

Abstract
The Phusion assembler has assembled the mouse genome from the whole-genome shotgun (WGS) dataset collected by the Mouse Genome Sequencing Consortium, at ∼7.5× sequence coverage, producing a high-quality draft assembly 2.6 gigabases in size, of which 90% of these bases are in 479 scaffolds. For the mouse genome, which is a large and repeat-rich genome, the input dataset was designed to include a high proportion of paired end sequences of various size selected inserts, from 2–200 kbp lengths, into various host vector templates. Phusion uses sequence data, called reads, and information about reads that share common templates, called read pairs, to drive the assembly of this large genome to highly accurate results. The preassembly stage, which clusters the reads into sensible groups, is a key element of the entire assembler, because it permits a simple approach to parallelization of the assembly stage, as each cluster can be treated independent of the others. In addition to the application of Phusion to the mouse genome, we will also present results from the WGS assembly of Caenorhabditis briggsae sequenced to about 11× coverage. The C. briggsae assembly was accessioned through EMBL, http://www.ebi.ac.uk/services/index.html, using the series CAAC01000001–CAAC01000578, however, the Phusion mouse assembly described here was not accessioned. The mouse data was generated by the Mouse Genome Sequencing Consortium. The C. briggsae sequence was generated at The Wellcome Trust Sanger Institute and the Genome Sequencing Center, Washington University School of Medicine.
Whole-genome shotgun (WGS) sequencing is an approach used since the early 1980s (Sanger et al. 1982); what has changed since then is the size of genome one considers reasonable for the technology available at the time (Staden 1980, 1982). During the 1980s, the optimal size developed from the successful WGS of bacteriophage λ at 49 kb (Sanger et al. 1982) up to hundreds of kilobases for various viral genomes by the end of the decade. Workstation class computers during the 1980s grew from submegabytes of random-access-memory (RAM) and submillions-of-instructions-per-second (MIPS) to a few Megabytes of RAM and a few MIPS. Cloning of target DNA into host vectors, such as cosmids, and creating a physical map, provided a means to tackle larger genomes with a hierarchical approach; for a brief review, see Olson (2001). Thus, WGS could continue to apply to these vector genomes that contained mapped segments of larger target genomes, for example, yeast and worm. The hierarchical approach effectively diagonalizes these larger genomes into groups of sequences from smaller regions through the biological process of cloning. However, cloning and mapping do add extra steps to the overall goal of determining the sequence of a target genome, and in the early 1990s, the WGS approach achieved a major milestone with Haemophilus influenzae Rd. (Fleischmann et al. 1995) at 1.8 Mbp in size. By the mid 1990s, workstation class computers became available with hundreds of Megabytes of RAM, and the TIGR ASSEMBLER used 30 h of CPU time on a Sun Microsystems SPARCenter 2000 computer with 512 Mbyte RAM to assemble the 24 k WGS sequencing reads collected from the H. influenzae Rd. genome. Through the second half of the 1990s, other megabase size genomes were determined with the WGS method, and the possibility of applying this approach to mammalian size genomes, and in particular the human genome, was hotly debated (Green 1997; Weber and Myers 1997). By the end of the decade, the WGS and assembly of the Drosophila genome (Myers et al. 2000) showed that this approach works for a genome of 120 Mbps (the euchromatic portion). Again, this went in stride with increasing computer memory and processing speed, which is necessary to keep track of the large number of reads and the multitude of pair-wise associations made between reads. The Drosophila assembly took less than a week, running on an 8 Compaq Alpha ES40s with 32 Gbytes of memory. As described in the 2001 publication, the Celera Assembler was applied to their human WGS data plus shredded public data, requiring 20,000 CPU hours, and the largest machine used had 64 Gbytes of RAM (Venter et al. 2001). Had they applied the software built for Drosophila, a computer with 600 Gbytes RAM would have been required. So, as computers have increased in speed and amounts of RAM over the last 20 yr by ∼5 orders of magnitude, so too have the size of genomes considered tractable using a WGS approach.
Over the last few years, many groups have become involved in developing WGS assemblers specifically for genome, or selected portions of genomes, for example, single chromosomes or groups of chromosomes, from larger than a few megabases up to multiple gigabases. All of these assemblers use paired-end sequencing of various sized insert templates to detect and avoid misassemblies, join contigs together, and guide the scaffolding of contigs. For a hybrid theory/simulation analysis of the power of paired end sequencing, see Siegel et al. (2000). The Celera Assembler, as described in Myers et al. (2000), carefully prepares the input reads by trimming back the ends, such that the remaining portion of each read was at least 98% accurate, followed by masking known contaminants, a hard screen, and identification of known repetitive elements, a soft screen. To find overlaps among the trimmed and unscreened portions of the reads, all reads were compared with all others by use of a very fast seed-and-extend method, which processed 32 million pairs of reads per second. The unitigger stage converted overlap information into consistent subassemblies, and identified the subassemblies as either containing unique regions (U-unitigs) or overcollapsed repetitive regions. These U-unitigs were ordered and oriented using read-pair information, requiring a minimum of two mate pairs between U-unitigs in this scaffolding stage. Inter- and intrascaffold gaps were then filled using three levels of increasingly aggressive repeat resolution. At the final stage, the consensus sequence was generated. The ARACHNE assembler was designed for paired end WGS data and their paper (Batzoglou et al. 2002) describes how reads were initially aligned to each other by a sorting method based on 24-bp long words, 24-mers, which were extended into longer alignments and together with read pair information, contigs were assembled. Repeat boundaries were detected both by excessive depth, as in Myers et al. (2000), and where conflicting read pair links to other contigs were found. Contig scaffolds were created requiring a minimum of two forward-reverse links between contigs. An updated version of ARACHNE was used to assemble the mouse genome (Jaffe et al. 2003). The JAZZ assembler was applied to the Fugu rubripes genome with WGS coverage at 5.7× (Aparicio et al. 2002), generating an assembly containing 332.5 Mbps. In the Malign module of the JAZZ assembler, reads were initially associated to each other by use of a hash table to find a minimum of 10 exactly matching 16-mers and followed by a banded Smith-Waterman alignment method. To avoid unnecessary alignments, 16-mers that occurred frequently were not used in the initial read-association step. Read layout and contig scaffolding follows an approach very similar to the ARACHNE and Celera assemblers using a module called Graphy. Consensus generation from the read layout applies base quality values from the reads, resulting in contig assemblies that include quality values; note that consensus sequence quality values were also generated by the ARACHNE, Celera, and the following RePS assembler. The RePS assembler (Wang et al. 2002) was applied to the 4.2× coverage WGS sequence data of the 466 Mbp rice genome (Yu et al. 2002). RePS starts by masking all 20-mers in the input data that occur more than a multiple of the depth of shotgun sequence coverage, these are called mathematically defined repeats (MDRs), grouping these masked reads using BLAST (Altschul et al. 1990) and assembling the groups with PHRAP (http://www.phrap.org/). All reads were then unmasked, and the reads within each PHRAP contig were processed with PHRAP again to recover the complete consensus sequence for each contig. Read pairing information was used to merge contigs and fill gaps with unassembled reads by use of the read-pair insert size information. Scaffolding orders and orients contigs using a minimum of two paired reads shared between contigs.
