

Abstract
Our recent study showed that SAG/RBX2 E3 ubiquitin ligase regulates apoptosis and vasculogenesis by promoting degradation of NOXA and NF1, and co-operates with Kras to promote lung tumorigenesis by activating NFκB and mTOR pathways via targeted degradation of tumor suppressive substrates including IκB, DEPTOR, p21 and p27. Here we investigated the role of Sag/Rbx2 E3 ligase in cellular senescence and immortalization of mouse embryonic fibroblasts (MEFs) and report that Sag is required for proper cell proliferation and KrasG12D-induced immortalization. Sag inactivation by genetic deletion remarkably suppresses cell proliferation by inducing senescence, which is associated with accumulation of p16, but not p53. Mechanistically, Sag deletion caused accumulation of Jun-B, a substrate of Sag-Fbxw7 E3 ligase and a transcription factor that drives p16 transcription. Importantly, senescence triggered by Sag deletion can be largely rescued by simultaneous deletion of Cdkn2a, the p16 encoding gene, indicating its causal role. Furthermore, KrasG12D-induced immortalization can also be abrogated by Sag deletion via senescence induction, which is again rescued by simultaneous deletion of Cdkn2a. Finally, we found that Sag deletion inactivates KrasG12D activity and block the MAPK signaling pathway, together with accumulated p16, to induce senescence. Taken together, our results demonstrated that Sag is a KrasG12D-cooperating oncogene required for KrasG12D-induced immortalization and transformation, and targeting SAG-SCF E3 ligase may, therefore, have therapeutic value for senescence-based cancer treatment.
Introduction
CRL (Cullin-RING ligase) is the multi-complex E3 ubiquitin ligase with SCF (Skp1-Cullin1-F box protein), also known as CRL1, as its founding member. CRL consists of four components: an adaptor protein (e.g. SKP1), one of seven cullin family members (e.g., Cul-1), a substrate recognizing receptor (e.g., F-box protein Skp2), and one of two small RING family proteins: RBX1/ROC1 and SAG/RBX2/ROC2. While the receptor protein determines the substrate specificity, the cullin-RING components constitute the core ubiquitin ligase activity. Activity of CRL also requires cullin neddylation. By promoting the ubiquitylation of various regulatory proteins for targeted degradation by 26S proteasome, CRL regulates many biological processes, including apoptosis, cell cycle progression, signal transduction, DNA replication, embryogenesis, and tumorigenesis [1], [2].
SAG (Sensitive to Apoptosis Gene), also known as RBX2 (RING box protein-2), ROC2 (regulator of cullins-2), or RNF7 (RING finger protein 7), an evolutionarily conserved small RING-containing protein with 113 amino acids, is the second member of the ROC/RBX/RING component of the CRL E3 ubiquitin ligases. In response to various stimuli (e.g., ROS, mitogen and hypoxia), SAG is induced at the transcriptional level by transcription factors AP-1 and HIF1α, respectively. Induced SAG then recruits other components of CRL E3s to promote the ubiquitylation and degradation of various substrates, including c-Jun [3], HIF-1α [4], IκBα [5], [6], Nf-1 [7], NOXA [8], p27 [9], and pro-caspase-3 [10] in a cell context, temporal, and spatial dependent manner. In human tissues, SAG overexpression was detected in carcinomas of lung, colon, stomach and liver, which is associated with poor prognosis in lung cancer patients [8], [11], [12]. In whole animals, SAG over-expression via injection of SAG expressing recombinant adenovirus or transduction of a Tat-SAG fusion protein protected mouse brain tissues from ischemia/hypoxia-induced damage [13], [14]. SAG transgenic expression in mouse skin inhibited tumor formation at the early stage by targeting c-Jun/AP1, but enhanced tumor growth at the later stage by targeting IκBα to activate NFκB in a DMBA-TPA carcinogenesis model [6], and promoted UVB-induced skin hyperplasia by targeting p27 [9]. Targeted Sag deletion in mouse caused embryonic lethality at E11.5-12.5, which is associated with overall growth retardation, induction of apoptosis, and poor vasculogenesis [7]. Conditional Sag deletion in endothelial cells also caused embryonic lethality around E15.5 [15]. Finally, we recently showed that Sag is required for lung tumorigenesis triggered by KrasG12D [16]. However, it is unknown whether Sag is required for proper cell proliferation, or for immortalization, induced by a mutant Kras.
In this study, we investigated these roles of Sag E3 ligase and report here that Sag is required for proliferation and Kras-induced immortalization of mouse embryonic fibroblasts (MEFs). We showed that genetic deletion of Sag remarkably suppresses proliferation and abrogates immortalization by inducing senescence. Mechanistically, we found that Jun-B, a transcription factor, that drives p16 expression, is a novel substrate of Sag E3. Targeted Sag deletion causes accumulation of Jun-B to transactivate p16, which in turn induces senescence. Significantly, simultaneous deletion of Cdkn2a, a gene encoding p16, completely rescued senescence phenotype, regardless of KrasG12D status. We further found that Sag deletion significantly inactivates Mapk signaling pathway by directly inhibiting constitutively active KrasG12D activity. Taken together, our results demonstrated that Sag is a growth essential gene required for cell proliferation as well as for Kras-induced immortalization, indicating that Sag plays a role at very early stage of neoplastic transformation. Thus, Sag targeting may have a value for chemoprevention, as well as for senescence-based cancer therapy, particularly in human cancers harboring a mutant Kras.
