

Abstract
Numerous studies by our lab and others demonstrate that epidermal growth factor receptor (EGFR) plays critical roles in primary breast cancer (BC) initiation, growth and dissemination. However, clinical trials targeting EGFR function in BC have lead to disappointing results. In the current study we sought to identify the mechanisms responsible for this disparity by investigating the function of EGFR across the continuum of the metastatic cascade. We previously established that overexpression of EGFR is sufficient for formation of in situ primary tumors by otherwise nontransformed murine mammary gland cells. Induction of epithelial-mesenchymal transition (EMT) is sufficient to drive the metastasis of these EGFR-transformed tumors. Examining growth factor receptor expression across this and other models revealed a potent downregulation of EGFR through metastatic progression. Consistent with diminution of EGFR following EMT and metastasis EGF stimulation changes from a proliferative to an apoptotic response in in situ versus metastatic tumor cells, respectively. Furthermore, overexpression of EGFR in metastatic MDA-MB-231 BC cells promoted their antitumorigenic response to EGF in three dimensional (3D) metastatic outgrowth assays. In line with the paradoxical function of EGFR through EMT and metastasis we demonstrate that the EGFR inhibitory molecule, Mitogen Induced Gene-6 (Mig6), is tumor suppressive in in situ tumor cells. However, Mig6 expression is absolutely required for prevention of apoptosis and ultimate metastasis of MDA-MB-231 cells. Further understanding of the paradoxical function of EGFR between primary and metastatic tumors will be essential for application of its targeted molecular therapies in BC.
Abbreviations: Mig6, mitogen-induced gene-6; BC, breast cancer; EGFR, epidermal growth factor receptor; Mig6-EBR, EGFR binding region of Mig6; TGF-β, transforming growth factor-β; WT, wild type; NMuMG, normal murine mammary gland cells; NME, NMuMG cells transformed by EGFR overexpression; CCLE, Cell Line Encyclopedia; MET, mesenchymal-epithelial transition
Introduction
Epidermal growth factor receptor (EGFR) is a well-established proto-oncogene. Indeed, we demonstrated that the overexpression of EGFR is capable of transforming mammary epithelial cells, and that transforming growth factor β (TGF-β)–mediated epithelial-mesenchymal transition (EMT) in concert with EGFR facilitates primary mammary tumor cell invasion and dissemination [1], [2]. Along these lines, primary breast tumors expressing elevated levels of EGFR display decreased clinical prognosis [3]. These and other findings point to the targeting of EGFR as an effective means by which to combat breast cancer (BC) metastasis. However, the clinical application of EGFR-targeting agents to treat BC has been largely unsuccessful [4], [5], [6]. The molecular mechanisms that underlie this inherent resistance of BC to EGFR-targeted therapies remain undefined.
An emerging topic in the field of EGFR signaling is the role of inhibitory proteins that function to mitigate signaling following EGFR activation. One such protein is Mitogen Inducible Gene-6 (Mig6), also known as receptor-associated late transducer (RALT), Gene33, or ErbB receptor feedback inhibitor (ERRFI). Cells utilize Mig6 and other feedback inhibitory molecules to balance physiological signaling emanating from EGFR in terms of both time and intensity [7]. Mechanistically, Mig6 binds to the intracellular domain of EGFR via its EGFR binding Region (EBR) and inhibits downstream signaling to various effector molecules such as Erk1/2 and Akt [8], [9]. Furthermore, Mig6 is genetically altered and transcriptionally silenced in lung cancer, a cancer that can be driven by constitutive activation of EGFR [9]. Genetic deletion of Mig6 results in tumor formation in mice due to hyper-activation of EGFR [10]. Finally, in addition to EGFR, Mig6 has more recently been shown to bind to and facilitate c-Abl nuclear translocation leading to induction of apoptosis [11].
While the studies referenced above point to Mig6 as a growth inhibitory, pro-apoptotic and overall tumor suppressive molecule, contrasting studies have demonstrated its function in tumor progression and inhibition of apoptosis. For instance, several breast cancer cell lines express high levels of Mig6 and overexpression of Mig6 in MCF-7 BC cells inhibits thier apoptosis [12]. Similarly, while genetic depletion of Mig6 does lead to hyperplasia in several organs it is also associated with enhanced apoptosis in endothelial cells [13]. Correspondingly, depletion of the Mig6 in bronchiolar epithelial cells does lead to increased cell proliferation, but similar measures in endothelial cells leads to apoptosis [13]. Along these lines, EGF stimulation can induce apoptosis in EGFR-amplified BC cells [14], [15], [16], [17]. We recently established that EGFR expression is diminished in fully metastatic BC cells as compared to their systemically invasive but dormant counterparts, and loss of high level EGFR expression can be brought about during recovery from TGF-β–induced epithelial-mesenchymal transition (EMT) [18]. Thus, the precise role of Mig6 and its regulation of pro versus antitumorigenic EGFR signaling during BC metastasis remain incompletely understood.
In the current study we delineate a paradoxical shift in EGFR function through the metastatic progression of BC. Using recombinant gene overexpression we clearly demonstrate that Mig6 acts as a tumor suppressor in EGFR-driven in situ mammary tumors. In contrast, interrogating several models of metastatic progression and clinical datasets uniformly indicate that as BCs progress from in situ to metastatic disease, EGFR expression is diminished. Consistent with an antitumorigenic function of EGFR during late-stage metastatic progression, overexpression of EGFR or depletion of Mig6 in MDA-MB-231 cells promoted their apoptosis and dramatically reduced their outgrowth in 3D culture and formation of pulmonary tumors in mice. Collectively, our data delineate a paradoxical shift in EGFR function through the metastatic progression of BC. These findings demonstrate a plausible mechanism to explain the inherent resistance of metastatic BC to EGFR-targeted therapies.
Materials and Methods
Cell lines and Cell Culture
Murine NMuMG, human MDA-MB-231, and human MDA-MB-468 cells were purchased from ATCC and cultured as described previously [2], [19]. Construction of NMuMG cells expressing human wild type (WT)-EGFR (NME) and their metastatic variants are described elsewhere [1], [2]. Cellular depletion of Mig6 in MDA-MB-231 was accomplished by VSVG lentiviral transduction of pLKO.1 shRNA vectors as previously described (Thermo Scientific), sequences of shRNAs can be found in Supplementary Table 1 [2]. The human MCF10A parental cell line and its increasingly tumorigenic variants T1k, Ca1h and Ca1a were kindly provided by Dr. Fred Miller (Wayne State University) and were cultured as described previously [20]. A list of the chemical inhibitors used throughout the study can be found in Supplementary Table 2.