The Phusion assembler was used to assemble the mouse genome at ∼7.5× WGS coverage and the C. Briggsae genome at ∼11× WGS coverage. Table 1 lists the stages of the Phusion assembler along with a short description. Results of these assemblies are given below, followed by a Discussion section and then a detailed description of the methods used. In the Methods section, some novel approaches to WGS assembly are illustrated. The Phusion assembler is modular, like JAZZ and RePS, and perhaps most similar to RePS in the way it uses concept of MDRs to identify repeats and PHRAP as its assembly engine.
Table 1.
An Overview of the Phusion Assembler Stages
| Stage | Brief description |
| Data Preparation | Contamination screening, vector and quality clipping. |
| Phusion Read Grouping | Reads that share low copy number words are grouped together into clusters that can be assembled independently of each other. |
| RPphrap | Each cluster of reads, and their associated read pairs, is assembled with PHRAP in an iterative way that allows for contigs to be extended and broken using read pair information. |
| RPjoin | Joins together contigs based on shared reads and sequence overlap as indicated by read pair information. |
| RPono | Scaffolding of contigs based on read pair information. |
| Contamination Screen | Contig and scaffold rejection based on an imbalance of read template origins. |
RESULTS
Mouse Assembly
For the mouse assembly, the initial set of reads from the Mouse Genome Sequencing Consortium consisted of those listed in the file ftp://ftp.ncbi.nih.gov/pub/TraceDB/mus_musculus/Feb_1_Freeze_Ti_List.gz and BACend reads from TIGR ftp://ftp.tigr.org/pub/data/m_musculus/bac_end_sequences/mbends. The Feb_1_Freeze_Ti_List file lists the trace identifiers (Ti's) as known to the NCBI Trace Archive Data Base (http://www.ncbi.nlm.nih.gov/Traces/trace.cgi?). Of the 40,793,320 reads from the freeze list, 32,042,831 (78.6%) passed the screens for contamination, for example, sequencing vectors, Escherichia coli, phage, etc., and minimum acceptable quality, that is, >99 clipped bases with <5% base call errors within the clipped region. The BACend reads from TIGR were used without quality values and clipping and contamination screening was not applied. Bad plate pairing detection (see Methods) was applied, which decoupled pairing information for ∼500 k templates, or about 3% of the total passed reads. Table 2 shows the distribution of reads over the different insert size ranges. Unpaired reads from all libraries are grouped together, as a read without a mate only contributes its sequence to the assembly.
Table 2.
Insert Sizes, Number of Reads and Effective Clone Coverage for the Mouse WGS Data Set
| Insert size range | Millions of reads | Percent of total | Effective clone coveragea |
| Less than 3 kb | 3.16 | 9.7% | 1.3 |
| 3 kb–7 kb | 19.32 | 59.5% | 15.3 |
| 7 kb–12 kb | 2.73 | 8.4% | 5.2 |
| 12 kb–50 kb | 1.05 | 3.2% | 7.4 |
| >50 kb | 0.39 | 1.2% | 12.7 |
| Total paired reads | 26.65 | 82.0% | 41.9 |
| Unpaired reads | 5.85 | 18.0% | |
| Total reads | 32.50 | 100.0% |
Assuming a 2.75 Gbp genome.
The 32,496,031 clipped reads comprise 19.3 Gbp, giving a 595-bp average read length, and cover the mouse genome to an average depth of approximately seven. The Phusion read grouping algorithm used a k-mer of 17 bases, ignored words that occurred more than D = 13 times, and the minimum number of matching k-mers to group reads together was set to M = 11. The Phusion algorithm automatically increased M to 20 to satisfy the maximum cluster size of 120,000 reads. Phusion clustered 28.7 M reads into 424 k groups, with 50% of the reads in groups of 287 or more reads. The largest cluster contained 70,059 reads. This grouping stage took 36 h of CPU time, running on one processor and used 97 Gbytes of memory of a Compaq Alpha GS320 equipped with 128 Gbytes of memory. This quick turnaround from input reads to clusters allowed tuning of the grouping parameters to find optimal settings. Less optimal settings were D=12 and D=14, both producing more clusters and incorporating fewer reads. A compute farm of 400 CPUs, Compaq Alpha DS10s with 1 Gbyte of memory each, assembled the clusters using RPphrap in ∼9 h elapsed time, using a total of 132 CPU days. RPjoin took 24 h to complete and used 70 Gbytes of memory, RPono took 28 h and 60 Gbytes of memory. The released assembly, which can be found at ftp://ftp.sanger.ac.uk/pub/image/tmp/ssahaAssemble/mouse/2002.02.01/, consists of 2.51 Gbps in 311,577 contigs with an N50 size of 20,121 bps and a total scaffold size of 2.62 Gbps in 70,427 scaffolds with an N50 size of 6.5 Mbps. Of the starting set of 32.5M reads, 29.3M (90.1%) are located in this assembly (N50 is a measure of the contig size at which 50% of the assembled bases are in contigs of this size or larger). Of the reads not in the assembly, 2.5 M were not clustered by the Phusion read grouping stage, 87 k reads were excluded by RPphrap stage, and 569 k were removed because they formed scaffolds that were smaller than 1 kb or contained fewer than three reads. There are 241,150 captured gaps in the scaffolds, totaling 117 Mbp, with an average size of 486 bps (rounding of contig and scaffold sizes lead to the apparent mismatch with the total gap size).
