

Abstract
The Mouse Genome Analysis Consortium aligned the human and mouse genome sequences for a variety of purposes, using alignment programs that suited the various needs. For investigating issues regarding genome evolution, a particularly sensitive method was needed to permit alignment of a large proportion of the neutrally evolving regions. We selected a program called BLASTZ, an independent implementation of the Gapped BLAST algorithm specifically designed for aligning two long genomic sequences. BLASTZ was subsequently modified, both to attain efficiency adequate for aligning entire mammalian genomes and to increase its sensitivity. This work describes BLASTZ, its modifications, the hardware environment on which we run it, and several empirical studies to validate its results.
One of the goals set by the Mouse Genome Analysis Consortium (Waterston et al. 2002) was to study mutation and selection, the main forces shaping the mouse and human genomes. Specific aims included: (1) Estimating the fraction of the human genome that is under selection (F. Chiaromonte, R. Weber, K.M. Roskin, M. Diekhans, W.J. Kent, and D. Haussler, in prep.), (2) determining the degree to which genome comparisons can pinpoint the regions under selection (Elnitski et al. 2003), and (3) measuring regional variation in the rate and pattern of neutral evolution (Hardison et al. 2003). Attaining these aims required an alignment program with higher sensitivity than needed for other Consortium goals, such as predicting novel protein-coding segments or identifying large genomic intervals in which gene order is conserved.
Ideally, our alignment program would identify orthologous regions of the human and mouse genomes, whether or not they are under selection. That is, it would determine correspondences between genomic positions that are descended from the same position in the ancestral genome, allowing nucleotide substitutions. In practice, success in reaching that goal is measured by the program's sensitivity (fraction of orthologous positions that it aligns) and specificity (fraction of the aligned positions that are orthologous). Many of our aims could have been addressed by a program that aligned neutrally evolving regions with a modest degree of sensitivity. For instance, regional variations (aim 3) could be assessed from a relatively small sample, (say, 1 of 10 orthologous regions), provided that there were no critical biases in the sampling process. Demands on specificity were higher, but it was acceptable for, say, 5% of the aligned positions to be nonorthologous.
To meet our needs, we enhanced the BLASTZ alignment program (Schwartz et al. 2000). Here, we describe the alignment program, the hardware environment, and several validation studies. Our results indicate that we have correctly determined the majority of what can be aligned between the human and mouse genomes. The C-language source code for BLASTZ and the code for extracting lineage-specific repeats (see below) can be downloaded freely from http://bio.cse.psu.edu/. Source code for axtBest, described below, can be obtained from Jim Kent (jim_kent@pacbell.net). Currently, axtBest-processed BLASTZ alignments of the human and mouse genomes are at http://genome.cse.ucse.edu/goldenPath/28jun2002/vsMm2/ and future versions will be made available at the USCS Genome Browser (http://genome.ucsc.edu/).
RESULTS
Software Design Issues
Our goal was to align an appreciable fraction of the neutrally evolving DNA in the human and mouse genomes. This sensitivity requirement disqualified several existing programs (Ning et al. 2001; Kent 2002) that sacrifice sensitivity to attain very short running times. Preliminary studies indicated that an appropriate level of sensitivity and specificity was attained by a program called BLASTZ, which is used by the PipMaker webserver (Schwartz et al. 2000) to align genomic sequences. BLASTZ follows the three-step strategy used by Gapped BLAST (Altschul et al. 1997), that is, find short near-exact matches, extend each short match without allowing gaps, and extend each gap-free match that exceeds a certain threshold by a dynamic programming procedure that permits gaps. The BLASTZ algorithm, with the additions described in this work, is summarized in Figure 1.
Figure 1.

BLASTZ in a nutshell.