Immunoblotting and Immunofluorescent Analyses
For immunoblot assays, equal aliquots of total cellular protein were resolved by SDS-PAGE and transferred to PVDF membranes using standard methods as described [21]. Immunofluorescent assays were conducted using primary antibodies in combination with a biotinylated secondary antibodies (Jackson) and Texas-Red conjugated avidin (Vector) as described [1]. Antibody concentrations and suppliers are listed in Supplementary Table 3.
Cell Biological Assays
DNA synthesis was measured by [3H]thymidine incorporation as previously described [18]. Caspase 3/7 activity was quantified using the Caspase 3/7 Glo reagent (Promega) according to the manufacturer’s instructions. Visualization of the actin cytoskeleton was perform by staining fixed cells with FITC-conjugated Phalloidin according to the manufactures instructions (Thermo Scientific).
Three-Dimensional (3D) Organotypic Growth Assays
Ninety-six well plates were coated with Cultrex (50 μl/well) and cells were resuspended in DMEM supplemented with 10% FBS and 4% Cultrex (150 μl/well). Luciferase-expressing cells were seeded at a density of 1 × 103 cells/well. Media containing the indicated inhibitors or growth factors was replaced every 4 days and organoid outgrowth was detected by the addition of D-luciferin potassium salt (Gold Biotechnology) to induce bioluminescence, which was quantified using a GloMax-Multi detection system (Promega).
Tumor Growth and Metastasis Analysis
Orthotopic NME tumors were established and tumor volume was quantified as previously described [2]. Pulmonary tumor development was assessed by injection of parental (scrambled shRNA) and Mig6-deficient cells into the lateral tail vein of nu/nu mice (1 × 106 cells/mouse). Where indicated tumor growth and metastasis was monitored by in vivo bioluminescent imaging as previously described [1], [2], [18], [20], [22]. Bioluminescent images were captured on a Xenogen IVIS-200 (PerkinElmer). All animal procedures were performed in accordance to protocols approved by the Institutional Animal Care and Use Committee for Case Western Reserve University (Cleveland, OH) and Purdue University (West Lafayette, IN).
In Silico Analyses
The Cancer Cell Line Encyclopedia contains a repository of log2 expression data derived from Affymetrix U133+2.0 Arrays for 947 unique human cancer cell lines. GEO Dataset GSE3744 contains expression data using the Affymetrix U133+2.0 for clinical samples classified as basal-like and non-basal-like [23]. This dataset was analyzed using the NCBI curated dataset browser tool. The TCGA [24] dataset was accessed via cBioPortal and analyzed using R.
Statistical Analyses
Statistical analyses were carried out using an unpaired Student’s T-test. P values < 0.05 were considered statistically significant. P values for all experiments are indicated. Correlation coefficients and P values were generated using Prizm-Graph Pad.
Results
Mig6 is Tumor Suppressive in an EGFR-Driven In Situ Model of Breast Cancer
Mig6 has previously been described as a tumor suppressor [10], findings that are contingent upon EGFR acting as a protumorigenic molecule. To verify the ability of Mig6 to suppress EGFR-induced tumorigenesis, we sought to heterologously overexpress Myc-tagged Mig6 constructs in our in situ model of normal murine mammary gland (NMuMG) cells transformed by overexpression of EGFR (referred to here as NME cells) (Figure 1A). Importantly, expression of these constructs did not appreciably affect EGFR levels in our recombinant system (Figure 1A). However, when these cells were cultured on 2D plastic substrates under a mixture of extracellular matrix, expression of the EGFR-binding region (EBR) or WT Mig6 decreased the formation of filopodia (arrows), an event we have previously attributed to the function of EGFR in these cells (Figure 1B; [25]). Furthermore, when these cells were cultured under 3D organotypic conditions expression of EBR and WT Mig6 significantly enhanced the formation of hollow, acinar structures, characteristic of normal mammary epithelial cells (Figure 1, B and C). Furthermore, expression of EBR and WT Mig6 decreased the ability of EGF to induce phosphorylation of EGFR at Y845 (Figure 1D). We recently demonstrated that the ability of EGFR to transform NMuMG cells is dependent upon downstream activation of Stat3 [19]. Importantly, this critical signaling event was potently inhibited by expression of EBR and WT Mig6 (Figure 1D). To focus specifically on the ability of the EGFR-directed function of Mig6 to inhibit tumor initiation we engrafted the NME cells expressing GFP or Mig6-EBR onto the fat pad of mice and primary tumor formation was monitored by caliper measurements (Figure 1E). Mig-EBR dramatically reduced the ability of NME cells to form primary tumors (Figure 1, F and G). Collectively, these findings indicate that EGFR-initiated tumorigenesis can be normalized by the inhibitory actions of Mig6.
Figure 1.
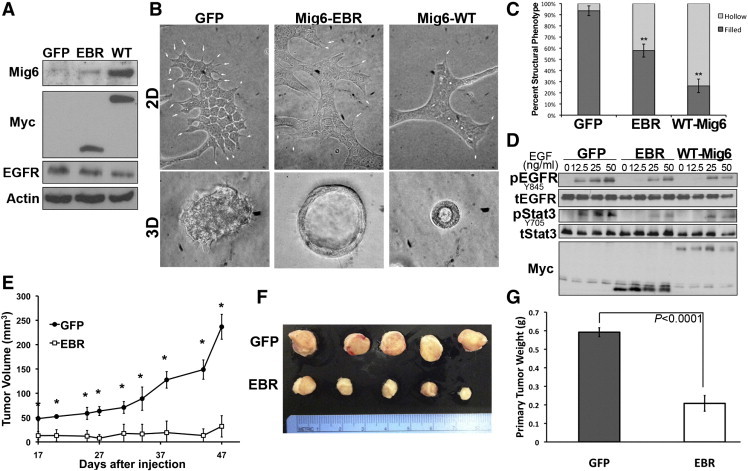
Recombinant expression of Mig6 is capable of reversing EGFR-mediated transformation. (A) NME cells were constructed to express Myc-tagged WT or the EGFR binding region (EBR) of Mig6. Expression of endogenous and recombinant WT-Mig6 were analyzed by immunoblot (EBR lacks the epitope of the Mig6 antibody), Myc and EGFR were also analyzed in these same lysates. Actin was assessed as a positive control. (B) Control (GFP) and Mig6 expressing cells were cultured on plastic under a cultrex-containing medium (arrows denoted filopodia) and within 3D organotypic culture conditions. (C) Filled (transformed) versus hollowed (nontransformed) 3D cell structures as shown in panel B were quantified for control and Mig6 expressing NME cells. Data are the mean percentages (± SE) of hollow versus filled structures per 10 high powered fields where ** indicates P < .01 as compared to the GFP control cells. (D) Control and Mig6 expressing NME cells were stimulated with the indicated concentrations of EGF for 30 minutes. Cells were subsequently analyzed by immunoblot for phosphorylation of EGFR (pEGFR) and Stat3 (pStat3). These blots were striped and reprobed for total EGFR (tEGFR), Stat3 (tStat3) and Myc as loading controls. Data in panels A and D are representative of at least 3 independent analyses. (E) NME cells expressing GFP as a control (GFP) or Mig6-EBR (EBR) were engrafted (2x106 cells per mouse) onto the mammary fatpad of mice and tumor growth was monitored by caliper measurements. * Indicates P < .01, n = 5 mice per group. (F) Visualization of control (GFP) and Mig6-EBR (EBR) primary tumor excised from mice 50 days after engraftment. (G) Mean and SE of wet weights of tumors shown in panel F, P value is indicated.
