

Abstract
New antibacterials need new approaches to overcome the problem of rapid antibiotic resistance. Here we review the development of potential new antibacterial drugs that do not kill bacteria or inhibit their growth, but combat disease instead by targeting bacterial virulence.
Keywords: antibacterial, antivirulence, bacterial infection, pilicide, quorum sensing
Introduction
In the ongoing battle between people and pathogens, the pendulum seems to be swinging in favour of the bugs. The rapid increase in resistance to antibiotics combined with the slowing to a trickle of new antibiotics progressing through the pipeline over the past decades has led to this point. The situation has been described by the Infectious Diseases Society of America as a looming ‘public health crisis’ 1.
There are any number of reasons why pathogenic bacteria acquire antibiotic resistance, and why resistance is growing at such an alarming rate. The question is, given where we are now, how can we ensure that the pendulum swings back in our favour? One school of thought is that we need to change the way we discover new antibiotics. Historically, antibiotics have been identified by their ability to kill or inhibit growth of bacteria. A prime example is penicillin, originally identified by Fleming's serendipitous discovery that a penicillium mould inhibited bacterial growth on an agar plate. Ever since, screening approaches have been engineered to find chemicals that do the same thing, and molecular approaches have focused on identifying essential genes to target for drug intervention. The problem with therapeutic approaches that target viability is that they induce a high selection pressure. A bacterium exhibiting resistance to the antibiotic will have an enormous selective advantage over its competitors in a bacterial population, so that resistance will develop rapidly in the presence of that antibiotic.
An alternative to killing bacteria or stopping their growth, is to search for drugs that disarm bacteria. This idea focuses on developing drugs that inhibit bacterial virulence 2–4 rather than bacterial viability (Figure 1). Targeting virulence offers several potential advantages including:
an increased repertoire of pharmacological targets
generating antimicrobials with new mechanisms of action
reducing resistance development due to decreased selective pressure 3
and potentially preserving gut microbiota.
Figure 1.
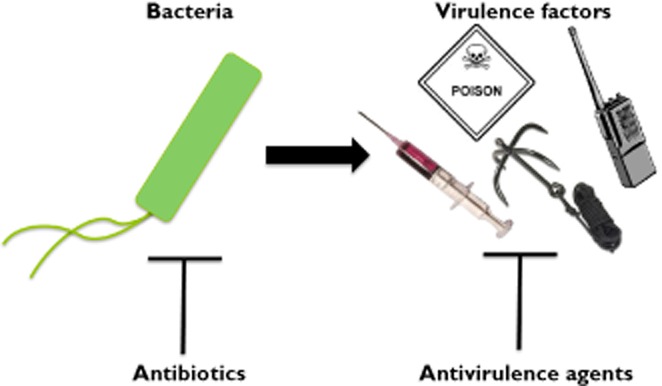
Antibiotics and antivirulence agents. Antibiotics (left) kill bacteria or prevent their growth. Antivirulence agents (right) render bacteria harmless by blocking the activity of virulence factors. Virulence factors can include toxins (denoted by poison sign), secretion systems (syringe), adhesion factors (grappling hook) or quorum sensing (walkie-talkie), amongst others
On the other hand, development of antivirulence therapies presents its own unique challenges. We can no longer use established screening systems that identify compounds that kill or inhibit growth of bacteria and minimal inhibitory concentration measures are obsolete in this scenario. Specific in vitro and in vivo assays will need to be developed to screen for compounds that inhibit specific virulence processes. Given that virulence mechanisms vary from one bacteria to another, antivirulence drugs are likely to have a narrow spectrum of activity. Their success in the clinic may well depend on development of real time diagnostics that identify the causative organism and enable therapy personalized to the infectious agent.
In this review we highlight several virulence pathways currently being targeted for the development of antivirulence drugs, including adhesion, secretion and toxin production (Figure 2, summarized in Table 1). We also highlight two master virulence targets that coordinate deployment of entire arsenals of virulence factors either by communicating information (quorum sensing) (Figure 2, Table 1) or by assembling an armoury of bacterial weapons (oxidative folding) (Figure 3). Drugs that block these master systems may have a broader spectrum of activity.
Figure 2.
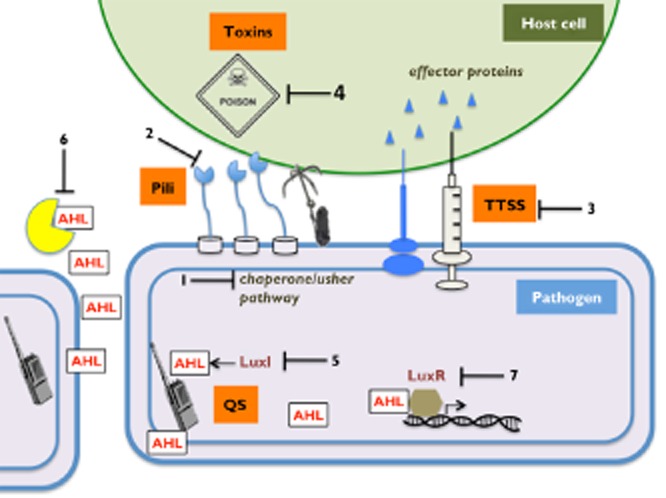
Examples of bacterial virulence pathways that have been targeted for antimicrobial development. Bacterial adhesion to the host cell; 1. Inhibitors of pili biosynthetic machineries (e.g. chaperone/usher pathway); 2. Inhibitors of the carbohydrate-binding sites in the adhesin molecules. Bacterial secretion systems; 3. Inhibitors of the type three secretion system (TTSS) to block injection of effector proteins to the host cell. Toxin production; 4. Toxin neutralization to inhibit damage to the host. Acyl-homoserine lactone (AHL) mediated quorum sensing (QS): 5. Inhibitors of AHL synthase LuxI. 6. AHL degrading enzymes (e.g. lactonase and acylase). 7. Inhibitors of AHL binding to transcriptional regulator LuxR
Table 1.
