

Abstract
We report a second-generation gene annotation of human chromosome 22. Using expressed sequence databases, comparative sequence analysis, and experimental verification, we have extended genes, fused previously fragmented structures, and identified new genes. The total length in exons of annotation was increased by 74% over our previously published annotation and includes 546 protein-coding genes and 234 pseudogenes. Thirty-two potential protein-coding annotations are partial copies of other genes, and may represent duplications on an evolutionary path to change or loss of function. We also identified 31 non-protein-coding transcripts, including 16 possible antisense RNAs. By extrapolation, we estimate the human genome contains 29,000–36,000 protein-coding genes, 21,300 pseudogenes, and 1500 antisense RNAs. We suggest that our revised annotation criteria provide a paradigm for future annotation of the human genome.
[Supplemental material is available online at www.genome.org. The sequence data from this study have been submitted to GenBank under accession nos. AL009266, AL021682-3, AL021708, AL022729, AL035081-2, AL035364, AL035366, AL035545, AL049654, AL050253-8, AL050345-6, AL079310, AL096779-81, AL096879-81, AL096883, AL096886, AL138578, AL157851, AL159142-3, AL160111-2, AL160131-2, AL160311, AL355092, AL355192, AL355841, AL359401, AL359403, AL365511-5, AL442116, AL449243, AL449244, AL450314, AL589866-7, AL590120, AL590887-8, BU583989–BU585359. The following individuals kindly provided reagents, samples, or unpublished information as indicated in the paper: J. Seilhamer, L. Stuve, H. Roest-Crollius, A. Levine, G. Slater, and J. Kent.]
Completion of the working draft sequence was a major milestone toward understanding the human genome (Lander et al. 2001; Venter et al. 2001). While work continues toward a highly accurate complete genome sequence, attention has also turned to identifying and annotating genes. The initial annotation stage involves providing detailed and accurate descriptions of gene structures at the nucleotide level. This is crucial to the general utility of the genome sequence because subsequent functional experiments are best served by standardized data sets, for instance, the set of protein-coding genes. Furthermore, as the rate of data generation increases, there is potential for increased noise in the data unless well-curated annotation is used.
There are three main approaches for gene annotation. First, a variety of programs use statistical methods to predict potential gene structures (Solovyev et al. 1995; Burge and Karlin 1997). Despite considerable success, prediction methods cannot give highly accurate human gene structures on their own, and are susceptible to overprediction and false negatives (Dunham et al. 1999b; Guigo et al. 2000). Second, identification of similarity between known expressed sequences and the genomic sequence can be used either to indicate direct evidence of expression or similarity to an expressed gene. This has the advantage that annotated genes are based on mRNAs, but can be confounded by artefactual or unprocessed clones in cDNA libraries and by restricted spatial or temporal expression. Finally, genomic sequence from other vertebrates enables identification of conserved sequences (Hardison et al. 1997; Dubchak et al. 2000; Mayor et al. 2000; Korf et al. 2001) that are likely to be genes. Unfortunately, the ideal combination of genomes to compare is not yet clear, and human/mouse comparisons have shown conservation outside genes (Deloukas et al. 2001; Frazer et al. 2001; Kondrashov and Shabalina 2002). In practice, a combination of approaches can be used to overcome the limitations of single methods, but the perfect mixture has still to be defined. Although there has been much activity in human gene annotation, it is indicative of the complexities of the process that there is still considerable uncertainty in estimates of the number of protein-coding genes (Ewing and Green 2000; Liang et al. 2000; Roest Crollius et al. 2000; Wright et al. 2001), as well as doubts about the accuracy of some gene identifications (Hogenesch et al. 2001). There are many methodological reasons for this imprecision, not the least of which is fragmentation of gene structures because of sequence gaps, alternate splicing, or alternate cDNA ends. However, we believe that two considerations may be key to prospects for developing high-quality annotation. First, any high throughput, automated application of gene-finding procedures will necessarily rely on a simplified “one size fits all” approach and therefore will always be hampered by inaccuracies or inadequacies in the methods. In this respect, the current generation of annotation databases should be considered as first-pass views. Problems in draft gene structures must be identified by quality control and resolved by manual intervention or additional experimentation. Second, there is increasing evidence that the human genome contains a complex mixture of protein-coding genes, pseudogenes (Harrison et al. 2002), non-protein-coding RNA transcripts (Mattick 2001) including antisense RNAs (Kumar and Carmichael 1998), and genomic duplications involving genes (Bailey et al. 2002). Any comprehensive annotation scheme should attempt to identify and differentiate between these categories. Therefore, we chose to create a highly curated and categorized set of gene structures on human chromosome 22 that might serve as a model for mammalian genome annotation.
The Annotation Process
Since our initial gene annotation of human chromosome 22 (Dunham et al. 1999b), the working draft sequence of the human genome has been completed (Lander et al. 2001) and the number of expressed sequences in dbEST has increased more than threefold (from 2.9 million entries in release 60 to 9.7 million entries in release 69). In addition, numerous studies have been reported that used the annotation and indicated that more genes might be present (de Souza et al. 2000; Penn et al. 2000; Roest Crollius et al. 2000; Das et al. 2001; Shoemaker et al. 2001; Wiemann et al. 2001; Kapranov et al. 2002). The finished sequence of chromosome 22 had also been updated, including closure of 1 of the 11 gaps and an additional sequence covering a region that was deleted in Bacterial Artificial Chromosome (BAC) AL022330. Full details of updates to the sequence are given at http://www.sanger.ac.uk/HGP/Chr22/cwa_archive/version_changes.html. Therefore, it was clear that a revised annotation was timely. Furthermore the reevaluation process would allow us to apply new annotation criteria.
As a basis for the revised annotation, we subjected the finished genomic sequence to a series of computational analyses. The sequences were analyzed for repeats and the repeats masked. The masked sequence was compared for similarity to public domain EST sequences, a set of vertebrate transcripts, and predicted protein databases. Genomic sequences from mouse and Tetraodon nigroviridis were matched to the chromosome 22 sequence using exonerate and Blat (Kent 2002), and Exofish (Roest Crollius et al. 2000), respectively. Gene structures were predicted by GenScan (Burge and Karlin 1997) and fgenesh (Solovyev et al. 1995) on assembled masked genomic sequence. Unmasked sequence was used to predict the presence of CpG islands. The complete analysis was imported into an implementation of ACEDB (Durbin and Thierry-Mieg 1991). In addition, we also obtained a set of 1436 EST and cDNAs from the Incyte Genomics database that either extended annotations or provided new spliced annotations (a kind gift from J. Seilhamer and L. Stuve [Incyte Genomics, Palo Alto, CA] in December 2000).