Results
Sag Disruption Suppresses Growth of MEFs
We recently found that Sag inactivation via the gene trap (gt) approach induced embryonic lethality at E11.5-12.5 stage, which is associated with growth retardation, induction of apoptosis, and poor vasculogenesis, and attributable at least in part to inactivation of Ras-MAPK signals via Nf1 accumulation.[7] To further define the role of Sag in cell proliferation, we generated primary MEFs and found that while early passage MEF cells from Sag+/+ or Saggt/+ (not shown) mice proliferated well with a doubling time of ~ 48 hours, Saggt/gt MEF cells underwent a complete growth arrest (Figure 1, A and B). To further confirm this finding, we generated MEFs from conditional Sagfl/fl mice. [15], [16] Sagfl/fl MEFs upon infection with Ad-Cre, but not Ad-GFP control, showed a complete elimination of Sag protein (Figure 1C) and complete growth arrest, as measured by ATP-lite proliferation assay (Figure 1D) and BrdU incorporation assay (Figure 1E). Thus, Sag is required for proper growth of MEFs.
Figure 1.

Sag disruption suppresses cell growth: (A and C). MEF cells were generated from E10.5 embryos after intercrossing of Saggt/+ mice (A) or from E13.5 embryos after intercrossing of Sagfl/+ mice, followed by Ad-Cre or Ad-GFP infection (C). MEF cells were then subjected to western blotting for Sag and actin. (B and D) Primary MEFs with indicated genotypes were cultured in a 96-well plate for up to 5 days. The ATP-lite proliferation assay was used to measure cell growth every 24 hours. Shown is mean ± SEM (n = 3). (E) BrdU-based proliferation assay. Data are shown mean ± SEM from three independent experiments. *P < .05. Scale bar represents 100 μm.
Sag Disruption Induces Premature Senescence
Significantly, both Saggt/gt MEFs and Sagfl/fl MEFs after Ad-Cre infection to eliminate Sag demonstrated a flattened and enlarged morphology with a positive p16 staining (Figure 2, A and B), reminiscent of premature senescence [17], [18]. Furthermore, at passage 6, about 55% of Sag-null MEFs was stained positively for senescence-associated β-Gal (SA-β-Gal), as compared to only 2.4% of wild-type MEFs (Figure 2C). Finally, we found that Sag deletion caused accumulation of p16, but not of p15, nor p53/p21 (Figure 2D). Thus, Sag inactivation induces a premature senescence phenotype via the p16 pathway, rather than the p15 pathway, nor the p53/p21 axis.
Figure 2.

Sag disruption induces senescence with p16 accumulation: (A-C). MEF cells with indicated genotypes were grown on coverslips and photographed (top panel) or stained with anti-p16 Ab (bottom panel) (A and B), or SA-β-Gal staining (C). Two independent Sagfl/fl MEFs were either infected with Ad-Cre or Ad-GFP control for 72 hours, followed by IB analysis with indicated Abs (D). **P < .01. Scale bar represents 100 μm.
Sag Disruption Causes Jun-B Accumulation to Transcriptionally Activate p16
Given the fact that p16 contains no lysine residue, it is unlikely that p16 is a direct ubiquitylation substrate of Sag E3 ligase. We then determined if accumulation of p16 protein upon Sag deletion is due to enhanced transcription of p16 mRNA. Indeed, semi-quantitative RT-PCR analysis in two lines of primary MEFs isolated from two independent Sagfl/fl embryos showed that p16 mRNA levels were much higher in Ad-Cre infected MEFs, as compared to MEFs infected with Ad-GFP control (Figure 3A). Thus, regulation of p16 occurred at the transcriptional level.
Figure 3.
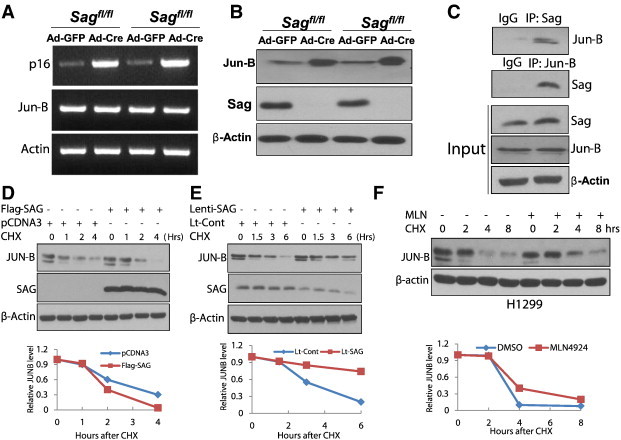
Sag disruption causes Jun-B accumulation to transcriptionally activate p16: Jun-B is a novel substrate of Sag E3. (A-C) Expression of Sag, p16 and Jun-B, and Sag-JunB binding: Two independent Sagfl/fl MEFs were either infected with Ad-Cre or Ad-GFP as control for 72 hours. One portion of cells was subjected to RT-PCR analysis for mRNA expression (A), second portion for IB for protein expression (B). wild-type MEFs were used for immunoprecipitation, followed by IB with indicated Abs (C). (D) SAG overexpression shortens the protein half-life of Jun-B: Flag-SAG plasmid was transfected in H1299 cells. Cells were treated with cycloheximide (CHX; 100 μg/ml) to block new protein synthesis for the indicated time periods and subjected to IB analysis. (E) SAG silencing extends the protein half-life of JunB: H1299 cells were infected with Lenti-SAG or Lenti-GFP as a control, and then treated with CHX (100 μg/ml), cells were harvested at the indicated time points and subjected to IB analysis. (F) MLN4924 extends the Jun-B protein half-life: H1299 cells were pretreated with MLN4924 (1 μM) for 24 hours, and then treated with CHX (100 μg/ml), cells were harvested at the indicated time points and subjected to IB analysis. Densitometry quantification was performed using ImageJ software with β-actin as the loading control.