Selected examples of virulence factor inhibitors
| Mode of action | Selection of studied pathogens | Reference | |
|---|---|---|---|
| Adhesion inhibitors | |||
| Aaptamines | Natural product sortase A inhibitors | S. aureus | 8 |
| Pyridazinone and pyrazolethione derivatives | Synthetic sortase A inhibitors | S. aureus, B. anthracis | 11 |
| Pilicides | Regulate pilus biogenesis by blocking the chaperone/usher assembly pathway | E. coli | 12 |
| Toxin inhibitors | |||
| Virstatin | Inhibits ToxT transcription factor blocking expression of cholera toxin | V. cholerae | 30 |
| ABthrax, Valortim | Antibodies; inhibit anthrax toxins | B. anthracis | 38–41 |
| TTSS inhibitors | |||
| Thiazolidinone derivatives | Prevent translocation of effector molecules | Yersinia, Salmonella, Francisella, Pseudomonas | 24 |
| Salicylidene acylhydrazides | Prevent translocation of effector molecules | Escherichia, Yersinia, Chlamydia, Pseudomonas, Salmonella, Shigella | 28 |
| QS inhibitors | |||
| Furanone derivatives | Inhibitors mimic AHLs; bind LuxR receptor and inhibit QS-regulated gene expression | E. coli, P. aeruginosa, Proteus mirabilis, Staphylococci | 53 |
| Lactonase and acylase | ‘Quorum quenching’ enzymes degrade AHL, to block the quorum sensing response | Bacillus ssp, Erwinia carotovora, P. aeruginosa, Pectobacterium carotovorum | 47,53 |
Figure 3.
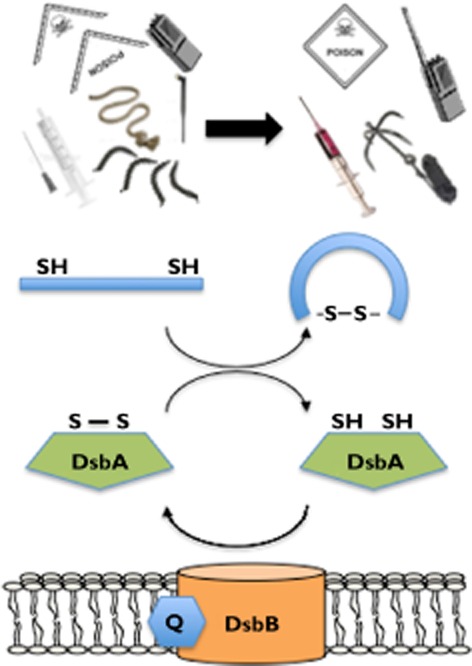
Schematic representation of the DSB catalytic cycle. Quinones (labelled Q) generate disulfides in DsbB (orange), which are transferred to DsbA (green), which catalyzes oxidative protein folding in substrate virulence factors (blue; and indicated above). In concert, the disulfide in DsbA (labelled S-S) is reduced to two thiols (labelled SH) to complete the catalytic cycle
Targeting occupation
A crucial first step in colonization by bacteria is adhesion to host cells. Blocking this process may prevent establishment and maintenance of infection 4. Adhesion is mediated by surface proteins (adhesins, autotransporters etc.) and multi-protein scaffolds (e.g. pili) protruding from bacteria that interact specifically with carbohydrates on the host cell surface.
In Gram-positive organisms, adhesion depends on sortases, a family of cysteine transpeptidases that covalently anchor adhesin proteins to the bacterial cell wall 5,6. Sortases have been targeted in several antibacterial drug discovery programmes and screening against Staphylococcus aureus and Bacillus anthracis have identified hits which could potentially be developed into potent sortase inhibitor drugs 7–11.
By contrast, cell adhesion and invasion in Gram-negative organisms generally relies on the production of pili 4. Two strategies have been developed to block pilus-mediated adhesion. One is the identification of pilicides, molecules that prevent pili biogenesis by interfering with the underlying usher-chaperone pathway 12. Pilicides have been shown to reduce production of several components of this pathway in uropathogenic E. coli including type I and P pili fimbrial proteins 13, Dr family adhesins 14 and curli 15, with some also preventing biofilm formation 12,15. Significantly, the usher-chaperone pili assembly machinery is present in many species including Escherichia, Salmonella, Klebsiella, Yersinia and Pseudomonas 16 suggesting that pilicides may have a broad spectrum of activity 17.
A second strategy to inhibit pili-based adhesion relies on physically blocking the interaction between the adhesin and the host cell. The carbohydrate binding site is localized at the very tip of the pili. Carbohydrate derivatives and molecules mimicking mammalian glycans dramatically reduce the adhesive properties of bacterial pili 18–20. Importantly, one such inhibitor prevented acute infection in vivo and also treated chronic cystitis caused by a multi-resistant E. coli in an animal model 18.
Targeting weapons delivery
Bacteria have evolved complex machineries to deliver proteins and toxins into a host cell across membranes and cell walls and these machineries play a central role in pathogenesis. The system attracting most attention is the type III secretion system (TTSS). This syringe-like multiprotein apparatus injects bacterial effector proteins and toxins directly into the host cell cytosol and thereby hijacks a wide range of cellular processes 21. Many components of the TTSS are specific to prokaryotes and several studies have explored TTSS inhibitors as potential therapeutics (recently reviewed in 22). Importantly, the TTSS machinery is present in many pathogens including Escherichia, Shigella, Salmonella, Pseudomonas, Chlamydia and Yersinia spp., so that targeting common elements could result in broad-spectrum TTSS inhibitors 23.
