

Abstract
Aims
This study aimed at describing adalimumab pharmacokinetics (PK) and the concentration–effect relationship of adalimumab using pharmacokinetic–pharmacodynamic (PK–PD) modelling in patients with rheumatoid arthritis (RA).
Methods
Adalimumab PK and PK–PD data were obtained from a multicentric observational study. Adalimumab (40 mg) was administered subcutaneously every other week, and its pharmacokinetics was described using a one-compartment model. The relationship between adalimumab concentration and C-reactive protein (CRP) concentration was described using an indirect response model with inhibition of CRP input, whereas the relationship between adalimumab concentration and disease activity score in 28 joints (DAS28) was described using a direct inhibition model. Dose regimens that included a loading dose of adalimumab were simulated.
Results
Thirty patients treated for RA were analysed. The following pharmacokinetic and PK–PD parameters were estimated (interidividual coefficient of variation): apparent volume of distribution (Vd/F) = 10.8 l (92%); apparent clearance (CL/F) = 0.32 l day−1 (17%); first-order absorption rate (ka) = 0.28 day−1; CRP input (kin) = 22.0 mg l−1 day−1 (65%); adalimumab concentration leading to a 50% decrease in kin (C50) = 3.6 mg l−1 (88%); baseline DAS28 (DAS0) = 5.5 mg l−1 (11%); and adalimumab concentration leading to 50% decrease of DAS0 (IC50) = 11.0 mg l−1 (71%). Simulations showed that a 160 mg loading dose should reduce the time to reach efficacy in terms of both CRP and DAS28 after the first injection.
Conclusions
This is the first study to describe adalimumab pharmacokinetics and the concentration–effect relationship in RA. A 160 mg loading dose may lead to an increased benefit from treatment in RA patients.
Keywords: adalimumab, pharmacokinetic–pharmacodynamic modelling, pharmacodynamics, pharmacokinetics, rheumatoid arthritis
What is Already Known about this Subject
The pharmacokinetics of adalimumab administered intravenously to rheumatoid arthritis (RA) patients has been described.
The response to adalimumab increases with its serum concentration.
A 160 mg loading dose leads to less frequent primary nonresponse and longer sustained clinical benefit in Crohn's disease patients.
What this Study Adds
This study is the first to describe adalimumab pharmacokinetics and the concentration–effect relationship in RA patients following subcutaneous administration.
Both adalimumab pharmacokinetics and the concentration–effect relationship vary widely between patients.
Simulations predict that a loading dose may lead to an increased benefit of adalimumab in RA patients.
Introduction
Adalimumab is a human monoclonal IgG1 antibody targeting tumour necrosis factor α (TNF-α). It belongs to the class of anti-TNF-α biopharmaceuticals, which has profoundly modified the treatment of several inflammatory diseases. Adalimumab is currently approved for rheumatoid arthritis (RA), ankylosing spondylitis, psoriatic arthritis, Crohn's disease, ulcerative colitis and psoriasis 1.
Approved doses of adalimumab lead to highly variable serum concentrations between patients 2–5. For therapeutic antibodies administered intravenously, such as infliximab, interindividual variability of serum concentrations is due to variability in antibody distribution and elimination 6–10. Subcutaneous administration of adalimumab constitutes an additional source of variability in concentrations. Adalimumab pharmacokinetics has been investigated using compartment modelling in only one study, in which adalimumab was administered intravenously. In that study, adalimumab steady-state volume of distribution, clearance and elimination half-life were 5.6 l, 0.22 l day−1 and 21 days, respectively 11. No study has reported pharmacokinetic parameters after subcutaneous infusions using modelling. However, these parameters were estimated using noncompartmental methods; the apparent volume of distribution, clearance, elimination half-life, time to maximal adalimumab concentration (tmax) and bioavailability (F) were ∼12 l, 0.48 l day−1, 17.3 days, 4–7 days and 60%, respectively 12. To date, no study has reported adalimumab pharmacokinetics following subcutaneous injections using compartmental modelling. Notably, the absorption kinetics of adalimumab was never reported.
The probability of clinical response to adalimumab was reported to increase with its trough serum concentrations 2–5,13,14. However, to our knowledge, an adalimumab concentration–effect relationship has never been described using pharmacokinetic–pharmacodynamic (PK–PD) modelling. A sound description of both adalimumab pharmacokinetics (after subcutaneous administration) and the PK–PD relationship is, however, a prerequisite to the development of therapeutic drug monitoring of this anti-TNF-α biopharmaceutical.
The approved dosing regimen of adalimumab in RA patients is 40 mg every other week, whereas a 160 mg loading dose is recommended in Crohn's disease patients 1. Karmiris et al. observed that Crohn's disease patients treated with a high loading dose had not only significantly higher trough concentrations 4 weeks after initiating adalimumab treatment, but also less frequent primary nonresponse and longer sustained clinical benefit than the other patients 13. The decrease in risk of primary nonresponse in patients treated with a higher loading dose may be explained by the fact that these patients reached adalimumab steady-state concentrations more rapidly than other patients. Given that the elimination half-life (t1/2) of adalimumab is long (∼20 days 11), the steady state should be attained 4 months after initiation of regular injections 11. As for Crohn's disease patients, RA patients may therefore benefit from a loading dose of adalimumab.
Our objectives were to analyse the dose–concentration–effect relationship of adalimumab in RA patients and to simulate the consequences of the use of a loading dose.
Methods
Patients
This study is a post hoc analysis of a prospective, observational, open, multicentric (Amiens, Caen, Lille, Rouen and Berck, France) 52 week study 15. The primary objective was to determine predictive factors of the response to TNF-α blockers. This study was approved by the regional ethics committee (CPP Nord-Ouest 1, France) and was registered at ClinicalTrials.gov under the number NCT00234234. All participants gave written informed consent at the time of enrolment. Thirty patients with active RA were eligible for this post hoc analysis. These patients received 40 mg adalimumab subcutaneously every other week combined with methotrexate, and follow-up was done for 1 year. Patients were assessed at baseline and at weeks 6, 12, 24 and 52. At each visit, patients were evaluated for disease activity score in 28 joints (DAS28), and blood samples were collected.