Given that Sag is the RING component of SCF E3 ubiquitin ligase required for its ligase activity, Sag disruption would be expected to cause the accumulation of its substrates [19]. We next focused on a known substrate of SCFFbxw7 E3 ligase, Jun-B, [20] which is also a transcription factor known to positively regulate p16. [21] Indeed, we found that Sag deletion in MEFs increased Jun-B protein (Figure 3B), but not Jun-B mRNA (Figure 3A), indicating that Sag-mediated regulation occurred likely at the posttranslational level. It has been previously shown that Fbxw7 is the F-box protein that mediated the ubiquitylation and degradation of Jun-B, [20] and that Fbxw7 is associated with Sag for targeted degradation of NF-1[7]. We, therefore, determined whether Sag-Jun-B could form a complex in vivo and found that indeed, endogenous Sag is associated with endogenous Jun-B (Figure 3C). Furthermore, we found Sag overexpression shortened the protein half-life of endogenous JUN-B (Figure 3D), whereas Sag silencing extended it (Figure 3E). Finally, we found that MLN4924, a small molecule inhibitor of NEDD8 activating enzyme, which indirectly inhibits SCF E3 ubiquitin ligase by cullin deneddylation [1], [22], effectively extended the protein half-life of JUN-B (Figure 3F). Taken together, our results demonstrated that Jun-B is a novel substrate of Sag E3 ubiquitin ligase and Jun-B accumulation, as a result of Sag deletion, transactivates p16 to induce senescence.
Senescence Induced by Sag Deletion can be Rescued by Simultaneous Jun-B Silencing or Cdkn2a Deletion
We next investigated whether accumulated Jun-B or p16 plays a causal role in senescence induced by Sag deletion. Indeed, we found that lentivirus-based Jun-B silencing rescued the effect of Sag deletion, as evidenced by abrogation of p16 increase and suppression of senescence (Figure 4, A and B). We further crossed Sagfl/fl mice with Cdkn2a-/- mice and generated Sagfl/fl;Cdkn2a-/- mice. MEFs were generated from these mice, along with Sagfl/fl;Cdkn2a+/+ control mice, and infected with Ad-Cre to delete Sag or Ad-GFP as the control (Figure 4C). Remarkably, simultaneously deletion of Cdkn2a, a gene encoding p16, completed rescued senescence induced by Sag deletion, as measured by SA-β-Gal staining (Figure 4D), as well as by 3T9 protocol which measures cumulative population doubling time by serial passaging [23] (Figure 4E).
Figure 4.

Senescence induced by Sag can be rescued by simultaneous Jun-B silencing or Cdkn2a deletion. MEF cells with indicated genotypes were infected with Ad-Cre or Ad-GFP, followed by transfection with Lenti-Jun-B or Lenti-Cont (scrambled control) [8]. After 3 days, one portion of cells was subjected to IB to measure the levels of Jun-B, p16 and Sag (A), another portion was for SA-β-Gal staining (B). MEF cells with indicated genotypes were infected with Ad-Cre or Ad-GFP, followed by IB for Sag, p16 and actin (C). MEF cells grown on coverslips were photographed after SA-β-Gal staining (D). MEF cells were cultured and passaged according to the 3T9 protocol to measure cellular immortalization and senescence (E). **P < .01. Scale bar represents 100 μm.
Sag Disruption Inhibits KrasG12D-Induced Immortalization by Inducing Senescence
To determine the effect of Sag inactivation on immortalization, triggered by mutant KrasG12D [24], we generated MEFs from compound mice with genotypes of LSL-KrasG12D [25] and Sagfl/+ or Sagfl/fl, respectively. We first confirmed that a) KrasG12D was activated in MEFs after infection of Ad-Cre, but not Ad-GFP control, through the Cre-recombinase mediated removal of Lox-STOP-Lox (LSL) fragment (Figure 5A), and b) Sag was inactivated in MEFs with genotype of Sagfl/fl, but not Sagfl/+ after Ad-Cre infection (Figure 5B). We further observed that upon Ad-Cre infection, MEF cells with KrasG12D;Sagfl/fl genotype showed a reduced growth rate (Figure 5C) with a premature senescence phenotype starting at passage 8, as evidenced by a significant increase of SA-β-Gal positive population (Figure 5D) and by failure in cumulative population doubling, measured by 3T9 protocol (Figure 5E). In contrast, KrasG12D;Sagfl/+ MEF cells proliferated well and showed no sign of senescence up to passage 18 (Figure 5, C–E), indicating an immortalized phenotype, consistent with what was reported for KrasG12D;Sag+/+ MEFs [24]. More specifically, MEFs at passage 8 with Sag inactivation and even in the presence of KrasG12D activation showed a flattened morphology with positive senescence-associated β-Gal (SA-β-Gal) staining [26] (Figure 5D). Thus, Sag is required for KrasG12D-mediated immortalization. Mechanistically, we found that Sag inactivation again fails to cause accumulation of p15 or p53/p21, but does trigger accumulation of p16 (Figure 5F), which is likely contributing to senescence induction [27].
Figure 5.

Sag disruption inhibits KrasG12D-induced immortalization by inducing senescence. (A and B) MEFs with indicated genotypes were infected with Ad-Cre, or Ad-GFP control, followed by PCR-based genotyping and IB to show activation of KrasG12D (A) and inactivation of Sag (B). (C-F) MEF cells following Ad-Cre infection were tested for proliferation by ATP-lite assay. Shown is mean ± SEM (n = 3) (C), senescence by morphological observation and SA-β-Gal staining (D) or by a series dilution using 3T9 protocol (E) and protein expression by IB with indicated Abs in two independent pairs of MEFs (F). **P < .01. Scale bar represents 100 μm.