Two differences between BLASTZ and Gapped BLAST were exploited in our whole-genome alignments. First, BLASTZ has an option to require that the matching regions that it reports must occur in the same order and orientation in both sequences. Second, BLASTZ uses an alignment-scoring scheme derived and evaluated by Chiaromonte et al. (2002). Nucleotide substitutions are scored by the matrix
 |
and a gap of length k is penalized by subtracting 400 + 30 k from the score. To determine whether a gap-free alignment is sufficiently promising to warrant initiation of a dynamic programming extension step, the sum of scores for its columns is multiplied by a value between 0 and 1 that measures sequence complexity, as described in detail by Chiaromonte et al. (2002). This makes it harder for a region of extremely biased nucleotide content to trigger a gapped alignment.
Two changes to BLASTZ significantly improved its execution speed for aligning entire genomes. First, when the program realizes that many regions of the mouse genome align to the same human segment, that segment is dynamically masked, that is, marked so that it will be ignored in later steps of the alignment process. Second, we adapted a very clever idea of Ma et al. (2002) for determining the initial short match that may seed an alignment. Formerly, BLASTZ looked for identical runs of eight consecutive nucleotides in each sequence. Ma et al. (2002) propose looking for runs of 19 consecutive nucleotides in each sequence, within which the 12 positions indicated by a 1 in the string 1110100110010101111 are identical. To increase sensitivity, we allow a transition (A–G, G–A, C–T or T–C) in any one of the 12 positions.
Later, BLASTZ was further modified to utilize the increasing contiguity of the available mouse sequence. For the earliest alignments, the mouse sequence was presented in unassembled reads of ∼500 bp each. A gap-free alignment was required to score at least 3000 (relative to the above substitution scores as adjusted by the measure of sequence complexity) before the dynamic programming step was initiated. Such a high threshold was needed to attain reasonable specificity. Availability of longer mouse segments suggested looking for pairs of alignments that connect nearby regions in both human and mouse genomes; the alignment procedure can be run with a lower threshold in the regions between the alignments. If the separation between the two alignments is <50 kb in both sequences, then BLASTZ recursively searches the intervening regions for 7-mer exact matches and requires a threshold of 2200 for initiating dynamic programming (without the adjustment for sequence complexity). If the separation is <10 kb, the threshold is lowered to 2000. In either case, the higher-sensitivity matches are required to occur with an order and orientation consistent with the stronger flanking matches.
Although the fee for initiating a gapped alignment is steep (e.g., 3000), once started, the alignment keeps extending as long as the average score of the added alignment remains positive. This observation suggests a strategy of physically removing lineage-specific interspersed repeats (i.e., that inserted after the human–mouse split), earlier utilized by Lee et al. (1998). Then, an alignment that is initiated on one side of the insertion point can freely jump to the other side, where it may detect alignable nucleotides that may not meet the steep initiation fee on their own. We now remove recent repeat elements, run BLASTZ, then adjust positions in the alignment to refer to the original sequences. These last two improvements, that is, recursive application of BLASTZ between adjacent alignments and excision of lineage-specific repeats, increased alignment coverage from 32% of the human genome to 40%.
The modified BLASTZ was used to compare all of the human sequence with all of the mouse, that is, to produce a complete catalog of matching regions. Frequently, more than one region of the mouse sequence aligned to the same region of the human sequence. This is a natural consequence of duplications in the mouse genome and in the human/mouse common ancestor. These duplications include paralogous genes, processed and unprocessed pseudogenes, tandem repeats, simple repeats, etc. For many purposes, one wants the single best, orthologous match for each human region. Typically, when looking at a region spanning several thousand bases, it is clear which alignment is the ortholog and which are the paralogs. The orthologous alignment usually is longer, and overall has a greater sequence identity. On the other hand, over a small region by chance, a paralog may have greater sequence identity than the ortholog. To automatically separate ortholog from paralog, we created a program, axtBest, which filters out all but the best alignment within a sliding window of 10,000 bases.