Comparing the assembly to 40 Mbps of finished clones from the same mouse strain C57BL/6J shows the assembly covers 94% of the bases, whereas the scaffolds cover 99.7%. There are three global scaffolding errors indicated from these 40 Mbps. These 40 Mbps also illustrate the sequence accuracy of the assembly. Because PHRAP is at the heart of the assembly process, quality values are assigned via this commonly used assembler, and are expected to be accurately determined (Ewing and Green 1998; Ewing et al. 1998). Given that this is an inbred mouse strain, single nucleotide polymorphisms (SNPs) should not occur. By use of these 40 Mbps of finished sequence and the Phusion mouse assembly as a reference, ssahaSNP (Ning et al. 2001) detected, on average, 1 variation every 87 kbps. This is well below the finishing criteria of 1 error in 10,000 bases, thus confirming the sequence accuracy of this assembly. There are 2.43 Gbps (96.9% of contig bases) with a quality value of 40 (1/10,000 error rate) or higher.
Because a fingerprint map was built from the same BAC clones, see http://genome.wustl.edu/projects/mouse/index.php?fpc=1, as the BAC end sequences from TIGR, integration of this map and the assembly allows placement of the scaffolds onto chromosomal positions. There are 393,470 BACend reads contained in the assembly, and 9775 scaffolds contain one or more BACends. This allowed 95.7% of the bases in the scaffolds to be linked to the FPC map. In some cases, the map and the BACend reads within the scaffolds show conflicting information. This would be expected at locations of global scaffolding error in the assembly. Therefore, when positioning scaffolds onto map coordinates, conflicts were resolved by breaking the scaffolds at the nearest contig boundary.
Caenorhabditis briggsae Assembly
For the C. briggsae assembly, the set of 2,354,875 WGS reads, available at the Trace Archives http://trace.ensembl.org/ and http://www.ncbi.nlm.nih.gov/Traces/trace.cgi?, and an additional 21,720 BACend reads formed the input to the Phusion assembler. Reads that were identified as purely contamination or low quality were rejected; however, reads that passed these screens were left untrimmed of low quality ends. Both the Phusion read grouping stage and the PHRAP assembler handle low-quality data very well, thus, it should be advantageous to leave these data in the assembly. Bad plate pairing detection decoupled ∼10,000 templates, or about 1% of the passed reads. Table 3 shows the distribution of reads over the different insert size ranges, along with effective clone coverage.
Table 3.
Insert Sizes, Number of Reads and Effective Clone Coverage for the C. Briggsae WGS Data Set
| Insert size range | Thousands of reads | Percent of total | Effective clone coverage |
| <3 kb | 905.9 | 43.4% | 9.3 |
| 3 kb–7 kb | 834.4 | 40.0% | 15.4 |
| 7 kb–12 kb | 76.1 | 3.7% | 3.8 |
| >50 kb | 16.3 | 0.8% | 5.7 |
| Total paired reads | 1832.7 | 87.9% | 34.2 |
| Unpaired reads | 252.6 | 12.1% | |
| Total reads | 2085.2 | 100.0% |
Overall sequence coverage is estimated at 11-fold, thus presenting a different challenge to the Phusion assembler compared with the mouse assembly. The main problem is that at this depth and with various repeat structures of the genome, the clustering algorithm tends to group all of the reads together. This is not desirable, as the assembly problem is not reduced to manageable sized groups. PHRAP can assemble groups of reads as large as a few hundred thousand at a time, but not two million reads within a sensible amount of time and memory. As shown earlier, M was increased to 20 for the 7.5× assembly of mouse to achieve cluster sizes below 120,000 reads. For C. briggsae, the Phusion parameters were set to use a k-mer of 16 bases, ignore words that occur more than D = 15 times, and the minimum number of matching k-mers to group reads together was set to M = 8. The cluster size dropped below 120,000 reads once M reached 34, and continued up to 44 to satisfy the maximum cluster size of 20,000 reads. Therefore, for C. briggsae, D was set lower relative to the depth of sequencing and M incremented to a higher level than for the mouse assembly.
Results of the assembly are as follows. Of the 2,085,214 decontaminated but unclipped reads, Phusion clustered 1,932,906 reads into 16,206 groups, with 50% of the reads in groups of 394 or more reads. The largest cluster contained 19,292 reads. This grouping stage took 75 min of CPU time and 10 Gbytes of memory running on a single processor of a Compaq Alpha ES40 equipped with 32 Gbytes of memory. These groups were assembled in 2 h elapsed time with RPphrap using 145 nodes of the 400-node CPU farm. Total CPU time used was about 9 d. The RPjoin and RPono stages also proceeded very quickly, taking about 2 h as well. Like the mouse assembly, an FPC fingerprint map of C. briggsae was generated, see http://genome.wustl.edu/projects/cbriggsae/index.php, and the BACends added to the assembly allowed integration of the two data sets. The assembly contains 1,945,314 reads (93.3% of the starting set of reads), in the cb25.agp8 assembly, see ftp://ftp.sanger.ac.uk/pub/wormbase/cbriggsae/cb25.agp8/. The N50 contig size is 41 kb and the N50 scaffold size is 1450 kb. On the basis of comparison to the 12 Mb of previously finished sequence, we estimate that the whole-genome shotgun assembly achieved 98% coverage of the C. briggsae genome at the contig level, and no global scaffolding errors were found. The size of the assembled and FPC mapped genome is 102 Mbp in 142 ultracontig pieces, with an additional 6 Mbp not placed on the FPC map, which is in 436 pieces (many highly repetitive). In the final sequence, 270 kb finished fosmid data from 155 accessions were incorporated to bridge scaffold gaps. Because of the absence of dense chromosomal maps for C. briggsae, we cannot assign the ultracontigs to chromosomal locations, and, therefore, cannot give draft chromosome sequences. This assembly was accessioned through EMBL, http://www.ebi.ac.uk/services/index.html, using the series CAAC01000001–CAAC01000578.
This assembly used unclipped reads, whereas the mouse assembly used clipped reads. An earlier assembly of C. briggsae starting with the same set of reads with quality trimming applied, resulted in contigs that had a 31-kb N50 measure. Thus, working with unclipped reads improved the assembly in terms of contig length by 32%. This was also tried with the mouse data, and using untrimmed reads increased the contig N50 size from 20 to 25kb.