Blockage of Kras-Induced Immortalization by Sag Disruption can be Rescued by Simultaneous Deletion of Cdkn2a
Given that p16 is accumulated in senescent KrasG12D;Sagfl/fl MEFs, we next determined whether p16 again plays a causal role. We generated MEFs with the following 4 genotypes: 1) KrasG12D;Sagfl/+;Cdkn2a+/+, 2) KrasG12D;Sagfl/+;Cdkn2a-/-, 3) KrasG12D;Sagfl/fl;Cdkn2a+/+, and 4) KrasG12D;Sagfl/fl;Cdkn2a-/- and found Cdkn2a deletion, which caused completely depletion of p16 protein (Figure 6A), rescued decreased growth rate of KrasG12D;Sagfl/fl;Cdkn2a+/+ MEFs (Figure 6B). More importantly, Cdkn2a deletion rescued senescence phenotype seen in KrasG12D;Sagfl/fl;Cdkn2a+/+ MEFs, completely, as measured by the SA-β-Gal staining (Figure 6C), and partially, as measured by the 3T9 protocol (Figure 6D). It is worth noting that the rescue effect appears to be less effective in the presence of KrasG12D, which extended the senescence occurring time from passage 6 to 9 in Sag-null MEFs (compare Figure 4E vs. Figure 6D). Nevertheless, p16 plays a key role in senescence triggered by Sag deletion, largely independent of KrasG12D, although the senescence process is being delayed if KrasG12D is present.
Figure 6.

Cdkn2a deletion rescues the senescence phenotype of KrasG12D;Sag-/- MEFs. MEF cells with indicated genotypes were infected with Ad-Cre, followed by IB for the levels of indicated proteins (A), by ATP-lite assay for proliferation. Data are shown mean ± SEM (n = 3) (B), by SA-β-Gal staining for senescence (C), or by 3T9 protocol for immortalization and senescence (D). **P < .01. Scale bar represents 100 μm.
Sag is Required for the Maintenance of Active Ras-Raf-Erk Signaling Pathway
Finally, we investigated the potential mechanism by which Sag deletion abrogates the ability of active KrasG12D to immortalize primary MEFs. We focused directly on the Kras activity by a classic RBD (Ras-binding domain of Raf-1) pull-down assay and found that Sag deletion significantly reduced Kras activity with no effect on total Ras levels (Figure 7A). Consistent with inactivation of Kras, Ras-Mapk signaling pathway was also inactivated, as evidenced by markedly reduction of Erk phosphorylation (Figure 7A). Few recent studies have shown that Ras/Erk activation is associated with ROS (reactive oxygen species) generation, which could triggers senescence [28], [29], [30], and we have previously shown that Sag has antioxidant activity [5], [31], [32]. We, therefore, used DCFHDA staining to measure ROS levels and found that Sag deletion had no significant effect on ROS generation (Figure 7B), excluding the involvement of ROS in the process. Finally, we measured the levels of several natural occurring inhibitors of Ras signalling pathway, including Erbin [33], Nf1 [7], Rkip [34], Spred2 [35], and Spry2 [36], and found that Sag deletion caused a moderate increase of Nf1, Rkip and Spred2, but not Erbin and Spry2 (Figure 7A), suggesting that Nf1, Rkip and Spred2 may contribute to inactivation of KrasG12D pathway.
Figure 7.

Sag is required for the maintenance of active Ras-Raf-Erk signaling pathway. MEF cells with indicated genotypes were infected with Ad-Cre. One portion of cells was subjected to Ras activity assay and IB analysis (A), another portion of cells for ROS measurement. Data are shown mean ± SEM (n = 3) (B).
Discussion
Sag, a stress inducible protein, has been shown to play a significant role in a variety of cellular processes, including embryogenesis, vasculogenesis, and apoptosis (for review see [19]). Our recent study has shown that Sag is involved in tumor angiogenesis [15] and required for KrasG12D-induced lung tumorigenesis [16]. Here we report a novel finding that Sag genetic inactivation in MEFs induces senescence, as evidenced by flattened cell morphology, enhanced staining of SA-β-Gal and p16. Mechanistically, we found that senescence triggered by Sag deletion is caused by p16, not by the p15 pathway, nor the p53/p21 axis. We further showed that Jun-B is a novel substrate of Sag E3 ubiquitin ligase, as evidenced by a) the binding between Sag and Jun-B under physiological conditions, likely through Fbxw7, since Sag directly binds to Fbxw7 [7], and Fbxw7 binds to Jun-B [20], b) Sag overexpression shortens protein half-life of Jun-B, whereas Sag silencing extends it; and c) pharmaceutical inactivation of Sag E3 by MLN4924 extends Jun-B half-life. Jun-B, upon accumulated following Sag deletion, transactivates p16 expression to induced senescence. Finally we showed by a genetic rescuing experiment that p16 accumulation is the cause, not the consequence, of premature senescence, triggered by Sag deletion. Thus, we identified that the Sag-JunB-p16 axis regulates proper proliferation of MEFs by preventing senescence.