Metastatic Breast Cancer Cells Become Inherently Resistant to Targeted Inhibition of EGFR
NME tumors can be driven to metastasize by a transient induction of EMT via treatment with TGF-β1 prior to allograft onto the mammary fat pad [2]. Isolation of the resulting pulmonary metastases yielded the highly aggressive NME-LM cell line. This cell line undergoes robust primary tumor formation and spontaneous pulmonary metastasis upon mammary fat pad engraftment, independent of TGF-β pretreatment [2]. Isolation of these resultant pulmonary metastases yielded the NME-LM2 cell line. These isogenic cell lines now constitute a progression series of cells ranging from nontransformed (NMuMG) to carcinoma in situ (NMuMG cells transformed by EGFR overexpression, NME) to highly metastatic (NME-LM and NME-LM2) [2]. Given the ability of Mig6-EBR to prevent NME tumor formation we next sought to verify inhibition of primary tumor development via pharmacological inhibition of EGFR. Indeed, similar to expression of Mig6-EBR, daily oral administration of Erlotinib (50 mg/kg) to mice bearing orthotopic NME cell engraftments dramatically reduced tumor formation (Figure 2, A, C and D). In line with our previous report, no tumor recurrence or metastases were observed by these cells irrespective of EGFR-directed therapy (Figure 2A; [2]). In stark contrast to the NME cells, tumor formation by the NME-LM cells (described above) was only minimally delayed by Erlotinib treatment (Figure 2B), and no significant differences were observed in primary tumor weight at the time of primary tumor excision (Figure 2E). Also, following primary tumor excision, NME-LM tumor recurrence was actually enhanced by Erlotinib treatment (Figure 2B). Consistent with the role of EGFR in primary tumor cell dissemination [1], [26], pulmonary dissemination of NME-LM cells was delayed by Erlotinib (Figure 2F), but at the time of necropsy robust pulmonary metastases were observed in NME-LM tumor bearing mice irrespective of treatment (Figure 2G). Taken together, these data strongly suggest that through the processes of EMT and metastasis even BC cells whose initial transformation is driven by EGFR are capable of inherently developing resistance to EGFR-targeted therapy.
Figure 2.
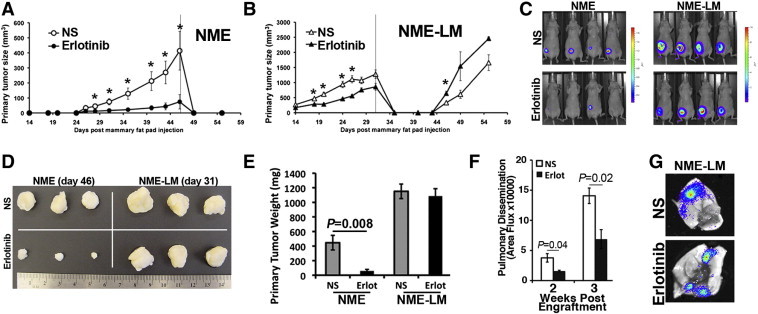
EGFR transformed mammary epithelial cells become inherently resistant to Erlotinib following EMT-induced metastasis. (A and B) NME and NME-LM (1x106) cells were engrafted onto the mammary fat pad of female nu/nu mice and 14 days thereafter mice were treated daily with Erlotinib (50 mg/kg). At the indicated time points (line on graph), primary tumors were surgically removed and mice were monitored for recurrent tumors. (C) Bioluminescent images showing the sensitivity (NME) and resistance (NME-LM) of the indicated tumors described in panels A and B to systemic Erlotinib treatment. (D) Ex-vivo visualization and (E) weight of indicated tumors from control and Erlotinib-treated mice. (F) Pulmonary dissemination of NME-LM tumors in control and Erlotinib-treated mice was determined by bioluminescence at the indicated time points following fat pad engraftment. For panel A–B and E–F data are the mean values ± SE (n = 4 mice per group) where * indicates P < .05. (G) Representative ex vivo bioluminescence images demonstrating robust metastasis in the lungs from control and Erlotinib-treated NME-LM tumor bearing mice.