DISCUSSION
The development of the Phusion assembler utilized the sequence from the mouse WGS data set and the C. briggsae data set to test and challenge all parts of the code from its inception in August ,2001. At that time, the C. briggsae data set was at ∼4.5× coverage and provided a good test set for developing Phusion because it was very quick to run; about a 2-h turnaround time. By October, 2001, the mouse data set had reached about that level of coverage ∼4×, but the mouse data set was a large amount of data, thus posing new challenges for memory use. The arrival of the GS320, with 128 Gbytes of memory, let those concerns fade away for a while, but the turnaround time was much longer, typically a few days. The Whitehead Institute Center for Genomic Research (WICGR) was also actively applying their ARACHNE assembler to the mouse data, and early on we agreed to use common assembly output formats to make comparison of the results easier. We also agreed to use common starting sets so that the assembly results would not be influenced by different numbers of input reads. All assemblies can be found at ftp://ftp.sanger.ac.uk/pub/image/tmp/ssahaAssemble/mouse for the Sanger Institute, and at ftp://wolfram.wi.mit.edu/pub/mouse_contigs/ for WICGR. The friendly competition that this dual effort instilled drove both assemblers to achieve the best possible results. Along the way, assemblies were selected for further annotation work. The November, 2001 Phusion assembly was selected, and can be seen at http://genome.cse.ucsc.edu/cgi-bin/hgGateway?db=mm1, whereas for the February 2002 data set assembly, the ARACHNE assembly was selected. The differences between the Phusion and ARACHNE assemblies based on the February, 2002 data set were small when looking at coverage, but at that time, the ARACHNE assembly had a longer N50 contig size and scaffold size, and no detectable global scaffold errors. Thus, the ARACHNE mouse assembly was selected as the basis of the MGSC version 3 assembly for analysis and comparison to the human genome in the main mouse paper (Mouse Genome Sequencing Consortium 2002).
All of the assemblers that have been applied to large genomes, >100 Mbps, and use read-pair information, that is, Celera, JAZZ, ARACHNE, RePS, and Phusion, use similar methods for the scaffolding stages. More differences arise in how each assembler initially clusters reads, forms alignments, and detects repeat-induced misassemblies. However, these assemblers can be grouped into two classes for this stage. Celera, JAZZ, and ARACHNE assemblers all compute local alignments between selected reads, whereas RePS and Phusion prepare and select reads such that the alignment problem can be solved by PHRAP. As mentioned in the introduction section, RePS and Phusion are quite similar in approach, although there are differences. One of the defining differences between RePS and Phusion is the way MDRs are applied. In RePS, the MDRs are hard-masked, such that PHRAP initially does not see the sequence in these regions. For Phusion, these regions are not masked. Another difference is that Phusion directly clusters the reads as an integral part of the histogram word analysis, whereas RePS uses a subsequent BLAST stage to cluster reads.
As mentioned in the Results section, one of the ways we improved on the contig N50 size was to use unclipped reads. This works because the unclipped, low-quality ends of the reads do contain many valid k-mer words, and leaving these in allows associations to be made between reads that would not have occurred if these ends were removed. The trade off is an increase in erroneous k-mer words, which adds predominantly to the number of words seen once, see Figure 2, below. Because the Phusion clustering stage requires a number of shared words between reads to make an association, these erroneous words would need to occur in a quite improbable way for the untrimmed ends to make new read–read associations. Even if that were to occur, PHRAP would not assemble these incorrectly associated reads, because the bulk of the good portion of the read would have come from different portions of the genome. Thus, not trimming the low-quality ends of the reads has an overall desirable effect.
Figure 2.

Word use distribution for the mouse ∼7.5-fold sequence data. The top curve is measured from the prepared dataset, and the bottom curve shows a Poisson distribution with a mean value of 7.
The computer requirements for Phusion are substantial. As presented in the Results section, using a k-mer, which represents >10 times the number of words than bases in the genome is desirable, and storage is needed for all bases, quality values, the sort arrays, and the read relationship matrix. For the mouse genome, the peak memory use was 97 Gbytes, which, for today, is quite a large amount of memory and places the use of this algorithm out of reach for most labs. Then, the next stage, RPphrap, used 132 CPU days of compute time, which again is quite extreme. However, one should keep in mind that computer specifications keep improving, and at some point, what may seem extreme today will be within reach by more labs in the near future. Also, the cost of the computers used for this assembly effort is still a small fraction of the cost to produce the sequence for the mouse genome. The benefit of this approach was a modular system with fast turnaround time for any of the stages, thus, improvements to the algorithms and different initial settings could be tested in a reasonable amount of time.
So far, only the ARACHNE and Phusion assemblers can be compared in a direct way, having used the same initial dataset. As the Trace Archives collect more complete datasets for additional organisms, for example, C briggsae, Ciona savignyi, Anopheles gambiae, etc., this will allow more comparisons to be made between assemblers. However, it is a major undertaking to commit to assembling large genomes, and often a lot of knowledge about the processes used in collecting the data needs to be known. Fortunately, there are numerous auxiliary information fields to describe this information, thus, much of this knowledge can be stored with the data. The number of genomes that will be sequenced using the WGS approach will surely increase, possibly quite dramatically if sequencing costs continue to drop, thus, the continued improvements in assembly algorithms and comparisons among them will remain an active field of research for quite some time.
METHODS
Data Preparation: Clip and Screen Reads, Remove Contaminants
The reads from the sequencing process are first screened to remove bad data prior to the start of the assembly process. Reads that are primarily of poor quality are removed completely from the data set. Also, the end portions of reads that are of poor quality are removed. This removal of reads and portions of reads is typically referred to as clipping. Poor quality is determined directly from the sequence quality information as generated by the PHRED (Ewing and Green 1998; Ewing et al. 1998) base-calling algorithm. The PHRED quality values give probability of error measures, and the trim points are selected such that the sum of the error probabilities within the clip window does not exceed 5% of the windowed bases, and that all 20-base-long segments within the window have a sum of error probabilities less than one. Base calls of N are considered a probability of error equal to one. Any sequence portions at the end of reads that relate to a vector used in the sequencing process are removed. Moreover, the reads are scanned for the presence of sequence portions that match the sequence of known contaminants, such as E. Coli or bacteriophage, and these sequence portions are also removed.
Phusion Read Grouping Stage
First Pass: Select Word Length, Prepare Histogram, and Select Cut-Off
The input data set, as prepared above, comprises a large number of reads, typically several hundreds of thousands to tens of millions. This data set is analyzed to form a histogram. The histogram analysis determines how many times words of a length k occur in the data set. A word is defined as a sequence portion in the read of k bases. These words are referred to as k-mers. The word length k should be selected so that it satisfies the following condition:
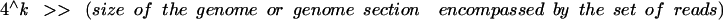 |
The effect of k-mer size on the assembly of C. briggsae is shown in Figure 1. Note that although the N50 contig size drops off substantially for k equal to 13 and 14, only at a k of 13 is there a drop off in assembly coverage by 1.5%.
Figure 1.

This graph shows the effect of k-mer on relative contig N50 size for C. briggsae assemblies. At k = 15, 4∧15 is about 10 times the genome size.