Indeed, high-throughput screening identified thiazolidinone derivatives that block TTSS from Gram-negative pathogens including S. Typhimurium and Yersinia enterocolitica, reduced the virulence of Pseudomonas syringae and inhibited other secretion systems such as the type II in Pseudomonas and the type IV in Francisella 24. Similarly, small molecule screening identified a series of salicylidene acylhydrazides capable of inhibiting the TTSS of intracellular (Chlamydia trachomatis) and extracellular pathogens (Yersinia ssp.) 25–27. Some of these compounds showed protective activity against the sexually transmitted pathogen C. trachomatis in mouse infection models 28.
Targeting toxins
Toxins are the primary virulence factors of many bacterial pathogens. Examples include botulinum and tetanus neurotoxins, cholera, anthrax, diphtheria and Shiga toxins. All are proteins delivered into the host to cause mass cell destruction and tissue damage 29. Their extreme toxicity and critical role in pathogenesis makes inhibition of toxin production an obvious approach for development of antivirulence antimicrobials. This can be achieved by targeting toxin transcription and expression. Virstatin inhibits the transcription factor ToxT that regulates expression of cholera toxin and cholera co-regulated pilus, and blocks intestinal colonization by this pathogen in murine models 30. Similarly, a small molecule inhibitor of toxin TcdA and TcdB expression by Clostridium difficile, has shown efficacy in a hamster model of gastrointestinal infection 31.
Antibodies have been developed to neutralize toxins and are already used to treat bacterial diseases such as tetanus, diphtheria and botulism 32. For example, botulism toxin neutralizing antibodies from horse sera are used to treat adult botulism and a human-derived botulism antitoxin has been used to treat infants 33. These outcomes provide clinical evidence validating the use of antitoxin drugs after infection.
Other antibody therapies are at different stages of development 34. For example, antibodies against Shiga toxin were shown to protect against Shiga toxin-producing E. coli (STEC) in a piglet model of acute gastroenteritis 35. Similarly, efficacy was demonstrated in mouse and hamster infection models by combining human antibodies against C. difficile toxins A and B 36. The potential use of B. anthracis as a bioweapon has made this and other high threat pathogens the focus of intense efforts to develop antibodies and vaccines 37. Antibodies that inhibit anthrax toxins (ABthrax, Valortim among others) have shown promising protection in a range of animal models and are now in clinical development 38–41.
Targeting communication systems
Bacterial cell-to-cell communication is essential for microbes to adapt to changing environments and this communication is regulated by quorum sensing (QS) networks. Gram-positive and Gram-negative bacteria both use complex regulatory QS circuits to sense their population densities and regulate the expression of virulence factors, allowing successful establishment of infection 42. The canonical QS pathways consist of secreted signal molecules known as autoinducers (AI, e.g. acyl-homoserine lactones (AHLs) in many Gram-negative bacteria, autoinducing peptides (AIPs) in Gram-positive bacteria). Upon reaching a threshold concentration, AI molecules interact with cognate sensor receptors (e.g. LuxR and LuxS receptors) to induce the expression of virulence genes.
Given the central role of QS systems in bacterial pathogenesis, many efforts have focused on interfering with these pathways (recently reviewed in 43–48). Quorum quenching is a term that has been used to describe ‘any approach that interferes with microbial QS signalling’ 49. QS networks have been quenched or modulated at three points (reviewed in 49) by (i) inhibiting signal generation (e.g. by blocking synthesis of AHL in vitro using AHL analogues 50,51), (ii) degrading the signal molecule (AHLs can be destroyed chemically by increasing the pH 2 or by use of ‘quorum quenching’ enzymes 47, or inactivated with antibodies 52) and (iii) blocking the interaction of the QS signal molecule with the receptor. The last is the most popular approach. Screening of natural and synthetic compounds has produced potent antagonists of sensing receptors for many bacteria (e.g. enterobacteria, Pseudomonas, Staphylococci) with some antagonists being protective in animal models of infection (reviewed in 53). Furthermore, inhibitors capable of blocking QS networks in several Gram-negative pathogens open the possibility of QS inhibitors with broad spectrum activity 54.
The increasing number of patent applications for QS inhibitors clearly reflects the interest in this approach 55. Notably, targeting QS has yielded potent molecules that prevent biofilm formation, a major hurdle in treating many bacterial infections 47. Although a lower risk of resistance development was predicted for QS-regulating molecules, recent data indicate that bacteria can develop resistance to these compounds 56. For example, the QS inhibitor C-30 57 had no effect on the growth of Pseudomonas aeruginosa in rich media, but in minimal media it did affect bacterial growth and selected for resistance 58.
Targeting weapons assembly
Virulence factors produced by bacteria are generally proteins and these virulence proteins need to be assembled correctly to function. An important feature of many virulence factor proteins produced by Gram-negative bacteria is the requirement for structural bracing in the form of disulfide bonds. Disulfide formation between pairs of cysteine residues increases the chemical and physical stability of proteins. Conversely, failure to form native disulfide bonds results in degradation and loss of activity.
Oxidative protein folding, the process of introducing disulfide bonds into folding proteins, is a rate-limiting step in the assembly of many virulence factors and requires the activity of specific enzymes 59. The classic bacterial disulfide bond (DSB) machinery, first characterized in E. coli K-12 60, comprises a soluble periplasmic enzyme DsbA and an integral membrane protein DsbB (Figure 3). DsbB and its quinone cofactor together generate disulfides de novo, and transfer them to DsbA 61 which introduces disulfides directly into folding proteins 62.