EGFR Expression is Diminished Following EMT and Metastasis
Given the inherent resistance of the NME-LM cells to EGFR targeted therapy, we next sought to utilize this isogenic system to identify mediators of this process. As noted above, NME cells can be driven to metastasize via TGF-β–induced EMT prior to tumor cell engraftment [2]. Our previous in vitro studies demonstrate that EGFR is not modulated during induction of EMT by TGF-β1, but EGFR is diminished following withdrawal of exogenous TGF-β1 [18]. To examine this phenomenon in vivo, we conducted single cell analysis of cell surface EGFR levels in cells subcultured from control (NS) or TGF-β1 pretreated NME primary tumors. Figure 3A shows flow cytometry using an antibody specific for the human EGFR transgene (Figure 3A). These two ex vivo primary tumors displayed similar EGFR profiles barring a small population of EGFR-negative cells that was only detectable in the TGF-β1 pretreated NME tumors (Figure 3A). In contrast to our in vitro data fluorescence-activated cell sorting for this ex vivo EGFR(-) population yielded a very mesenchymal cell population (Figure 3A; inset) that failed to thrive when cultured alone. Immunofluorescent staining verified the presence of a highly mesenchymal population of cells that lacked expression of Ecad and displayed a very filamentous actin cytoskeleton (Figure 3B). More importantly, although resistant to puromycin, EGFR was undetectable in this mesenchymal cell population (Figure 3B). Similar comparison of cell surface EGFR levels between the parental NME cells and their metastatic and isogenic NME-LM counterparts also displayed a marked reduction in cell surface EGFR levels (Figure 3C). Analysis of total and phosphorylated levels of EGFR similarly revealed a marked diminution in the NME-LM cells as compared to their parental NME cells (Figure 4D, F). Given that EGFR is recombinantly overexpressed in the NME system we sought to identify possible mechanisms of negative selection that could be altering the observed EGFR expression levels in NME-LM cells. Consistent with a negative selection against high level EGFR expression, prolonged stimulation of NME-LM cells with exogenous EGF results in apoptosis (Figure 3D) and drastic inhibition of outgrowth when cultured under 3D organotypic conditions (Figure 3E). These findings contrast sharply with those obtained in the parental NME cell line in which apoptosis and growth inhibition results upon treatment with the EGFR inhibitor AG1478 (Figure 3D and E). Furthermore, the maintenance of 3D growth of the NME-LM cells in the presence of AG1478 (Figure 3E) is consistent with our in vivo data (Figure 2), as well as with the clinical inability of EGFR-targeted therapies to effectively treat metastatic BC [4].
Figure 3.
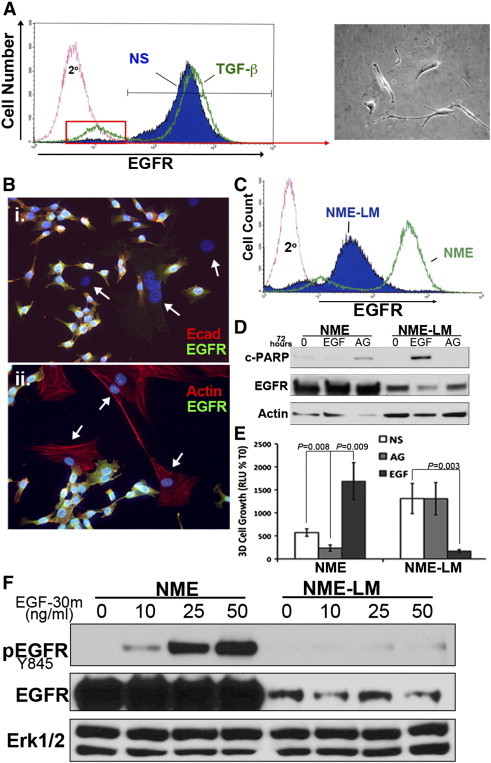
EGFR expression is diminished following EMT-induced metastasis. (A) Following primary tumor removal, control (NS) and TGF-β1–pretreated (TGF-β) NME primary tumors were disassociated and subcultured in the presence of Puromycin (5 μg/ml). The resultant cultures were analyzed by flow-cytometry for cell surface EGFR. Isolation of the distinct EGFR-negative population within the post-EMT tumors by FACS analysis (denoted by the red box) resulted in a highly mesenchymal cell population. (B) Ex-vivo subcultured, post-EMT primary tumor cells were co-stained with antibodies against Ecad and EGFR (i), or co-stained with phalloidin and antibodies against EGFR (ii). Arrows denote cells with a highly mesenchymal phenotype (either Ecad negative or F-actin positive) that fail to express EGFR. (C) NME-LM cells and their parental NME counterparts were analyzed by flow cytometry for cell surface expression of EGFR. (D) NME and NME-LM cells were treated for 48 hours with EGF (50 ng/ml) or the EGFR inhibitor, AG1478 (1 μM). Afterward, whole cell lysates were analyzed by immunoblot for cleavage of PARP and expression levels of EGFR. (E) The outgrowth of NME and NME-LM cells grown under 3D-culture conditions in the presence of EGF or AG1478 was quantified by bioluminescence. Data are the mean (± SE) of 3 independent experiments completed in triplicate resulting in the indicated P values. (F) NME and NME-LM cells were serum starved for 6 hours and subsequently stimulated with the indicated concentrations of EGF for 30 minutes. Whole cell lysates were analyzed for phosphorylated EGFR (pEGFR). This blot was striped and reprobed for total EGFR (tEGFR). Erk1/2 served as a loading control. Data in panels A-D and F are representative of at least two independent experiments yielding similar results.
Figure 4.
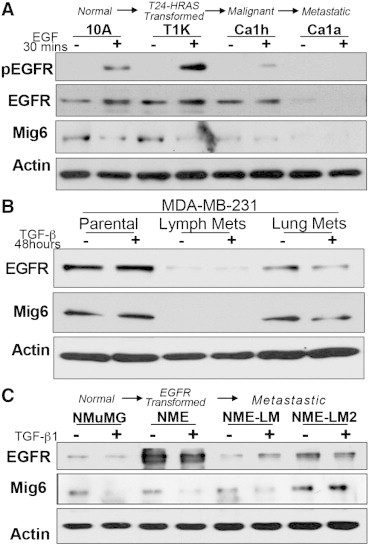
Expression of EGFR is diminished during the metastatic progression of human BCs. (A) The human MCF10A progression series consisting of normal (MCF10A), transformed (T1k), malignant (Ca1h), and metastatic (Ca1a) cells were serum deprived and stimulated with EGF for 30 minutes and subsequently analyzed for EGFR phosphorylation (pEGFR) and total expression of EGFR and Mig6. Actin served as a loading control. (B) Human MDA-MB-231 BC cells were engrafted onto the mammary fat pad of female nu/nu mice and resulting metastases from the lymph node and lungs were harvested and subcultured. These two independent cell lines and the parental cells were stimulated with TGF-β1 for 48 hours and analyzed for expression of EGFR and Mig6. Actin served as a loading control. (C) The murine NME progression series was analyzed by immunoblot for the expression EGFR and Mig6 under nonstimulated conditions (−) and following a 48 hour treatment with TGF-β1 (5 ng/ml). Data in A-C are representative of at least 3 independent experiments yielding similar results.
