

Abstract
The recently discovered seventh order of methanogens, the Methanomassiliicoccales (previously referred to as “Methanoplasmatales”), so far consists exclusively of obligately hydrogen-dependent methylotrophs. We sequenced the complete genome of “Candidatus Methanoplasma termitum” from a highly enriched culture obtained from the intestinal tract of termites and compared it with the previously published genomes of three other strains from the human gut, including the first isolate of the order. Like all other strains, “Ca. Methanoplasma termitum” lacks the entire pathway for CO2 reduction to methyl coenzyme M and produces methane by hydrogen-dependent reduction of methanol or methylamines, which is consistent with additional physiological data. However, the shared absence of cytochromes and an energy-converting hydrogenase for the reoxidation of the ferredoxin produced by the soluble heterodisulfide reductase indicates that Methanomassiliicoccales employ a new mode of energy metabolism, which differs from that proposed for the obligately methylotrophic Methanosphaera stadtmanae. Instead, all strains possess a novel complex that is related to the F420:methanophenazine oxidoreductase (Fpo) of Methanosarcinales but lacks an F420-oxidizing module, resembling the apparently ferredoxin-dependent Fpo-like homolog in Methanosaeta thermophila. Since all Methanomassiliicoccales also lack the subunit E of the membrane-bound heterodisulfide reductase (HdrDE), we propose that the Fpo-like complex interacts directly with subunit D, forming an energy-converting ferredoxin:heterodisulfide oxidoreductase. The dual function of heterodisulfide in Methanomassiliicoccales, which serves both in electron bifurcation and as terminal acceptor in a membrane-associated redox process, may be a unique characteristic of the novel order.
INTRODUCTION
Methanogenesis is catalyzed exclusively by members of the archaeal domain. Methanogenic archaea occur only in the phylum Euryarchaeota and are phylogenetically diverse. The species described to date fall into seven orders that differ both in the biochemistry of their catabolic pathways and in their ecological niches (1, 2).
Methanogens from all basal orders (Methanopyrales, Methanococcales, and Methanobacteriales) are hydrogenotrophs. They reduce CO2 to CH4 via the C1 pathway, using H2 or sometimes formate as an electron donor (1, 2). The hydrogenotrophic pathway is found also in most of the derived lineages of methanogens (Methanomicrobiales and Methanocellales) and was most probably present already in the common ancestor of the Euryarchaeota (3). Hydrogenotrophic methanogens typically lack cytochromes and conserve energy with the methyltetrahydromethanopterin (methyl-H4MPT):coenzyme M methyltransferase complex (Mtr), which uses the free energy of methyl transfer to establish a Na+-motive force across the membrane (4). The low-potential reducing equivalents for CO2 reduction are provided by electron bifurcation at the cytoplasmic heterodisulfide reductase complex (HdrABC) (5, 6).
Members of the order Methanosarcinales are the only methanogens that possess cytochromes (2). They have an entirely different mode of energy conservation, which involves a membrane-bound electron transport chain that couples heterodisulfide reduction to the generation of an electrochemical proton gradient (7), which is more efficient than electron bifurcation and allows a higher growth yield. In this way, they can grow (i) on H2 and CO2, (ii) on the methyl groups of methanol or methylamines, which are partially oxidized to CO2 in order to provide reducing equivalents for methyl reduction, and (iii) by disproportionation of acetate, in which methyl groups are reduced to methane with electrons derived from the oxidation of the carbonyl group to CO2 (1, 2).
A third group of methanogens is restricted to growth on methanol and methylamines but lacks the ability to oxidize the latter to CO2, which makes methanogenesis obligately dependent on molecular hydrogen. The group is phylogenetically and biochemically heterogeneous, comprising Methanosphaera stadtmanae (Methanobacteriales) (8, 9), Methanomicrococcus blatticola (Methanosarcinales) (10, 11), and members of the recently discovered seventh order of methanogens (12–14), for which we had originally suggested the provisional name “Methanoplasmatales” based on their close phylogenetic relationship to the nonmethanogenic Thermoplasmatales (12). Although this provisional name had been readily adopted by numerous authors, the Bacteriological Code (15) dictates that the taxonomic name, no matter how unwieldy, must be derived from the genus name of the first isolate in the order, in this case Methanomassiliicoccus (Mmc.) luminyensis (13). Therefore, this publication uses the validly published name Methanomassiliicoccales (16) for the seventh order of methanogens.
So far, the Methanomassiliicoccales comprise only the type species, Mmc. luminyensis, and several, in part highly enriched, cultures from the intestinal tracts of termites (12), humans (17, 18), and an anaerobic digester (19). The consistent presence of the mcrA gene (encoding the alpha-subunit of methyl coenzyme M [methyl-CoM] reductase) indicates that also the lineages without cultured representatives are methanogenic (12) (Fig. 1).
FIG 1.

Phylogenetic tree of the major lineages in the class Thermoplasmata, illustrating the relationship among the mostly uncultivated members of the order Methanomassiliicoccales and to other lineages of the Euryarchaeota. Methanogenic lineages are shaded in gray. Strains with published genomes or draft genomes are in bold. The original tree is based on an unambiguous alignment of more than 300 16S rRNA genes (>1,250 nucleotide positions) and was reconstructed using a maximum-likelihood algorithm (RAxML). The tree was rooted using representatives of other methanogenic lineages. Nodes not supported by neighbor-joining and maximum-parsimony (MP) analyses are shown as multifurcations; highly supported nodes (100 bootstraps, MP) are marked (solid circles, >95%; open circles, >70%). Scale bar indicates 0.1 substitutions per site.