Gene structures were annotated with assistance from a variety of software tools. We aimed to identify genes, and their genomic structures, supported by evidence from transcribed sequences across their entire length. Full-length cDNA sequences or assembled ESTs were aligned to genomic DNA to resolve their splice sites and confirm their 3′ ends. A 3′ end was judged confirmed if it had a run of at least four A residues at the 3′ end of the cDNA/EST not present in the genomic sequence. ESTs alone were annotated either if they were shown to be spliced compared with genomic DNA sequence at consensus donor and acceptor sites (Levine and Durbin 2001) or they had a confirmed 3′ end. Other unspliced ESTs were not annotated. Solitary unspliced cDNA sequences were not annotated if they had a stretch of poly(A) in the genomic sequence directly 3′ because we considered these stretches to arise from artefactual priming from genomic DNA or unspliced RNA (Wolfsberg and Landsman 1997) in the cDNA library construction. A further set of transcripts were rejected from the final annotations because they spliced within a series of interspersed repeat elements and did not contain an open reading frame (ORF). Alternative splicing was not comprehensively annotated in this analysis, although exons that were clearly part of alternative transcripts are included in the accompanying data files. Our rationale for this exclusion was that, because we aimed to describe the genomic structures of genes supported by transcribed sequence across their full length, we did not believe it helpful to the community to provide a long list of speculative alternatively spliced structures based on individual ESTs. Only spliced exons found in individual ESTs or cDNAs can be definitively shown to arise from the same transcript. All other structures must make assumptions about which exons are likely to form part of a complete transcript, as they are not physically linked on the same experimental evidence. There is still considerable work to do to verify the full complexity of alternative transcription across human genes by extensive resequencing of multiple cDNA clones from the same gene; although other investigators have started with bioinformatics-based identification of alternatively spliced ESTs on human chromosome 22 (Lander et al. 2001; Hide et al. 2001), a full description was beyond the scope of this work. Therefore, a proportion of unannotated intragenic EST matches may represent parts of the estimated 35% of genes with more than one transcript (Mironov et al. 1999). Where no transcribed sequence information was available, preliminary gene structures were predicted using matches to paralogous or orthologous sequences. Pseudogenes were detected and annotated by identifying non-exact matches to protein sequence, and, where possible, matches to 5′ and 3′ untranslated regions of the presumed functional gene. A small number of non-exact matches to cDNAs without a defined ORF were also annotated as pseudogenes. Data from subsequent searches of DNA and protein databases were added to the analysis and a semiautomated system was used to detect matches that might extend an existing annotation or identify a new gene. The annotation was then manually updated where necessary. The Incyte Genomics data provided additional information on 232 annotations. This information comprised 181 genes, with 8 derived from Incyte data alone; 61 were extended 5′ by at least one base, 39 were extended 3′, and 125 filled gaps in preliminary structures. This extra information was also used to extend and confirm 51 pseudogene structures. The 1436 Incyte sequences that provide this additional analysis have been submitted to the dbEST database.
Although some genes on chromosome 22 had complete cDNA sequence, for many, only partial cDNA sequence was available. Generally incomplete genes were EST clusters or cDNA sequences representing 3′ ends, occasionally with 5′ ESTs indicating potential upstream exons. In other cases without matching ESTs or cDNAs, paralogous or orthologous sequence had been used to define a preliminary gene structure. In order to extend genes, to join 5′ and 3′ EST clusters, and to confirm expression of preliminary gene structures, cDNA sequence was obtained by directed approaches. Genes with suspected additional 5′ or 3′ exons were extended using an adapted vectorette PCR cDNA library screening method (Riley et al. 1990). 5′ and 3′ EST clusters were joined and preliminary gene structures confirmed by amplifying fragments between presumed exons using PCR from cDNA. All amplified PCR fragments were sequenced and aligned to chromosome 22 and the annotation was updated. Overall, 36% of genes and 24% of exons had at least one of these directed cDNA sequence contributing to the annotation. However, ultimately <1% of the annotated bases are covered by directed sequencing outside public EST or cDNA matches. This is because new data continue to enter the databases and confirm earlier experimental work, although it should be pointed out that this includes our own cDNA submissions.
For each gene, the largest ORF of 300 bases or more was predicted and annotated. This cutoff for ORF size was arbitrary and could be set lower, but at this level no previously described protein-coding gene on chromosome 22 is excluded. In >90% of annotations, this ORF was initiated at a strong or adequate Kozak consensus sequence (Kozak 1999) or GNNATGA that we considered adequate based on empirical observation. In all except five of the remainder, a Kozak sequence was found downstream in the same reading frame and may represent the major translation initiation site. In the remaining five structures there was either no suitable Kozak sequence or it was in an alternate shorter reading frame.
Finally we had identified 936 structures supported by expressed sequence evidence, or by similarity to a known gene or protein (including the immunoglobulin λ locus variable [IGLV] and joining [IGLJ]) gene segments [Kawasaki et al. 1995; Lefranc 2001]). It remains possible that we will miss genes expressed at low levels or with limited tissue distributions, although several lines of evidence indicate that there are unlikely to be many. First, we investigated whether further genes could be identified from GenScan-predicted exons outside the annotation. Oligonucleotides were designed from 77 randomly chosen GenScan predicted exons, 40 of which coaligned with mouse genomic matches. These were used to search for clones in the 13 cDNA libraries. Any cDNA inserts found were amplified and sequenced. The small number of resulting sequences that matched chromosome 22 did not splice relative to the genomic sequence and therefore did not provide sufficient information to annotate new genes. Second, analysis of 1912 chromosome 22 sequences conserved between human, mouse, and Tetraodon showed 1900 covered by annotated structures, indicating that >99% of strongly conserved exons have already been identified. Of the 12 conserved sequences outside the annotation, 8 align with CpG islands or short stretches of low complexity sequence and are unlikely to represent genes, 1 resided in the intron of an annotated gene and was not tested further, 2 were too small to be tested by PCR, and the last could not be detected in the cDNA libraries used here. Finally, we examined the distribution of all sequences conserved in mouse. Within the span of annotated genes (intragenic), the mean conserved sequence density is 199 “hits” per Mb as detected by Blat. When annotated exon matches are removed, the intronic hit density is 30 hits per Mb, representing the residual level of sequence conservation due to either functional conservation or insufficient evolutionary divergence time. Intriguingly, the conserved sequence density between gene annotation (intergenic) is also 30 hits per Mb, indicating that there is a background level of “conservation” that may not have functional significance and that there are few, if any, conserved genes remaining to be found.
Genome annotation is a continuous process. As new data enter public databases and directed cDNA sequencing adds to the analysis, the annotation must evolve. However, we believe we have now identified the vast majority of the coding genes and have extensively reevaluated the original chromosome 22 annotations. The data describing the complete annotation set and the associated reference sequence can be found at http://www.sanger.ac.uk/HGP/Chr22. The annotation is also available as specific tracks on the NCBI 30 human genome build through the UCSC genome browser (http://genome.ucsc.edu) and the Ensembl annotation server (http://www.ensembl.org )
Annotation Classification
We categorized the annotated gene structures on the basis of the following structural features (for simplicity, IGLV and IGLJ gene segments were excluded):
A complete protein-coding gene had sequence identity to human cDNAs or ESTs across its entire length, and a predicted ORF of at least 300 bases. The probable 5′ end of the protein coding region was declared established if there was either an in-frame stop 5′ to the annotated ATG, or the ORF start matched a published protein, or the 5′ end of the structure overlapped a predicted CpG island. The 3′ end of the gene was considered established if there was a run of at least four A residues at the 3′ end of the cDNA or ESTs not present in the genomic sequence, or if there was an AATAAA or ATTAAA polyadenylation signal within 60 bases of the end. Structures from cDNAs with complete ORFs but lying partially within one of the genomic sequence gaps were also counted as coding genes.
