction in lymphoid tissue inducer cells, may be important to the therapeutic effects of DAC HYP.16,17
There are limitations to the current analysis. Because the CD56bright NK cell counts predictive of year 1 outcomes were measured after randomization, it is possible that as-yet-unidentified baseline differences in MS prognosis might have contributed to differences in outcomes among DAC HYP–treated patients. However, the finding that CD56bright NK cell counts were not predictive of outcome in the placebo group during the first year supports the interpretation that CD56bright NK cells are not a marker of MS prognosis overall but rather reflect biologically relevant variability in DAC HYP treatment response. Given that CD56bright NK cell expansion is a dynamic process over time, there are multiple potential ways to assess its predictive utility (e.g., measurements at different time points, relative changes vs absolute counts, etc.). In this analysis, the absolute numbers of CD56bright NK cells measured at early time points after treatment initiation were the prespecified predictors of response based on findings from previous studies and their potential for having greater clinical utility than measurements made at later time points. Additional analyses may identify other longitudinally defined predictors related to changes in CD56bright NK cells that could be informative of DAC HYP response over longer time periods. Finally, the results of these analyses and the relationship of CD56bright NK cell numbers to clinical outcomes should be confirmed prospectively in larger datasets with sufficient power to provide definitive evidence.
Personalizing treatment decisions in RRMS based on biologic differences in disease and/or treatment response requires independent validation of a clinically meaningful relationship between biomarker(s) and outcomes. While this analysis confirms that individual variability in CD56bright NK cell counts after treatment with DAC HYP contains some prognostic information, their relationship to DAC HYP response is complex and does not appear to translate directly into simple classifications such as “responders” and “nonresponders.” Nevertheless, these findings support the biological relevance of these cells to the effects of DAC HYP on MS, and additional phenotypic characterization of CD56bright NK cells should be considered as a way to better understand individual variation in the treatment response to DAC HYP.
Supplementary Material
ACKNOWLEDGMENT
The authors thank the SELECT and SELECTION study investigators for their contributions in the performance of the original studies. Elizabeth Cassell of Excel Scientific Solutions copyedited and styled the manuscript per journal requirements for which funding was provided by Biogen Idec and AbbVie Biotherapeutics Inc.
GLOSSARY
- ARR
annualized relapse rate
- CI
confidence interval
- DAC
daclizumab
- HYP
high-yield process
- IL
interleukin
- ITT
intention-to-treat
- IQR
interquartile range
- MFI
mean fluorescence intensity
- MS
multiple sclerosis
- NK
natural killer
- RRMS
relapsing-remitting MS
Footnotes
Supplemental data at Neurology.org/nn
AUTHOR CONTRIBUTIONS
J. Elkins, J. Sheridan, L. Amaravadi, K. Riester, K. Selmaj, B. Bielekova, and G. Giovannoni participated in the conception/design of the study and analysis/interpretation of the data, revised the manuscript, and approved the final version. Biogen Idec and AbbVie Biotherapeutics Inc. provided funding for editorial support in the development of this manuscript. E. Parr wrote the first draft of the manuscript with direction from coauthors and approved the final version. Biogen Idec and AbbVie Biotherapeutics Inc. reviewed and provided feedback on the manuscript to the authors. The authors had full editorial control of the manuscript and provided their final approval of all content.
STUDY FUNDING
This study was funded by Biogen Idec and AbbVie Biotherapeutics Inc.
DISCLOSURE
J. Elkins is employed by Biogen Idec. J. Sheridan is employed by AbbVie Biotherapeutics Inc. L. Amaravadi has received travel funding from the American Association of Pharmaceutical Scientists, holds a patent for evaluating immune responses to a therapeutic agent, and is an employee of Biogen Idec. K. Riester is employed by Biogen Idec. K. Selmaj is on the scientific advisory board for Genzyme, Biogen Idec, Novartis, Synthon, and Roche; has received funding from Biogen Idec, Novartis, Teva, Bayer, and Roche; is on the editorial board for Journal of Neuroimmunology; is a consultant for Genzyme, Novartis, Biogen Idec, Roche, Synthon, and Merck Serono; and is on the speakers' bureau for Novartis, Merck Serono, Biogen Idec, Bayer, and Genzyme. B. Bielekova holds NIH patents related to daclizumab and receives royalty payments from NIH and receives research support from National Institute of Neurological Disorders and Stroke, Biogen, Abbvie, and Santhera Pharmaceuticals. E. Parr is employed by Excel Scientific Solutions. G. Giovannoni is on the scientific advisory board for Biogen Idec, Fiveprime, Genzyme, GW Pharma, Ironwood, Merck Serono, Novartis, Roche, Sanofi-Aventis, Synthon BV, Teva, Vertex Pharmaceuticals, Abbvie, and Canbex; receives speaker honoraria from Biogen Idec, Genzyme, GW Pharma, Merck Serono, Novartis, Roche, and Teva; is on the editorial board for Multiple Sclerosis and Related Disorders; has consulted for Biogen Idec, Fiveprime, Genzyme, GW Pharma, Ironwood, Merck Serono, Novartis, Roche, Sanofi-Aventis, Synthon BV, Teva, Vertex Pharmaceuticals, Abbvie, and Canbex; is on the speakers' bureau for Novartis and Teva; and receives research support from Genzyme and Merck. Go to Neurology.org/nn for full disclosures.