Data
Adalimumab concentrations
Adalimumab concentrations were measured in the Pilot Centre for Therapeutic Antibody Monitoring (CePiBAc) of Tours University Hospital, France. Adalimumab concentrations were measured using a validated enzyme-linked immunosorbent assay adapted from the one developed for infliximab 8. Briefly, recombinant human TNF-α was coated on the solid phase, to recognize adalimumab present in the sera. A therapeutic monoclonal antibody was detected by an anti-human immunoglobulin G Fcγ-specific antibody conjugated to horseradish peroxidase. The limit of detection of the assay was 0.04 mg l−1, and the lower and upper limits of quantification were 0.1 and 4.9 mg l−1, respectively. Sera exceeding the upper limit of quantification were diluted 1:10. The intraday precision estimates of the enzyme-linked immunosorbent assay were 4.2, 7.5 and 6.8% for the 0.1, 2.0 and 4.9 mg l−1 quality controls, respectively. The corresponding biases were −10.8, −9.1 and −2.7%, respectively. The interday precision estimates were −10.2, 5.6 and 10.0%, respectively. Corresponding bias were −12.9, 1.3 and 2.1%, respectively.
Other laboratory analyses
At each visit, erythrocyte sedimentation rate (ESR) and C-reactive protein (CRP) concentrations were measured locally in the laboratory of the recruiting centres. Antibodies to adalimumab (ATA) were detected in the CePiBAc of Tours University Hospital, France, using double-antigen enzyme-linked immunosorbent TNF-α adalimumab-coated plates and their detection by peroxidase-conjugated IgG. Owing to the interference of circulating adalimumab, ATA could not be measured if the adalimumab concentration was >2 mg l−1. Above this value, the ATA test may lead to false-negative results. The cut-off value (for false-positive ATA), determined using untreated samples of patients with an autoimmune disease, was 0.128 mg l−1 (obtained from the 99th percentile). However, this applies to the first sample in each patient for which the interpretation cannot be based on adalimumab concentration.
Clinical end-points
At each visit, treatment efficacy was assessed by a trained rheumatologist through the measurement of the disease activity score score in 28 joints (DAS28) 16. Briefly, DAS28 is an index that measures the disease activity in patients with RA. This score includes ESR (in millimetres per hour), the tender joint count (TJC), the swollen joint count (SJC) and the visual analog scale general health patient (VAS, in millimetres). The DAS28 is calculated as follows: 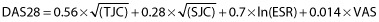
Pharmacokinetic and pharmacokinetic–pharmacodynamic analysis
Software
Pharmacokinetic and CRP data were analysed with a population approach using the nonlinear mixed-effects modelling software MONOLIX 4.2.2, which combines the stochastic expectation-maximization (SAEM) algorithm and a Markov chain Monte-Carlo procedure for likelihood maximization. To ensure the best possible convergence, a large number of iterations (700 for K1 and 300 for K2) were performed, with K1 and K2 being ‘iteration kernels’ referring to the SAEM procedure of Monolix. During K1, the sequence of step sizes is constant, which allows exploration of the parameter space. During K2, the step sizes decrease to ensure convergence. Two Markov chains were used, and simulated annealing was applied to improve the convergence of the SAEM algorithm towards the global maximum of the likelihood. The Fisher information matrix and likelihood were computed using stochastic approximation and importance sampling, respectively. These techniques are longer to compute than linearization, but provide more reliable results. The random seed was changed between each run. All pharmacokinetic and PK–PD models were run simultaneously.
Structural models
Pharmacokinetic model
Adalimumab concentrations were described using compartmental pharmacokinetic modelling. One and two mammillary models with first-order absorption, distribution and elimination constants were tested. Estimated pharmacokinetic parameters were apparent volumes of distribution and clearances. These values were apparent because adalimumab administration is extravascular (subcutaneous). Structural models were compared using Akaike's information criterion (AIC), defined as follows: AIC = −2LL + 2p, where −2LL is the −2 × ln-likelihood and p is the number of model parameters to estimate. The model with the lowest AIC was selected.
Pharmacokinetic–pharmacodynamic models
C-Reactive protein and DAS28 are considered to be the key relevant variables for evaluation. Given that normalization of CRP is associated with sustained clinical response, CRP may be considered as a clinically relevant biomarker 13,17–20. In addition, CRP is increased by TNF-α 21. Usage of adalimumab leads to a decrease in CRP production. Therefore, the relationship between adalimumab and CRP was described using an indirect response model with inhibition of CRP input (Figure 1), as in previous studies 17,22,23. Using this model, the estimated parameters were as follows: zero-order CRP input (kin; in milligrams per litre per day); first-order CRP output (kout; per day); and adalimumab concentration leading to 50% of maximal kin inhibition (C50; in milligrams per litre). The parameter kout, which was poorly identifiable, had to be fixed. Given that the CRP elimination half-life (t1/2CRP) was reported to be 19 h 24, we fixed kout as ln(2)/t1/2CRP = 0.875 day−1.
Figure 1.
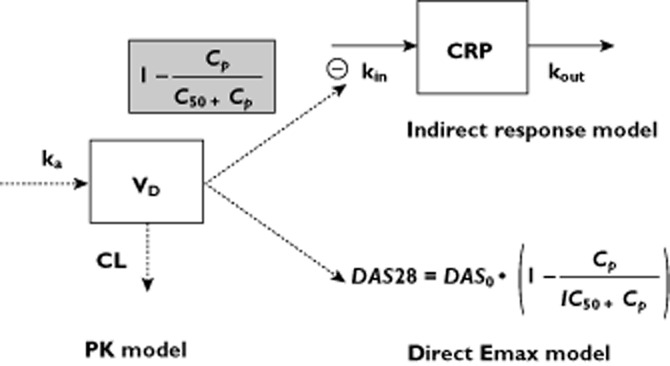
Pharmacokinetic and pharmacokinetic–pharmacodynamic (PK–PD) models. Adalimumab pharmacokinetics was described using a one-compartment model with first-order absorption and elimination rates. The relationship between adalimumab concentrations and C-reactive protein (CRP) levels was described using an indirect model with inhibition of CRP input. The relationship between adalimumab concentrations and the disease activity score in 28 joints (DAS28) was described using a direct Emax inhibitory model. Abbreviations: CL, clearance; Cp, model-predicted adalimumab concentrations; CRP, serum C-reactive protein concentrations; kin, zero-order production rate constant; ka, first-order absorption rate constant; kout, first-order elimination rate constant; C50, adalimumab concentration leading to a 50% decrease of kin; DAS0, DAS28 at baseline, IC50, adalimumab concentration leading to a 50% decrease of DAS0; VD, volume of distribution
The relationship between adalimumab concentrations and DAS28 was analysed using a direct inhibitory Emax model (Figure 1). We used direct rather than indirect PK–PD modelling, as done in other studies, because DAS28 is a continuous measurement 25. Estimated parameters were the value of DAS28 at baseline (DAS280) and adalimumab concentration leading to a 50% decrease in DAS280 (EC50; in milligrams per litre).