Whilst some variation exists in the DSB enzymes in different bacteria (reviewed in 63), there is now overwhelming evidence that the DSB oxidative protein folding machinery is a master regulator of bacterial virulence. Recent compelling evidence comes from a study using an animal model of melioidosis in which mice infected with the causative agent of melioidosis, Burkholderia pseudomallei, all died within 42 days whereas mice infected with B. pseudomallei lacking the gene for DsbA all survived 64. Similarly, animal infection models have demonstrated that deletion of dsbA or dsbB in uropathogenic E. coli (UPEC) severely attenuated its ability to colonize the bladder 65, and that dsbA mutants in Salmonella enterica serovar Typhimirium were avirulent 66.
Indeed, many bacteria lacking a functional DsbA have been shown to have reduced virulence, increased sensitivity to antibiotics and diminished capacity to cause infection. These include uropathogenic E. coli (UPEC), enteropathogenic E. coli (EPEC), Bordetella pertussis (whooping cough), Vibrio cholerae (cholera), P. aeruginosa (opportunistic human pathogen), Haemophilus influenzae (opportunistic human pathogen), S. flexneri (diarrhoea) and Neisseria meningitidis (bacterial meningitis) amongst others 67–74.
The loss of virulence can be attributed to the misfolding of a (normally) disulfide-containing protein substrate of DsbA. For example, E. coli dsbA mutants are non-motile. The loss of motility is a consequence of the misfolding of protein FlgI, a component of the periplasmic ring of the flagellar motor and a DsbA substrate 75. DsbA is also required for the correct folding of virulence proteins involved in bacterial adhesion, secretion and toxicity. Similarly, mutational inactivation of dsbB also affects bacterial virulence 76.
These observations point to a major regulatory role in virulence and identify the DSB enzymes as key targets for the development of anti-virulence agents. The DSB machinery offers a number of advantages as antibacterial drug targets including:
DSB inhibition affects multiple bacterial virulence pathways;
DSB enzymes are localized to the outer compartment of bacteria making drug delivery more amenable than cytoplasmic targets;
DsbAs are more highly conserved than the virulence factors they assemble, so that inhibitors are likely to be effective against multiple pathogens and
Structures of DSB enzymes are in the public domain, so that structure-based approaches for drug discovery are supported.
However, whereas DSB systems are conserved and required for pathogenicity in Gram-negative bacteria, the link between DSB systems and virulence in Gram-positive organisms is not confirmed. Moreover, to our knowledge, there are no reports of small molecule inhibitors of DSB enzymes that are effective in vivo. Indeed, they represent challenging targets for drug design. Structures of DsbA and DsbB reveal that their interaction surfaces lack deep binding cavities, which is often an impediment to inhibitor design. Nevertheless, there are an increasing number of examples of small molecules designed to block protein–protein interactions against other targets 77. Furthermore, screening of small molecule ‘fragments’ identified compounds that interacted with DsbB and led to a series of compounds capable of inhibiting DsbB in vitro 78. If we can develop inhibitors of DSB-mediated oxidative protein folding, these would have enormous value as antivirulence agents by potentially blocking the assembly of multiple bacterial virulence factors.
Conclusion
In the search for future antibacterials to overcome antibiotic resistance, antivirulence agents promise more than a glimmer of hope. Several strategies have been put forward, and target validation and preliminary screening have been performed to identify important virulence pathways and master virulence machineries. However, aside from antibodies that inactivate specific bacterial toxins, none of these compounds with new mechanisms of actions has yet reached the clinic. So it remains to be seen whether all or some of these antivirulence approaches will live up to expectations. We eagerly await studies showing how new generation antivirulence antibacterials perform, whether they will reduce resistance development, whether they will need to be combined with traditional antibiotics, or whether they can resurrect antibiotics made obsolete by bacterial resistance mechanisms.
Competing Interests
All authors have completed the Unified Competing Interest form at http://www.icmje.org/coi_disclosure.pdf (available on request from the corresponding author) and declare JLM had support from the Australian Research Council for the submitted work, MJS and JLM had funded grants from the Australian Research Council in the previous 3 years; BH and MJS had support from the Australian National Health and Medical Research Council in the previous 3 years and MJS and JLM had grants and non-financial support from Biota Pty Ltd over the past 3 years. MJS had grants and non-financial support from Biota Pty Ltd outside the submitted work over the past 3 years.
JLM is an ARC Australian Laureate Fellow (FL0992138). BH is supported by a La Trobe Institute for Molecular Science Fellowship.