A partial gene had sequence similarity to cDNA, EST, or peptide sequence but did not comply with the complete gene criteria. We have also annotated a new subcategory for 32 of these partial genes: partial gene duplications. These annotations match part of a coding gene elsewhere in the human genome, do not have an exact cDNA match, and do not have a disrupted ORF. This set partially overlaps with the segmental duplications previously described (Bailey et al. 2002). Where draft sequence was available, the surrounding intron sequences of the partial gene and the coding gene were compared and shown to have a high degree of similarity (Fig. 1). Because these partial gene duplications have no evidence of expression and are incomplete copies of the original genes, it is possible that they represent a form of pseudogene that we term “prepseudogenes”, in which the function of a duplicated gene or gene fragment has been disrupted perhaps at the level of transcription, but the sequence still retains an intact ORF. A similar set of genes has also recently been proposed in Caenorhabditis elegans (Mounsey et al. 2002).
Noncoding RNA genes included small RNAs, and published or complete genes that did not contain an ORF of at least 300 bases. Annotations with ORFs <300 bases that were on the opposite strand to a coding gene and were either overlapping with at least one exon or within 2 kb of the 5′ end were considered potential antisense transcripts. We observed 6 small RNA genes, 9 genes with no ORF, and 16 potential antisense genes. Intriguingly, a further 13 protein-coding genes also partially overlap with another protein-coding gene and may be antisense transcripts. A further set of rejected transcripts spliced together a series of interspersed repeat elements without an ORF representing additional noncoding transcripts.
A pseudogene had similarity to a known gene or protein but had evidence of disrupted function, either from a disrupted ORF relative to the presumed functional gene, generally in the form of one or more reading frame shifts, or because the structure was disrupted relative to a related expressed cDNA and there was no evidence of expression of RNA from the annotated structure. In addition, structures previously identified in the literature as pseudogenes were included. Pseudogenes are generally considered to arise by either retrotransposition or gene duplication. Of the 234 pseudogenes (not including the IGL locus), 168 do not contain introns and are potentially produced by retrotransposition. Of these, 27 contain sufficient sequence conservation at the 3′ end and a stretch of at least five As in the genomic DNA preceded by a polyadenylation signal to indicate relatively recent processing. Of the 66 pseudogenes believed to be the result of gene duplications, most have intron/exon structure. They include 23 low-copy-repeat sequences in 22q11 derived from a genomic duplication (Bailey et al. 2002). If the partial gene duplications do represent prepseudogenes, this could further increase the number of pseudogenes at the expense of protein-coding genes.
Figure 1.

Demonstration of a partial gene duplication using DOTTER analysis. Part of the adaptin gene (AP1B1) has been duplicated and rearranged 2.8 Mb telomeric on chromosome 22, to leave a structure in which the duplicated exon 6 (ADTB1L1) is followed 3′ by the duplicated exon 1 (ADTB1L2) and there is conservation of the surrounding intron sequences. Genomic sequence accession number and portion of the sequence used is indicated (note AC000026 has been reversed). Gene structures are shown by a black line (introns) and hashed boxes (exons), and an arrow pointing toward the 3′ end of the gene. Within the dot matrix, diagonal lines indicate regions of sequence identity and dashed lines show where exon sequences align.
Using these classification criteria, the 936 structures annotated comprised 393 complete protein-coding genes, 153 partial genes, 31 noncoding transcripts, 234 pseudogenes, and 125 IGLV and J gene segments (Table 1). Since our original annotation (Dunham et al. 1999b), we have added 198 new annotations comprising 40 complete coding genes, 66 partial genes, 25 noncoding RNAs, and 67 pseudogenes. The overall genomic span of the annotation has increased from 13.0 Mb to 18.6 Mb (43% increase), and the coverage in exonic bases has increased from 1.04 Mb to 1.81 Mb (74%) so that >50% of the chromosome 22 sequence is occupied by genes, and 5% by exons. However, the overall protein-coding gene number has only increased from 545 to 546. This is because extension of annotations resulted in 83 original gene structures being merged into current genes, new classification allowed rejection of 13 original genes, and reassessment of the remainder of the original annotations resulted in a change of category. In contrast, the pseudogene count has increased from 134 to 234 because of the availability of additional genomic and expressed sequence.
Table 1.
Gene Number and Categories
| Gene categories | Total | Subcategories | Total | Genes with ORF | Probable 5′-end | Confirmed 3′ end | Antisense features | Single exon | Multi exon | No. of exons |
| Coding gene | 393 | Full | 387 | 387 | 380 | 371 | 2 | 19 | 368 | 3749 |
| Partially in gap | 6 | 6 | 1 | 5 | 6 | 30 | ||||
| Partial gene | 153 | Potential coding | 121 | 68 | 10 | 56 | 33 | 88 | 523 | |
| Gene duplication | 32 | 8 | 24 | 98 | ||||||
| Noncoding gene | 31 | Small RNA | 6 | 6 | 6 | |||||
| Possible antisense | 16 | 12 | 16 | 9 | 7 | 29 | ||||
| Gene—no ORF | 9 | 3 | 5 | 4 | 19 | |||||
| Pseudogene | 234 | Retrotransposon | 168 | 168 | 168 | |||||
| Gene duplication | 66 | 5 | 61 | 348 | ||||||
| IGLV | 118 | |||||||||
| IGLJ | 7 | |||||||||
| Total | 936 | 811 | 461 | 391 | 447 | 18 | 253 | 558 | 4970 |
(IGLV) Immunoglobulin λ variable; (IGLJ) immunoglobulin λ joining.
The Characteristics of the Annotation Set
Next, we assessed the contributions of different data sources to the annotation using an approach widely used to assess gene prediction software (Burset and Guigo 1996). The final annotation coordinates were extracted and compared with the bases matched by each data source. We calculated sensitivity (a measure of the ability to detect true positives), specificity (a measure of the ability to discriminate against false positives), and the fraction of the annotation detected by gene and by exon, for each data source, over both the full annotation and by category (Table 2, Fig. 2).
Table 2.