To verify that diminution of EGFR is not an artifact of our overexpression system, we assessed EGFR expression across the human MCF-10A BC progression series. Consistent with the results of our NME progression series, the fully metastatic MCF-Ca1a cells expressed very little EGFR (Figure 4A). Consistent with our previous studies in the NME cells, endogenous EGFR expression was not affected upon in vitro TGF-β1 stimulation of the MDA-MB-231, but was markedly lower in ex vivo cells that had metastasized from the mammary fat pad to axillary lymph nodes and lungs (Figure 4B). Expression of Mig6 mirrored that of EGFR and was similarly diminished in increasingly metastatic BC cells (Figure 4, A and B). Consistent with the notion that EGFR and Mig6 expression levels track together, the slight increase in EGFR expression observed in the NME-LM2 cells as compared to the NME-LM cells correlated with a corresponding increase in Mig6 (Figure 4C). Furthermore, Mig6 is maintained upon TGF-β treatment of the NME-LM2 cells, which is contrast to their nonmetastatic counterparts in which Mig6 is decreased during TGF-β-induced EMT (Figure 4C). The precise mechanisms that lead to the paradoxical function and diminution of EGFR following EMT and metastasis are likely to be highly complex. Nevertheless, our data clearly indicate that the processes of metastasis support the inherent resistance of BC to EGFR-targeted therapies via a shift in EGFR signaling from pro- to antitumorigenic.
There is a Direct Relationship Between EGFR and Mig6 Expression in Basal-Like Breast Cancer
To expand our findings beyond our in vitro model systems we sought to further examine the regulation of EGFR and Mig6 in clinical BC samples. Therefore, we analyzed the TCGA BC dataset for the expression relationship between EGFR and Mig6 [24]. Consistent with our findings in Figure 4, there was a significant direct correlation between EGFR and Mig6 expression (Figure 5A). Next, we analyzed the GEO data set GSE3744 in which patient BCs are classified as basal like and non-basal like as characterized by microarray analysis [23]. Consistent with EGFR being a major defining factor of basal-like BC [27] and there being a direct correlation between EGFR and Mig6, Mig6 expression was significantly higher in this more aggressive subtype (Figure 5B). Finally, we analyzed the cell line encyclopedia (CCLE) to establish the ratio of Mig6 to EGFR across the 58 available BC cell lines (Figure 5C). This analysis indicated that similar to the clinical datasets there is a robust direct relationship between Mig6 and EGFR expression (Figure 5C). Two basal-like cell lines, the MDA-MB-231 (MDA-231) and the EGFR-amplified MDA-MB-468 (MDA-468) cells expressed the second highest and lowest Mig6/EGFR expression ratio, respectively. In contrast to the MDA-231 cells, EGF stimulation of the MDA-468 cells is known to induce apoptosis [14]. Along these lines, EGF stimulation results in a progressive decrease in [3H]Thymidine incorporation in the MDA-468 cells, but not in the MDA-231 cells (Figure 5D). In an attempt to manipulate the Mig6/EGFR ratio in the MDA-231 cells we overexpressed EGFR (Figure 5E). However, consistent with all of the correlative data above, recombinant overexpression of EGFR in the MDA-231 cells lead to a concomitant increase in Mig6 expression levels (Figure 5E). These data support our conclusion that there is a direct relationship between expression levels of EGFR and Mig6 in metastatic BC (Figure 5E). Finally, outgrowth of the MDA-231 cells under 3D pulmonary organotypic conditions confirmed an antitumorigenic effect of EGF in these metastatic BC cells, a result that was significantly enhanced upon overexpression of EGFR (Figure 5F).
Figure 5.
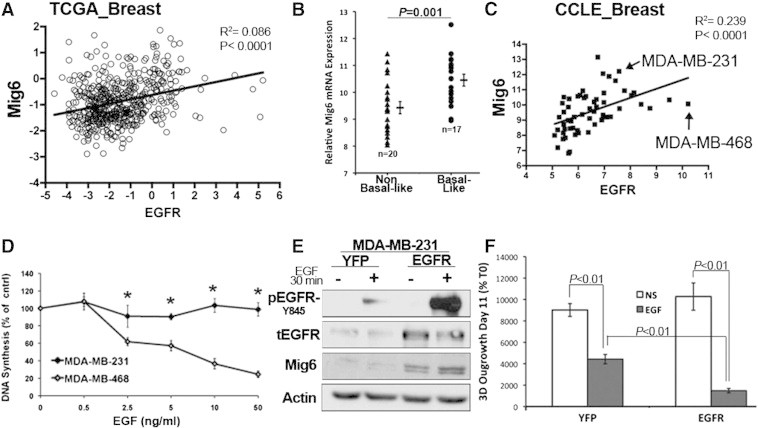
Expression of Mig6 and EGFR are directly related and required at a balanced ratio for basal-like BC survival in response to EGF. (A) Analysis of the TCGA BC data set comparing expression of Mig6 versus EGFR. (B) Geo data set GSE3744 was analyzed for expression levels of Mig6 between non basal-like and basal-like BCs. Data are the Mig6 expression values for individual patient samples resulting in the indicated means (± SE) and P value. (C) Expression levels of EGFR in relation to Mig6 across 58 human BC cell lines recorded in the CCLE database. The data points for MDA-MB-231 and MDA-MB-468 cells are noted. (D) MDA-MB-231 and MDA-MB-468 were treated with the indicated concentrations of EGF for 48 hours and DNA synthesis was quantified by 3H Thymidine incorporation. Data are the mean (± SE) of at least 2 independent experiments carried out in triplicate where * indicates a P ≤ .01. (E) MDA-MB-231 cells were stably constructed to overexpress EGFR or YFP as a control. Phosphorylation of EGFR in response to EGF as well total expression of EGFR and Mig6 were analyzed by western blot. Actin served as a loading control. (F) Control (YFP) and EGFR overexpressing MDA-MB-231 cells were cultured under 3D growth conditions in the presence or absence of EGF (50 ng/ml) for 11 days and 3D outgrowth was quantified by bioluminescence. Data are the mean of three independent experiments conducted in triplicate resulting in the indicated P values.