Meanwhile, genome sequences of three members of the Methanomassiliicoccales have been reported. While the genome sequences of the enrichment cultures of “Candidatus Methanomethylophilus alvus” (17) and “Candidatus Methanomassiliicoccus intestinalis” (18) are complete and annotated, that of the type strain, Mmc. luminyensis (20), remains to be finished. All strains lack the genes encoding the entire C1 pathway for the reduction of CO2 to methyl-CoM but possess the complete gene sets for the utilization of methanol and methylamines (14). This explains the strict dependence of methanogenesis on the simultaneous presence of hydrogen and methanol or trimethylamine documented for Mmc. luminyensis (21).
However, the fundamental consequences of the absence of the C1 pathway for the energy metabolism of the Methanomassiliicoccales have so far escaped attention. Without formylmethanofuran dehydrogenase and an energy-converting Mtr complex, the reoxidation of reduced ferredoxin formed during heterodisulfide reduction and the strategy for energy conversion in Methanomassiliicoccales must differ fundamentally from that in other methanogens.
In this study, we analyzed the genome of “Candidatus Methanoplasma termitum” strain MpT1, which we previously enriched from a termite gut, and compared it to the genomes of its three distant relatives of the order Methanomassiliicoccales that stem from the human intestinal tract. Shortly before submission of this work, Borrel et al. (22) reported a comprehensive analysis of the three previously published genomes that also addressed the evolution of methanogenesis and reevaluates the core genome of methanogens. Our report focuses on the energy metabolism of Methanomassiliicoccales, which differs fundamentally from that of the other orders. The genome analysis is supported by new physiological data documenting differences in the methanogenic substrates of Mmc. luminyensis and “Ca. Methanoplasma termitum.” Moreover, we present ultrastructural data for “Ca. Methanoplasma termitum” that provide new information on the unusual cell envelope of Methanomassiliicoccales.
MATERIALS AND METHODS
Strains.
The highly enriched cultures of “Ca. Methanoplasma termitum” strain MpT1 and the closely related strain MpM2 were obtained in a previous study (12). Mmc. luminyensis (DSMZ 25720) was purchased from the German Collection of Microorganism and Cell Cultures (http://www.dsmz.de/).
Cultivation.
Cultures were grown in anoxic, bicarbonate-buffered mineral medium (AM5) (23) under an atmosphere of N2-CO2 (80:20 [vol/vol]) with dithiothreitol (1 mM) as a reducing agent (12). The basal medium was supplemented with Casamino Acids (2 g/liter), yeast extract (2 g/liter), 2-mercaptoethanesulfonate (10 mg/liter), acetate (1 mM), and formate (0.5 mM). The medium (4.5 ml) was dispensed into 15-ml rubber-stoppered glass vials. Substrates were added from sterile stock solutions, and hydrogen gas (5 ml) was added to the headspace. Tubes were inoculated (0.3 ml) with methanol-starved precultures and incubated at 30°C in the dark. At regular intervals, aliquots of the headspace (0.2 ml) were sampled with a gas-tight syringe, and the methane content was analyzed using a gas chromatograph equipped with a packed column (Porapak Q, 80/100 mesh, 274 cm by 3.18 mm [inside diameter]) and a flame ionization detector.
Light microscopy.
Cells in 300-μl culture aliquots were concentrated by centrifugation at 10,000 × g for 10 min and routinely inspected by phase-contrast microscopy using an Axiophot epifluorescence microscope (Zeiss, Wetzlar, Germany). Autofluorescence of cofactor F420 was tested using an HC filter set (F36-544; AHF Analysentechnik, Tübingen, Germany) with bandpass filters (wavelength/bandwidth: excitation, 438/24 nm, beam splitter, 458/− nm; emission, 483/32 nm).
Electron microscopy.
For negative stains, fresh cultures were chemically fixed with 1.25% glutaraldehyde and concentrated by centrifugation (see above). Aliquots (5 μl) were applied to carbon-coated copper grids and stained as previously described (24). For ultrastructural characterization, 2 μl of concentrated but unfixed cells was frozen under high pressure, freeze substituted, embedded in Epon resin, ultrathin sectioned, and poststained as described previously (25). Freeze substitution was performed with acetone containing 0.2% OsO4, 0.25% uranyl acetate, and 5% water. Transmission electron microscopy was carried out on a JEOL JEM2100 (JEOL, Tokyo, Japan) equipped with an LaB6 cathode and operated at 120 kV. Images were recorded using a 2k × 2k fast-scan charge-coupled-device (CCD) camera F214 in combination with the EM-Menu4 software package (TVIPS, Gauting, Germany).
Phylogenetic analysis.
16S rRNA gene sequences were retrieved from GenBank (http://www.ncbi.nlm.nih.gov/) and imported into the current Silva database (version 115; http://www.arb-silva.de) (26) using the ARB software package (27). The automatic alignment was manually refined, and a 30% consensus filter was used to exclude highly variable positions. Phylogenetic trees of near-full-length sequences (>1,250 bp) were calculated using PhyML (28), a maximum-likelihood method implemented in ARB. Tree topology and node support (100 bootstraps) were tested using the maximum-parsimony method (DNAPARS) implemented in ARB.