Implementation Issues and Hardware Environment
We divide the human genome into ∼3000 segments of typical length 1.01 Mb (the end of each chromosome has a shorter piece), with a 10-kb overlap between adjacent segments. Any alignment that extends for 10 kb is almost certain to contain a gap-free segment scoring >3000, therefore, 10-kb overlaps should be adequate to guarantee that no alignments will be lost by the segmentation. We precompute a list of jobs, each of which is to align one of these human segments to one of approximately one-hundred 30-Mb segments of mouse. The scheduling software available on the 1024 CPU cluster that we use doesn't support processor affinity of any sort; each job is entirely independent. Oblivious scheduling makes it unattractive to reduce disk space requirements by devoting each node to a particular genomic region, which would involve significant administrative overhead. Fortunately, we have sufficient local disk on each node to provide a copy of all of the input that might be required. The output was stored via NFS in a central server. The processes were not in general I/O bound, spending only 7% of their time waiting on I/O.
Dynamic masking was invented to handle cases like human chromosome 19, in which zinc finger or olfactory receptor genes match a huge number of times, but are not flagged as repeats. Oblivious scheduling partially defeats this optimization, however, as each human segment is compared many times with different parts of mouse, and no efficient mechanism is available to share dynamic masking information.
To align 2.8 Gb of human sequence versus 2.5 Gb of mouse sequence took 481 days of CPU time and a half day of wall clock time on a cluster of 1024 833-Mhz Pentium III CPUs. This produced 9 Gbytes of output in a relatively space-efficient format that describes the alignments by coordinates within the sequences. These are translated to a textual representation, called axt, which includes the actual bases. Whereas axt files are large, for many post-processing steps, the improved locality of reference (avoiding the need to retrieve parts of multigigabyte datasets) is a clear necessity. The initial axt files were 20 Gbytes, but running axtBest reduced them to 2.5 Gbytes. Only 3.3% of the human genome is covered by multiple alignments (assuming proper masking of interspersed repeats and low-complexity regions), but some of these places, particularly on chromosome 19, are covered to a great depth.
Software Evaluation
Like other alignment programs, BLASTZ permits adjustment of numerous parameters and thresholds, such as gap penalties and the threshold of 3000 for initiating a gapped alignment based on a gap-free alignment. It is a tricky business to test whether a particular combination of values is doing a good job. Moreover, different classes of parameters and thresholds might be best tested in different ways. For instance, with seeding strategies, it may work to use simulations that avoid running the actual code on real data (Ma et al. 2002; Kent 2002), whereas for some purposes, it may be best to run the actual software on large amounts of real data, such as in the tests described below. Many other classes of sequence analysis software benefit from availability of an experiment-based gold standard; protein alignments are checked against X-ray crystal structures and gene predictions are compared with cDNA sequences. On the other hand, when trying to correctly align neutrally evolving sequences, it is not clear how one can determine the correct answer. The maxim “it is an order of magnitude easier to design two good programs than to tell which one is better” seems appropriate here.
Our aim was to determine all homologous matches between human and mouse genomic regions, which are then filtered and examined in a variety of ways for a variety of purposes, such as identifying regions of conserved synteny. Early in the project, when we were working with unassembled mouse reads, there was no choice but to compare all of the human sequence with all of the mouse. Later, when reliable assemblies were produced and syntenic segments and blocks had been identified at moderate resolution, it became feasible to avoid an all-vs-all computation (although this would preclude some of the studies that we want to perform). Computational efficiency would increase dramatically if each region of the human sequence were compared with a small segment of the mouse genome rather than to all of it. Another benefit would be to substantially decrease the likelihood of a match to unrelated sequence occurring by chance.
However, experimental evidence indicates that the level of spurious matches in our all-vs-all comparisons is quite low. We reversed the soft-masked mouse sequence (without complementing it) and aligned it to human. (Soft-masking means using lower-case letters for nucleotides in interspersed repeats, but not entirely obliterating them, so that BLASTZ can align them if they lie adjacent to an aligning single-copy region.) The reversed mouse sequence has precisely the same size and local compositional complexity as mouse; for instance, a microsatellite sequence cacaca … in mouse is preserved in the reversed sequence. Thus, the quantity of matches to the reversed mouse should approximate the quantity of spurious matches to mouse. A more refined analysis shows that alignments with reversed mouse will tend to slightly underestimate spurious human–mouse matches, as mammalian genome sequences exhibit significant asymmetries in dinucleotide (and higher order) composition. The strongest dinucleotide asymmetry is that CpG occurs much less frequently than GpC; the excess of CpG over GpC in the reversed mouse, together with lesser effects from other dinucleotides, will make matches with human less frequently than they would be with equal dinucleotide frequencies. [The approach of Chiaromonte et al. (2002) circumvents this problem, but has other difficulties.]