To generate the histogram, the complete data set is scanned for all k-mer words at all base locations in every read. The reverse complement of each k-mer is also computed at each location. Only k-mers that contain exclusively A, C, G, or T's are considered. For convenience during processing, these bases are converted to the binary values 00, 01, 10, and 11 for A, C, G, and T, respectively. Thus, a k-mer once converted to this binary representation can take on any value from 0 to 4∧k-1. For each k-mer and its reverse complement, only the minimum value of these two words is used. Otherwise, a 2.7 Gbp genome would appear to be 5.4 Gbp if both strands are taken into consideration.
The k-mer histogram is stored as an array that is 4∧k long and comprised of a byte (8 bits) for each element. Because some words will occur more times than can be accumulated in a byte, that is, >255 times, the accumulation is stopped if the element has reached the maximum value, that is, 255 in the case of a byte. By using one byte for each word—occurrence value means the 17-mer array requires 16 Gbytes of memory.
After compiling the k-mer histogram to record the number of times each k-mer occurs in the data set, the results are condensed into a second histogram showing the k-mer word-use distribution, indicating the statistical distribution of the number of times k-mer words occur in the data set.
Figure 2 shows in its upper curve an example of such a word-use distribution histogram for a data set obtained from ∼7.5-fold sequencing of the mouse genome. The histogram is generated with 17-mers and shows that a very large number of k-mer words occurred only once, reflecting bad data caused by sequencing errors. Discounting the bad data peak at a value of 1, the distribution shows that the k-mer words occurred most often 6 times in the data set, which is close to the estimated 7.5-fold redundancy of the sequencing. The long tail indicates that many k-mer words occurred a large number of times in the data set. These words are indicative of repetitive elements in the genome, and are termed mathematically defined repeats (MDRs) as in Wang et al. (2002). This is characteristic of most data sets, as genomes generally contain repetitive elements.
For comparison, the lower curve in the figure is a Poisson distribution with a mean value of 7. This is the form that the word-use distribution would have if the genome did not contain repeats and if the sequencing were error free. At 7× coverage with no sequencing errors, words that occur once should be 1/23 the number of words that occur 7 times. The measured value of words seen once is 0.5 G words, which is off scale in Figure 2, and indicates that the number of erroneous words is ∼3% of the total number of words, which reflects the number of erroneous 17-mers left after read-clipping thresholds used at the preprocessing stage. At a word occurrence level of 12, the graph shows that approximately half the words arise from unique regions of the genome and the other half arise from repetitive regions of the genome, with the assumption that the reads are truly randomly distributed.
The compilation of the word-use distribution described above is highly valuable, as it identifies all of those reads that relate to repetitive sequence portions. The highly repetitive sequence portions should not be used for determining which reads overlap, as they are not specific to a unique portion of the genome and will thus be a source of spurious alignment attempts in the assembler. The next stage of the process thus excludes from consideration all of the words in the data set that occur more than a certain number of times, D, based on the word-use distribution analysis, in which D will typically be higher than n, the sequencing redundancy factor.
The value of D is set so as to capture most of the underlying Poisson distribution of the unique regions of the genome, thereby excluding the repetitive sequence portions that do not uniquely identify any particular portion of the genome or genome section. The value of D may be set automatically or manually from the word-use distribution. Automatic setting may be performed on the basis of exclusion of all words with an occurrence more than a given factor of the distribution peak, or on the basis of a certain fraction of the distribution that captures a given proportion of the words in the Poisson distribution. With the word-use distribution shown in Figure 2, a value for D of 13 captures 97% of the unique and error-free words in the input set.
Second Pass: Create Sorted List of Read Associations
Another pass is then made through the data set of all reads to fill a new array with the k-mers that occur less than or equal to D times. Each k-mer word is tabulated along with the read index of each of its occurrences. For example, if k = 10, one of the k-mer words may be ACAGAAAAGC. Its read index may relate to a read named, for example, 10h06.p1c. The occurrences of each k-mer word may be collated in a variety of ways. One convenient way is to fill a table or array with pairs of numbers using high and low bits of an appropriately size word, for example, 8 bytes, with the k-mer occupying the high bits and the read index occupying the low bits. The array can be sorted conveniently so as to group reads that share common words, as in Table 4, below. The two selected k-mers in the example have eight reads sharing the first k-mer and six reads sharing the second k-mer. Table 4 shows a small part of a sorted list of selected words and their associated read indices. The binary values for both fields have been converted back to the text they represent for ease of understanding. The sorted list of read indices of each k-mer is now used to generate a further array that groups all of the reads that are deduced to belong to a contiguous section of genome. Each such group of reads identified in this way is referred to as a cluster.
Table 4.
Sorted List of Each k-Mer and Its Read Indices
| High bits | Low bits |
| ACAGAAAAGC | 10h06.p1c |
| ACAGAAAAGC | 12a04.q1c |
| ACAGAAAAGC | 13d01.p1c |
| ACAGAAAAGC | 16d01.p1c |
| ACAGAAAAGC | 26g04.p1c |
| ACAGAAAAGC | 33h02.q1c |
| ACAGAAAAGC | 37g12.p1c |
| ACAGAAAAGC | 40d06.p1c |
| ACAGAAAAGG | 16a02.p1c |
| ACAGAAAAGG | 20a10.p1c |
| ACAGAAAAGG | 22a03.p1c |
| ACAGAAAAGG | 26e12.q1c |
| ACAGAAAAGG | 30e12.q1c |
| ACAGAAAAGG | 47a01.p1c |
Third Pass: Read Clustering by Creating Read-Relation Matrix
The sorted list is now processed to fill a read-relation matrix. In this step, it is determined for each read all of the other reads that share any of the same k-mers. Moreover, for each pair of reads associated by at least one common k-mer word, it is determined how many times common k-mers occur. The read-relation data is created by filling a matrix that contains one row for each read. The row location is the index value of the read. The columns are filled with all other reads that share common k-mer words with the row's read together with the number of times, m, the association was made.
Table 5 below shows some example rows from this matrix. The first row in this example relates to the read 25a08.p1c that shares 232 k-mers with the read 25c12.q1c, 163 with 19c12.q1c, and 135 with 1c05.q1c. It is noted that the 232 common k-mers between 25a08.p1c and 25c12.q1c may relate to <232 different words. This will be the case if words occur more than once in the two reads. It is also noted that sequencing errors will cause a background level of random associations between reads. Fortunately, these random associations will be limited to a relatively small number of words and can, therefore, be filtered out by ignoring associations between reads that do not occur above a certain number of times. This filtering is achievable by setting a threshold value of M. The association is thus cancelled from the matrix if the number of shared k-mers m between the row's read and the other read is less than M. A value of M = 11 is used in this example. As the value of M is set higher, it becomes increasingly likely that some true associations are removed. As explained further below, removal of weak true associations are in some cases beneficial to the assembly overall assembly process.