For phylogenetic analysis of the large subunit of the 11-subunit complex, complex 1 of the respiratory chain, F420H2 dehydrogenases, and [NiFe] hydrogenases, sequences were retrieved from the IMG database (https://img.jgi.doe.gov/cgi-bin/w/main.cgi) and analyzed using the Mega5 software package (http://www.megasoftware.net/). Sequences were automatically aligned with the ClustalW function implemented in Mega5. The alignment was manually refined in ARB. Trees were calculated based on the deduced amino acid sequence using PhyML. Tree topology and node support (100 bootstraps) were tested using the maximum-parsimony method (PROTPARS) implemented in ARB.
Genome sequencing.
The genome of strain MpT1 was sequenced using a combined 454 pyrosequencing and Sanger sequencing approach. DNA was isolated from the enrichment culture by detergent extraction (cetyltrimethylammonium bromide [CTAB] method) (29) and used to generate a 454 shotgun library according to the GS Rapid Library protocol, which was sequenced with the Genome Sequencer FLX+ system (454 Life Sciences, Roche Applied Science, Branford, CT) using titanium chemistry. In total, 107,475 shotgun reads were generated and assembled de novo into 72 large contigs (>500 bp) with an average coverage of 37-fold using Roche Newbler assembler software 2.6. Sequences were edited and final gaps were closed as previously described (30).
Sequence annotation.
All genome sequences were uploaded to the Integrated Microbial Genomes Expert Review (IMG/ER) platform (31, https://img.jgi.doe.gov/cgi-bin/er/main.cgi). In the case of Ca. “Methanomassiliicoccus intestinalis” and “Ca. Methanomethylophilus alvus,” the original RAST annotations in the GenBank entry were preserved. In the case of “Ca. Methanoplasma termitum” and Mmc. luminyensis, coding sequences were predicted and annotated using the automated pipeline of IMG/ER. Briefly, protein-coding genes were identified with GeneMark, and candidate homolog genes of the genomes were computed using BLASTp. Automated annotations of coding sequences were verified and curated by comparing various annotations based on functional resources, such as COG clusters (32), Pfam (33), TIGRfam (34), and Gene Ontology (35). In addition, genes were associated with gene product names in the Swiss-Prot database (36), EC numbers (37), KEGG orthology terms (38), COG functional categories, KEGG categories (38), and MetaCyc pathway collections (39). The annotated genome sequences of “Ca. Methanoplasma termitum” (Gi21292) and Mmc. luminyensis (Gi17673) are available in the Genomes Online database (http://www.genomesonline.org/).
Nucleotide sequence accession number.
The annotated genome of “Ca. Methanoplasma termitum” was deposited in GenBank under accession number CP010070.
RESULTS AND DISCUSSION
Genome characteristics.
The genome of “Candidatus Methanoplasma termitum” strain MpT1 is the fourth genome sequence reported for a member of the Methanomassiliicoccales. A fifth genome, for strain BRNA1, has been obtained from a rumen enrichment culture (S. E. Denman, P. Evans, L. Bragg, J. Padmanahba, M. McKenzie, D. Edwards, J. Krzycki, C. McSweeney, and M. Morrison, unpublished data). The sequence is available in GenBank (CP002916) but was not included in our analysis since the publication is still pending.
“Ca. Methanoplasma termitum” has a circular genome with a size of 1.49 Mbp; there is no evidence for the presence of plasmids. The genome is even smaller than that of “Ca. Methanomethylophilus alvus” (1.66 Mbp) and slightly larger than that of strain BRNA1 (1.46 Mbp), which are close relatives in the intestinal cluster (rumen cluster C) (Fig. 1). The more distantly related members of the genus Methanomassiliicoccus have considerably larger genomes (1.93 Mbp in “Ca. Methanomassiliicoccus intestinalis” and >2.62 Mbp in the unfinished genome of Mmc. luminyensis). The coding densities of all genomes are similar, with about 1,000 bp per gene. The G+C contents of the strains differ strongly but do not correlate with phylogenetic distance. Details are presented in Table 1.
TABLE 1.
Comparison of the genome features of “Ca. Methanoplasma termitum” and other members of the order Methanomassiliicoccalesa
| Parameter | Intestinal cluster |
Methanomassiliicoccaceae |
||
|---|---|---|---|---|
| “Ca. Methanoplasma termitum” strain MpT1 | “Ca. Methanomethylophilus alvus” strain Mx1201 | Mmc. luminyensis strain B10 | “Ca. Methanomassiliicoccus intestinalis” strain Mx1 | |
| Isolation source | Termite gut (12) | Human feces (17) | Human feces (13) | Human feces (18) |
| Accession no. (GenBank) | CP010070 | CP004049 | CAJE01000001 to -26b | CP005934 |
| GOLD IDc | Gi21292 | Gc0042696 | Gi17673 | Gc0050196 |
| Genome size (bp) | 1,488,669 | 1,666,795 | >2,620,233b | 1,931,651 |
| G+C content (mol%) | 49.2 | 55.6 | 60.5 | 41.3 |
| No. of protein-coding genes | 1,415 | 1,653 | 2,625 | 1,826 |
| No. of rRNA genes | 3 | 4d | 4d | 4d |
| No. of tRNA genes | 46 | 48e | 48f | 46 |
Genome features of “Ca. Methanomethylophilus alvus,” Mmc. luminyensis, and “Ca. Methanomassiliicoccus intestinalis” are based on the latest report by Borrel et al. (22).
The draft genome consists of 26 scaffolds.
Accession number in the Genomes Online database (http://genomesonline.org).
Genome contains two copies of the 5S rRNA gene.
Three of the 48 tRNA genes are pseudogenized.