References
- Infectious Disease Society of America. Bad Bugs, No Drugs: As Antibiotic Discovery Stagnates, A Public Health Crisis Brews. Alexandria, VA: Infectious Disease Society of America; 2004. [Google Scholar]
- Zucca M, Scutera S, Savoia D. New antimicrobial frontiers. Mini Rev Med Chem. 2011;11:888–900. doi: 10.2174/138955711796575498. [DOI] [PubMed] [Google Scholar]
- Escaich S. Novel agents to inhibit microbial virulence and pathogenicity. Expert Opin Ther Pat. 2010;20:1401–1418. doi: 10.1517/13543776.2010.511176. [DOI] [PubMed] [Google Scholar]
- Cegelski L, Marshall GR, Eldridge GR, Hultgren SJ. The biology and future prospects of antivirulence therapies. Nat Rev Microbiol. 2008;6:17–27. doi: 10.1038/nrmicro1818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suree N, Jung ME, Clubb RT. Recent advances towards new anti-infective agents that inhibit cell surface protein anchoring in Staphylococcus aureus and other gram-positive pathogens. Mini Rev Med Chem. 2007;7:991–1000. doi: 10.2174/138955707782110097. [DOI] [PubMed] [Google Scholar]
- Maresso AW, Schneewind O. Sortase as a target of anti-infective therapy. Pharmacol Rev. 2008;60:128–141. doi: 10.1124/pr.107.07110. [DOI] [PubMed] [Google Scholar]
- Chan AH, Wereszczynski J, Amer BR, Yi SW, Jung ME, McCammon JA, Clubb RT. Discovery of Staphylococcus aureus sortase A inhibitors using virtual screening and the relaxed complex scheme. Chem Biol Drug Des. 2013;82:418–428. doi: 10.1111/cbdd.12167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang KH, Chung SC, Shin J, Lee SH, Kim TI, Lee HS, Oh KB. Aaptamines as sortase A inhibitors from the tropical sponge Aaptos aaptos. Bioorg Med Chem Lett. 2007;17:5366–5369. doi: 10.1016/j.bmcl.2007.08.007. [DOI] [PubMed] [Google Scholar]
- Hu P, Huang P, Chen MW. Curcumin reduces Streptococcus mutans biofilm formation by inhibiting sortase A activity. Arch Oral Biol. 2013;58:1343–1348. doi: 10.1016/j.archoralbio.2013.05.004. [DOI] [PubMed] [Google Scholar]
- Hu P, Huang P, Chen WM. Curcumin inhibits the sortase A activity of the Streptococcus mutans UA159. Appl Biochem Biotechnol. 2013;171:396–402. doi: 10.1007/s12010-013-0378-9. [DOI] [PubMed] [Google Scholar]
- Suree N, Yi SW, Thieu W, Marohn M, Damoiseaux R, Chan A, Jung ME, Clubb RT. Discovery and structure-activity relationship analysis of Staphylococcus aureus sortase A inhibitors. Bioorg Med Chem. 2009;17:7174–7185. doi: 10.1016/j.bmc.2009.08.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aberg V, Almqvist F. Pilicides-small molecules targeting bacterial virulence. Org Biomol Chem. 2007;5:1827–1834. doi: 10.1039/b702397a. [DOI] [PubMed] [Google Scholar]
- Pinkner JS, Remaut H, Buelens F, Miller E, Aberg V, Pemberton N, Hedenstrom M, Larsson A, Seed P, Waksman G, Hultgren SJ, Almqvist F. Rationally designed small compounds inhibit pilus biogenesis in uropathogenic bacteria. Proc Natl Acad Sci U S A. 2006;103:17897–17902. doi: 10.1073/pnas.0606795103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piatek R, Zalewska-Piatek B, Dzierzbicka K, Makowiec S, Pilipczuk J, Szemiako K, Cyranka-Czaja A, Wojciechowski M. Pilicides inhibit the FGL chaperone/usher assisted biogenesis of the Dr fimbrial polyadhesin from uropathogenic Escherichia coli. BMC Microbiol. 2013;13:131. doi: 10.1186/1471-2180-13-131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cegelski L, Pinkner JS, Hammer ND, Cusumano CK, Hung CS, Chorell E, Aberg V, Walker JN, Seed PC, Almqvist F, Chapman MR, Hultgren SJ. Small-molecule inhibitors target Escherichia coli amyloid biogenesis and biofilm formation. Nat Chem Biol. 2009;5:913–919. doi: 10.1038/nchembio.242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busch A, Waksman G. Chaperone-usher pathways: diversity and pilus assembly mechanism. Philos Trans R Soc Lond B Biol Sci. 2012;367:1112–1122. doi: 10.1098/rstb.2011.0206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chorell E, Pinkner JS, Phan G, Edvinsson S, Buelens F, Remaut H, Waksman G, Hultgren SJ, Almqvist F. Design and synthesis of C-2 substituted thiazolo and dihydrothiazolo ring-fused 2-pyridones: pilicides with increased antivirulence activity. J Med Chem. 2010;53:5690–5695. doi: 10.1021/jm100470t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Totsika M, Kostakioti M, Hannan TJ, Upton M, Beatson SA, Janetka JW, Hultgren SJ, Schembri MA. A FimH inhibitor prevents acute bladder infection and treats chronic cystitis caused by multidrug-resistant uropathogenic Escherichia coli ST131. J Infect Dis. 2013;208:921–928. doi: 10.1093/infdis/jit245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouckaert J, Berglund J, Schembri M, De Genst E, Cools L, Wuhrer M, Hung CS, Pinkner J, Slattegard R, Zavialov A, Choudhury D, Langermann S, Hultgren SJ, Wyns L, Klemm P, Oscarson S, Knight SD, De Greve H. Receptor binding studies disclose a novel class of high-affinity inhibitors of the Escherichia coli FimH adhesin. Mol Microbiol. 2005;55:441–455. doi: 10.1111/j.1365-2958.2004.04415.x. [DOI] [PubMed] [Google Scholar]
- Touaibia M, Roy R. Glycodendrimers as anti-adhesion drugs against type 1 fimbriated E. coli uropathogenic infections. Mini Rev Med Chem. 2007;7:1270–1283. doi: 10.2174/138955707782795610. [DOI] [PubMed] [Google Scholar]