REFERENCES
- 1.Waldmann TA. The IL-2/IL-15 receptor systems: targets for immunotherapy. J Clin Immunol 2002;22:51–56. [DOI] [PubMed] [Google Scholar]
- 2.Martin JF, Perry JSA, Jakhete NR, Wang X, Bielekova B. An IL-2 paradox: blocking CD25 on T cells induces IL-2–driven activation of CD56bright NK cells. J Immunol 2010;185:1311–1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sheridan JP, Zhang Y, Riester K, et al. Intermediate-affinity interleukin-2 receptor expression predicts CD56bright natural killer cell expansion after daclizumab treatment in the CHOICE study of patients with multiple sclerosis. Mult Scler 2011;17:1441–1448. [DOI] [PubMed] [Google Scholar]
- 4.Bielekova B, Catalfamo M, Reichert-Scrivner S, et al. Regulatory CD56bright natural killer cells mediate immunomodulatory effects of IL-2Rα-targeted therapy (daclizumab) in multiple sclerosis. Proc Natl Acad Sci U S A 2006;103:5941–5946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wynn D, Kaufman M, Montalban X, et al. ; CHOICE investigators. Daclizumab in active relapsing multiple sclerosis (CHOICE study): a phase 2, randomised, double-blind, placebo-controlled, add-on trial with interferon beta. Lancet Neurol 2010;9:381–390. [DOI] [PubMed] [Google Scholar]
- 6.Gold R, Giovannoni G, Selmaj K, et al. ; SELECT study investigators. Daclizumab high-yield process in relapsing-remitting multiple sclerosis (SELECT): a randomised, double-blind, placebo-controlled trial. Lancet 2013;381:2167–2175. [DOI] [PubMed] [Google Scholar]
- 7.Giovannoni G, Gold R, Selmaj K, et al. ; SELECTION study investigators. Daclizumab high-yield process in relapsing-remitting multiple sclerosis (SELECTION): a multicentre, randomised, double-blind extension trial. Lancet Neurol 2014;13:472–481. [DOI] [PubMed] [Google Scholar]
- 8.Zhang B, Yamamura T, Kondo T, Fujiwara M, Tabira T. Regulation of experimental autoimmune encephalomyelitis by natural killer (NK) cells. J Exp Med 1997;186:1677–1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hao J, Campagnolo D, Liu R, et al. Interleukin-2/interleukin-2 antibody therapy induces target organ natural killer cells that inhibit central nervous system inflammation. Ann Neurol 2011;69:721–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Xu W, Fazekas G, Hara H, Tabira T. Mechanism of natural killer (NK) cell regulatory role in experimental autoimmune encephalomyelitis. J Neuroimmunol 2005;163:24–30. [DOI] [PubMed] [Google Scholar]
- 11.Benczur M, Petrányl GG, Pálffy G, et al. Dysfunction of natural killer cells in multiple sclerosis: a possible pathogenetic factor. Clin Exp Immunol 1980;39:657–662. [PMC free article] [PubMed] [Google Scholar]
- 12.Merrill J, Jondal M, Seeley J, Ullberg M, Sidén A. Decreased NK killing in patients with multiple sclerosis: an analysis on the level of the single effector cell in peripheral blood and cerebrospinal fluid in relation to the activity in the disease. Clin Exp Immunol 1982;47:419–430. [PMC free article] [PubMed] [Google Scholar]
- 13.Neighbour PA, Grayzel AI, Miller AE. Endogenous and interferon-augmented natural killer cell activity of human peripheral blood mononuclear cells in vitro. Studies of patients with multiple sclerosis, systemic lupus erythematosus or rheumatoid arthritis. Clin Exp Immunol 1982;49:11–21. [PMC free article] [PubMed] [Google Scholar]
- 14.Oger J, Kastrukoff LF, Li DK, Paty DW. Multiple sclerosis: in relapsing patients, immune functions vary with disease activity as assessed by MRI. Neurology 1988;38:1739–1744. [DOI] [PubMed] [Google Scholar]
- 15.Jiang W, Chai NR, Maric D, Bielekova B. Unexpected role for granzyme K in CD56bright NK cell-mediated immunoregulation of multiple sclerosis. J Immunol 2011;187:781–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Perry JS, Han S, Xu Q, et al. Inhibition of LTi cell development by CD25 blockade is associated with decreased intrathecal inflammation in multiple sclerosis. Sci Transl Med 2012;4:145ra106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wuest SC, Edwan JH, Martin JF, et al. A role for interleukin-2 trans-presentation in dendritic cell–mediated T cell activation in humans, as revealed by daclizumab therapy. Nat Med 2011;17:604–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.