Interindividual and error models
The interindividual variability in pharmacokinetic and PK–PD parameters was described using an exponential model, as follows: θi = θTV × exp(ηi), where θi is the estimated individual parameter, θTV is the typical value of the parameter and ηi is the random effect for the ith patient. The values of ηi were assumed to be normally distributed, with a mean of zero and variance ω2. This variance was fixed to zero for parameters for which ηi could not be estimated. Correlations between random effects were tested. Additive, proportional and mixed additive–proportional residual error models were tested. For example, the combined additive–proportional model was implemented as follows: YO,ij = YP,ij × (1 + εprop,ij) + εadd,ij, where YO,ij and YP,ij are observed and predicted jth measurements for the ith patient, respectively, and εadd,ij and εprop,ij are additive and proportional errors, with a mean of zero and respective variances σadd2 and σprop2.
Covariates
Owing to the relatively small number of patients, only three covariates were tested. Binary covariates were sex and corticosteroid cotreatment. The continuous covariate was bodyweight. The influence of binary covariate on θTV was implemented as ln(θTV) = ln(θCAT=0) + βCAT=1, where θCAT=0 is the value of θ for the reference category and βCAT=1 is a parameter which provides the value of θTV for the other category. Reference categories were women and no corticosteroid treatment for sex and corticosteroid cotreatment, respectively. The continuous covariate (COV) was centred on its median, as follows: θi = θ0 × (COV/med(COV))βcov, where θ0 is the value of θ for the median value of COV [med(COV)] and βCOV quantifies the influence of COV on θ.
Model comparison and covariate selection
Interindividual, residual and covariate models were compared using −2LL and AIC. Of two models, that with the lowest significant −2LL value, assessed by a likelihood ratio χ2 test (LRT), and the lowest AIC, was selected. First, the individual influence of each covariate on each pharmacokinetic and PK–PD parameter was tested using the likelihood ratio test with α = 0.1. When covariates were redundant, the most significant were kept in the final model. Given that the number of selected covariates at the first step was low, no stepwise forward/backward covariate selection was needed; the combination of covariates which influenced parameters was tested to build the final model. The covariates were kept in the final model if their influence was significant for α = 0.02. The goodness of covariate description was assessed by visual inspection of the plot of random effects (i.e. η) vs. covariate plots.
Model goodness of fit and evaluation
In general, the goodness of fit for a given model was assessed by plots of population-predicted (PRED) and individual-predicted (IPRED) measurements vs. observed measurements, IPRED and observed concentrations (DV) vs. time, and by evaluating the residuals via graphical inspection of population (PWRES) and individual (IWRES) weighted residual distributions and normalized prediction distribution errors (NPDE) 26. Given that NPDE should be normally distributed, their distribution was tested using the Kolmogorov–Smirnov test at the level of α = 0.05. We concluded that there was a departure from normality if the P value was <0.05.
Simulations
Three dosing regimens were simulated: (i) no change in dosing scheme (40 mg every other week); (ii) a single 80 mg loading dose followed by 40 mg every other week; and (iii) a single 160 mg loading dose followed by 40 mg every other week. The dosing regimen that led to the earliest time to achieve a steady state for adalimumab concentrations, CRP and DAS28 values was considered to be the best. Simulations were made using typical pharmacokinetic and PK–PD parameters that were estimated in the modelling step. Neither covariates nor interindividual distributions of pharmacokinetic or PK–PD parameters were taken into account.
Results
Patients
Of the 30 patients analysed in this study, 23 were women (77%), 17 were cotreated with corticosteroids, and their median bodyweight was 67 kg (Table 1). The presence of ATA was tested in 10 samples from four patients, in whom adalimumab concentrations were <2 mg l−1, but ATA were detected in none of them.
Table 1.
Patient characteristics at baseline
| Patients (n = 30) | |
|---|---|
| Sex, women [n (%)] | 23 (77) |
| Age (years) | 55 [24–77] |
| Bodyweight (kg) | 67 [45–115] |
| Comedication | |
| Methotrexate [n (%)] | 30 (100) |
| Corticosteroids [n (%)] | 17 (56.7) |
| NSAIDs [n (%)] | 14 (46.7) |
| ATA+ | 0 |
| CRP (mg l−1) | 22 [5–139] |
| DAS28 | 5.6 [3.7–7.8] |
Results are presented as the absolute number (%) or as the median [range]. Abbreviations are as follows: ATA, antibodies toward adalimumab; CRP, C-reactive protein; DAS28, disease activity score in 28 joints; NSAIDs, nonsteroidal anti-inflammatory drugs.
Pharmacokinetic and pharmacokinetic–pharmacodynamic analysis
A total of 129 serum adalimumab concentration, 139 CRP and 141 DAS28 measurements were available for analysis.
Adalimumab concentrations were best described using a one-compartment model with first-order absorption rate (Figure 1). Parameters describing a peripheral compartment were not identifiable. The best residual error model was proportional. For indirect response and direct Emax models describing concentration–CRP and concentration–DAS28 relationships, respectively, best residual error models were mixed additive–proportional and additive, respectively. Plots of predicted vs. observed measurements (i.e. adalimumab and CRP or DAS28) showed that pharmacokinetic and PK–PD models described the data satisfactorily (Figure 2). Pharmacokinetic and PK–PD parameters were estimated with satisfactory accuracy (Table 2). All diagnostic plots were obtained from the final model. Population (PWRES) and individual residuals (IWRES) and normalized prediction distribution error (NPDE) plots showed that there was no obvious model misspecification (Figure 3). Notably, no NPDE distribution was significantly different from Gaussian distributions, with the Kolmogorov–Smirnov statistic being 0.0465 (P = 0.13), 0.0747 (P = 0.059) and 0.0710 (P = 0.078) for adalimumab concentrations, CRP and DAS28 values, respectively. Although CRP showed extreme values that could not be described by an indirect PK–PD model, graphical analysis of residuals showed no obvious departure from normality (Figure 3).