- Chatterjee S, Chaudhury S, McShan AC, Kaur K, De Guzman RN. Structure and biophysics of type III secretion in bacteria. Biochemistry. 2013;52:2508–2517. doi: 10.1021/bi400160a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan MC, Linington RG, Auerbuch V. Chemical inhibitors of the type three secretion system: disarming bacterial pathogens. Antimicrob Agents Chemother. 2012;56:5433–5441. doi: 10.1128/AAC.00975-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkar-Tyson M, Atkins HS. Antimicrobials for bacterial bioterrorism agents. Future Microbiol. 2011;6:667–676. doi: 10.2217/fmb.11.50. [DOI] [PubMed] [Google Scholar]
- Felise HB, Nguyen HV, Pfuetzner RA, Barry KC, Jackson SR, Blanc MP, Bronstein PA, Kline T, Miller SI. An inhibitor of gram-negative bacterial virulence protein secretion. Cell Host Microbe. 2008;4:325–336. doi: 10.1016/j.chom.2008.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf K, Betts HJ, Chellas-Gery B, Hower S, Linton CN, Fields KA. Treatment of Chlamydia trachomatis with a small molecule inhibitor of the Yersinia type III secretion system disrupts progression of the chlamydial developmental cycle. Mol Microbiol. 2006;61:1543–1555. doi: 10.1111/j.1365-2958.2006.05347.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enquist PA, Gylfe A, Hagglund U, Lindstrom P, Norberg-Scherman H, Sundin C, Elofsson M. Derivatives of 8-hydroxyquinoline – antibacterial agents that target intra-and extracellular Gram-negative pathogens. Bioorg Med Chem Lett. 2012;22:3550–3553. doi: 10.1016/j.bmcl.2012.03.096. [DOI] [PubMed] [Google Scholar]
- Ur-Rehman T, Slepenkin A, Chu H, Blomgren A, Dahlgren MK, Zetterstrom CE, Peterson EM, Elofsson M, Gylfe A. Pre-clinical pharmacokinetics and anti-chlamydial activity of salicylidene acylhydrazide inhibitors of bacterial type III secretion. J Antibiot (Tokyo) 2012;65:397–404. doi: 10.1038/ja.2012.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slepenkin A, Chu H, Elofsson M, Keyser P, Peterson EM. Protection of mice from a Chlamydia trachomatis vaginal infection using a salicylidene acylhydrazide, a potential microbicide. J Infect Dis. 2011;204:1313–1320. doi: 10.1093/infdis/jir552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt CK, Meysick KC, O'Brien AD. Bacterial toxins: friends or foes? Emerg Infect Dis. 1999;5:224–234. doi: 10.3201/eid0502.990206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hung DT, Shakhnovich EA, Pierson E, Mekalanos JJ. Small-molecule inhibitor of Vibrio cholerae virulence and intestinal colonization. Science. 2005;310:670–674. doi: 10.1126/science.1116739. [DOI] [PubMed] [Google Scholar]
- Ochsner UA, Bell SJ, O'Leary AL, Hoang T, Stone KC, Young CL, Critchley IA, Janjic N. Inhibitory effect of REP3123 on toxin and spore formation in Clostridium difficile, and in vivo efficacy in a hamster gastrointestinal infection model. J Antimicrob Chemother. 2009;63:964–971. doi: 10.1093/jac/dkp042. [DOI] [PubMed] [Google Scholar]
- Keller MA, Stiehm ER. Passive immunity in prevention and treatment of infectious diseases. Clin Microbiol Rev. 2000;13:602–614. doi: 10.1128/cmr.13.4.602-614.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnon SS, Schechter R, Maslanka SE, Jewell NP, Hatheway CL. Human botulism immune globulin for the treatment of infant botulism. N Engl J Med. 2006;354:462–471. doi: 10.1056/NEJMoa051926. [DOI] [PubMed] [Google Scholar]
- Bebbington C, Yarranton G. Antibodies for the treatment of bacterial infections: current experience and future prospects. Curr Opin Biotechnol. 2008;19:613–619. doi: 10.1016/j.copbio.2008.10.002. [DOI] [PubMed] [Google Scholar]
- Sheoran AS, Chapman-Bonofiglio S, Harvey BR, Mukherjee J, Georgiou G, Donohue-Rolfe A, Tzipori S. Human antibody against shiga toxin 2 administered to piglets after the onset of diarrhea due to Escherichia coli O157:H7 prevents fatal systemic complications. Infect Immun. 2005;73:4607–4613. doi: 10.1128/IAI.73.8.4607-4613.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babcock GJ, Broering TJ, Hernandez HJ, Mandell RB, Donahue K, Boatright N, Stack AM, Lowy I, Graziano R, Molrine D, Ambrosino DM, Thomas WD., Jr Human monoclonal antibodies directed against toxins A and B prevent Clostridium difficile-induced mortality in hamsters. Infect Immun. 2006;74:6339–6347. doi: 10.1128/IAI.00982-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell PK. Project BioShield: what it is, why it is needed, and its accomplishments so far. Clin Infect Dis. 2007;45(Suppl 1):S68–72. doi: 10.1086/518151. [DOI] [PubMed] [Google Scholar]
- Subramanian GM, Cronin PW, Poley G, Weinstein A, Stoughton SM, Zhong J, Ou Y, Zmuda JF, Osborn BL, Freimuth WW. A phase 1 study of PAmAb, a fully human monoclonal antibody against Bacillus anthracis protective antigen, in healthy volunteers. Clin Infect Dis. 2005;41:12–20. doi: 10.1086/430708. [DOI] [PubMed] [Google Scholar]