Results of some whole-genome runs that measured coverage by outer alignments (Step 2 of Figure 1) are given in Table 1. Placing a lower bound of 3000 on scores for gapped alignments (which should eliminate no outer alignments), 39.154% of the human sequence aligned to mouse, and only 0.164% aligned to reversed mouse. This confirms the high specificity of our approach even before axtBest is applied. Imposing the requirement that gapped alignments score at least 5000 reduced coverage by only 0.221% of human, but halved coverage by bogus alignments from 0.164% to 0.075%. Requiring a score of 10,000 and keeping only regions that align to just one place in the mouse genome, we still align 36.831% of human, whereas only 0.007% aligns to reversed mouse. Of course, for some applications, for example, exploring gene duplications, that strategy for attaining extremely high specificity would throw out the baby with the bath water.
Table 1.
Coverage by Outer Alignments
| Score | 1 Mus | >1 Mus | 1 Rev | >1 Rev |
| 3000 | 0.36814 | 0.02340 | 0.00084 | 0.00080 |
| 4000 | 0.36859 | 0.02230 | 0.00040 | 0.00074 |
| 5000 | 0.36958 | 0.01975 | 0.00016 | 0.00059 |
| 6000 | 0.36992 | 0.01829 | 0.00013 | 0.00051 |
| 7000 | 0.36997 | 0.01697 | 0.00011 | 0.00043 |
| 8000 | 0.36966 | 0.01586 | 0.00010 | 0.00037 |
| 9000 | 0.36911 | 0.01490 | 0.00008 | 0.00033 |
| 10000 | 0.36831 | 0.01405 | 0.00007 | 0.00030 |
The columns have the following meanings: (1) score threshold for a gapped outer alignment (Step 2.2.2 of Fig. 1); (2) fraction of the genome covered by exactly one alignment; (3) fraction of the genome covered by more than one alignment; (4) fraction of the genome covered by exactly one alignment with reversed mouse; (5) fraction of the genome covered by more than one alignment with reversed mouse.
These whole-genome tests distinguished outer alignments (all-vs-all) from inner alignments (higher sensitivity searches between two adjacent outer alignments). BLASTZ's inner alignment steps searched 26% of the human sequence and added 2% of it to reported alignments. The ratio of true positives to false positives was ∼1000:1, suggesting that sensitivity in inner alignment steps can be safely increased. The average length of a human region searched during an inner-alignment computation was 3 kb, hence, the alignment space for the all-vs-all outer alignments is 3000 times bigger than for inner alignments (2.9 Gb-by-2.5 Gb vs. 0.8 Gb-by-3 kb). This suggests that inner alignments can be performed at relatively low efficiency, say, with a Smith-Waterman algorithm, without appreciably affecting the total computation time.
Another test of the specificity of BLASTZ alignments is based on conservation of synteny. Human Chromosome 20 is considered to be completely homologous to parts of mouse Chromosome 2, that is, the synteny observed on human Chromosome 20 is conserved on parts of mouse Chromosome 2. After filtering through axtBest, only 3.3% of the aligned bases on human Chromosome 20 were found to align outside of mouse chromosome 2. A certain degree of alignment to nonhomologous chromosomes is to be expected from processed pseudogenes. Because only ∼96% of the mouse genome is sequenced, in some cases, a paralog on another chromosome will fill in for a nonsequenced ortholog on chromosome 2 as well.
We used human chromosome 20 to compare the fraction of the human sequence and various gene-related features that are aligned by BLASTZ, PatternHunter (Ma et al. 2002) and translated BLAT (Kent 2002); see Tables 2 and 3. Although translated BLAT and Pattern Hunter are both relatively sensitive to finding alignments of coding exons, BLASTZ is still more sensitive, and by taking an appropriate subset can be relatively specific as well. BLASTZ is significantly more sensitive aligning UTRs and upstream regions. This sensitivity helps avoid missing alignments in regulatory regions.