Figure 2.

Observed values of adalimumab concentrations, CRP concentrations and DAS28 measurements vs. population model-predicted values (PRED) and individual predicted values (IPRED). DV, dependent value
Table 2.
Parameter estimates
| Parameter (units) | Estimate | RSE (%) |
|---|---|---|
| V/F (l) | 10.8 | 20 |
| CL/F (l day−1) | 0.32 | 5 |
| SX_CL | 0.32 | 34 |
| WT_CL | 0.81 | 28 |
| ka (day−1) | 0.28 | 4 |
| kin (mg l−1 day−1) | 22 | 15 |
| kout (day−1) | 0.875 | (Fixed) |
| C50 (mg l−1) | 3.6 | 26 |
| DAS280 | 5.5 | 3 |
| IC50 (mg l−1) | 11.0 | 16 |
| ωVd (%) | 92 | 16 |
| ωCL (%) | 17 | 27 |
| ωKin (%) | 65 | 18 |
| ωKout (%) | – | – |
| ωC50 (%) | 88 | 27 |
| ωImax (%) | 11 | 32 |
| ωIC50 (%) | 71 | 17 |
| σprop_PK (%) | 24 | 9 |
| σadd_CRP (mg l−1) | 1.6 | 29 |
| σprop_CRP (%) | 52 | 10 |
| σadd_DAS | 68 | 8 |
The value of kout was fixed, and no interindividual variability was estimated for this parameter.
The RSE (%) was obtained as follows:RSE = (estimate/standard error) × 100. Abbreviations are as follows: C50, adalimumab concentration leading to a 50% decrease of kin; CL/F, apparent clearance; CRP, C-reactive protein; DAS280, estimated baseline disease activity score in 28 joints; IC50, adalimumab concentration leading to a decrease in DAS280 of 50%; ka, first-order absorption rate constant; kin, zero-order CRP input; kout, first-order CRP output; PK, pharmacokinetics; RSE, relative standard error; SX, sex; Vd/F, apparent volume of distribution; WT, bodyweight; σadd, additive error standard deviation; σprop, proportional error standard deviation; ω, interindividual standard deviation.
Figure 3.

Distribution of population (WRES) and individual weighted residuals (IWRES) vs. individual predictions, and of NPDE for adalimumab concentrations, CRP concentrations and DAS28 measurements
A large interindividual variability in pharmacokinetics and PK–PD parameters was observed. This was notably the case for estimated apparent volume of distribution (V/F), C50 and IC50, for which interindividual standard deviations were 92, 88 and 71%, respectively. During the univariate step, CL/F was found to be influenced by both sex and bodyweight, and a tendency was observed for a relationship between kin and bodyweight. In the final model, the only affected parameter was CL/F, which increased with weight (LRT = 13.1, P < 0.01) and was higher in men than in women (LRT = 8.7, P < 0.01). The interindividual standard deviation of CL/F, corresponding to unexplained variability, was 17%. Of note, without covariates, this standard deviation was 35%. (Figure 4). The elimination half-life (t1/2) for median-weighted women and men was 24.1 and 17.4 days, respectively. The time to maximal adalimumab concentration (tmax) derived from typical values was 9.1 days after injection. Adalimumab steady-state concentrations and maximal response (in terms of CRP and DAS28) were reached around 20 weeks after the beginning of adalimumab treatment (Figure 5).
Figure 4.
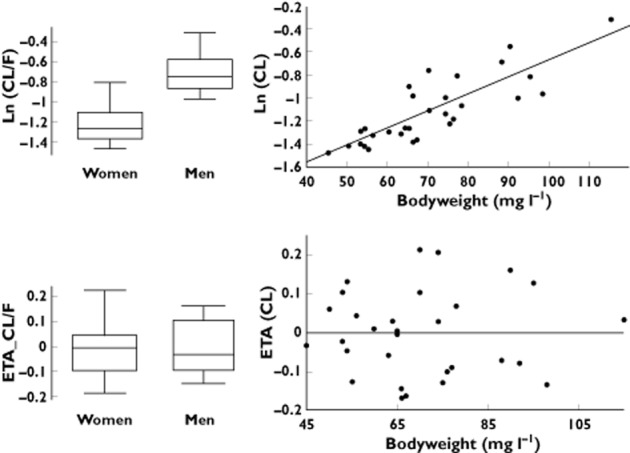
Apparent clearance (CL/F) vs. sex and bodyweight (above) and random effect for apparent clearance (ETA_CL/F) vs. sex and bodyweight (below)
Figure 5.

Observed (filled circles) and model-predicted adalimumab concentrations (continuous lines), observed (open diamonds) and model-predicted CRP concentrations (dotted lines), observed (open squares) and model predicted DAS28 measurements (dashed lines) vs. time in four representative patients
Simulations
Our simulation showed that, for a typical patient, a single 80 mg loading dose leads to increased adalimumab concentrations but has only a limited effect on the time to reach the steady state. In contrast, a dosing regimen including a single 160 mg loading dose leads to concentrations above steady-state values between the first and the fifth injection and allows the maximal response in terms of CRP and DAS28 to be reached before the second injection (Figure 6).
Figure 6.