Table 5.
Selected Example Rows from a Read Relation Matrix.
| 25a08.p1c | 232 25a12.q1c | 163 19c12.q1c | 135 1c05.q1c |
| 25a12.q1c | 232 25a08.p1c | 103 19c12.q1c | 5 15d02.p1c |
| 19c12.q1c | 163 25a08.p1c | 103 25a12.q1c | |
| 1c05.q1c | 135 25a08.p1c | 17 16b09.q1c | |
| 16b09.q1c | 17 1c05.q1c |
The example in Table 5 also shows all other rows that are listed on the first row and an additional row, 16b09.q1c, which is linked from 1c05.q1c. One read, 15d02.p1c, is not followed because it only has five shared k-mers with 25a12.q1c. Table 5 is a very simple example of traversing all links branching from the read 25a08.p1c. The contents of Table 5 are thus a closed set of five reads that are deduced to collectively define a potentially contiguous section of the genome being sequenced. An important point to note here is that multiple sequence alignment is not performed at this stage to create this closed set of reads. The k-mer association approach described above has allowed determination that these five reads may concatenate somehow to form a contiguous section, without having to go through the computational complexity of an alignment process. Therefore, any cluster of reads is defined as the simply connected components of the undirected graph of reads that have an edge between them, in which the edges are defined by pairs of reads that share M or more selected k-mers.
A key advantage of grouping the reads into contiguous groups in this way by use of read associations is that it makes subsequent alignment computationally easy. This is because the reads of each cluster can be aligned independently of the reads of any other cluster. The isolated groups of reads can thus be passed onto any assembly algorithm as independent sets. This allows multiple sequence alignment processing of the different clusters to proceed in parallel. Moreover, it means that the assembler is given an alignment problem that is known to be easily soluble because the reads are associated with each other through mostly unique words. Reads that are primarily made up of repetitive words, which cause assembly algorithms the most difficulty, will not become associated with any cluster. The cluster size is also controllable, as described below, which allows cluster size to be optimized to the cluster size most efficiently processed by the assembler.
The clustering of data prior to alignment means that the process of alignment is confined to groups of reads that are already known to fit together, that is, contiguous read groups. Taking a jigsaw analogy, the clustering may be considered to be the step of creating piles of jigsaw pieces with common patterns and/or colors before trying to fit any individual pieces together. The step of fitting the pieces together may be considered to be analogous to alignment and is only attempted within each pile.
Deliver Clusters to Assembler
Each group of associated reads, that is, each cluster, can then be assembled independently. For example, if 17 clusters are identified in the preassembly described above, the set can be assembled in parallel on 17 different CPUs. The preassembly has been designed in such a way that it is compatible with any assembler and in such a way that it does not duplicate the alignment process carried out by conventional assemblers. This approach has been followed deliberately, as it is very powerful to separate the preassembly stage from the subsequent alignment steps, which can be carried out by any conventional assembler. The approach described thus provides preprocessing to allow conventional assemblers to be supplied with easy-to-assemble contiguous groups of reads.
Iterative Recomputation to Adjust Cluster Size
The values of D or M can be adjusted depending on the genome characteristics to obtain the desired level of clustering. In an extreme case, the initial analysis may associate all of the reads with each other in one large cluster, which is clearly not a useful result. This can occur when the depth of sequencing is sufficiently large (large sequencing redundancy factor n) to cause a high level of linking between all the reads. This can be countered using a higher value of M and/or a lower value for D.
At the other extreme, it may also occur that the initial analysis results in a large number of very small clusters. In this case, the initial analysis is not optimized for alignment and recomputation is desirable. This can be countered using a lower value of M and/or a higher value for D.
Considering the case of there being only one large cluster, or an undesirably low number of large clusters, increasing the value for M can be implemented within the algorithm whenever a given maximum cluster size, C, is exceeded. For these clusters, M is incremented iteratively, until the cluster sizes drop below C. In a typical example, M starts initially at a value of 11 and is then incremented to 50 in steps of 2, until a desired maximum cluster size C is no longer exceeded.
Using M as the adjustable parameter for varying cluster size keeps recomputation to a minimum, as recompilation of the read indices of each common k-mer is not necessary, and as the recomputation can be confined to breaking down only the large clusters. All clusters smaller than C reads do not need to be recomputed.
If D is used as an adjustable parameter for varying cluster size, this will require more recomputation than adjusting M, as it will necessitate returning back to the second pass stage of creating the lists of all read indices of each k-mer that occur less than the new value for D, and then recomputing the read relation matrix.
RPphrap
So far, the Phusion assembly process has grouped reads on the basis of sequence content only. From this point on, however, information about read pairing is used extensively. As described in the Results section, reads are generated by sequencing off of both ends of a double-stranded plasmid, fosmid, or BAC template. The genomic DNA is size selected prior to ligation, and both the size and standard deviation of the size are used when sequence reads from both ends of a template are found in the assembly stages. We refer to these paired ends as read pairs, and for a given pair, we will also refer to the association between these reads as mates.
Once the read clusters are formed, each cluster, along with read-pair information, read-sequence data, both ends if available, and quality values, are assembled independently using PHRAP at the heart of a master program that applies read-pair information in an iterative way. The version of PHRAP that we use, version 0.990319, is not capable of using read-pair information. This master program, RPphrap, uses read-pair information to split PHRAP-generated contigs at locations that show read-pair insert size consistency violations. For PHRAP-generated contigs that contain one but not both reads of a read pair, the missing read is projected out from its mate to its expected position, and if that position is within 1 standard deviation of overlapping the contig, then that read is added to the set of reads in the contig. This process of splitting and extending is shown in Figure 3. This process is applied to all reads in each contig and all new groups of reads are reassembled at the next RPphrap iteration. We add mates into contigs in this controlled way, so that the contig extension is controlled. If all mates were added without regard to estimated placement, then disconnected groups of reads may form new contigs that are isolated from the current contig. For example, if a contig is 2-kb long to start with, and let's say all reads were from read pairs that span 10 kb, then an uncontrolled inclusion of these mates could form two new contigs ∼8 kb away in both directions from the starting contig.
Figure 3.