Mig6 is Required for the Metastatic Outgrowth of MDA-MB-231 Cells
Our findings in Figure 5 suggest that the 3D outgrowth inhibitory effects of EGFR may be partially masked in MDA-231 cells upon EGFR overexpression due to concomitant upregulation of Mig6 (Figure 5E). To identify the role of Mig6 in empowering metastatic BCs with the ability to subvert growth inhibitory EGFR signaling, we utilized shRNAs to specifically deplete Mig6 in the MDA-231 cells (Figure 6A). Consistent with our previous data in Figures 4 and 5, depletion of Mig6 using two independent shRNAs led to a corresponding decrease in EGFR levels, however receptor levels were still readily detectable in these cells (Figure 6A). Depletion of Mig6 dramatically inhibited the 3D-outgrowth of MDA-231 cells independent of addition of exogenous EGF (Figure 6B and C). Examination of these 3D cultures revealed the appearance of several apoptotic bodies (arrows, Figure 6B). This observation was verified by increased levels of Caspase3/7 activity in Mig6-depleted MDA-231 cells as compared to their control scrambled shRNA-expressing counterparts (Figure 6D). Consistent with increased levels of apoptosis, we found that MDA-231 cells lacking Mig6 expression immediately failed to thrive within the pulmonary microenvironment after being injected into the lateral tail vein mice (Figure 6, E and F), thereby dramatically inhibiting the formation of pulmonary tumors by Mig6-deficient MDA-231 cells as compared to their Mig6 proficient counterparts (Figure 6, E–G).
Figure 6.
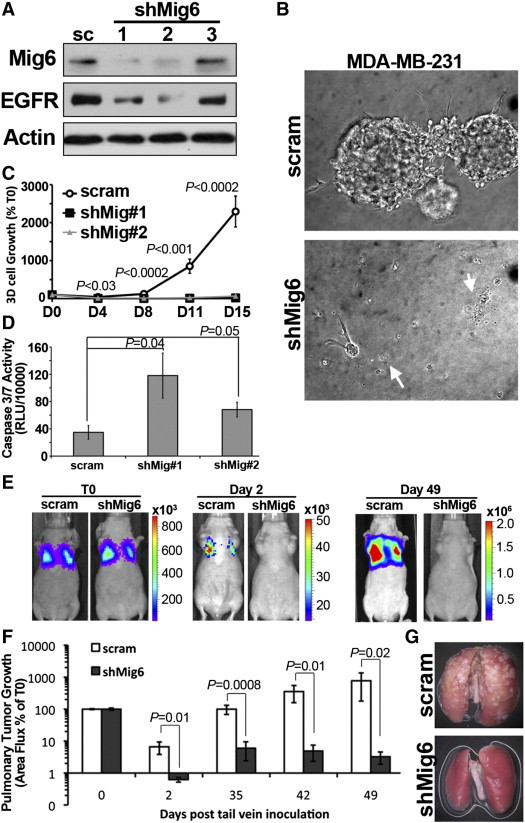
Mig6 depletion induces apoptosis and prevents pulmonary metastasis of MDA-MB-231 cells. (A) MDA-MB-231 cells stably expressing a scrambled (sc) shRNA or three independent Mig6 targeted shRNAs were analyzed for Mig6 and EGFR expression by immunoblot. Actin served as a loading control. (B and C) Luciferase expressing, Mig6-depleted MDA-MB-231 cells were grown under 3D culture conditions and cellular outgrowth was quantified by bioluminescence. Photomicrographs in panel B are representative structures formed by control (scram) and Mig6-depleted (shMig6) cells. Arrows indicate apoptotic cell morphologies. Data in panel C are the average bioluminescent (± SE) values normalized to values measured immediately after plating (T0) from two independent experiments carried out in triplicate. (D) The floating cell fraction of control and Mig6-depleted MDA-MB-231 cells were collected and assayed for Caspase 3/7 activity. (E) Control (scram) and Mig6-depleted (shMig6) MDA-MB-231 cells were injected into the lateral tail vein of nu/nu mice. Longitudinal bioluminescent images from the same mice are shown immediately following injection (T0) and at the indicated time points thereafter. (F) Data are the mean (± SE, n = 5 mice per group) pulmonary bioluminescent values, normalized to the injected value (T0). (G) Representative ex vivo lungs isolated from mice 49 days after injection with control (scram) or Mig6-depleted (shMig6) MDA-MB-231 cells.
Discussion
The current study delineates the evolution of EGFR function over the metastatic progression of BC. Our previous studies demonstrate the transforming capabilities of EGFR overexpression in mammary epithelial cells and the ability of EMT to drive EGFR-mediated invasion and dissemination [1]. However, our findings herein have gone on to establish a proapoptotic switch in EGFR function in the later stages of metastatic outgrowth, a process that can be regulated by corresponding expression of Mig6.
The notion that EGFR can play an anti-tumorigenic role in late-stage BC is supported by our recent study in which we observed loss of EGFR expression in metastatic outgrowth proficient D2.A1 BC cells as compared to their invasive but systemically dormant D2.OR counterparts [18]. We also previously demonstrated the progressive loss of EGFR expression in the NME cells during in vitro recovery from TGF-β-induced EMT [18]. This progressive down-regulation of EGFR during oncogenic mesenchymal-epithelial transition (MET) is further supported herein by our data utilizing the MCF-10A progression series where EGFR expression is diminished in Ca1a cells as compared to their Ca1h counterparts. Indeed, separate studies demonstrate a MET reaction characterizes the metastatic progression of these two cell lines [28]. Along these lines, EGFR is undetectable in the highly metastatic 4T1 model of BC (unpublished observation), a cell line we have recently characterized to undergo EMT:MET reactions during tumor growth and metastasis [2]. The importance of this bifurcated expression of EGFR is born out by clinical data that indicate BCs expressing very high or low levels of EGFR have similarly poorer outcomes as compared to BCs expressing intermediate levels of EGFR [29]. Our findings herein expand upon these primary tumor-derived data to suggest that even high-level EGFR expression is diminished during formation of the metastatic lesion. These data are consistent with clinical findings that have demonstrated dowregulation of EGFR2 (Her2) in metastatic versus primary tumors [30]. Whether this discordance in receptor expression results due active downregulation of EGFR expression or selection of a preexisting EGFRlow subpopulation remains to be established.