It is well-known that immortalization of primary fibroblasts can be induced by a single oncogene, whereas transformation will require collaboration of two oncogenes or one oncogene in combination with the loss of a tumor suppressor gene [37], [38]. It is also known that overexpression of an active Ras oncogene (Hras or Kras) in primary MEFs induces premature senescence [18], [24], whereas expression of KrasG12D at physiological levels immortalizes MEFs and induces proliferation [24]. In this study, we showed that Sag deletion blocks KrasG12D-induced immortalization again through inducing senescence, although appearance of senescence induced by Sag deletion was delayed. Mechanistically, we showed that Sag is required for the maintenance of KrasG12D activity, since Sag deletion significantly reduces such an activity, leading to inactivation of Ras-Raf-Mapk signaling pathway. Among few naturally occurring inhibitors of Ras, we identified that Sag deletion caused moderate increase of Nf1, Rkip and Spred2. However, accumulated Nf1 is unlikely to inactivate KrasG12D, since Nf1 with GAP activity is a naturally occurring inhibitor of wild-type Ras, but not mutant constitutively active Ras, which has very low intrinsic GTPase activity and insensitive to RasGAPs [39], [40]. Similarly, other two Ras inhibitors, Rkip and Spred2, are not direct inhibitors of KrasG12D, although they block Ras mediated signaling pathway and contribute to its inactivation. Thus, exactly mechanism by which Sag deletion inactivates KrasG12D activity remains elusive. Finally, we showed that abrogation of Kras-induced immortalization by inducing senescence as a result of Sag deletion is causally related to p16, but not to ROS generation, and it can be largely rescued by simultaneous depletion of p16 encoding gene Cdkn2a.
In summary, we showed here that on one hand, Sag promotes cell proliferation by inducing ubiquitylation and degradation of Jun-B to prevent p16 transactivation. On the other hand, Sag cooperates with KrasG12D to trigger immortalization by maintaining an active Ras-Raf-Mapk signaling pathway. Likewise, Sag inactivation by genetic deletion causes Jun-B accumulation to induce p16 expression and subsequent premature senescence. Sag inactivation also blocks the Ras-Raf-Mapk signaling pathway to inhibit proliferation (Figure 8). Future study is directed to elucidate the mechanism by which Sag maintains KrasG12D in a constitutively active form.
Figure 8.

Mechanism of Sag action in senescence and immortalization: Sag prevents senescence by promoting Jun-B ubiquitylation and degradation to block p16 expression, and Sag cooperates with KrasG12D to induce immortalization by maintaining active Kras activity and Mapk signaling pathway. Sag deletion induces senescence by causing Jun-B accumulation to increase p16 expression, and Sag deletion abrogates immortalization by inactivating Kras activity and Mapk signaling pathway (see text for details).
Materials and Methods
Mouse Studies
The Sagfl/fl conditional KO mouse model was generated with exon 1 flanked with loxP sites [16]. Cdkn2a-/- mice were purchased from Jackson laboratories. All procedures were approved by the University of Michigan Committee on Use and Care of Animals. Animal care was provided in accordance with the principles and procedures outlined in the National Research Council Guide for the Care and Use of Laboratory Animals.
Generation and Maintenance of MEFs
MEFs were isolated from day E10.5 or E13.5 embryos, as described [16]. Briefly, the embryos were washed with DMEM supplemented with 25 mM HEPES buffer and PBS. The tissue was then minced with a scalpel and digested with 0.05% trypsin solution containing 0.53 mM EDTA (Invitrogen Life Technologies, Carlsbad, CA) for 20 min at 37°C with vigorous shaking. The tissue mixture was then passed 3 times through an 18G needle to further dissociate any remaining clumps. The cells from each embryo were put into a 100-mm dish and incubated at 37°C in a 5% CO2 humidified incubator. MEF media contained 15% FBS, 2mM L-glutamine, 0.1 mM MEM non-essential amino acids and 10 μg/ml gentamycin.
PCR-Based Genotyping
Genomic DNA was isolated from mouse tail tips and was genotyped using the primer set of PSag-KO-F: 5′-TTCTGGCCAGGTGTGGTGATATC-3′, and PSag-KO-G: 5′-CTTAGCCTT GGTTGTGTAGAC-3′ to detect floxed allele (140 bp) and wild-type allele (105 bp). The primer set for detecting the removal of the Sag targeting fragment (1.3 kp) is PSAG-KO-Seq-B: 5′-GTAACTCCAGACAATGCTCGCT′-3 and PSAG-KO-Seq-R: 5′-TGAGTTCCAGGACAGCCA GGG-3′ with Sag deletion (275 bp) or without Sag deletion (1.6 kb). The primer set for KrasG12D activation is Kras-CreF: 5′-TCCGAATTCAGTGACTACAGA-3′ and Kras-CreR: 5′-CTAGCC ACCATGGTCTGAGT-3′. Unrecombined 2 loxP band is approximately 500 bp, whereas wildtype is 620 bp. Upon recombination, the 500 bp is lost and a 650 bp 1 loxP band is present which represents the recombined Kras mutant allele [16].
ATPlite Based Cell Proliferation Assay
Cells were seeded in 96-well plates in triplicates and cell proliferation was measured with an ATPlite kit (Perkin Elmer, Boston, MA) [41].
BrdU Incorporation Assay
MEFs after 48-hours of infection with Ad-GFP or Ad-Cre were serum starved for 18 hours to allow cells arrested at the G0 phase. BrdU (100 μg/ml) was then added into the culture medium. BrdU incorporation assay was performed as described previously [42] with the following modifications. Cells were fixed in 4% PFA-PBS and BrdU positive cells detected with a 5-Bromo-2′-deoxy-uridine labeling and detection Kit II (Roche, Indianapolis, IN) and counterstained with Eosin-Y.
Immunoblotting Analysis
MEFs Cells were harvested, lysed in a Triton X-100 lysis buffer and subjected to immunoblotting analysis [41]. SAG monoclonal antibody was raised against the RING domain (AA44-113) [8]. Other antibodies were purchased commercially as follows: p21 (BD Transduction Labs, Gibbstown, NJ), p53, Jun-B, p-Erk, Erk, and Rkip (Cell Signaling, Danvers, MA), p16, p15, Nf1, and Spry2 (Santa Cruz Biotechnology, Santa Cruz, CA), and Spred2 and β-Actin (Sigma, St. Louis, MO). Anti-Erbin antibody was a gift from Dr. Lin Mei [33].