Five of the 48 tRNA genes are pseudogenized. The original genome announcement (13) reported 42 tRNAs.
rRNA operon structure.
“Ca. Methanoplasma termitum” has a single set of rRNA genes, which are located in different regions of the chromosome. The rRNA genes in most other methanogens are organized in an operon, but a separation of 5S, 16S, and 23S rRNA genes has been reported also for other members of the Methanomassiliicoccales (17, 18, 20). Since the same feature is present also in Thermoplasma acidophilum (40) and the deep-branching “Candidatus Aciduliprofundum boonei” strain T469 (GenBank accession number CP001941.1), it may be a trait shared by all members of the class Thermoplasmata (Fig. 1). A second copy of the 5S rRNA gene, which is encountered in all other Methanomassiliicoccales, is absent in “Ca. Methanoplasma termitum.”
tRNAs.
The genome of “Ca. Methanoplasma termitum” encodes 46 tRNAs for all amino acids (see Table S1 in the supplemental material). The same number of tRNAs is present in “Ca. Methanomassiliicoccus intestinalis” (18). “Ca. Methanomethylophilus alvus” and Mmc. luminyensis have two additional tRNA genes, but several tRNA genes are pseudogenized, and the draft genome of Mmc. luminyensis lacks a tRNA for lysine (Table 1). Like the three other strains, “Ca. Methanoplasma termitum” possesses a tRNA for pyrrolysine, the corresponding tRNAPyl synthetase, and all enzymes for the biosynthesis of pyrrolysine (41), which is required for growth on methylamines (see below). Pyrrolysine operon structure and position of the tRNAPyl gene are the same as in Mmc. luminyensis and “Ca. Methanomassiliicoccus intestinalis” (42). A tRNA gene for selenocysteine is not present in any of the genomes.
The methyl-reducing pathway.
Like all other members of Methanomassiliicoccales, “Ca. Methanoplasma termitum” possesses the complete gene sets encoding methanol transferase (MtaABC) and methyl-CoM reductase (McrABCDG), the key enzymes of the methyl reduction pathway (43). Also, a methyl viologen-dependent hydrogenase/heterodisulfide reductase complex (MvhADG/HdrABC), which is required for the regeneration of CoM, is encoded by the genome (Fig. 2). The genes required for the reduction of CO2 to the methyl level, however, are lacking in all strains.
FIG 2.

Energy metabolism of “Ca. Methanoplasma termitum” and other members of the order Methanomassiliicoccales. Black arrows indicate reactions whose enzymes are encoded in all genomes; red arrows indicate the proposed reaction of the heterodisulfide reductase (HdrD) coupled to the Fpo-like complex (Fig. 6). Blue-green arrows indicate that the enzymes are not present in “Ca. Methanoplasma termitum” but are present in the genomes indicated by colored dots (blue, Methanomassiliicoccus luminyensis; green, “Ca. Methanomassiliicoccus intestinalis,” red, “Ca. Methanomethylophilus alvus”). Abbreviations: Mta, methanol:CoM methyltransferase; Mvh, non-F420-reducing hydrogenase; Hdr, heterodisulfide reductase; Mcr, methyl-CoM reductase; Fpo-like, F420H2-dehydrogenase-like complex; MtbA, methylcobamide:CoM methyltransferase; Mtm, monomethylamine methyltransferase; Mtb, dimethylamine methyltransferase; Mtt, trimethylamine methyltransferase; Aha, AoA1-ATP synthase.
The conspicuous absence of the CO2 reduction pathway in all Methanomassiliicoccales (14; also this study) is in agreement with the obligate dependence of methanogenesis on methanol, which has been experimentally confirmed for Mmc. luminyensis (21) (Fig. 3A) and “Ca. Methanoplasma termitum” (Fig. 3B). The absence of this pathway also precludes the oxidation of methyl groups to CO2, which explains the obligate hydrogen requirement of methanogenesis in Mmc. luminyensis (21), a trait that is corroborated by the present study (Fig. 3A). Moreover, it substantiates the proposal that methane formation from methanol alone by “Ca. Methanoplasma termitum” (Fig. 3B) is driven by an internal hydrogen production of the clostridia present in the enrichment culture (12).
FIG 3.

Time course of methane accumulation in the culture headspace of Methanomassiliicoccus luminyensis (A and C) and “Ca. Methanoplasma termitum” (B and D) incubated in bicarbonate-buffered medium supplemented with H2 (ca. 50 kPa), methanol (50 mM), or acetate (30 mM) (A and B) or H2 combined with different methylamines (10 mM) (C and D). To avoid a transfer of residual methanol with the inoculum, the precultures were grown under methanol limitation. The values are means of three replicate cultures; standard deviations are shown only if they are larger than the symbols.
The inability to disproportionate methanol is found also in Methanosphaera stadtmanae (Methanobacteriales). In contrast to Methanomassiliicoccales, M. stadtmanae possesses all genes required for the reduction of CO2 to methane and for the oxidation of methanol to CO2 (9), but the activities of the corresponding enzymes in cell extracts are either low or absent (44, 45). It has been speculated that the absence of Mtr activity indicates that the enzyme is not required for methanogenesis from methanol and H2, and the low specific activities of formylmethanofuran dehydrogenase, together with the apparent molybdopterin auxotrophy of M. stadtmanae, may be related to an exclusively anabolic function of this enzyme (9). Also in the obligately methylotrophic Methanomicrococcus blatticola (Methanosarcinales), the low activities of F420-dependent enzymes indicate an inability to oxidize methyl groups (11). Further insights into the pathway will be possible when a genome sequence is available also for this strain.