Assessment of the Annotation Set
| Total annotation set | Coding genes only | ORF of coding genes | Pseudogenes | |||||||||||||
| Sn | Sp | G | E | Sn | Sp | G | E | Sn | Sp | G | E | Sn | Sp | G | E | |
| cDNA | ||||||||||||||||
| dbEST | 0.77 | 0.42 | 0.97 | 0.92 | 0.80 | 0.32 | 0.98 | 0.92 | 0.85 | 0.17 | 0.98 | 0.93 | 0.71 | 0.08 | 0.97 | 0.93 |
| EMBL_vertrna | 0.69 | 0.79 | 0.88 | 0.87 | 0.74 | 0.62 | 0.88 | 0.87 | 0.87 | 0.36 | 0.92 | 0.91 | 0.62 | 0.14 | 0.92 | 0.87 |
| Human | 0.68 | 0.79 | 0.82 | 0.83 | ||||||||||||
| Mouse | 0.10 | 0.96 | 0.49 | 0.28 | ||||||||||||
| Other | 0.11 | 0.93 | 0.38 | 0.24 | ||||||||||||
| Protein databases | 0.55 | 0.31 | 0.91 | 0.89 | 0.58 | 0.24 | 0.93 | 0.90 | 0.94 | 0.19 | 0.98 | 0.94 | 0.55 | 0.06 | 0.91 | 0.87 |
| Comparative | ||||||||||||||||
| Mus blat | 0.29 | 0.66 | 0.77 | 0.59 | 0.33 | 0.55 | 0.81 | 0.60 | 0.60 | 0.49 | 0.88 | 0.64 | 0.24 | 0.11 | 0.73 | 0.59 |
| Mus exo | 0.26 | 0.57 | 0.79 | 0.54 | 0.28 | 0.45 | 0.80 | 0.53 | 0.49 | 0.40 | 0.88 | 0.56 | 0.25 | 0.11 | 0.79 | 0.60 |
| Exofish | 0.13 | 0.92 | 0.59 | 0.38 | 0.15 | 0.76 | 0.64 | 0.38 | 0.29 | 0.75 | 0.72 | 0.42 | 0.12 | 0.16 | 0.54 | 0.45 |
| Prediction | ||||||||||||||||
| Genscan | 0.39 | 0.68 | 0.79 | 0.79 | 0.46 | 0.59 | 0.90 | 0.82 | 0.88 | 0.56 | 0.95 | 0.87 | 0.24 | 0.08 | 0.61 | 0.57 |
| fgenesh | 0.35 | 0.76 | 0.70 | 0.75 | 0.42 | 0.67 | 0.82 | 0.79 | 0.82 | 0.63 | 0.89 | 0.85 | 0.19 | 0.08 | 0.48 | 0.51 |
| Multiple | ||||||||||||||||
| GS+blata | 0.27 | 0.95 | 0.62 | 0.49 | 0.32 | 0.85 | 0.75 | 0.52 | 0.65 | 0.83 | 0.85 | 0.58 | 0.13 | 0.09 | 0.38 | 0.30 |
| Fish + mouseb | 0.11 | 0.95 | 0.54 | 0.31 | 0.12 | 0.79 | 0.59 | 0.31 | 0.25 | 0.78 | 0.67 | 0.34 | 0.09 | 0.16 | 0.47 | 0.35 |
Note that for the subdivided data, the specificity calculation includes all matches and so will be reduced relative to the total annotation. Comparisons of specificity are therefore only fair within columns.
GenScan exons aligned with a blat mouse match.
Exofish matches that align with a mouse match from either method.
(Sn) Sensitivity (including all immunoglobulin λ [IGL] segments); (Sp) specificity (including all IGL segments); (G) gene hits (only counting [GLC] segments); (E) exon hits (only counting immunoglobulin λ constant [IGLC] segments).
Figure 2.

Plot of sensitivity versus specificity for each data source. Sensitivity and specificity were calculated over the full annotation set as described in Methods; see Table 2. Each data source is indicated by a different color and symbol.
The cDNA sequence databases each have high sensitivity and provide the bulk of the annotation coverage. This is expected, given our emphasis on validation of annotation by expressed sequence data. However the difference in specificity between the cDNA and EST data sets is informative. dbEST sequences identified 97% of the annotation, but with low specificity because over half the matched bases were considered unsuitable for annotation due to the requirement for ESTs to splice or have confirmed 3′ ends. Thus, a high level of intervention is required to filter the noise from this large data set. However, although the EMBL_vertrna database detects slightly fewer annotations, it has almost double the specificity. This indicates that extracting sequence records containing vertebrate cDNA sequences provides a higher quality of annotation. The effort required to produce “full-length” cDNA sequences is well justified and should play a major part in future human genome annotation. Coverage of the annotation in cDNA sequences is also remarkably even and deep. Hence, if we require two or more EMBL_vertrna sequences to match annotation before declaring a true positive match, then sensitivity and specificity remain quite high at 0.52 and 0.77, respectively (compared with 0.69 and 0.79 for a single EMBL_vertrna match), whereas the gene and exon hits both drop only slightly to 0.84 (data not shown). Examining the species of origin of the cDNA sequences in this set reveals that, of 6048 EMBL_vertrna sequences used in support of the annotation, the major contribution (64%) is from human clones, whereas 19% are murine clones, and the remainder are from a variety of other species. This reflects the contribution of the extensive programs aimed at producing full-length human cDNA sequences (Strausberg et al. 1999; Nagase et al. 2000; Wiemann et al. 2001) and latterly the RIKEN mouse cDNA sequencing program (Kawai et al. 2001). However, in terms of producing the annotation, human cDNAs detect 83% of the annotated exons, whereas murine cDNAs detect only 28%, compared with 87% detected by the complete EMBL_vertmrna set (Table 2). Clearly, human cDNAs contributed the majority of the information at the time of analysis.
On the other hand, careful filtering of EST data can provide the maximum annotation, as judged by the high sensitivity. These observations may also partly explain why approaches that aim to measure transcription directly using microarrays indicate more transcription than accounted for by annotated exons (Kapranov et al. 2002) because the cDNA used in these experiments may contain similar unspliced or artefactual transcripts. The raw specificity score is not completely appropriate for cDNA-derived data sources, because it takes no account of the weight of evidence supporting a particular annotation or false positive. For instance, a single EST containing an unspliced intron contributes as much to the false-positive score as 50 ESTs associated with the correct exon structure contribute to the true positive score. We attempted to compensate for this effect by either weighting the contribution of each base to the specificity according to the number of matches involving that base (“weighted specificity”) or by setting a threshold so that true- and false-positive bases only contribute if they have at least N matches (“threshold specificity”). Application of these measures raises specificity with some reduction in sensitivity and could form the basis of automated strategies to annotate from EST data (the behavior of each of these measures for the full annotation is explored in the Supplementary information).
The protein databases show high sensitivity and gene detection rate particularly if only the ORF of coding genes are considered. This is partly expected because the translations of public cDNAs are regularly added to databases such as Swiss Prot and Trembl, but also reflects matching of orthologs and paralogs, as well as conserved protein domains. In contrast, specificity is very low with reflecting nonspecific protein matches because of the relatively low percentage identity cutoff we have used. For instance, simple sequences and CG-rich predicted CpG islands are frequently matched with fragments of collagen genes.
The T. nigroviridis (Exofish) and mouse genomic sequence analysis allows us to test the utility of comparative analysis for gene annotation. Exofish was highly specific, illustrating that similarity between human and Tetraodon sequences, as filtered through the exofish algorithm, is very likely to indicate part of a gene. However, low sensitivity indicates that the exofish approach cannot give full coverage, and indeed the 59% gene-detection rate indicates many structures were missed. This may partly reflect lack of sequence conservation, and the 1.1 × coverage of the sequence data used. Mouse genomic sequence, however, shows double the sensitivity and high gene and exon detection, at the expense of much lower specificity. This is consistent with the observation that a quarter of mouse sequences matched using Blat lay outside annotated exons. Taking exofish regions that align to mouse matches, the specificity increases to 95%, 3% above exofish alone. This combination is the most accurate gene identification method assessed, but at the expense of sensitivity. It is important to note that, although comparative sequencing methods identify regions to be annotated, they do not provide enough information on their own to accurately annotate gene structures.