Immunohistochemistry Staining
For p16 staining, MEF cells were seeded on a cover slid and grown for few days before being subjected to p16 immunostaining using anti-p16 Ab (Santa Cruz Biotechnology, Santa Cruz, CA) with ABC kits (Vector Labs, Burlingame, CA). The sections were developed with DAB and counterstained with haematoxylin. Normal goat serum was used for negative controls.
SA-β-Galactosidase Staining for Senescence
The MEF cells with various indicated genotypes at passages of 4-7 were seeded in 6-well plate. Cells were grown for 2-days to reach sub-confluency, then subjected to SA-β-gal staining, as described [43].
RT-PCR Analysis
The total RNA was isolated from MEFs after infection with Ad-GFP or Ad-Cre using Trizol (Invitrogen, Carlsbad, CA) and cDNA was made with random primers and SuperScript III reverse transcriptase (Invitrogen, Carlsbad, CA). The sequence of primers used is as follows: Jun-B-F: 5′-GCAGCTACTTTTCGGGTCAG-3′, and Jun-B-R: 5′-TTCATCTTGTGCAGGT CGTC-3′. P16-F: 5′-GAACTCTTTCGGTCGTACCC-3′, and P16-R: 5′-CGAATCTGCACCG TAGTTGA-3′. Actin-F: 5′-CACAGCTTCTTTGCAGCTCCTT-3′ and Actin-R: 5′-CGTCATCC ATGGCGAACTG-3′.
Ras Activity Assay
Ras activity was measured by using Ras binding domain of Raf (RBD) pull-down assay kit (Millipore, Billerica, MA), following manufacturer's instruction.
ROS Measurement
The MEF cells with various indicated genotypes at passage 4 to 7 were seeded in 60-mm dish. Cells were grown for 2-days to reach sub-confluency and stained with 20 μM DCFHDA for 30 min in the dark and analyzed using the flow cytometer equipped with a 488-nm argon laser as a light source to determine the DCF fluorescence intensity. The green fluorescence was measured in the FL1 (FITC) channel. The mean fluorescence intensity (MFI) of 10,000 cells was analyzed by WinMDI 2.8 software. The MFI data were normalized to control levels and expressed as relative fluorescence intensity (DCF) [44].
Statistical Analysis
Statistical analysis was performed using two-tailed Student's t-test. All statistical analyses were carried out using the GraphPad Prism software version 5.01 (GraphPad, San Diego, CA). Data were expressed as mean ± SEM of at least 3 independent experiments. P < .05 was considered statistically significant.
Acknowledgements
This work is supported by the NCI grants (CA118762, CA156744 and CA171277) to YS.
Contributor Information
Mingjia Tan, Email: mjtan@umich.edu.
Yi Sun, Email: sunyi@umich.edu, yisun@zju.edu.cn.
References
- 1.Zhao Y., Sun Y. Cullin-RING ligases as attractive anti-cancer targets. Curr Pharm Des. 2013;19:3215–3225. doi: 10.2174/13816128113199990300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Deshaies R.J. SCF and Cullin/Ring H2-based ubiquitin ligases. Annu Rev Cell Dev Biol. 1999;15:435–467. doi: 10.1146/annurev.cellbio.15.1.435. [DOI] [PubMed] [Google Scholar]
- 3.Gu Q., Tan M., Sun Y. SAG/ROC2/Rbx2 is a novel activator protein-1 target that promotes c-Jun degradation and inhibits 12-O-tetradecanoylphorbol-13-acetate-induced neoplastic transformation. Cancer Res. 2007;67:3616–3625. doi: 10.1158/0008-5472.CAN-06-4020. [DOI] [PubMed] [Google Scholar]
- 4.Tan M., Gu Q., He H., Pamarthy D., Semenza G.L., Sun Y. SAG/ROC2/RBX2 is a HIF-1 target gene that promotes HIF-1alpha ubiquitination and degradation. Oncogene. 2008;27:1404–1411. doi: 10.1038/sj.onc.1210780. [DOI] [PubMed] [Google Scholar]
- 5.Tan M., Zhu Y., Kovacev J., Zhao Y., Pan Z.Q., Spitz D.R., Sun Y. Disruption of Sag/Rbx2/Roc2 induces radiosensitization by increasing ROS levels and blocking NF-kB activation in mouse embryonic stem cells. Free Radic Biol Med. 2010;49:976–983. doi: 10.1016/j.freeradbiomed.2010.05.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gu Q., Bowden G.T., Normolle D., Sun Y. SAG/ROC2 E3 ligase regulates skin carcinogenesis by stage-dependent targeting of c-Jun/AP1 and IkappaB-alpha/NF-kappaB. J Cell Biol. 2007;178:1009–1023. doi: 10.1083/jcb.200612067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tan M., Zhao Y., Kim S.J., Liu M., Jia L., Saunders T.L., Zhu Y., Sun Y. SAG/RBX2/ROC2 E3 ubiquitin ligase is essential for vascular and neural development by targeting NF1 for degradation. Dev Cell. 2011;21:1062–1076. doi: 10.1016/j.devcel.2011.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jia L., Yang J., Hao X., Zheng M., He H., Xiong X., Xu L., Sun Y. Validation of SAG/RBX2/ROC2 E3 ubiquitin ligase as an anticancer and radiosensitizing target. Clin Cancer Res. 2010;16:814–824. doi: 10.1158/1078-0432.CCR-09-1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.He H., Gu Q., Zheng M., Normolle D., Sun Y. SAG/ROC2/RBX2 E3 ligase promotes UVB-induced skin hyperplasia, but not skin tumors, by simultaneously targeting c-Jun/AP-1 and p27. Carcinogenesis. 2008;29:858–865. doi: 10.1093/carcin/bgn021. [DOI] [PubMed] [Google Scholar]