In addition to depletion of EGFR, our data in Figure 4, Figure 5 suggest that metastatic BCs can also neutralize EGFR-driven apoptosis via Mig6. This exciting finding is contrasted sharply by the widely assumed notion that Mig6 functions solely as a tumor suppressor, a supposition that holds to the idea that EGFR signaling universally functions in an oncogenic manner [10], [31]. However, it should be noted that several recent studies have established the ability of EGFR to induce apoptosis in BCs [15], [16], [17], [32], while others have established the ability of Mig6 to protect human BC cells from apoptosis [33]. Indeed, genetic inactivation of Mig6 results in pulmonary endothelial cell apoptosis [13]. Likewise, a recent study demonstrated that EGF-induced apoptosis in MDA-468 cells, which scored the lowest Mig6:EGFR ratio in our 58 cell line analysis, occurs upon EGFR internalization [17]. Therefore, it is tempting to speculate that unbalanced expression levels of Mig6 with relation to EGFR leads to unabated activation of internalized EGFR, initiating a pro-apoptotic event. Why this does not occur in normal and premalignant cells in which overexpression of EGFR is strictly protumorigenic is a subject currently under investigation in our laboratory.
In summary, our study has delineated a paradoxical shift in which the potent growth promoting properties of EGFR are converted to antitumorigenic mechanisms during the later stages of metastatic progression. Furthermore, we show that the antitumorigenic properties of EGFR in metastatic BC cells are held at bay by the EGFR inhibitory molecule Mig6. This EGFR paradox presents a plausible explanation for the inherent resistance of metastatic BC to EGFR-targeted therapies. Understanding the evolution in EGFR and other RTK signaling across the metastatic cascade will be essential for the proper application of their respective targeted therapies for the treatment of metastatic BC.
Acknowledgements
The authors would like to thank members of the Wendt laboratory for their critical reading of this manuscript. Research support was provided in part by the National Institutes of Health (CA166140) to M.K.W; the National Institutes of Health (GM081498) to C.R.C.; the National Institutes of Health (CA129359 and CA177069) to W.P.S.; and a National Institutes of Health (T32 HL007653) to N.B. We also acknowledge the expertise of the personnel within the Case Comprehensive Cancer Center (NIH project P30 CA043703) and the Purdue University Center for Cancer Research (NIH project P30 CA023168) small animal imaging core facilities.
Footnotes
Supplementary data to this article can be found online at http://dx.doi.org/10.1016/j.neo.2014.11.009.
Appendix A. Supplementary Data
Supplementary Tables.
References
- 1.Wendt MK, Smith JA, Schiemann WP. Transforming growth factor-beta-induced epithelial-mesenchymal transition facilitates epidermal growth factor-dependent breast cancer progression. Oncogene. 2010;29(49):6485–6498. doi: 10.1038/onc.2010.377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wendt MK, Taylor MA, Schiemann BJ, Sossey-Alaoui K, Schiemann WP. Fibroblast growth factor receptor splice variants are stable markers of oncogenic transforming growth factor- beta1 signaling in metastatic breast cancers. Breast Cancer Res. 2014;16(2):R24. doi: 10.1186/bcr3623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tischkowitz M, Brunet J-S, Begin L, Huntsman D, Cheang M, Akslen L, Nielsen T, Foulkes W. Use of immunohistochemical markers can refine prognosis in triple negative breast cancer. BMC Cancer. 2007;7(1):134. doi: 10.1186/1471-2407-7-134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dickler MN, Cobleigh MA, Miller KD, Klein PM, Winer EP. Efficacy and safety of erlotinib in patients with locally advanced or metastatic breast cancer. Breast Cancer Res Treat. 2009;115(1):115–121. doi: 10.1007/s10549-008-0055-9. [DOI] [PubMed] [Google Scholar]
- 5.Dickler MN, Rugo HS, Eberle CA, Brogi E, Caravelli JF, Panageas KS, Boyd J, Yeh B, Lake DE. A phase II trial of erlotinib in combination with bevacizumab in patients with metastatic breast cancer. Clin Cancer Res. 2008;14(23):7878–7883. doi: 10.1158/1078-0432.CCR-08-0141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tredan O, Campone M, Jassem J, Vyzula R, Coudert B, Pacilio C, Prausova J, Hardy-Bessard AC, Arance A. Ixabepilone Alone or With Cetuximab as First-Line Treatment for Advanced/Metastatic Triple-Negative Breast Cancer. Clin Breast Cancer. 2014 doi: 10.1016/j.clbc.2014.07.007. [pii: S1526-8209(14)00162-1] [DOI] [PubMed] [Google Scholar]
- 7.Segatto O, Anastasi S, Alema S. Regulation of epidermal growth factor receptor signalling by inducible feedback inhibitors. J Cell Sci. 2011;124(Pt 11):1785–1793. doi: 10.1242/jcs.083303. [DOI] [PubMed] [Google Scholar]
- 8.Anastasi S, Baietti MF, Frosi Y, Alema S, Segatto O. The evolutionarily conserved EBR module of RALT/MIG6 mediates suppression of the EGFR catalytic activity. Oncogene. 2007;26(57):7833–7846. doi: 10.1038/sj.onc.1210590. [DOI] [PubMed] [Google Scholar]
- 9.Zhang YW, Staal B, Su Y, Swiatek P, Zhao P, Cao B, Resau J, Sigler R, Bronson R. Evidence that MIG-6 is a tumor-suppressor gene. Oncogene. 2007;26(2):269–276. doi: 10.1038/sj.onc.1209790. [DOI] [PubMed] [Google Scholar]
- 10.Ferby I, Reschke M, Kudlacek O, Knyazev P, Pante G, Amann K, Sommergruber W, Kraut N, Ullrich A. Mig6 is a negative regulator of EGF receptor-mediated skin morphogenesis and tumor formation. Nat Med. 2006;12(5):568–573. doi: 10.1038/nm1401. [DOI] [PubMed] [Google Scholar]
- 11.Hopkins S, Linderoth E, Hantschel O, Suarez-Henriques P, Pilia G, Kendrick H, Smalley MJ, Superti-Furga G, Ferby I. Mig6 is a sensor of EGF receptor inactivation that directly activates c-Abl to induce apoptosis during epithelial homeostasis. Dev Cell. 2012;23(3):547–559. doi: 10.1016/j.devcel.2012.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xu J, Keeton AB, Wu L, Franklin JL, Cao X, Messina JL. Gene 33 inhibits apoptosis of breast cancer cells and increases poly(ADP-ribose) polymerase expression. Breast Cancer Res Treat. 2005;91(3):207–215. doi: 10.1007/s10549-005-1040-1. [DOI] [PubMed] [Google Scholar]