- Mohamed N, Clagett M, Li J, Jones S, Pincus S, D'Alia G, Nardone L, Babin M, Spitalny G, Casey L. A high-affinity monoclonal antibody to anthrax protective antigen passively protects rabbits before and after aerosolized Bacillus anthracis spore challenge. Infect Immun. 2005;73:795–802. doi: 10.1128/IAI.73.2.795-802.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riddle V, Leese P, Blanset D, Adamcio M, Meldorf M, Lowy I. Phase I study evaluating the safety and pharmacokinetics of MDX-1303, a fully human monoclonal antibody against Bacillus anthracis protective antigen, in healthy volunteers. Clin Vaccine Immunol. 2011;18:2136–2142. doi: 10.1128/CVI.05059-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitale L, Blanset D, Lowy I, O'Neill T, Goldstein J, Little SF, Andrews GP, Dorough G, Taylor RK, Keler T. Prophylaxis and therapy of inhalational anthrax by a novel monoclonal antibody to protective antigen that mimics vaccine-induced immunity. Infect Immun. 2006;74:5840–5847. doi: 10.1128/IAI.00712-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller MB, Bassler BL. Quorum sensing in bacteria. Annu Rev Microbiol. 2001;55:165–199. doi: 10.1146/annurev.micro.55.1.165. [DOI] [PubMed] [Google Scholar]
- Sintim HO, Smith JA, Wang J, Nakayama S, Yan L. Paradigm shift in discovering next-generation anti-infective agents: targeting quorum sensing, c-di-GMP signaling and biofilm formation in bacteria with small molecules. Future Med Chem. 2010;2:1005–1035. doi: 10.4155/fmc.10.185. [DOI] [PubMed] [Google Scholar]
- Mitchell RJ, Lee SK, Kim T, Ghim CM. Microbial linguistics: perspectives and applications of microbial cell-to-cell communication. BMB Rep. 2011;44:1–10. doi: 10.5483/BMBRep.2011.44.1.1. [DOI] [PubMed] [Google Scholar]
- Lazar V. Quorum sensing in biofilms – how to destroy the bacterial citadels or their cohesion/power? Anaerobe. 2011;17:280–285. doi: 10.1016/j.anaerobe.2011.03.023. [DOI] [PubMed] [Google Scholar]
- Stevens AM, Queneau Y, Soulere L, von Bodman S, Doutheau A. Mechanisms and synthetic modulators of AHL-dependent gene regulation. Chem Rev. 2011;111:4–27. doi: 10.1021/cr100064s. [DOI] [PubMed] [Google Scholar]
- Tay SB, Yew WS. Development of quorum-based anti-virulence therapeutics targeting gram-negative bacterial pathogens. Int J Mol Sci. 2013;14:16570–16599. doi: 10.3390/ijms140816570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaSarre B, Federle MJ. Exploiting quorum sensing to confuse bacterial pathogens. Microbiol Mol Biol Rev. 2013;77:73–111. doi: 10.1128/MMBR.00046-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, Kaufmann GF. Quo vadis quorum quenching? Curr Opin Pharmacol. 2013;13:688–698. doi: 10.1016/j.coph.2013.07.003. [DOI] [PubMed] [Google Scholar]
- Rasmussen TB, Givskov M. Quorum-sensing inhibitors as anti-pathogenic drugs. Int J Med Microbiol. 2006;296:149–161. doi: 10.1016/j.ijmm.2006.02.005. [DOI] [PubMed] [Google Scholar]
- Morkunas B, Galloway WR, Wright M, Ibbeson BM, Hodgkinson JT, O'Connell KM, Bartolucci N, Della Valle M, Welch M, Spring DR. Inhibition of the production of the Pseudomonas aeruginosa virulence factor pyocyanin in wild-type cells by quorum sensing autoinducer-mimics. Org Biomol Chem. 2012;10:8452–8464. doi: 10.1039/c2ob26501j. [DOI] [PubMed] [Google Scholar]
- Miyairi S, Tateda K, Fuse ET, Ueda C, Saito H, Takabatake T, Ishii Y, Horikawa M, Ishiguro M, Standiford TJ, Yamaguchi K. Immunization with 3-oxododecanoyl-L-homoserine lactone-protein conjugate protects mice from lethal Pseudomonas aeruginosa lung infection. J Med Microbiol. 2006;55:1381–1387. doi: 10.1099/jmm.0.46658-0. [DOI] [PubMed] [Google Scholar]
- Hirakawa H, Tomita H. Interference of bacterial cell-to-cell communication: a new concept of antimicrobial chemotherapy breaks antibiotic resistance. Front Microbiol. 2013;4:1–14. doi: 10.3389/fmicb.2013.00114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasko DA, Moreira CG, Li R, Reading NC, Ritchie JM, Waldor MK, Williams N, Taussig R, Wei S, Roth M, Hughes DT, Huntley JF, Fina MW, Falck JR, Sperandio V. Targeting QseC signaling and virulence for antibiotic development. Science. 2008;321:1078–1080. doi: 10.1126/science.1160354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero M, Acuna L, Otero A. Patents on quorum quenching: interfering with bacterial communication as a strategy to fight infections. Recent Pat Biotechnol. 2012;6:2–12. doi: 10.2174/187220812799789208. [DOI] [PubMed] [Google Scholar]
- Garcia-Contreras R, Martinez-Vazquez M, Velazquez Guadarrama N, Villegas Paneda AG, Hashimoto T, Maeda T, Quezada H, Wood TK. Resistance to the quorum-quenching compounds brominated furanone C-30 and 5-fluorouracil in Pseudomonas aeruginosa clinical isolates. Pathog Dis. 2013;68:8–11. doi: 10.1111/2049-632X.12039. [DOI] [PubMed] [Google Scholar]
- Hentzer M, Wu H, Andersen JB, Riedel K, Rasmussen TB, Bagge N, Kumar N, Schembri MA, Song Z, Kristoffersen P, Manefield M, Costerton JW, Molin S, Eberl L, Steinberg P, Kjelleberg S, Hoiby N, Givskov M. Attenuation of Pseudomonas aeruginosa virulence by quorum sensing inhibitors. EMBO J. 2003;22:3803–3815. doi: 10.1093/emboj/cdg366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda T, Garcia-Contreras R, Pu M, Sheng L, Garcia LR, Tomas M, Wood TK. Quorum quenching quandary: resistance to antivirulence compounds. ISME J. 2012;6:493–501. doi: 10.1038/ismej.2011.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardwell JC, Lee JO, Jander G, Martin N, Belin D, Beckwith J. A pathway for disulfide bond formation in vivo. Proc Natl Acad Sci U S A. 1993;90:1038–1042. doi: 10.1073/pnas.90.3.1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardwell JC, McGovern K, Beckwith J. Identification of a protein required for disulfide bond formation in vivo. Cell. 1991;67:581–589. doi: 10.1016/0092-8674(91)90532-4. [DOI] [PubMed] [Google Scholar]