Table 2.
Comparison of Genome Coverage
| chr20 | CDS | 3′UTR | 5′UTR | upstream | |
| Blastz all | 40.5% | 98.5% | 87.1% | 89.0% | 87.2% |
| Blastz tight | 5.6% | 92.5% | 26.0% | 39.6% | 28.3% |
| PH all | 29.7% | 95.5% | 55.0% | 59.3% | 52.5% |
| PH tight | 5.0% | 91.2% | 25.1% | 36.3% | 25.2% |
| transl. BLAT | 5.8% | 90.3% | 29.2% | 38.4% | 27.2% |
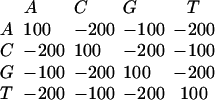 |
a gap open score of −2000, a gap extension score of −50, and a threshold of 3400.
Table 3.
Comparison of Covered Regions
| All | Tight | |
| BLASTZ only | 54.1% | 12.2% |
| PH only | 10.2% | 3.3% |
| Both | 35.7% | 85.5% |
Venn Diagram of PatternHunter and BLASTZ alignment coverage. The amount of human bases covered by one program but not the other and the amount covered by both programs are shown as a percentage of the amount covered by either program.
DISCUSSION
The Mouse Genome Analysis Group used a variety of programs to align the mouse and human sequences, including BLAST (Altschul et al. 1997), Blast2sequences (Tatusova and Madden 1999), BLAT (Kent 2002), and PatternHunter (Ma et al. 2002). However, none of these programs was suited to our goal of investigating fine-scale features of genome evolution, primarily because they were tuned for aligning protein-coding regions, whereas our focus was neutral evolution. Hence, we chose to develop a new program. An empirical comparison of available tools indicated that the BLASTZ program was a good place to start.
Two aspects of BLASTZ's design philosophy proved valuable. First, BLASTZ is intended for use at all stages of genome sequencing, including the initial period when only small contigs, or even simply unassembled 500-bp reads, are available. BLASTZ permitted us to begin our analyses quite early. This capability is, of course, essential in cases in which sequencing of a second genome stops with light shotgun coverage.
Another tenant of the BLASTZ philosophy is that the alignment program should not enforce critical a priori assumptions about which alignments are important; rather, it should be fairly inclusive. The task of processing and filtering the initial alignments in various ways is left to downstream programs, which can be made quite flexible and efficient (e.g., Huang et al. 1994; Zhang et al. 1999).
We expect that further work on BLASTZ will soon yield a 10-fold reduction in execution time for all-vs-all genome comparisons with no degradation of sensitivity and specificity. For finished mammalian genomes, reliable coarse-grained mapping of homologous regions will make it possible to update a genomic alignment on a single workstation. This can be accomplished by use of the Gapped BLAST design; different approaches may do even better.
A number of difficult tasks remain before the problem of aligning two mammalian genome sequences is adequately solved. Echoing the sentiments of Miller (2001), we look forward to progress in the intertwined areas of producing higher-sensitivity alignments and of evaluating their correctness, and hope that investigators will adopt a cooperative spirit reflecting the highest ideals of the Genome Project.
WEB SITE REFERENCES
http://bio.cse.psu.edu/; BLASTZ source code.
http://genome.cse.ucse.edu/goldenPath/28jun2002/vsMm2/; mouse–human alignments made using the June 2002 human assembly (also known as build 30) vs. the Feb. 2002 mouse assembly (also known as MGSCv3 or mm2).
http://genome.ucsc.edu/; in the future, whole-genome alignments will be available from this site.
Acknowledgments
We appreciate the feedback given by Krishna Roskin, Ryan Weber, Angie Hinrichs, and Mark Diekhans. Phil Green pointed out the effect of asymmetries in dinucleotide frequencies on aligning with reversed mouse. S.S., R.H., and W.M. are supported by grant HG-02238 from the National Human Genome Research Institute, with additional support to R.H. coming from NIH grant RO1 DK27635. W.J.K., R.B., and D.H. are supported by NHGRI Grant 1P41HG02371.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
E-MAIL webb@bio.cse.psu.edu; FAX (814) 865-3176.