- 10.Tan M., Gallegos J.R., Gu Q., Huang Y., Li J., Jin Y., Lu H., Sun Y. SAG/ROC-SCFbeta-TrCP E3 ubiquitin ligase promotes pro-caspase-3 degradation as a mechanism of apoptosis protection. Neoplasia. 2006;8:1042–1054. doi: 10.1593/neo.06568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Huang Y., Duan H., Sun Y. Elevated expression of SAG/ROC2/Rbx2/Hrt2 in human colon carcinomas: SAG does not induce neoplastic transformation, but its antisense transfection inhibits tumor cell growth. Mol Carcinog. 2001;30:62–70. [PubMed] [Google Scholar]
- 12.Sasaki H., Yukiue H., Kobayashi Y., Moriyama S., Nakashima Y., Kaji M., Fukai I., Kiriyama M., Yamakawa Y., Fujii Y. Expression of the sensitive to apoptosis gene, SAG, as a prognostic marker in nonsmall cell lung cancer. Int J Cancer. 2001;95:375–377. doi: 10.1002/1097-0215(20011120)95:6<375::aid-ijc1066>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- 13.Yang G.Y., Pang L., Ge H.L., Tan M., Ye W., Liu X.H., Huang F.P., Wu D.C., Che X.M., Song Y. Attenuation of ischemia-induced mouse brain injury by SAG, a redox- inducible antioxidant protein. J Cereb Blood Flow Metab. 2001;21:722–733. doi: 10.1097/00004647-200106000-00010. [DOI] [PubMed] [Google Scholar]
- 14.Kim D.W., Lee S.H., Jeong M.S., Sohn E.J., Kim M.J., Jeong H.J., An J.J., Jang S.H., Won M.H., Hwang I.K. Transduced Tat-SAG fusion protein protects against oxidative stress and brain ischemic insult. Free Radic Biol Med. 2010;48:969–977. doi: 10.1016/j.freeradbiomed.2010.01.023. [DOI] [PubMed] [Google Scholar]
- 15.Tan M., Li H., Sun Y. Endothelial deletion of Sag/Rbx2/Roc2 E3 ubiquitin ligase causes embryonic lethality and blocks tumor angiogenesis. Oncogene. 2013 doi: 10.1038/onc.2013.473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li H., Tan M., Jia L., Wei D., Zhao Y., Chen G., Xu J., Zhao L., Thomas D., Beer D.G. Inactivation of SAG/RBX2 E3 ubiquitin ligase suppresses KrasG12D-driven lung tumorigenesis. J Clin Invest. 2014;124:835–846. doi: 10.1172/JCI70297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Campisi J., d'Adda di Fagagna F. Cellular senescence: when bad things happen to good cells. Nat Rev Mol Cell Biol. 2007;8:729–740. doi: 10.1038/nrm2233. [DOI] [PubMed] [Google Scholar]
- 18.Serrano M., Lin A.W., McCurrach M.E., Beach D., Lowe S.W. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell. 1997;88:593–602. doi: 10.1016/s0092-8674(00)81902-9. [DOI] [PubMed] [Google Scholar]
- 19.Sun Y., Li H. Functional characterization of SAG/RBX2/ROC2/RNF7, an antioxidant protein and an E3 ubiquitin ligase. Protein Cell. 2013;4:103–116. doi: 10.1007/s13238-012-2105-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Perez-Benavente B., Garcia J.L., Rodriguez M.S., Pineda-Lucena A., Piechaczyk M., Font de Mora J., Farras R. GSK3-SCF(FBXW7) targets JunB for degradation in G2 to preserve chromatid cohesion before anaphase. Oncogene. 2013;32:2189–2199. doi: 10.1038/onc.2012.235. [DOI] [PubMed] [Google Scholar]
- 21.Passegue E., Wagner E.F. JunB suppresses cell proliferation by transcriptional activation of p16(INK4a) expression. EMBO J. 2000;19:2969–2979. doi: 10.1093/emboj/19.12.2969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Soucy T.A., Smith P.G., Milhollen M.A., Berger A.J., Gavin J.M., Adhikari S., Brownell J.E., Burke K.E., Cardin D.P., Critchley S. An inhibitor of NEDD8-activating enzyme as a new approach to treat cancer. Nature. 2009;458:732–736. doi: 10.1038/nature07884. [DOI] [PubMed] [Google Scholar]
- 23.Hayflick L., Moorhead P.S. The serial cultivation of human diploid cell strains. Exp Cell Res. 1961;25:585–621. doi: 10.1016/0014-4827(61)90192-6. [DOI] [PubMed] [Google Scholar]
- 24.Tuveson D.A., Shaw A.T., Willis N.A., Silver D.P., Jackson E.L., Chang S., Mercer K.L., Grochow R., Hock H., Crowley D. Endogenous oncogenic K-ras(G12D) stimulates proliferation and widespread neoplastic and developmental defects. Cancer Cell. 2004;5:375–387. doi: 10.1016/s1535-6108(04)00085-6. [DOI] [PubMed] [Google Scholar]
- 25.Jackson E.L., Willis N., Mercer K., Bronson R.T., Crowley D., Montoya R., Jacks T., Tuveson D.A. Analysis of lung tumor initiation and progression using conditional expression of oncogenic K-ras. Genes Dev. 2001;15:3243–3248. doi: 10.1101/gad.943001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Itahana K., Campisi J., Dimri G.P. Methods to detect biomarkers of cellular senescence: the senescence-associated beta-galactosidase assay. Methods Mol Biol. 2007;371:21–31. doi: 10.1007/978-1-59745-361-5_3. [DOI] [PubMed] [Google Scholar]