- Inaba K, Murakami S, Suzuki M, Nakagawa A, Yamashita E, Okada K, Ito K. Crystal structure of the DsbB-DsbA complex reveals a mechanism of disulfide bond generation. Cell. 2006;127:789–801. doi: 10.1016/j.cell.2006.10.034. [DOI] [PubMed] [Google Scholar]
- Bader M, Muse W, Ballou DP, Gassner C, Bardwell JC. Oxidative protein folding is driven by the electron transport system. Cell. 1999;98:217–227. doi: 10.1016/s0092-8674(00)81016-8. [DOI] [PubMed] [Google Scholar]
- Heras B, Shouldice SR, Totsika M, Scanlon MJ, Schembri MA, Martin JL. DSB proteins and bacterial pathogenicity. Nat Rev Microbiol. 2009;7:215–225. doi: 10.1038/nrmicro2087. [DOI] [PubMed] [Google Scholar]
- Ireland PM, McMahon RM, Marshall LE, Halili M, Furlong E, Tay S, Martin J, Sarkar-Tyson M. Disarming Burkholderia pseudomallei: structural and functional characterisation of a disulfide oxidoreductase (DsbA) required for virulence in vivo. Antioxid Redox Signal. 2014;20:606–617. doi: 10.1089/ars.2013.5375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Totsika M, Heras B, Wurpel DJ, Schembri MA. Characterization of two homologous disulfide bond systems involved in virulence factor biogenesis in uropathogenic Escherichia coli CFT073. J Bacteriol. 2009;191:3901–3908. doi: 10.1128/JB.00143-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miki T, Okada N, Danbara H. Two periplasmic disulfide oxidoreductases, DsbA and SrgA, target outer membrane protein SpiA, a component of the Salmonella pathogenicity island 2 type III secretion system. J Biol Chem. 2004;279:34631–34642. doi: 10.1074/jbc.M402760200. [DOI] [PubMed] [Google Scholar]
- Jacob-Dubuisson F, Pinkner J, Xu Z, Striker R, Padmanhaban A, Hultgren SJ. PapD chaperone function in pilus biogenesis depends on oxidant and chaperone-like activities of DsbA. Proc Natl Acad Sci USA. 1994;91:11552–11556. doi: 10.1073/pnas.91.24.11552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang HZ, Donnenberg MS. DsbA is required for stability of the type IV pilin of enteropathogenic Escherichia coli. Mol Microbiol. 1996;21:787–797. doi: 10.1046/j.1365-2958.1996.431403.x. [DOI] [PubMed] [Google Scholar]
- Stenson TH, Weiss AA. DsbA and DsbC are required for secretion of pertussis toxin by Bordetella pertussis. Infect Immun. 2002;70:2297–2303. doi: 10.1128/IAI.70.5.2297-2303.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peek JA, Taylor RK. Characterization of a periplasmic thiol: disulfide interchange protein required for the functional maturation of secreted virulence factors of Vibrio cholerae. Proc Natl Acad Sci USA. 1992;89:6210–6214. doi: 10.1073/pnas.89.13.6210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha UH, Wang Y, DsbA JS. of Pseudomonas aeruginosa is essential for multiple virulence factors. Infect Immun. 2003;71:1590–1595. doi: 10.1128/IAI.71.3.1590-1595.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomb JF. A periplasmic protein disulfide oxidoreductase is required for transformation of Haemophilus influenzae Rd. Proc Natl Acad Sci USA. 1992;89:10252–10256. doi: 10.1073/pnas.89.21.10252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watarai M, Tobe T, Yoshikawa M, Sasakawa C. Disulfide oxidoreductase activity of Shigella flexneri is required for release of Ipa proteins and invasion of epithelial cells. Proc Natl Acad Sci USA. 1995;92:4927–4931. doi: 10.1073/pnas.92.11.4927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tinsley CR, Voulhoux R, Beretti JL, Tommassen J, Nassif X. Three homologues, including two membrane-bound proteins, of the disulfide oxidoreductase DsbA in Neisseria meningitidis: effects on bacterial growth and biogenesis of functional type IV pili. J Biol Chem. 2004;279:27078–27087. doi: 10.1074/jbc.M313404200. [DOI] [PubMed] [Google Scholar]
- Dailey FE, Berg HC. Mutants in disulfide bond formation that disrupt flagellar assembly in Escherichia coli. Proc Natl Acad Sci USA. 1993;90:1043–1047. doi: 10.1073/pnas.90.3.1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin A, Scott DW, Mann BJ. Francisella tularensis subsp. tularensis Schu S4 disulfide bond formation protein B, but not an RND-type efflux pump, is required for virulence. Infect Immun. 2008;76:3086–3092. doi: 10.1128/IAI.00363-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jubb H, Higueruelo AP, Winter A, Blundell TL. Structural biology and drug discovery for protein-protein interactions. Trends Pharmacol Sci. 2012;33:241–248. doi: 10.1016/j.tips.2012.03.006. [DOI] [PubMed] [Google Scholar]
- Fruh V, Zhou Y, Chen D, Loch C, Ab E, Grinkova YN, Verheij H, Sligar SG, Bushweller JH, Siegal G. Application of fragment-based drug discovery to membrane proteins: identification of ligands of the integral membrane enzyme DsbB. Chem Biol. 2010;17:881–891. doi: 10.1016/j.chembiol.2010.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]