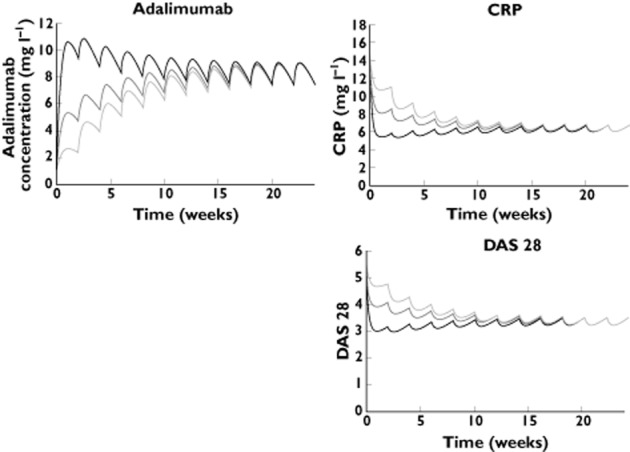
Simulations of three adalimumab dosing regimens using typical pharmacokinetic and PK–PD parameters: no loading dose (40 mg every other week); 80 mg loading dose (80 mg, then 40 mg every other week); and 160 mg loading dose (160 mg, then 40 mg every other week). Using a 160 mg loading dose, adalimumab concentrations after the loading dose are superior to steady-state concentrations, and maximal effect is reached between the first and the second injections. —, no loading dose; —, 80 mg loading dose; —, 160 mg loading dose
Discussion
We used pharmacokinetic and PK–PD modelling to investigate adalimumab dose–concentration–effect relationship in RA patients administered with adalimumab via the subcutaneous route. To our knowledge, adalimumab pharmacokinetics has been analysed using compartmental modelling in only one study, in which adalimumab was administered intravenously to RA patients 11. Therefore, adalimumab pharmacokinetics has not described in the case of administration via the subcutaneous route and, notably, the absorption kinetics of adalimumab following administration via the subcutaneous route has never been reported. In addition, the adalimumab concentration–effect relationship has never described using PK–PD modelling.
A total of 30 patients were analysed in the present study. Adalimumab pharmacokinetics was best described using a one-compartment model with first-order elimination rate, whereas Weisman et al. used a two-compartment model 11. The pharmacokinetics of therapeutic antibodies administered intravenously is often described using a two-compartment model. However, when the subcutaneous route is used, e.g. for omalizumab, an anti-IgE monoclonal antibody, one-compartment models are appropriate 27,28. An exception was efalizumab, an anti-CD11a antibody previously used in psoriatic patients. Its pharmacokinetics following subcutaneous administration was described using a two-compartment model by Ng et al. 29, but the authors used a population approach based on data from both subcutaneous and intravenous administrations.
We described adalimumab absorption kinetics using a first-order absorption rate (ka), as previously done for omalizumab 27,28. We estimated a mean ka of 0.28 day−1. This value is lower than that reported for omalizumab (∼0.45 day−1 [27, 28]) but similar to that reported for efalizumab (∼0.25 day−1 29). The calculated time to reach adalimumab maximal concentration after injection (tmax) is therefore 9.1 days, a value greater than that reported in the adalimumab approval document (5.5 ± 2.3 days) 12. The estimated apparent volume of distribution (V/F) is 12.4 l. Given that adalimumab bioavailability (F) is 64% 12, the adalimumab volume of distribution (V) in our study would be 6.9 l, a value which is slightly higher than the value reported in the adalimumab approval document (4.7–6.0 l) 12. We observed an increase in apparent adalimumab clearance with bodyweight and a higher value in men than in women. The influence of patient characteristics on pharmacokinetic parameters has not previously been reported for adalimumab, but has been reported for other monoclonal antibodies, such as infliximab 7 or rituximab 30. The value of clearance estimated in the present study, corrected according to bioavailability, is 0.20 l day−1, a value similar to that reported by Weisman et al. (0.22 l day−1 11). The interindividual variance of ka was not identifiable, probably due to the scarceness of sampling times. This variability may be large; for other antibodies administered subcutaneously, the value of ωka was 50–150% 27–29. In our study, ωV/F was surprisingly large (92%), and greater than what is commonly found for other monoclonal antibodies (∼30–40% 31). The non-estimation of ωka might have resulted in an overestimation of ωV/F. Prospective studies with intensive blood sampling would be needed in order to analyse the interindividual variability in adalimumab absorption kinetics more precisely.
No antibodies toward adalimumab were detected in the present study. However, three patients may have developed ATA; patients #19 and #23 had adalimumab concentrations around 2 mg l−1, and patient #30 had concentrations decreasing from 3.5 to 1.3 mg l−1 between week 6 and week 52 despite the continuation at the standard dose. For these patients, estimated values of CL/F were ∼0.70 l day−1, a value twice that of the typical value (0.31 l day−1). The lack of detection of ATA in these patients may be explained either by high concentrations of free adalimumab molecules, which are known to interfere with ATA detection, or by the fact that ATA molecules were engaged into adalimumab–ATA complexes, which may be cleared faster than free adalimumab or ATA and may not be detected by our ATA test.
We report, for the first time, the adalimumab concentration–effect relationship using PK–PD modelling. The effect of adalimumab was assessed by both CRP and DAS28. For this description, we first used CRP as a biomarker. Indeed, CRP has been shown repeatedly to correlate well with sustained clinical response 8,17,18,20, and the relationship between infliximab concentration and CRP was previously investigated in RA 32,33. Being an anti-TNF-α monoclonal antibody, adalimumab acts as a noncompetitive antagonist of TNF-α and does not act directly on CRP. This justifies the use of an indirect PK–PD model with inhibition of CRP production. This model was used by others to describe the relationship between concentrations of therapeutic antibodies and CRP concentrations 17,22. However, despite its relevance, this biomarker displayed unexpected fluctuations, probably because of inflammatory phenomena independent from RA activity (e.g. infections) and/or polymorphisms on the CRP gene that are responsible for an interindividual variability in CRP production 34. In addition, kout had to be fixed according to the value of CRP half-life reported in the literature, i.e. 19 h. This value is considered to be constant in all conditions of health and disease 24.
We described the relationship between adalimumab concentration and clinical efficacy assessed by DAS28 using a direct Emax model. Such a model was preferred to an indirect response model, which is not adapted to the description of a disease activity score because it requires the estimation a DAS28 ‘input’ and a DAS28 ‘output’, parameters that would be difficult to interpret. We previously described the relationship between adalimumab concentration and DAS28 using a direct inhibition Emax model but without pharmacokinetic modelling 35.