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.809403. Article published online before print in December 2002.
REFERENCES
- 1.Altschul S.F., Madden, T.L., Schaffer, A., Zhang, J., Zhang, Z., Miller, W., and Lipman, D.J. 1997. Gapped BLAST and PSI-BLAST—a new generation of protein database search programs. Nucleic Acids Res. 25: 3389-3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chiaromonte, F., Yap, V.-B., and Miller, W. 2002. Scoring pairwise genomic sequence alignments. Pacific Symp. Biocomput. 115–126. [DOI] [PubMed]
- 3.Elnitski, L., Hardison, R.C., Li, J., Yang, S., Kolbe, D., Eswara, P., O'Connor, M.J., Schwartz, S., Miller, W., and Chiaromonte, F. 2003. Distinguishing regulatory DNA from neutral sites. Genome Res. (this issue). [DOI] [PMC free article] [PubMed]
- 4.Hardison, R.C., Roskin, K., Yang, S., Diekhans, M., Kent, W.J., Weber, R., Elnitski, L., Li, J., O'Connor, M., Kolbe, D., et al. 2003. Covariation in frequencies of substitution, deletion, transposition, and recombination during eutherian evolution. Genome Res. (this issue). [DOI] [PMC free article] [PubMed]
- 5.Huang, X., Pevzner, P., and Miller, W. 1994. Parametric recomputing in alignment graphs. Combinatorial Pattern Matching (Springer Lecture Notes in Computer Science, 807), 87–101.
- 6.Kent W. J. 2002. BLAT—the BLAST-Like Alignment Tool. Genome Res. 12: 656-664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kent W.J., Sugnet, C., Furey, T., Roskin, K., Pringle, T., Zahler, A., and Haussler, D. 2002. The human genome browser at UCSC. Genome Res. 12: 996-1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lee I.Y., Westaway, D., Smit, A.F., Wang, K., Seto, J., Chen, L., Acharya, C., Ankener, M., Baskin, D., Cooper, C., et al. 1998. Complete genomic sequence and analysis of the prion protein gene region from three mammalian species. Genome Res. 8: 1022-1037. [DOI] [PubMed] [Google Scholar]
- 9.Ma B., Tromp, J., and Li, M. 2002. PatternHunter: Faster and more sensitive homology search. Bioinformatics 18: 440-445. [DOI] [PubMed] [Google Scholar]
- 10.Miller W. 2001. Comparison of genomic DNA sequences: Solved and unsolved problems. Bioinformatics 17: 391-397. [DOI] [PubMed] [Google Scholar]
- 11.Ning Z., Cox, A J., and Mullikin, J.C. 2001. SSAHA: A fast search method for large DNA databases. Genome Res. 11: 1725-1729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pruitt K. D. and Maglott, D. R. 2001. RefSeq and LocusLink: NCBI gene-centered resources. Nucleic Acids Res. 29: 137-140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schwartz S., Zhang, Z., Frazer, K.A., Smit, A., Riemer, C., Bouck, J., Gibbs, R., Hardison, R.C., and Miller, W. 2000. PipMaker—a Web server for aligning two genomic DNA sequences. Genome Res. 10: 577-586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tatusova T.A. and Madden, T.L. 1999. BLAST 2 Sequences, a new tool for comparing protein and nucleotide sequences. FEMS Microbiol. Lett. 174: 247-250. [DOI] [PubMed] [Google Scholar]
- 15.Waterston R.H., Lindblad-Toh, K., Birney, E., Rogers, J., Abril, J.F., Agarwal, P., Agarwala, R., Ainscough, R., Alexandersson, M., An, P., et al. 2002. Initial sequencing and comparative analysis of the mouse genome. Nature 420: 520-562. [DOI] [PubMed] [Google Scholar]
- 16.Zhang Z., Berman, B., Wiehe, T., and Miller, W. 1999. Post-processing long pairwise alignments. Bioinformatics 15: 1012-1019. [DOI] [PubMed] [Google Scholar]