Growth on methylamines.
The previously reported presence of the complete gene sets for the utilization of mono-, di-, and trimethylamine in all genomes of Methanomassiliicoccales (17, 18, 42) (Fig. 2) suggested that methylamines can be used as substrates by all strains investigated so far. However, experimental evidence for this trait is available only for Mmc. luminyensis (21), correcting a contradictory statement in the original species description concerning methane formation from trimethylamine (13). Our study extends these results with detailed time courses of H2-dependent methanogenesis from all three methylamines (Fig. 3C).
The genome of “Ca. Methanoplasma termitum,” however, contains only homologs encoding the substrate-specific monomethylamine methyltransferase (MtmB) and the monomethylamine corrinoid protein (MtmC) (Fig. 2). As in all other Methanomassiliicoccales (42), the mtmB gene of “Ca. Methanoplasma termitum” is interrupted by an in-frame amber codon, which indicates that the enzyme contains pyrrolysine (46, 47), a common feature of all methylamine methyltransferases that serves to activate and orient the methylamine for methyl transfer to the cobalt ion of the corrinoid protein (48). Although the genome lacks the gene for the methylcobamide:CoM methyltransferase (MtbA) present in all methylamine utilization complexes (49), the capacity to produce methane from monomethylamine (Fig. 3D) indicates that “Ca. Methanoplasma termitum” possesses a functional complex, in which MtbA is probably replaced by MtaA, its homolog in the methanol methyltransferase complex.
The lack of capacity of di- and trimethylamine utilization and the putative loss of the mtbA gene may be related to streamlining of the “Ca. Methanoplasma termitum” genome, which is even smaller than that of the closely related “Ca. Methanomethylophilus alvus.” Since also their relatives in the bovine rumen (rumen cluster C [50]) can utilize all three methylamines (51), the capacity to metabolize methylamines may have been lost because it does not provide an advantage in termite guts.
Energy metabolism.
It has remained entirely obscure how members of the Methanomassiliicoccales reoxidize reduced ferredoxin formed by electron bifurcation at the soluble heterodisulfide reductase (HdrABC) and how they couple this process with the generation of an electrochemical membrane potential. In the case of the obligately methyl-reducing M. stadtmanae, it has been speculated that both tasks are accomplished by the energy-converting [NiFe] hydrogenase Ehb (2), a homolog of the anaplerotic Eha complex of hydrogenotrophic methanogens (52, 53). However, homologs of the Eha and Ehb gene clusters are entirely absent in all Methanomassiliicoccales. Also, ferredoxin-dependent hydrogenases of the Ech type (54, 55), which are involved in energy conversion in most Methanosarcina species (56, 57), are absent in “Ca. Methanoplasma termitum” and “Ca. Methanomethylophilus alvus,” members of the intestinal cluster. The two complete gene sets in the genomes of Mmc. luminyensis and “Ca. Methanomassiliicoccus intestinalis” (see Table S2 in the supplemental material), which have highest sequence similarity to the Ech genes of Methanosarcina barkeri (Fig. 4) and the canonical NiFe-binding motif of [NiFe] hydrogenases (Fig. 5), are probably involved in the redox cycling of ferredoxin produced/consumed by the CO dehydrogenase/acetyl-CoA synthetase complex present only in the Methanomassiliicoccaceae (see below).
FIG 4.

Phylogenetic tree of Fpo, Nuo, and related 11-subunit complexes and [NiFe] hydrogenases of bacteria and archaea. The Fpo-like complexes of Methanomassiliicoccales do not cluster with the 11-subunit complexes of their closest relatives, Thermoplasmatales and “Ca. Aciduliprofundum boonei,” but share high sequence similarity with the Fpo and Fpo-like complexes of Methanosarcinales (strains indicated in bold). The tree is based on a translated amino acid alignment of the homologs encoding the large subunit of the respective complex and was reconstructed using a maximum-likelihood algorithm (PhyML). Nodes that were not supported by neighbor-joining and maximum-parsimony (MP) analyses are shown as multifurcations; highly supported nodes (1,000 bootstraps, MP) are marked (solid circles, >95%; open circles, >70%). Scale bar indicates 0.1 substitution per site.
FIG 5.
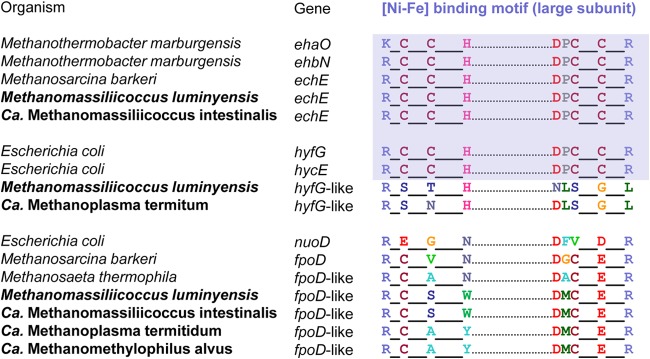
Comparison of the [NiFe]-binding motif in the large subunit of selected [NiFe] hydrogenases with the corresponding amino acid residues in the homologous complexes encoded in the genomes of Methanomassiliicoccales (in bold). Blue shading indicates the typical motif of bona fide hydrogenases. Like the homologous subunits in Fpo and Nuo, which do not contain a [NiFe] cofactor, both the Hyf-like and Fpo-like complexes of Methanomassiliicoccales deviate strongly from this consensus.