The typical value of C50 was 3.6 mg l−1, meaning that the adalimumab concentration necessary to decrease CRP production (and therefore CRP concentration) by 50% is 3.6 mg l−1 for a ‘typical’ patient (Table 2). A similar interpretation may be drawn from IC50; the adalimumab concentration leading to a decrease of initial disease activity (DAS280) by 50% is 11.0 mg l−1 for a ‘typical’ patient. The large interindividual variability of both C50 (ωC50 = 88%) and IC50 (ωIC50 = 71%) suggests that RA patients may have a highly variable sensitivity to adalimumab.
The current dose regimen for adalimumab in RA patients (40 mg every other week) results in a long delay in reaching steady-state concentrations and maximal effect (∼10–20 weeks). Simulations showed that using a loading dose may decrease this delay; maximal effect would be reached between the first and the second injection (Figure 6). The use of a loading dose of adalimumab has already been investigated in Crohn's disease, notably in the CLASSIC-I trial (299 patients) 4 and in the cohort (168 patients) of Karmiris et al. 13. These studies showed a better efficacy of adalimumab when a 160 mg loading dose is used, which may be due to higher concentrations at the beginning of the treatment 4,13. In addition, the frequency of occurrence of adverse side effects was similar with or without a loading dose 4. However, adverse side effects may be different in Crohn's disease patients and in RA patients. Therefore, a future study showing the benefit of a 160 mg loading dose in RA patients will have to assess the safety and tolerance of this loading dose.
Overall, this is the first study to provide a description of adalimumab pharmacokinetics and a concentration–effect relationship in RA patients. Notably, it provides the first estimation of adalimumab absorption kinetics in RA patients. Our model shows that a loading dose in RA patients may allow more rapid attainment of benefit from treatment in RA patients.
Competing Interests
All authors have completed the Unified Competing Interest form at http://www.icmje.org/coi_disclosure.pdf (available on request from the corresponding author) and declare:
DT, CV and HW declares no support from any organization for the submitted work; no financial relationships with any organizations that might have an interest in the submitted work in the previous 3 years; no other relationships or activities that could appear to have influenced the submitted work.
ED declare no support from any organization for the submitted work; no financial relationships with any organisations that might have an interest in the submitted work in the previous 3 years; was invited to attend international congresses by Roche and UCB; she has acted as a consultant and given lectures on behalf of her institution for BMS and Abbvie.
PF declares no support from any organization for the submitted work; no financial relationships with any organizations that might have an interest in the submitted work in the previous 3 years; was invited was invited to attend congresses by BMS and Roche.
TL declares no support from any organization of the submitted work; participated on behalf of his institution in clinical trials sponsored by BMS, Lilly, MSD, Novartis, Novo-Nordisk, Pfizer, Roche-Chugai, UCB; he has been a consultant for BMS, Pfizer, Roche-Chugai, SOBI, UCB; he has been invited to attend international congresses by MSD and Pfizer.
XLL declares no support from any organization of the submitted work; participated on behalf of his institution in clinical trials sponsored by Abbvie, AB Science, BMS, MSD, Novartis, Pfizer, Roche-Chugai, Schering Plough and UCB; he had been a consultant for Abbvie, Amgen, Novartis, Pfizer, Roche-Chugai and Schering Plough; he has been invited to attend international congresses by MSD, Pfizer and Roche Chugai; he has obtained on behalf of his institution grants for research projects from Abbvie, Amgen, MSD, Pfizer, Roche Schering Plough and UCB.
OV declares no support from any organization of the submitted work; participated on behalf of his institution in clinical trials sponsored by BMS, Roche, Roche-Chugai, Pfizer, UCB, AB Science, Schering Plough and Abbott; he is a member of the French advisory boards of BMS, SOBI and UCB; he has given lectures for BMS, Pfizer, MSD and Roche and has obtained on behalf of his institution grants for research projects from Pfizer, Roche and UCB.
PG declares no support from any organization of the submitted work; participated on behalf of his institution in clinical trials sponsored by Abbott, Roche, BMS, Lilly, Novartis, Pfizer, UCB and MSD; he has been a consultant and given lectures on behalf of his institution for Abbott, BMS, MSD, Pfizer, UCB; he has been invited to attend international congresses by MSD, Roche, BMS and Abbott.
DM declares no support from any organization of the submitted work; participated on behalf of his institution in clinical trials sponsored by Abbott, Roche, BMS, Pfizer, UCB and MSD; his hospital received a grant for research from Abbott in 2004; he has been a consultant and given lectures on behalf of his institution for MSD and Pfizer; he has been invited to attend international congresses by MSD, Roche, BMS and Abbott.
GP declares no support from any organization of the submitted work; is involved in clinical studies sponsored by Genzyme, Novartis and Roche Pharma; his research team has received financial support from Abbott Pharma, Chugai, Janssen, LFB (Laboratoire Français des Biotechnologies), Pierre-Fabre Laboratories, Wyeth and Merck Serono.
The authors thank Anne-Claire Duveau and Caroline Brochon for technical assistance with adalimumab concentration measurements and Soujanya Ratna Edupugantifor critical revision of the manuscript. This work was post hoc analysis of a previous work published elsewhere (clinicaltrials.gov, NCT00234234). Measurements of adalimumab serum concentrations were carried out within the CePiBAc platform. CePiBAc was cofinanced by the European Union. Europe is committed to the region Centre with the European Regional Development Fund. This work was partly supported by the French Higher Education and Research ministry under the program ‘Investissements d'avenir’ Grant Agreement: LabEx MAbImprove ANR-10-LABX-53-01.
References
- FDA. 2011. Adalimumab indications and usage. Accessed at http://www.fda.gov/downloads/Drugs/DevelopmentApprovalProcess/HowDrugsareDevelopedandApproved/ApprovalApplications/TherapeuticBiologicApplications/ucm092762.pdf (last accessed 16 October 2014).