Both GenScan and fgenesh predict only coding regions of genes. Hence, the fairest comparison of these data is with coding-gene ORFs. Sensitivity, specificity, and gene and exon detection rate are all high, with GenScan slightly more sensitive and fgenesh slightly more specific. However, neither method is able to detect every gene or to identify complete exon/intron structures. Separating each GenScan prediction into exons and examining only those exons that align to a mouse Blat match dramatically increases specificity, confirming that a combined approach reduces the problems of false positives from either method alone (Korf et al. 2001).
For pseudogene annotation, both the dbEST and vertrna databases have high sensitivity and detection. Mouse genomic sequence also scores well for pseudogene detection at the gene-hit level, but with low sensitivity, indicating that comparative genomic sequence analysis detects the pseudogenes but does not give full coverage of pseudogene structures. Gene prediction programs identify ORFs, and, not surprisingly, performed poorly for pseudogene detection.
It is also informative to compare the results of the detailed annotation approach performed here with annotation obtained from high-throughput approaches. One such approach is simply to align vertebrate cDNA sequences to the genomic sequence, as we have done. The resulting sensitivity and specificity is relatively high, as discussed earlier, contributing approximately 70% of the annotation. However this sensitivity is slightly inflated because we have been contributing additional cDNA sequences to the databases as we have confirmed them. To assess a different annotation approach, we also examined an example of one of the high-throughput annotation databases, Ensembl (build 28) (http://www.ensembl.org), by calculating sensitivity (0.50) and specificity (0.90) compared with the whole chromosome 22 annotation as before. These results indicate that, although Ensembl has relatively few false-positive predictions, the additional information gained by the detailed approach described here over and above the first pass approach is substantial. However, it should also be pointed out that simply measuring sensitivity and specificity alone does not provide detailed information on either the continuity of gene structures or their correctness. Indeed, as time has passed, much of the experimental cDNA sequencing we have done has been confirmed by new ESTs or cDNAs entering the databases, and the key value of human intervention and experimental sequencing comes in linking partial structures and resolving evidence so that annotations are accurate.
DISCUSSION
The updated set of public and proprietary EST and cDNA sequence databases as well as experimental verification has allowed the extent of protein-coding gene annotation to be greatly increased (Table 1). We have subdivided the set of 153 partial genes into 121 genes where we believe there is more cDNA sequence to identify and 32 where the annotation appears to be the result of a small genomic duplication that included only part of a coding gene. We propose that these partial gene duplications are prepseudogenes, as they do not have a disrupted ORF, but at the same time they have no evidence of expression and are not a complete copy of the original gene, as has recently been proposed in C. elegans (Mounsey et al. 2002). We have also improved the annotation of pseudogenes so that the pseudogene content of chromosome 22 is now almost one-third of the total annotation. This is primarily because of the availability of more human genome sequence. Intriguingly, the revised annotation criteria have allowed us to categorize 31 transcripts that do not appear to code for protein, 16 of which may be antisense RNAs.
Previous annotations of finished human chromosome sequence (Dunham et al. 1999b; Hattori et al. 2000; Deloukas et al. 2001) have followed similar strategies, on the basis of identification of transcribed sequences supported by cDNA or EST sequences. However, until now these annotations have not been updated to take account of new evidence and have not had extensive experimental confirmation. The present annotation involved several iterations of automatic analysis of the genomic sequence, followed by manual annotation and completion of genes with directed laboratory work. It follows a set of criteria as defined earlier and each annotation has been assessed individually according to the evidence. The gene prediction programs and the cross-species analysis have helped to identify expressed regions of the genome, but it is the cDNA-derived sequence data that have been used to define the final gene structures. In addition, in contrast to previous chromosome annotations including our own, we have chosen to categorize our gene structures according to the completeness and functionality of the structures rather than by the evidence that was used to find them. It seems to us that this has significant value for users of the annotation. The requirement for evidence of transcription in human tissue before a gene is considered complete should ensure that the genes are biologically active. The annotation cannot be final, as a proportion of the genes does not satisfy the complete gene criteria. There remains the possibility of further genes to be discovered and for movement between categories as further data become available, for instance by experimental confirmation of the 5′ and 3′ ends of partial genes. Additionally, with further sequence information, some unspliced ESTs currently outside of our criteria may span splice sites or gain confirmed 3′ ends, providing new gene annotations. However, overall we believe that this annotation provides a high-quality reference both for functional analysis of genes on chromosome 22, and for training the next generation of annotation programs. It also acts as a template for future human genome annotation, and we recommend consideration of a similar strategy and classification to provide a complete human gene index. In considering the practicality of this approach, it is noteworthy that our annotation of 1% of the genome has required ∼6 person years for bioinformatics, annotation, and experimental work, within the context of the bioinformatics infrastructure of a large genome center. Clearly, this would have to be streamlined for the remainder of the genome, but, with the experience gained and the increasing amount of cDNA sequence entering the database, this should be possible. It could be envisaged that a three-tier approach could be adopted, with initial annotation based on human intervention to resolve initial gene structures based on expressed sequence alignments, gene prediction, and comparative sequence data. This would be followed by a second tier of experimental cDNA sequencing to join or resolve gene structures, and then a final tier of detailed analysis gene by gene with experimentation to finish off 5′ ends, or fully describe alternative splices. The manpower required would increase through the tiers, and would become increasingly community based for the final tier. The initial part of this system is already the basis for annotation of finished genome sequence at the Sanger Institute.
Finally, our new annotation enables revision of the estimated number of protein-coding genes in the genome. Extrapolating from 546 protein-coding genes from 1.1% of the genome and correcting for gene density (http://www.ncbi.nlm.nih.gov/genemap99/page.cgi?F=GeneDistrib.html) gives 35,968 genes in the genome. Extending the analysis to include the published annotations of chromosomes 20 (Deloukas et al. 2001) and 21 (Hattori et al. 2000) and taking the mean gives a lower estimate of 30,137. If 32 chromosome 22 partial gene duplications are also excluded, the same calculation predicts 29,434 protein-coding genes. Assuming the pseudogene distribution within the genome is unbiased, the chromosome 22 data indicate there are ∼21,300 pseudogenes in the whole genome. Similarly, without distribution bias, there may be as many as 1500 antisense RNAs.