The only other putative hydrogenases in Methanomassiliicoccales are the Hyf-like complexes encoded in the genomes of “Ca. Methanoplasma termitum” and Mmc. luminyensis (see Table S3 in the supplemental material). Their large subunits are most closely related to the [NiFe] hydrogenases Hyc and Hyf of Escherichia coli (Fig. 4). However, genes for several subunits of Hyc and Hyf are missing (see Table S3), and the large subunit of the Hyf-like complex (HyfG) shows several deviations from the canonical [NiFe]-binding motif (Fig. 5). Even if the Hyf-like complexes were functional energy-converting hydrogenases, their absence in “Ca. Methanomethylophilus alvus” and “Ca. Methanomassiliicoccus intestinalis” makes them unlikely candidates for energy metabolism, which should be conserved among all Methanomassiliicoccales. It is also not possible that Ech and Hyf-like complexes substitute for each other, because none of the complexes are present in “Ca. Methanomethylophilus alvus.” Therefore, it seems safe to conclude that the reoxidation of ferredoxin in Methanomassiliicoccales does not involve an energy-converting hydrogenase.
The Fpo-like complex of Methanomassiliicoccales.
The genomes of all Methanomassiliicoccales have a gene cluster that encodes homologs of the 11 core subunits shared by the membrane-bound F420-methanophenazine oxidoreductase complex (Fpo) of methanogens and the NADH-ubiquinone oxidoreductase complex (Nuo) and its homologs in many bacteria. However, homologs of the subunits responsible for binding and oxidation of F420 (FpoFO) or NADH (NuoEFG) are lacking (Table 2). Phylogenetic analysis of the amino acid sequences of the large subunit revealed that the 11-subunit complex of Methanomassiliicoccales is more closely related to the Fpo and Fpo-like complexes of Methanosarcina and Methanosaeta spp. than to bacterial 11-subunit complexes (including Nuo) or the [NiFe] hydrogenases of methanogens (Fig. 4). The numerous deviations from the canonical [NiFe]-binding motifs in the large subunit (Fig. 5) make it unlikely that the new complex is an [NiFe] hydrogenase.
TABLE 2.
Genes encoding the different subunits of Fpo in Methanosarcina mazei and their homologs in the 11-subunit complex present in all Methanomassiliicoccalesa
| Protein(s) or subunit | Fpo, Methanosarcina mazei | 11-subunit complex |
Nuo, E. coli | |||
|---|---|---|---|---|---|---|
| “Ca. Methanoplasma termitum” | “Ca. Methanomethylophilus alvus” | Mmc. luminyensis | “Ca. Methanomassiliicoccus intestinalis” | |||
| Large subunit | fpoD | Mpt1_c12630 | MMALV_01980 | WP_019176180 | MMINT_02020 | nuoD |
| Small subunit | fpoB | Mpt1_c12650 | MMALV_01960 | WP_019176182 | MMINT_02000 | nuoB |
| 4Fe/4S-Fd | fpoI | Mpt1_c12610 | MMALV_02000 | WP_019176178 | MMINT_02040 | nuoI |
| Small protein | fpoC | Mpt1_c12640 | MMALV_01970 | WP_019176181 | MMINT_02010 | nuoC |
| Transmembrane proteins | fpoL | Mpt1_c12570 | MMALV_02040 | WP_019176174 | MMINT_02080 | nuoL |
| fpoM | Mpt1_c12560 | MMALV_02050 | WP_019176173 | MMINT_02090 | nuoM | |
| fpoN | Mpt1_c12550 | MMALV_02060 | WP_019176172 | MMINT_02100 | nuoN | |
| fpoH | Mpt1_c12620 | MMALV_01990 | WP_019176179 | MMINT_02030 | nuoH | |
| fpoK | Mpt1_c12580 | MMALV_02030 | WP_019176175 | MMINT_02070 | nuoK | |
| fpoJ | Mpt1_c12600 | MMALV_02010 | WP_019176177 | MMINT_02050 | nuoJ | |
| fpoA | Mpt1_c12660 | MMALV_01955 | WP_019176183 | MMINT_01985 | nuoA | |
| F420 and phenazine binding module | fpoFO | — | — | — | — | — |
| NADH-oxidizing module | — | — | — | — | — | nuoEFG |
The homologs in the Nuo complex of Escherichia coli are shown for comparison. —, not present.
It has been proposed that 11-subunit complexes are derived from [NiFe] hydrogenases that lost their [NiFe] cluster and gained new functions by association with additional electron-transferring subunits, such as NuoEFG or FpoFO (58). Although 11-subunit complexes are present in many bacteria and archaea, their interacting partner proteins or the redox process catalyzed by the respective complex are often unclear (58).
A novel mechanism of energy conversion.
Recently, Welte and Deppenmeier (59) provided strong evidence that the Fpo-like complex in the obligately aceticlastic Methanosaeta (Mt.) thermophila does not oxidize cofactor F420 but catalyzes the ferredoxin-dependent reduction of methanophenazine (Fig. 6). Unlike the aceticlastic Methanosarcina species, which reoxidize the ferredoxin produced during the cleavage of acetyl-CoA by using either an Ech hydrogenase or an Rnf complex, Mt. thermophila directly channels the electrons of ferredoxin into a membrane-bound electron transport chain consisting of a ferredoxin:methanophenazine oxidoreductase (the Fpo-like complex) and the canonical methanophenazine-dependent heterodisulfide reductase (HdrDE) (57). The assumption that the “headless” Fpo-like 11-subunit complex (lacking FpoF) does not interact with F420H2 but accepts electrons directly from Fdred is consistent with the absence of F420-dependent activities and the presence of ferredoxin-dependent heterodisulfide reductase activities in the membrane fraction of Mt. thermophila (59). There is also no evidence for the presence of F420-dependent enzymes in any of the four Methanomassiliicoccales genomes, and although autofluorescence at 420 nm is mentioned in the species description of Mmc. luminyensis (13), we could not detect the characteristic autofluorescence of cofactor F420 in Mmc. luminyensis or “Ca. Methanoplasma termitum” (12; also this study). Therefore, we assume that also the Fpo-like 11-subunit complex of Methanomassiliicoccales must interact directly with ferredoxin.