- Bartelds GM, Krieckaert CL, Nurmohamed MT, van Schouwenburg PA, Lems WF, Twisk JW, Dijkmans BA, Aarden L, Wolbink GJ. Development of antidrug antibodies against adalimumab and association with disease activity and treatment failure during long-term follow-up. JAMA. 2011;305:1460–1468. doi: 10.1001/jama.2011.406. [DOI] [PubMed] [Google Scholar]
- Bartelds GM, Wijbrandts CA, Nurmohamed MT, Stapel S, Lems WF, Aarden L, Dijkmans BA, Tak PP, Wolbink GJ. Clinical response to adalimumab: relationship to anti-adalimumab antibodies and serum adalimumab concentrations in rheumatoid arthritis. Ann Rheum Dis. 2007;66:921–926. doi: 10.1136/ard.2006.065615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanauer SB, Sandborn WJ, Rutgeerts P, Fedorak RN, Lukas M, MacIntosh D, Panaccione R, Wolf D, Pollack P. Human anti-tumor necrosis factor monoclonal antibody (adalimumab) in Crohn's disease: the CLASSIC-I trial. Gastroenterology. 2006;130:323–333. doi: 10.1053/j.gastro.2005.11.030. quiz 591. [DOI] [PubMed] [Google Scholar]
- van de Putte LB, Atkins C, Malaise M, Sany J, Russell AS, van Riel PL, Settas L, Bijlsma JW, Todesco S, Dougados M, Nash P, Emery P, Walter N, Kaul M, Fischkoff S, Kupper H. Efficacy and safety of adalimumab as monotherapy in patients with rheumatoid arthritis for whom previous disease modifying antirheumatic drug treatment has failed. Ann Rheum Dis. 2004;63:508–516. doi: 10.1136/ard.2003.013052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fasanmade AA, Adedokun OJ, Blank M, Zhou H, Davis HM. Pharmacokinetic properties of infliximab in children and adults with Crohn's disease: a retrospective analysis of data from 2 phase III clinical trials. Clin Ther. 2011;33:946–964. doi: 10.1016/j.clinthera.2011.06.002. [DOI] [PubMed] [Google Scholar]
- Fasanmade AA, Adedokun OJ, Ford J, Hernandez D, Johanns J, Hu C, Davis HM, Zhou H. Population pharmacokinetic analysis of infliximab in patients with ulcerative colitis. Eur J Clin Pharmacol. 2009;65:1211–1228. doi: 10.1007/s00228-009-0718-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ternant D, Mulleman D, Degenne D, Willot S, Guillaumin JM, Watier H, Goupille P, Paintaud G. An enzyme-linked immunosorbent assay for therapeutic drug monitoring of infliximab. Ther Drug Monit. 2006;28:169–174. doi: 10.1097/01.ftd.0000189901.08684.4b. [DOI] [PubMed] [Google Scholar]
- Ternant D, Mulleman D, Lauféron F, Vignault C, Ducourau E, Wendling D, Goupille P, Paintaud G. Influence of methotrexate on infliximab pharmacokinetics and pharmacodynamics in ankylosing spondylitis. Br J Clin Pharmacol. 73:55–65. doi: 10.1111/j.1365-2125.2011.04050.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Z, Seitz K, Fasanmade A, Ford J, Williamson P, Xu W, Davis HM, Zhou H. Population pharmacokinetics of infliximab in patients with ankylosing spondylitis. J Clin Pharmacol. 2008;48:681–695. doi: 10.1177/0091270008316886. [DOI] [PubMed] [Google Scholar]
- Weisman MH, Moreland LW, Furst DE, Weinblatt ME, Keystone EC, Paulus HE, Teoh LS, Velagapudi RB, Noertersheuser PA, Granneman GR, Fischkoff SA, Chartash EK. Efficacy, pharmacokinetic, and safety assessment of adalimumab, a fully human anti-tumor necrosis factor-alpha monoclonal antibody, in adults with rheumatoid arthritis receiving concomitant methotrexate: a pilot study. Clin Ther. 2003;25:1700–1721. doi: 10.1016/s0149-2918(03)80164-9. [DOI] [PubMed] [Google Scholar]
- FDA. 2002. Adalimumab Product Approval Information – Licensing Action 12/31/02. Accessed at http://www.fda.gov/downloads/Drugs/DevelopmentApprovalProcess/HowDrugsareDevelopedandApproved/ApprovalApplications/TherapeuticBiologicApplications/ucm092762.pdf (last accessed 12 February 2013).
- Karmiris K, Paintaud G, Noman M, Magdelaine-Beuzelin C, Ferrante M, Degenne D, Claes K, Coopman T, Van Schuerbeek N, Van Assche G, Vermeire S, Rutgeerts P. Influence of trough serum levels and immunogenicity on long-term outcome of adalimumab therapy in Crohn's disease. Gastroenterology. 2009;137:1628–1640. doi: 10.1053/j.gastro.2009.07.062. [DOI] [PubMed] [Google Scholar]
- Radstake TR, Svenson M, Eijsbouts AM, van den Hoogen FH, Enevold C, van Riel PL, Bendtzen K. Formation of antibodies against infliximab and adalimumab strongly correlates with functional drug levels and clinical responses in rheumatoid arthritis. Ann Rheum Dis. 2009;68:1739–1745. doi: 10.1136/ard.2008.092833. [DOI] [PubMed] [Google Scholar]
- Blache C, Lequerre T, Roucheux A, Beutheu S, Dedreux I, Jacquot S, Le Loet X, Boyer O, Vittecoq O. Number and phenotype of rheumatoid arthritis patients' CD4+CD25hi regulatory T cells are not affected by adalimumab or etanercept. Rheumatology. 2011;50:1814–1822. doi: 10.1093/rheumatology/ker183. [DOI] [PubMed] [Google Scholar]
- van Gestel AM, Prevoo ML, van 't Hof MA, van Rijswijk MH, van de Putte LB, van Riel PL. Development and validation of the European League Against Rheumatism response criteria for rheumatoid arthritis. Comparison with the preliminary American College of Rheumatology and the World Health Organization/International League Against Rheumatism Criteria. Arthritis Rheum. 1996;39:34–40. doi: 10.1002/art.1780390105. [DOI] [PubMed] [Google Scholar]