- 27.Deng Y., Chan S.S., Chang S. Telomere dysfunction and tumour suppression: the senescence connection. Nat Rev Cancer. 2008;8:450–458. doi: 10.1038/nrc2393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Qi M., Zhou H., Fan S., Li Z., Yao G., Tashiro S., Onodera S., Xia M., Ikejima T. mTOR inactivation by ROS-JNK-p53 pathway plays an essential role in Psedolaric acid B induced autophagy-dependent senescence in murine fibrosarcoma L929 cells. Eur J Pharmacol. 2013;715:76–88. doi: 10.1016/j.ejphar.2013.05.051. [DOI] [PubMed] [Google Scholar]
- 29.Zamkova M., Khromova N., Kopnin B.P., Kopnin P. Ras-induced ROS upregulation affecting cell proliferation is connected with cell type-specific alterations of HSF1/SESN3/p21Cip1/WAF1 pathways. Cell Cycle. 2013;12:826–836. doi: 10.4161/cc.23723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Moon E.J., Sonveaux P., Porporato P.E., Danhier P., Gallez B., Batinic-Haberle I., Nien Y.C., Schroeder T., Dewhirst M.W. NADPH oxidase-mediated reactive oxygen species production activates hypoxia-inducible factor-1 (HIF-1) via the ERK pathway after hyperthermia treatment. Proc Natl Acad Sci U S A. 2010;107:20477–20482. doi: 10.1073/pnas.1006646107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Swaroop M., Bian J., Aviram M., Duan H., Bisgaier C.L., Loo J.A., Sun Y. Expression, purification, and biochemical characterization of SAG, a RING finger redox sensitive protein. Free Radic Biol Med. 1999;27:193–202. doi: 10.1016/s0891-5849(99)00078-7. [DOI] [PubMed] [Google Scholar]
- 32.Duan H., Wang Y., Aviram M., Swaroop M., Loo J.A., Bian J., Tian Y., Mueller T., Bisgaier C.L., Sun Y. SAG, a novel zinc RING finger protein that protects cells from apoptosis induced by redox agents. Mol Cell Biol. 1999;19:3145–3155. doi: 10.1128/mcb.19.4.3145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dai P., Xiong W.C., Mei L. Erbin inhibits RAF activation by disrupting the sur-8-Ras-Raf complex. J Biol Chem. 2006;281:927–933. doi: 10.1074/jbc.M507360200. [DOI] [PubMed] [Google Scholar]
- 34.Hagan S., Garcia R., Dhillon A., Kolch W. Raf kinase inhibitor protein regulation of raf and MAPK signaling. Methods Enzymol. 2006;407:248–259. doi: 10.1016/S0076-6879(05)07021-7. [DOI] [PubMed] [Google Scholar]
- 35.Yoshida T., Hisamoto T., Akiba J., Koga H., Nakamura K., Tokunaga Y., Hanada S., Kumemura H., Maeyama M., Harada M. Spreds, inhibitors of the Ras/ERK signal transduction, are dysregulated in human hepatocellular carcinoma and linked to the malignant phenotype of tumors. Oncogene. 2006;25:6056–6066. doi: 10.1038/sj.onc.1209635. [DOI] [PubMed] [Google Scholar]
- 36.Guy G.R., Jackson R.A., Yusoff P., Chow S.Y. Sprouty proteins: modified modulators, matchmakers or missing links? J Endocrinol. 2009;203:191–202. doi: 10.1677/JOE-09-0110. [DOI] [PubMed] [Google Scholar]
- 37.Land H., Parada L.F., Weinberg R.A. Tumorigenic conversion of primary embryo fibroblasts requires at least two cooperating oncogenes. Nature. 1983;304:596–602. doi: 10.1038/304596a0. [DOI] [PubMed] [Google Scholar]
- 38.Serrano M., Lee H., Chin L., Cordon-Cardo C., Beach D., DePinho R.A. Role of the INK4a locus in tumor suppression and cell mortality. Cell. 1996;85:27–37. doi: 10.1016/s0092-8674(00)81079-x. [DOI] [PubMed] [Google Scholar]
- 39.Cichowski K., Jacks T. NF1 tumor suppressor gene function: narrowing the GAP. Cell. 2001;104:593–604. doi: 10.1016/s0092-8674(01)00245-8. [DOI] [PubMed] [Google Scholar]
- 40.Le L.Q., Parada L.F. Tumor microenvironment and neurofibromatosis type I: connecting the GAPs. Oncogene. 2007;26:4609–4616. doi: 10.1038/sj.onc.1210261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bockbrader K.M., Tan M., Sun Y. A small molecule Smac-mimic compound induces apoptosis and sensitizes TRAIL- and etoposide-induced apoptosis in breast cancer cells. Oncogene. 2005;24:7381–7388. doi: 10.1038/sj.onc.1208888. [DOI] [PubMed] [Google Scholar]
- 42.Tan M., Davis S.W., Saunders T.L., Zhu Y., Sun Y. RBX1/ROC1 disruption results in early embryonic lethality due to proliferation failure, partially rescued by simultaneous loss of p27. Proc Natl Acad Sci U S A. 2009;106:6203–6208. doi: 10.1073/pnas.0812425106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jia L., Li H., Sun Y. Induction of p21-dependent senescence by an NAE inhibitor, MLN4924, as a mechanism of growth suppression. Neoplasia. 2011;13:561–569. doi: 10.1593/neo.11420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Xie C.M., Chan W.Y., Yu S., Zhao J., Cheng C.H. Bufalin induces autophagy-mediated cell death in human colon cancer cells through reactive oxygen species generation and JNK activation. Free Radic Biol Med. 2011;51:1365–1375. doi: 10.1016/j.freeradbiomed.2011.06.016. [DOI] [PubMed] [Google Scholar]