FIG 6.
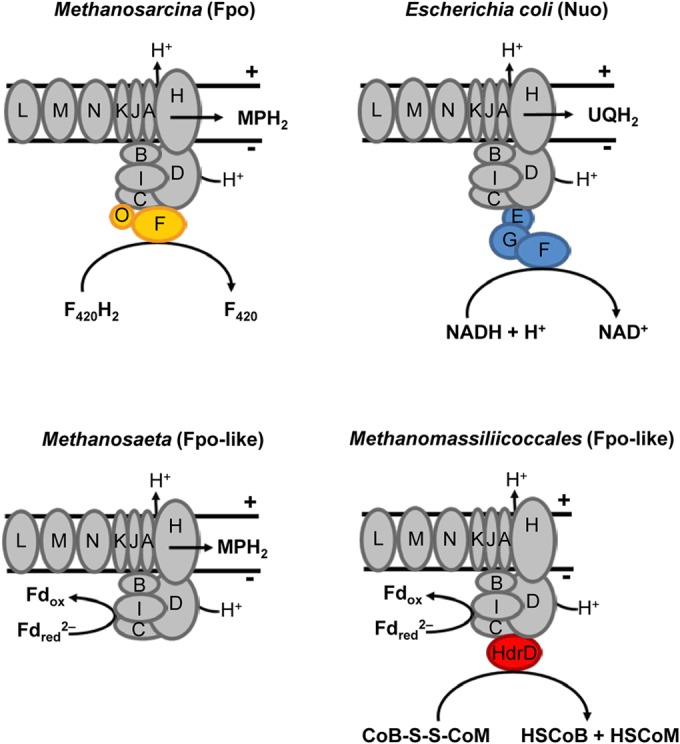
Redox processes catalyzed by the 11-subunit core complexes and their specific electron-transferring modules in Methanosarcina mazei (Fpo) and Escherichia coli (Nuo) and hypothetical processes and potential interaction partners of the Fpo-like complexes in Methanosaeta thermophila (59) and Methanomassiliicoccales (this study). The common core complex of 11 subunits is shown in gray, and specific subunits of the different complexes are indicated by different colors. In all cases, the complex serves as a redox-driven proton pump. For further explanations, see the text. F420, coenzyme F420; Fd, ferredoxin; MP, methanophenazine; UQ, ubiquinone.
It is striking that all Methanomassiliicoccales genomes encode a homolog of HdrD, the heterodisulfide-reducing subunit of the membrane-bound heterodisulfide reductase complex (HdrDE) in Methanosarcinales, but lack the hdrE gene, which encodes the cytochrome b-containing membrane anchor of the complex that accepts electrons from methanophenazine (60). Since there is also no evidence for other enzymes with cytochromes or cytochrome biosynthesis in any of the genomes (see below), the Fpo-like complex of Methanomassiliicoccales cannot couple ferredoxin oxidation to heterodisulfide reduction the same way as proposed for Mt. thermophila, i.e., via methanophenazine and a canonical heterodisulfide reductase (HdrDE) (59). Instead, we propose that it interacts directly with HdrD, imparting to the entire complex the function of an energy-converting ferredoxin:heterodisulfide oxidoreductase (Fig. 6). Also, a recent comparative analysis of the three previously published genomes came to a similar conclusion but assumed that the electrons of the Fpo-like complex are transferred to a (possibly heterodisulfide-reducing) membrane-bound protein complex via an unidentified membrane-soluble electron carrier (22), analogous to the situation in Mt. thermophila (59).
The subunit of the Fpo-like complex responsible for ferredoxin oxidation remains to be identified. It has been suggested that the unusual density of lysine at the extended C terminus of FpoI in Mt. thermophila (see Fig. S1 in the supplemental material) may serve for interaction with the acidic ferredoxin (57). Although also the C terminus of the FpoI subunit of Methanomassiliicoccales species is extended and rich in lysine, it is noteworthy that these features are not present in the homologous subunits of the ferredoxin-oxidizing [NiFe] hydrogenases (HycF and HyfH) (see Fig. S1).
Energetic aspects.
Welte and Deppenmeier (57) have pointed out that the more negative redox potential of ferredoxin (E0′ = −500 mV) compared to that of cofactor F420 (−360 mV) renders the reaction with methanophenazine catalyzed by the Fpo-like complex of Mt. thermophila more exergonic than that of the canonical F420-dependent Fpo of Methanosarcina species. In the case of the Methanomassiliicoccales, the direct reduction of the heterodisulfide via HdrD should be even more favorable, since the midpoint potential of the heterodisulfide (−140 mV) is slightly more positive than that of methanophenazine (−165 mV [57]). It would be premature to speculate on the number of protons translocated by the Fpo-like complex of Methanomassiliicoccales, but we want to point out that electron bifurcation at the soluble heterodisulfide reductase dictates that only the electrons of every second hydrogen oxidized by the Mvh/HdrABC complex will feed into the energy-converting ferredoxin:heterodisulfide oxidoreductase (Fpo-like/HdrD) complex (Fig. 2). This should negatively affect growth yield but may also increase the competiveness by decreasing the threshold for hydrogen (see below).