(A) Inconsistent read pair, the one with the wavy lines over it will break a contig, creating three independent groups that are reassembled on the next iteration of PHRAP. (B) Read pairs are used to extend contigs by adding mates' of reads that should fall near the ends of the contig to the set of reads assembled for that contig on the following PHRAP iteration.
Bad Plate-Pairing Detection
As RPphrap is running, if a read is assembled into a contig and its mate should also be within the limits of the contig, but is not there, a warning message is generated. After all clusters have passed through RPphrap, statistics are measured on pairing failure rates, and when these strongly correlate with potential laboratory tracking errors, then these sets of read pairs are decoupled, and the entire RPphrap assembly process is rerun. For example, some laboratories produce separate forward and reverse direction sequencing reaction plates for a given set of templates. If these two plates get incorrectly labeled so that they are no longer tracked as originating from the same template, then RPphrap will generate many warning messages for these reads, as the read pairs will most likely not be grouped into common contigs.
RPjoin
The final set of contigs from the RPphrap stage include many reads that are present more than once in the assembly. This is caused by the contig extension phase in which mates are added iteratively. For example, say a 15-kb contig, labeled C1, is made up of 5-kb insert read pairs. The iterative extension process could extend this contig outward by 5 kb on both ends, resulting in C1 growing into a 25-kb contig. If another independent contig, C2, happened to have been formed just a few tens or hundreds of bases away from this other 15-kb starting contig, then the growth of the C1 will extend over C2 and quite likely contain shared reads. Remember, the starting clusters are all processed independently so that RPphrap can be distributed over many CPUs, thus the overlap cannot be detected until after the RPphrap stage.
Therefore, this stage, RPjoin, first looks for shared reads among all contigs. For all pairs of overlapping contigs, a merging process intermeshes the reads and splices the sequence together. The locations of all of the reads are readjusted to their new contig location.
Because the assembly process is not perfect, some reads are assembled into the wrong locations. When RPjoin finds two contigs that share a common read, but the sequence data do not agree over the extent of the contigs if placed according to the contig locations of this read, the read is removed from the smaller of the two contigs.
Once the shared reads are completely depleted, that is, no read appears more than once in the assembly, a second type of contig merging is applied, which looks for inferred overlap based on read pairs that span contigs. For a given contig, all of the reads in that contig that have mates in another contig are measured for inferred placement of the other contigs. Because the location and orientation of each read is known, a contig–contig gap size is computed. As multiple read pairs may indicate an association between two contigs, and average gap size is computed. If this gap size is negative, this indicates a potential overlap and triggers RPjoin to look for sequence similarity at the overlapping ends. If this is found and consistent, then these contigs are merged in the same way as in the shared read stage above.
RPono
At this stage, no read is represented more than once in the assembly, and all overlapping contigs should be joined. Thus, what is left is a collection of contigs that are bounded repetitive regions that never formed clusters, were not extended into by the iterative RPphrap extension process, lack read coverage due to statistical sampling reasons, or regions that were not clonable within the template vectors used. This RPono stage continues to apply read-pair information to place contigs into an ordered and oriented set. It uses the second part of RPjoin to calculate average contig to contig gap sizes. Contigs are ordered and oriented in an iterative way, looking first for contig–contig gaps that are small, and working up to larger gaps in subsequent iterations. Only contigs that have more than one link to another contig are considered, which reduces the likelihood of making an incorrect join.
The iterative approach using increasing allowed gap sizes eliminates the need to fill in large gaps had this process attempted this in a purely greedy fashion. For example, two large contigs that span one small 2-kb contig may have many more links joining them together than the smaller contig, and a greedy method without a maximum gap size limit would join the large contigs together first. Using the maximum gap size, one of the two larger contigs will link in the smaller contig first, as its spacing would not exceed the maximum gap size. This method is applied with a typical maximum gap size increasing through 1, 2, 4, 10, 20, and 40 kb. The end point depends on the initial N50 contig size and the insert sizes used. For example, if only 2-kb inserts were used, advancing beyond a 2-kb maximum gap size would not change the outcome of this process.
Contamination Detection
Sequence contamination has always been an issue, and at most sequencing centers there are various methods for detecting and removal of contamination. For example, clones without an insert produce a sequence that is purely the clone's vector sequence. This is detected by sequence similarity to expected sources of contamination, like the sequencing vectors used. This early detection method works well for small sets of expected contamination, up to a few million bases. However, while WGS sequencing the mouse genome at the main sequencing centers, these centers were also sequencing human BAC clones along with many other genomes, making the contamination sources arise from many billions of bases. Fortunately, there is a very clear and detectable signal that can identify these other sources of contamination. If enough contaminant sequencing reads are present in the set of all reads presented to the assembler, then these will form contigs that are made up purely from one center's reads, and typically even from a particular ligation.
For example, if three centers produce equal amounts of WGS data, and one center improperly pooled DNA from a human clone together with whole-genomic mouse DNA, then the resulting sequence reads from that mixture would produce assemblies of the human clone that would lack sequence reads from the other centers. Thus, to detect suspected contamination-derived contigs, one only needs to count the origin of the reads in each contig, and if that is purely from one center, then the probability that the contig is contaminant is 1–0.3333∧n, in which n is the number of reads in the contig.
Availability
Phusion is undergoing a rewrite of the code to make this a portable package. It will be made available free of charge to academic sites, but requires licensing for commercial use. For more information please contact the authors.
WEB SITE REFERENCES
ftp://ftp.ncbi.nih.gov/pub/TraceDB/mus_musculus/Feb_1_Freeze_Ti_List.gz; Initial dataset for mouse.
ftp://ftp.sanger.ac.uk/pub/image/tmp/ssahaAssemble/mouse/; Phusion mouse assembly.
ftp://ftp.sanger.ac.uk/pub/wormbase/cbriggsae/cb25.agp8/; Phusion C.briggsae assembly.
ftp://ftp.tigr.org/pub/data/m_musculus/bac_end_sequences/mbends; Mouse BACend sequence.
http://genome.cse.ucsc.edu/cgi-bin/hgGateway?db=mm1; Phusion assembly.
http://genome.wustl.edu/projects/cbriggsae/index.php; Caenorhabditis Briggsae.
http://genome.wustl.edu/projects/mouse/index.php?fpc=1; Mouse Genome.
http://trace.ensembl.org/; Ensembl Genome Server.
ftp://wolfram.wi.mit.edu/pub/mouse_contigs/; ARACHNE mouse assembly.
http://www.ebi.ac.uk/services/index.html; EBI Services.
http://www.ncbi.nlm.nih.gov/Traces/trace.cgi?; Trace DB.
http://www.phrap.org/; Genome Software Development Page.