METHODS
Genomic Sequence Analysis
The assembled genomic DNA sequence of human chromosome 22 (Version3 http://www.sanger.ac.uk/HGP/Chr22/cwa_archive/Release_3_14–09–2001 identical to the NCBI 30 build of the human genome sequence [see http://www.ensembl.org/Homo_sapiens/stats/status.html]) was analyzed for repetitive sequences and repeats were masked using RepeatMasker (http://ftp.genome.washington.edu/RM/RepeatMasker.html). Masked sequence was analyzed using WU BLAST 2.0 (W. Gish, 1996–2002, http://blast.wustl.edu) against dbEST (version 112001; Boguski and Schuler 1995); a set of vertebrate mRNA sequences extracted from EMBL 68 (termed EMBL_vertmRNA); a set of 1436 Incyte Genomics EST and cDNA sequences received in December 2000 that matched chromosome 22 at 85% identity and either extended annotated genes or were new spliced structures (a kind gift from J. Seilhamer and L. Stuve); and the predicted protein databases Swissprot 40.6 (Bairoch and Apweiler 2000), sptrEMBL 15, Wormpep 25, and Gadfly release 2. Nucleotide and protein similarity searches were postprocessed using MSPcrunch (Sonnhammer and Durbin 1994) to exclude matches below either 90% or 30% identity, respectively. The masked sequence was also matched to T. nigroviridis genomic sequence (421 Mb of Tetraodon shotgun sequence reads, equivalent to 1.1 × genome coverage) using the Exofish algorithm (Roest Crollius et al. 2000; a kind gift from H. Roest-Crollius [Genoscope, Evry, France]) and to mouse whole genomic sequence using BLAT (Kent 2002; ∼8 Gb of raw mouse shotgun sequence reads equivalent to 2.0 × genome coverage, a kind gift from J. Kent [University of California, Santa Cruz, CA]) and exonerate (G. Slater, unpubl.; ∼5Gb of raw mouse shotgun sequence reads equivalent to 1.3 × genome coverage, a kind gift from G. Slater [Wellcome Trust Sanger Institute, Hinxton, UK]). Masked sequence was analyzed for putative exons using GenScan (Burge and Karlin 1997) and fgenesh (Solovyev et al. 1995). CpG islands of ≥400 bases were predicted in unmasked sequence using CPGFIND (G. Micklem, unpubl.), using minimum cutoffs of 50% G+C and 0.6 observed/expected CpG frequency. Potential U2-dependent introns were identified by scanning the sequence and identifying introns supported by spliced ESTs or cDNAs (Levine and Durbin 2001). Analyses were converted into ACEDB format and displayed in a chromosome 22-specific implementation of ACEDB (Durbin and Thierry-Mieg 1991; Dunham and Maslen 1996). To annotate gene structures and check annotation, we used a mixture of manual inspection, a variety of tools provided in ACEDB including Genefinder (Favello et al. 1995), alignment between cDNA and genomic sequences using EST_GENOME (Mott 1997), and a series of external Perl scripts written ad_hoc in Perl 5.004_04. Tools to extract and convert data formats were also written in Perl. Perl scripts are available on request from dmb@sanger.ac.uk. DOTTER was performed as previously described (Sonnhammer and Durbin 1995). The variable (IGLV) gene segments of the immunoglobulin λ locus were annotated based on previously published descriptions (Kawasaki et al. 1995; Lefranc 2001).
Gene names used in the annotation are either approved by the HUGO Nomenclature Committee (Wain et al. 2002), or, when an official name has not yet been assigned, are named by reference to the sequence in which they reside.
Directed cDNA Sequencing
To confirm and extend the cDNA sequence, we either sequenced fragments that were PCR amplified from primary cDNA or sequenced fragments that were PCR amplified from pools of DNA from cDNA clones modified with vectorette bubble linkers. For amplification from cDNA, PCR primers designed to bridge between potential exons were used to amplify from Human Universal QUICK-Clone cDNA (Clontech Cat# 7109–1). PCR was performed as described in Dunham et al. (1999a) using Advantage Taq Polymerase (Clontech), PerfectMatch PCR Enhancer (Stratagene), and TaqExtender PCR additive (Stratagene). PCR fragments were cleaned by either QIAquick Gel Extraction Kit or Shrimp alkaline phosphatase (1 unit, Amersham) and ExonucleaseI (1 unit, Amersham), sequenced and realigned to genomic sequence using EST_GENOME (Mott 1997).
For recovery of cDNA fragments by vectorette PCR, cDNA libraries from 13 tissues (Invitrogen: fetal brain, fetal liver, neuroblastoma, fetal lung, adult heart, peripheral blood HL60; Clontech: testis, adult lung; a gift from D. Simmonds [Institute of Molecular Medicine, Oxford, UK]: placenta, adult brain, monocyte U937, B lymphoma Daudi; a gift from M. Stammers [Wellcome Trust Sanger Institute, Hinxton, UK]: small intestine) were titrated and, for each library, 25 pools of 20,000 clones (a total of 6.5 million cDNA clones) were grown overnight in 25 mL of LB broth plus the appropriate antibiotic. DNA was extracted, the cDNA insert excised with an appropriate restriction enzyme, and vectorette bubble adaptors (Riley et al. 1990) ligated to the cDNA fragments. To identify pools containing useful cDNA clones, we screened primers designed from potential exon sequences across the pools by PCR. The complete cDNA clone inserts were then amplified from the positive pools of 20,000 clones using one gene specific primer and the bubble primer 224 by PCR using Ampli-Taq with manual hot start, PerfectMatch PCR Enhancer (Stratagene) and TaqExtender PCR additive (Stratagene). Amplified fragments were purified, sequenced, and realigned to the genomic DNA as described earlier.
Analysis of the Annotation
All analyses of the annotation set were performed using the genomic sequence analysis data and annotation extracted in gff format (http://www.sanger.ac.uk/Software/formats/GFF/index.shtml) from the ACEDB database. The coordinates of the annotation features and data sources relative to the genomic sequences were extracted and overall coverage in nucleotides calculated. The sensitivity and specificity of each annotation data source relative to the final annotation were determined as described in Burset and Guigo (1996). For the strand-specific data sources (protein databases, GenScan, and fgenesh), we calculated sensitivity and specificity on both a strand-specific and strand-independent basis. However, because there was no significant difference in these figures, only the strand-independent data are shown in Table 2.
In brief, the following values were calculated.
True positives (TP): The number of annotated nucleotides matched by a data source.
False negatives (FN): The number of annotated nucleotides not matched in the data source.
False positives (FP): The number of chromosome 22 sequence nucleotides aligned to the data source that do not form part of the annotation.
Sensitivity (Sn) and specificity (Sp) were defined as:
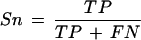 |
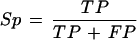 |
In addition, for the expressed sequence data sources (dbEST and vertebrate mRNAs), we calculated a weighted specificity and a threshold specificity. For the weighted specificity, we take into account the number of sequence matches in the data source to a true-positive or false-positive base by incrementing the contribution of that base by the number of times it is matched in the data source, either without limit or up to a limit. In the threshold specificity, we only allow a base to be included in the specificity calculation if it has been matched by the data source at least N times, where N is the threshold value. The behavior of both of these modified specificity values is examined in the supplementary information. We also determined whether any part of a gene or exon was detected by each data source, which we refer to as gene hits and exon hits, respectively. The IGLV gene segments were not included in the gene or exon detection analysis. The IGLJ gene segments were included in pairs with their respective Immunoglobulin λ constant (IGLC) gene segment. A variety of Perl scripts were used to analyze other features of the annotated genes, and these are available on request from dmb@sanger.ac.uk.
WEB SITE REFERENCES
http://blast.wustl.edu; W. Gish WU-BLAST 2.0.
http://ftp.genome.washington.edu/RM/RepeatMasker.html; A. Smit RepeatMasker.
http://www.ensembl.org; Ensembl annotation server.
http://www.ncbi.nlm.nih.gov/genemap99/page.cgi?F=GeneDistrib.html; Gene Map'99.
http://www.sanger.ac.uk/HGP/Chr22; Sanger Institute chromosome 22 home page.
http://www.sanger.ac.uk/HGP/Chr22/cwa_archive/Release_3_14–09–2001; Sanger Institute chromosome 22 sequence assembly.
http://www.sanger.ac.uk/Software/formats/GFF/index.shtml; GFF format rules.