- 13.Jin N, Cho SN, Raso MG, Wistuba I, Smith Y, Yang Y, Kurie JM, Yen R, Evans CM. Mig-6 is required for appropriate lung development and to ensure normal adult lung homeostasis. Development. 2009;136(19):3347–3356. doi: 10.1242/dev.032979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ettenberg SA, Rubinstein YR, Banerjee P, Nau MM, Keane MM, Lipkowitz S. cbl-b inhibits EGF-receptor-induced apoptosis by enhancing ubiquitination and degradation of activated receptors. Mol Cell Biol Res Commun. 1999;2(2):111–118. doi: 10.1006/mcbr.1999.0157. [DOI] [PubMed] [Google Scholar]
- 15.Hyatt DC, Ceresa BP. Cellular localization of the activated EGFR determines its effect on cell growth in MDA-MB-468 cells. Exp Cell Res. 2008;314(18):3415–3425. doi: 10.1016/j.yexcr.2008.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Reinehr R, Haussinger D. CD95 death receptor and epidermal growth factor receptor (EGFR) in liver cell apoptosis and regeneration. Arch Biochem Biophys. 2012;518(1):2–7. doi: 10.1016/j.abb.2011.12.004. [DOI] [PubMed] [Google Scholar]
- 17.Rush JS, Quinalty LM, Engelman L, Sherry DM, Ceresa BP. Endosomal accumulation of the activated epidermal growth factor receptor (EGFR) induces apoptosis. J Biol Chem. 2012;287(1):712–722. doi: 10.1074/jbc.M111.294470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wendt MK, Taylor MA, Schiemann BJ, Schiemann WP. Down-regulation of epithelial cadherin is required to initiate metastatic outgrowth of breast cancer. Mol Biol Cell. 2011;22(14):2423–2435. doi: 10.1091/mbc.E11-04-0306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Balanis N, Wendt MK, Schiemann BJ, Wang Z, Schiemann WP, Carlin CR. Epithelial-to-Mesenchymal Transition Promotes Breast Cancer Progression via a Fibronectin-Dependent Stat3 Signaling Pathway. J Biol Chem. 2013;288(25):17954–17967. doi: 10.1074/jbc.M113.475277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wendt MK, Smith JA, Schiemann WP. p130Cas is required for mammary tumor growth and transforming growth factor-beta-mediated metastasis through regulation of Smad2/3 activity. J Biol Chem. 2009;284(49):34145–34156. doi: 10.1074/jbc.M109.023614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wendt M, Schiemann W. Therapeutic targeting of the focal adhesion complex prevents oncogenic TGF-beta signaling and metastasis. Breast Cancer Res. 2009;11(5):R68. doi: 10.1186/bcr2360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wendt MK, Schiemann BJ, Parvani JG, Lee YH, Kang Y, Schiemann WP. TGF-beta stimulates Pyk2 expression as part of an epithelial-mesenchymal transition program required for metastatic outgrowth of breast cancer. Oncogene. 2013;32(16):2005–2015. doi: 10.1038/onc.2012.230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Richardson AL, Wang ZC, De Nicolo A, Lu X, Brown M, Miron A, Liao X, Iglehart JD, Livingston DM. X chromosomal abnormalities in basal-like human breast cancer. Cancer Cell. 2006;9(2):121–132. doi: 10.1016/j.ccr.2006.01.013. [DOI] [PubMed] [Google Scholar]
- 24.TCGA Comprehensive molecular portraits of human breast tumours. Nature. 2012;490(7418):61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Balanis N, Yoshigi M, Wendt MK, Schiemann WP, Carlin CR. beta3 integrin-EGF receptor cross-talk activates p190RhoGAP in mouse mammary gland epithelial cells. Mol Biol Cell. 2011;22(22):4288–4301. doi: 10.1091/mbc.E10-08-0700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wyckoff J, Wang W, Lin EY, Wang Y, Pixley F, Stanley ER, Graf T, Pollard JW, Segall J. A Paracrine Loop between Tumor Cells and Macrophages Is Required for Tumor Cell Migration in Mammary Tumors. Cancer Res. 2004;64(19):7022–7029. doi: 10.1158/0008-5472.CAN-04-1449. [DOI] [PubMed] [Google Scholar]
- 27.Cheang MC, Voduc D, Bajdik C, Leung S, McKinney S, Chia SK, Perou CM, Nielsen TO. Basal-like breast cancer defined by five biomarkers has superior prognostic value than triple-negative phenotype. Clin Cancer Res. 2008;14(5):1368–1376. doi: 10.1158/1078-0432.CCR-07-1658. [DOI] [PubMed] [Google Scholar]
- 28.Korpal M, Ell BJ, Buffa FM, Ibrahim T, Blanco MA, Celia-Terrassa T, Mercatali L, Khan Z, Goodarzi H. Direct targeting of Sec23a by miR-200s influences cancer cell secretome and promotes metastatic colonization. Nat Med. 2011;17(9):1101–1108. doi: 10.1038/nm.2401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kreike B, Hart G, Bartelink H, van de Vijver MJ. Analysis of breast cancer related gene expression using natural splines and the Cox proportional hazard model to identify prognostic associations. Breast Cancer Res Treat. 2010;122(3):711–720. doi: 10.1007/s10549-009-0588-6. [DOI] [PubMed] [Google Scholar]
- 30.Niikura N, Liu J, Hayashi N, Mittendorf EA, Gong Y, Palla SL, Tokuda Y, Gonzalez-Angulo AM, Hortobagyi GN. Loss of human epidermal growth factor receptor 2 (HER2) expression in metastatic sites of HER2-overexpressing primary breast tumors. J Clin Oncol. 2012;30(6):593–599. doi: 10.1200/JCO.2010.33.8889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hackel PO, Gishizky M, Ullrich A. Mig-6 is a negative regulator of the epidermal growth factor receptor signal. Biol Chem. 2001;382(12):1649–1662. doi: 10.1515/BC.2001.200. [DOI] [PubMed] [Google Scholar]
- 32.Armstrong DK, Kaufmann SH, Ottaviano YL, Furuya Y, Buckley JA, Isaacs JT, Davidson NE. Epidermal growth factor-mediated apoptosis of MDA-MB-468 human breast cancer cells. Cancer Res. 1994;54(20):5280–5283. [PubMed] [Google Scholar]
- 33.Xu D, Makkinje A, Kyriakis JM. Gene 33 is an endogenous inhibitor of epidermal growth factor (EGF) receptor signaling and mediates dexamethasone-induced suppression of EGF function. J Biol Chem. 2005;280(4):2924–2933. doi: 10.1074/jbc.M408907200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Tables.