- Ait-Oudhia S, Lowe PJ, Mager DE. Bridging clinical outcomes of canakinumab treatment in patients with rheumatoid arthritis with a population model of IL-1β kinetics. CPT Pharmacometrics Syst Pharmacol. 2012;2012:6. doi: 10.1038/psp.2012.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu YL, Rubin DT, Vermeire S, Louis E, Robinson AM, Lomax KG, Pollack PF, Paulson SK. Serum adalimumab concentration and clinical remission in patients with Crohn's disease. Inflamm Bowel Dis. 2013;19:1112–1122. doi: 10.1097/MIB.0b013e3182813242. [DOI] [PubMed] [Google Scholar]
- Jürgens M, Mahachie John JM, Cleynen I, Schnitzler F, Fidder H, van Moerkercke W, Ballet V, Noman M, Hoffman I, van Assche G, Rutgeerts PJ, van Steen K, Vermeire S. Levels of C-reactive protein are associated with response to infliximab therapy in patients with Crohn's disease. Clin Gastroenterol Hepatol. 2011;9:421–427. doi: 10.1016/j.cgh.2011.02.008. [DOI] [PubMed] [Google Scholar]
- Magro F, Rodrigues-Pinto E, Santos-Antunes J, Vilas-Boas F, Lopes S, Nunes A, Camila-Dias C, Macedo G. High C-reactive protein in Crohn's disease patients predicts nonresponse to infliximab treatment. J Crohns Colitis. 8:129–136. doi: 10.1016/j.crohns.2013.07.005. [DOI] [PubMed] [Google Scholar]
- Kravitz MS, Shoenfeld Y. Autoimmunity to protective molecules: is it the perpetuum mobile (vicious cycle) of autoimmune rheumatic diseases? Nat Clin Pract Rheumatol. 2006;2:481–490. doi: 10.1038/ncprheum0290. [DOI] [PubMed] [Google Scholar]
- Puchalski T, Prabhakar U, Jiao Q, Berns B, Davis HM. Pharmacokinetic and pharmacodynamic modeling of an anti-interleukin-6 chimeric monoclonal antibody (siltuximab) in patients with metastatic renal cell carcinoma. Clin Cancer Res. 2010;16:1652–1661. doi: 10.1158/1078-0432.CCR-09-2581. [DOI] [PubMed] [Google Scholar]
- Urien S, Bardin C, Bader-Meunier B, Mouy R, Compeyrot-Lacassagne S, Foissac F, Florkin B, Wouters C, Neven B, Treluyer JM, Quartier P. Anakinra pharmacokinetics in children and adolescents with systemic-onset juvenile idiopathic arthritis and autoinflammatory syndromes. BMC. 2013;14:40. doi: 10.1186/2050-6511-14-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pepys MB, Hirschfield GM. C-Reactive protein: a critical update. J Clin Invest. 2003;111:1805–1812. doi: 10.1172/JCI18921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levi M, Grange S, Frey N. Exposure-response relationship of tocilizumab, an anti-IL-6 receptor monoclonal antibody, in a large population of patients with rheumatoid arthritis. J Clin Pharmacol. 2013;53:151–159. doi: 10.1177/0091270012437585. [DOI] [PubMed] [Google Scholar]
- Comets E, Brendel K, Mentre F. Computing normalised prediction distribution errors to evaluate nonlinear mixed-effect models: the npde add-on package for R. Comput Methods Programs Biomed. 2008;90:154–166. doi: 10.1016/j.cmpb.2007.12.002. [DOI] [PubMed] [Google Scholar]
- Hayashi N, Tsukamoto Y, Sallas WM, Lowe PJ. A mechanism-based binding model for the population pharmacokinetics and pharmacodynamics of omalizumab. Br J Clin Pharmacol. 2007;63:548–561. doi: 10.1111/j.1365-2125.2006.02803.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe PJ, Tannenbaum S, Gautier A, Jimenez P. Relationship between omalizumab pharmacokinetics, IgE pharmacodynamics and symptoms in patients with severe persistent allergic (IgE-mediated) asthma. Br J Clin Pharmacol. 2009;68:61–76. doi: 10.1111/j.1365-2125.2009.03401.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng CM, Joshi A, Dedrick RL, Garovoy MR, Bauer RJ. Pharmacokinetic–pharmacodynamic–efficacy analysis of efalizumab in patients with moderate to severe psoriasis. Pharm Res. 2005;22:1088–1100. doi: 10.1007/s11095-005-5642-4. [DOI] [PubMed] [Google Scholar]
- Ng CM, Bruno R, Combs D, Davies B. Population pharmacokinetics of rituximab (anti-CD20 monoclonal antibody) in rheumatoid arthritis patients during a phase II clinical trial. J Clin Pharmacol. 2005;45:792–801. doi: 10.1177/0091270005277075. [DOI] [PubMed] [Google Scholar]
- Dirks NL, Meibohm B. Population pharmacokinetics of therapeutic monoclonal antibodies. Clin Pharmacokinet. 2010;49:633–659. doi: 10.2165/11535960-000000000-00000. [DOI] [PubMed] [Google Scholar]
- Mori S. A relationship between pharmacokinetics (PK) and the efficacy of infliximab for patients with rheumatoid arthritis: characterization of infliximab-resistant cases and PK-based modified therapy. Mod Rheumatol. 2007;17:83–91. doi: 10.1007/s10165-006-0544-9. [DOI] [PubMed] [Google Scholar]
- Mulleman D, Chu Miow Lin D, Ducourau E, Emond P, Ternant D, Magdelaine-Beuzelin C, Valat JP, Paintaud G, Goupille P. Trough infliximab concentrations predict efficacy and sustained control of disease activity in rheumatoid arthritis. Ther Drug Monit. 2010;32:232–236. doi: 10.1097/FTD.0b013e3181cc6fef. [DOI] [PubMed] [Google Scholar]
- Willot S, Vermeire S, Ohresser M, Rutgeerts P, Paintaud G, Belaiche J, De Vos M, Van Gossum A, Franchimont D, Colombel JF, Watier H, Louis E. No association between C-reactive protein gene polymorphisms and decrease of C-reactive protein serum concentration after infliximab treatment in Crohn's disease. Pharmacogenet Genomics. 2006;16:37–42. doi: 10.1097/01.fpc.0000182776.57437.d8. [DOI] [PubMed] [Google Scholar]
- Ducourau E, Ternant D, Lequerre T, Fuzibet P, Le Loet X, Watier H, Goupille P, Paintaud G, Vittecoq O, Mulleman D. Towards an individualised target concentration of adalimumab in rheumatoid arthritis. Ann Rheum Dis. 2014;2014:2013–204971. doi: 10.1136/annrheumdis-2013-204971. [DOI] [PubMed] [Google Scholar]