Acknowledgments
We thank Jeff Seilhamer and Laura Stuve for the gift of ESTs from the Incyte database, Hugues Roest-Crollius for exofish data, Aaron Levine for spliced EST analysis, the Mouse Genome Sequence Consortium for early access to mouse shotgun data, Guy Slater for use of the exonerate mouse matches, and Jim Kent for Blat mouse matches. Many thanks to Richard Glynne, Jim Kent, and Jane Rogers for helpful comments on the manuscript. This work was supported by the Wellcome Trust.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
E-MAIL id1@sanger.ac.uk; FAX +44 (0) 1223 494919
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.695703. Article published online before print in December 2002.
REFERENCES
- 1.Bailey J.A., Yavor, A.M., Viggiano, L., Misceo, D., Horvath, J.E., Archidiacono, N., Schwartz, S., Rocchi, M., and Eichler, E.E. 2002. Human-specific duplication and mosaic transcripts: The recent paralogous structure of chromosome 22. Am. J. Hum. Genet. 70: 83-100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bairoch A. and Apweiler, R. 2000. The SWISS-PROT protein sequence database and its supplement TrEMBL in 2000. Nucleic Acids Res. 28: 45-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Boguski M.S. and Schuler, G.D. 1995. ESTablishing a human transcript map. Nat. Genet. 10: 369-371. [DOI] [PubMed] [Google Scholar]
- 4.Burge C. and Karlin, S. 1997. Prediction of complete gene structures in human genomic DNA. J. Mol. Biol. 268: 78-94. [DOI] [PubMed] [Google Scholar]
- 5.Burset M. and Guigo, R. 1996. Evaluation of gene structure prediction programs. Genomics 34: 353-367. [DOI] [PubMed] [Google Scholar]
- 6.Das M., Burge, C.B., Park, E., Colinas, J., and Pelletier, J. 2001. Assessment of the total number of human transcription units. Genomics 77: 71-78. [DOI] [PubMed] [Google Scholar]
- 7.Deloukas P., Matthews, L.H., Ashurst, J., Burton, J., Gilbert, J.G., Jones, M., Stavrides, G., Almeida, J.P., Babbage, A.K., Bagguley, C.L., et al. 2001. The DNA sequence and comparative analysis of human chromosome 20. Nature 414: 865-871. [DOI] [PubMed] [Google Scholar]
- 8.de Souza S.J., Camargo, A.A., Briones, M.R., Costa, F.F., Nagai, M.A., Verjovski-Almeida, S., Zago, M.A., Andrade, L.E., Carrer, H., El-Dorry, H.F., et al. 2000. Identification of human chromosome 22 transcribed sequences with ORF expressed sequence tags. Proc. Natl. Acad. Sci. 97: 12690-12693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dubchak I., Brudno, M., Loots, G.G., Pachter, L., Mayor, C., Rubin, E.M., and Frazer, K.A. 2000. Active conservation of noncoding sequences revealed by three-way species comparisons. Genome Res. 10: 1304-1306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dunham I. and Maslen, G.L. 1996. Use of ACEDB as a database for YAC library data management. Methods Mol. Biol. 54: 253-280. [DOI] [PubMed] [Google Scholar]
- 11.Dunham I., Dewar, K., Kim, U.-J., and Ross, M.T. 1999a. Bacterial cloning systems. In Genome analysis: A laboratory manual series (ed. J. Roskams), pp. 1–86. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 12.Dunham I., Shimizu, N., Roe, B.A., Chissoe, S., Hunt, A.R., Collins, J.E., Bruskiewich, R., Beare, D.M., Clamp, M., Smink, L.J., et al. 1999b. The DNA sequence of human chromosome 22. Nature 402: 489-495. [DOI] [PubMed] [Google Scholar]
- 13.Durbin, R. and Thierry-Mieg, J. 1991. A.C. elegans database. http://www.sanger.ac.uk/Software/Acedb/.
- 14.Ewing B. and Green, P. 2000. Analysis of expressed sequence tags indicates 35,000 human genes. Nat. Genet. 25: 232-234. [DOI] [PubMed] [Google Scholar]
- 15.Favello A., Hillier, L., and Wilson, R.K. 1995. Genomic DNA sequencing methods. Methods Cell Biol. 48: 551-569. [DOI] [PubMed] [Google Scholar]
- 16.Frazer K.A., Sheehan, J.B., Stokowski, R.P., Chen, X., Hosseini, R., Cheng, J.F., Fodor, S.P., Cox, D.R., and Patil, N. 2001. Evolutionarily conserved sequences on human chromosome 21. Genome Res. 11: 1651-1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Guigo R., Agarwal, P., Abril, J.F., Burset, M., and Fickett, J.W. 2000. An assessment of gene prediction accuracy in large DNA sequences. Genome Res. 10: 1631-1642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hardison R.C., Oeltjen, J., and Miller, W. 1997. Long human-mouse sequence alignments reveal novel regulatory elements: A reason to sequence the mouse genome. Genome Res. 7: 959-966. [DOI] [PubMed] [Google Scholar]
- 19.Harrison P.M., Hegyi, H., Balasubramanian, S., Luscombe, N.M., Bertone, P., Echols, N., Johnson, T., and Gerstein, M. 2002. Molecular fossils in the human genome: Identification and analysis of the pseudogenes in chromosomes 21 and 22. Genome Res. 12: 272-280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hattori M., Fujiyama, A., Taylor, T.D., Watanabe, H., Yada, T., Park, H.S., Toyoda, A., Ishii, K., Totoki, Y., Choi, D.K., et al. 2000. The DNA sequence of human chromosome 21. Nature 405: 311-319. [DOI] [PubMed] [Google Scholar]
- 21.Hide W.A., Babenko, V.N., van Heusden, P.A., Seoighe, C., and Kelso, J.F. 2001. The contribution of exon-skipping events on chromosome 22 to protein coding diversity. Genome Res. 11: 1848-1853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hogenesch J.B., Ching, K.A., Batalov, S., Su, A.I., Walker, J.R., Zhou, Y., Kay, S.A., Schultz, P.G., and Cooke, M.P. 2001. A comparison of the Celera and Ensembl predicted gene sets reveals little overlap in novel genes. Cell 106: 413-415. [DOI] [PubMed] [Google Scholar]
- 23.Kapranov P., Cawley, S.E., Drenkow, J., Bekiranov, S., Strausberg, R.L., Fodor, S.P., and Gingeras, T.R. 2002. Large-scale transcriptional activity in chromosomes 21 and 22. Science 296: 916-919. [DOI] [PubMed] [Google Scholar]
- 24.Kawai J., Shinagawa, A., Shibata, K., Yoshino, M., Itoh, M., Ishii, Y., Arakawa, T., Hara, A., Fukunishi, Y., Konno, H., et al. 2001. Functional annotation of a full-length mouse cDNA collection. Nature 409: 685-690. [DOI] [PubMed] [Google Scholar]