Acknowledgments
We thank TIGR for providing the mouse BACend sequences, MGSC for funding, the participating sequencing centers for generating the WGS data, and the groups involved in producing the physical map of the mouse genome (Gregory et al. 2002). Dr. Tim Hubbard and Dr. Carol Scott at The Sanger Institute helped with integrating the mouse and C. briggsae assemblies to their physical maps. C. briggsae sequence data were produced at The Sanger Institute and at Genome Sequencing Center, Washington University School of Medicine. We thank Dr. David Jaffe and his assembly team for all of the helpful discussions that we had during the intensive efforts at assembling the mouse genome. Deanna Church and others at NCBI were most helpful in evaluating the results of the Phusion and ARACHNE assemblers. Thanks also go out to the WEB and analysis teams for posting the Phusion mouse assemblies in searchable and viewable forms at UCSC, NCBI, and Ensembl. Dr. Richard Durbin and others at the Sanger Institute made many helpful comments and suggestions throughout the development of Phusion. Funding for this work was provided by The Wellcome Trust.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
E-MAIL jcm@sanger.ac.uk; FAX 44-1223-494-919
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.731003. Article published online before print in December 2002.
REFERENCES
- 1.Altschul S.F., Gish, W., Miller, W., Myers, E.W., and Lipman, D.J. 1990. Basic local alignment search tool. J. Mol. Biol. 215: 403-410. [DOI] [PubMed] [Google Scholar]
- 2.Aparicio S., Chapman, J., Stupka, E., Putnam, N., Chia, J.M., Dehal, P., Christoffels, A., Rash, S., Hoon, S., Smit, A.F., et al. 2002. Whole-genome shotgun assembly and analysis of the genome of Fugu rubripes. Science 297: 1301-1310. [DOI] [PubMed] [Google Scholar]
- 3.Batzoglou S., Jaffe, D.B., Stanley, K., Butler, J., Gnerre, S., Mauceli, E., Berger, B., Mesirov, J.P., and Lander, E.S. 2002. ARACHNE: A whole-genome shotgun assembler. Genome Res. 12: 177-189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ewing B. and Green, P. 1998. Base-calling of automated sequencer traces using phred. II. Error probabilities. Genome Res. 8: 186-194. [PubMed] [Google Scholar]
- 5.Ewing B., Hillier, L., Wendl, M.C., and Green, P. 1998. Base-calling of automated sequencer traces using phred. I. Accuracy assessment. Genome Res. 8: 175-185. [DOI] [PubMed] [Google Scholar]
- 6.Fleischmann R.D., Adams, M.D., White, O., Clayton, R.A., Kirkness, E.F., Kerlavage, A.R., Bult, C.J., Tomb, J.F., Dougherty, B.A., Merrick, J.M., et al. 1995. Whole-genome random sequencing and assembly of Haemophilus influenzae Rd. Science 269: 496-512. [DOI] [PubMed] [Google Scholar]
- 7.Green P. 1997. Against a whole-genome shotgun. Genome Res. 7: 410-417. [DOI] [PubMed] [Google Scholar]
- 8.Gregory S.G., Sekhon, M., Schein, J., Zhao, S., Osoegawa, K., Scott, C.E., Evans, R.S., Burridge, P.W., Cox, T.V., Fox, C.A., et al. 2002. A physical map of the mouse genome. Nature 418: 743-750. [DOI] [PubMed] [Google Scholar]
- 9.Jaffe, D.B., Butler, J., Gnerre, S., Mauceli, E., Lindblad-Toh, K., Mesirov, J.P., Zody, M.C., and Lander. E.S., 2003. Whole-genome sequence assembly for mammalian genomes: Arachne 2. Genome Res.(this issue). [DOI] [PMC free article] [PubMed]
- 10.Mouse Genome Sequencing Consortium 2002. Initial sequencing and comparative analysis of the mouse genome. Nature 420: 520-562. [DOI] [PubMed] [Google Scholar]
- 11.Myers E.W., Sutton, G.G., Delcher, A.L., Dew, I.M., Fasulo, D.P., Flanigan, M.J., Kravitz, S.A., Mobarry, C.M., Reinert, K.H., Remington, K.A, et al. 2000. A whole-genome assembly of Drosophila. Science 287: 2196-2204. [DOI] [PubMed] [Google Scholar]
- 12.Ning Z., Cox, A.J., and Mullikin, J.C. 2001. SSAHA: A fast search method for large DNA databases. Genome Res. 11: 1725-1729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Olson M.V. 2001. The maps. Clone by clone by clone. Nature 409: 816-818. [DOI] [PubMed] [Google Scholar]
- 14.Sanger F., Coulson, A.R., Hong, G.F., Hill, D.F., and Petersen, G.B. 1982. Nucleotide sequence of bacteriophage λ DNA. J. Mol. Biol. 162: 729-773. [DOI] [PubMed] [Google Scholar]
- 15.Siegel A.F., van den Engh, G., Hood, L., Trask, B., and Roach, J.C. 2000. Modeling the feasibility of whole genome shotgun sequencing using a pairwise end strategy. Genomics 68: 237-246. [DOI] [PubMed] [Google Scholar]
- 16.Staden R. 1980. A new computer method for the storage and manipulation of DNA gel reading data. Nucleic Acids Res. 8: 3673-3694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Staden R. 1982. Automation of the computer handling of gel reading data produced by the shotgun method of DNA sequencing. Nucleic Acids Res. 10: 4731-4751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Venter J.C., Adams, M.D., Myers, E.W., Li, P.W., Mural, R.J., Sutton, G.G., Smith, H.O., Yandell, M., Evans, C.A., Holt, R.A., et al. 2001. The sequence of the human genome. Science 291: 1304-1351. [DOI] [PubMed] [Google Scholar]
- 19.Wang J., Wong, G.K., Ni, P., Han, Y., Huang, X., Zhang, J., Ye, C., Zhang, Y., Hu, J., Zhang, K., et al. 2002. RePS: A sequence assembler that masks exact repeats identified from the shotgun data. Genome Res. 12: 824-831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Weber J.L. and Myers, E.W. 1997. Human whole-genome shotgun sequencing. Genome Res. 7: 401-409. [DOI] [PubMed] [Google Scholar]
- 21.Yu J., Hu, S., Wang, J., Wong, G.K., Li, S., Liu, B., Deng, Y., Dai, L., Zhou, Y., Zhang, X., et al. 2002. A draft sequence of the rice genome (Oryza sativa L. ssp. indica). Science 296: 79-92. [DOI] [PubMed] [Google Scholar]