- 25.Kawasaki K., Minoshima, S., Schooler, K., Kudoh, J., Asakawa, S., de Jong, P.J., and Shimizu, N. 1995. The organization of the human immunoglobulin λ gene locus. Genome Res. 5: 125-135. [DOI] [PubMed] [Google Scholar]
- 26.Kent W.J. 2002. BLAT—The BLAST-like alignment tool. Genome Res. 12: 656-664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kondrashov A.S. and Shabalina, S.A. 2002. Classification of common conserved sequences in mammalian intergenic regions. Hum. Mol. Genet. 11: 669-674. [DOI] [PubMed] [Google Scholar]
- 28.Korf I., Flicek, P., Duan, D., and Brent, M.R. 2001. Integrating genomic homology into gene structure prediction. Bioinformatics 17 (Suppl 1): S140-148. [DOI] [PubMed] [Google Scholar]
- 29.Kozak M. 1999. Initiation of translation in prokaryotes and eukaryotes. Gene 234: 187-208. [DOI] [PubMed] [Google Scholar]
- 30.Kumar M. and Carmichael, G.G. 1998. Antisense RNA: Function and fate of duplex RNA in cells of higher eukaryotes. Microbiol. Mol. Biol. Rev. 62: 1415-1434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lander E.S., Linton, L.M., Birren, B., Nusbaum, C., Zody, M.C., Baldwin, J., Devon, K., Dewar, K., Doyle, M., FitzHugh, W., et al. 2001. Initial sequencing and analysis of the human genome. Nature 409: 860-921. [DOI] [PubMed] [Google Scholar]
- 32.Lefranc M.P. 2001. Nomenclature of the human immunoglobulin λ (IGL) genes. Exp. Clin. Immunogenet. 18: 242-254. [DOI] [PubMed] [Google Scholar]
- 33.Levine A. and Durbin, R. 2001. A computational scan for U12-dependent introns in the human genome sequence. Nucleic Acids Res. 29: 4006-4013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liang F., Holt, I., Pertea, G., Karamycheva, S., Salzberg, S.L., and Quackenbush, J. 2000. Gene index analysis of the human genome estimates approximately 120,000 genes. Nat. Genet. 25: 239-240. [DOI] [PubMed] [Google Scholar]
- 35.Mattick J.S. 2001. Non-coding RNAs: The architects of eukaryotic complexity. EMBO Rep. 2: 986-991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mayor C., Brudno, M., Schwartz, J.R., Poliakov, A., Rubin, E.M., Frazer, K.A., Pachter, L.S., and Dubchak, I. 2000. VISTA: Visualizing global DNA sequence alignments of arbitrary length. Bioinformatics 16: 1046-1047. [DOI] [PubMed] [Google Scholar]
- 37.Mironov A.A., Fickett, J.W., and Gelfand, M.S. 1999. Frequent alternative splicing of human genes. Genome Res. 9: 1288-1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mott R. 1997. EST_GENOME: A program to align spliced DNA sequences to unspliced genomic DNA. Comput. Appl. Biosci. 13: 477-478. [DOI] [PubMed] [Google Scholar]
- 39.Mounsey A., Bauer, P., and Hope, I.A. 2002. Evidence suggesting that a fifth of annotated Caenorhabditis elegans genes may be pseudogenes. Genome Res. 12: 770-775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nagase T., Kikuno, R., Ishikawa, K., Hirosawa, M., and Ohara, O. 2000. Prediction of the coding sequences of unidentified human genes. XVII. The complete sequences of 100 new cDNA clones from brain which code for large proteins in vitro. DNA Res. 7: 143-150. [DOI] [PubMed] [Google Scholar]
- 41.Penn S.G., Rank, D.R., Hanzel, D.K., and Barker, D.L. 2000. Mining the human genome using microarrays of open reading frames. Nat. Genet. 26: 315-318. [DOI] [PubMed] [Google Scholar]
- 42.Riley J., Butler, R., Ogilvie, D., Finniear, R., Jenner, D., Powell, S., Anand, R., Smith, J.C., and Markham, A.F. 1990. A novel, rapid method for the isolation of terminal sequences from yeast artificial chromosome (YAC) clones. Nucleic Acids Res. 18: 2887-2890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Roest Crollius H., Jaillon, O., Bernot, A., Dasilva, C., Bouneau, L., Fischer, C., Fizames, C., Wincker, P., Brottier, P., Quetier, F., et al. 2000. Estimate of human gene number provided by genome-wide analysis using Tetraodon nigroviridis DNA sequence. Nat. Genet. 25: 235-238. [DOI] [PubMed] [Google Scholar]
- 44.Shoemaker D.D., Schadt, E.E., Armour, C.D., He, Y.D., Garrett-Engele, P., McDonagh, P.D., Loerch, P.M., Leonardson, A., Lum, P.Y., Cavet, G., et al. 2001. Experimental annotation of the human genome using microarray technology. Nature 409: 922-927. [DOI] [PubMed] [Google Scholar]
- 45.Solovyev V.V., Salamov, A.A., and Lawrence, C.B. 1995. Identification of human gene structure using linear discriminant functions and dynamic programming. Proc. Int. Conf. Intell. Syst. Mol. Biol. 3: 367-375. [PubMed] [Google Scholar]
- 46.Sonnhammer E.L. and Durbin, R. 1994. A workbench for large-scale sequence homology analysis. Comput. Appl. Biosci. 10: 301-307. [DOI] [PubMed] [Google Scholar]
- 47.___, 1995. A dot-matrix program with dynamic threshold control suited for genomic DNA and protein sequence analysis. Gene 167: GC1-10. [DOI] [PubMed] [Google Scholar]
- 48.Strausberg R.L., Feingold, E.A., Klausner, R.D., and Collins, F.S. 1999. The mammalian gene collection. Science 286: 455-457. [DOI] [PubMed] [Google Scholar]
- 49.Venter J.C., Adams, M.C., Myers, E.W., Li, P.W., Mural, R.J., Sutton, G.G., Smith, H.O., Yandell, M., Evans, C.A., Holt, R.A., et al. 2001. The sequence of the human genome. Science 291: 1304-1351. [DOI] [PubMed] [Google Scholar]
- 50.Wain H.M., Lush, M., Ducluzeau, F., and Povey, S. 2002. Genew: The human gene nomenclature database. Nucleic Acids Res. 30: 169-171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wiemann S., Weil, B., Wellenreuther, R., Gassenhuber, J., Glassl, S., Ansorge, W., Bocher, M., Blocker, H., Bauersachs, S., Blum, H., et al. 2001. Toward a catalog of human genes and proteins: Sequencing and analysis of 500 novel complete protein coding human cDNAs. Genome Res. 11: 422-435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wolfsberg T.G. and Landsman, D. 1997. A comparison of expressed sequence tags (ESTs) to human genomic sequences. Nucleic Acids Res. 25: 1626-1632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wright F.A., Lemon, W.J., Zhao, W.D., Sears, R., Zhuo, D., Wang, J.P., Yang, H.Y., Baer, T., Stredney, D., Spitzner, J., et al. 2001. A draft annotation and overview of the human genome. Genome Biol. 2: RESEARCH0025. [DOI] [PMC free article] [PubMed] [Google Scholar]