All genomes of Methanomassiliicoccales encode an H+/Na+ antiporter and the typical AoA1-ATP synthase of archaea. The C subunit of the ATP synthase (AhaC) has the same conserved Na+-binding motif as in Methanosarcina acetivorans and Methanosarcina mazei, but also in those organisms the ion specificity of ATP synthase is not fully resolved (4). Since all 11-subunit complexes are considered to be proton pumps (61) and since the Fpo-like complex is the only energy-converting complex in Methanomassiliicoccales, it is likely that methanogenesis is coupled to ATP synthesis via a proton motive force.
Acetyl-CoA synthesis.
Like the other Methanomassiliicoccales, “Ca. Methanoplasma termitum” possesses a homolog of the acsA gene encoding an ADP-forming acetyl-CoA synthetase, which allows heterotrophic growth on acetate (Fig. 7). The presence of gene clusters encoding a CO dehydrogenase/acetyl-CoA synthase complex, two Ech hydrogenases, and a 5,10-methylenetetrahydrofolate reductase (MetF) in Mmc. luminyensis and “Ca. Methanomassiliicoccus intestinalis” suggests that the members of the Methanomassiliicoccaceae may be able to synthesize acetyl-CoA also from formate and CO2 (Fig. 7). However, it should be noted that the structure of the gene cluster encoding CO dehydrogenase/acetyl-CoA synthase differs from that in other methanogens (see Fig. S2 in the supplemental material). It lacks the gene encoding the epsilon subunit (CdhB) typical of methanogens, and the genes encoding the alpha and beta subunits (CdhA/C) seem to be fused and truncated compared to the cdhA gene of other methanogens. The fused gene shows highest sequence similarity to a homolog in the homoacetogenic Acetonema longum, although the latter possesses also a second, nontruncated cdhA gene. In addition, the beta-part of the cdhA/C gene of Mmc. luminyensis is interrupted by several insertions, which suggests that the gene may no longer encode a functional enzyme.
FIG 7.

Intermediary metabolism and glycolysis/gluconeogenesis of “Ca. Methanoplasma termitum” and other Methanomassiliicoccales. Black arrows indicate reactions whose enzymes are encoded by all genomes. Blue-green arrows indicate that the enzymes are not present in “Ca. Methanoplasma termitum” but are present in the genomes indicated by the colored dots. Purple arrows indicate link points to amino acid and nucleic acid biosynthesis. Gray arrows indicate important enzymes not encoded by any of the four genomes. *, “Ca. Methanomethylophilus alvus” has gpmA instead of apgM; #, acnA was not found in Mmc. luminyensis.
One-carbon metabolism.
All Methanomassiliicoccales possess the genes required to generate 5,10-methylenetetrahydrofolate from formate. However, the absence of formate dehydrogenase suggests that the pathway operates in the reverse direction, generating both 5,10-methylenetetrahydrofolate (for pyrimidine biosynthesis) and formate (for purine biosynthesis and as a cosubstrate of the ribonucleotide reductase) from serine (Fig. 7). The same anabolic role of the C1 pathway has been postulated for M. stadtmanae (9). The CO dehydrogenase/acetyl-CoA synthase of Methanomassiliicoccales, if at all functional, also may serve to generate C1 compounds from acetyl-CoA.
Gluconeogenesis and glycolysis.
All Methanomassiliicoccales possess the genes required for gluconeogenesis via pyruvate-ferredoxin oxidoreductase and a 2,3-bisphosphoglycerate-independent phosphoglycerate mutase (ApgM), which is characteristic of archaea (62). Only “Ca. Methanomethylophilus alvus” possesses the bacterial, bisphosphoglycerate-dependent variant (GpmA). Also the bifunctional fructose-1,6-bisphosphate aldolase/phosphatase present in all strains is typical for archaeal gluconeogenesis (63). The genome of “Ca. Methanoplasma termitum” lacks a homolog encoding phosphoglucoisomerase (Pgi), but since the pathway of gluconeogenesis is otherwise complete, this step may involve an unknown enzyme. Genes for glycogen biosynthesis or degradation are not present in any of the strains. Only Mmc. luminyensis, the strain with the largest genome, may be capable of glycolysis because it possesses a phosphofructokinase (PfkB) and an archaeal class I fructose-bisphosphate aldolase (FbaA) (Fig. 7).
Intermediary metabolism.
As in all methanogens, the tricarboxylic acid (TCA) cycle of Methanomassiliicoccales is incomplete. Mmc. luminyensis and “Ca. Methanomassiliicoccus intestinalis” both possess a phosphoenolpyruvate carboxykinase (PckA) for providing oxaloacetate, a Si-citrate synthase (GltA), and the remaining enzymes of the oxidative branch (a homolog encoding aconitase [AcnA] is missing in the draft genome of Mmc. luminyensis). Both Methanomassiliicoccus species should also have the capacity to synthesize succinyl-CoA, either via 2-oxoglutarate (“Ca. Methanomassiliicoccus intestinalis”) or via the reductive branch (Mmc. luminyensis), involving a cytochrome-free succinate dehydrogenase (ShdAB) and succinyl-CoA synthetase (SucCD). All four strains should be able to synthesize malate from pyruvate via malic enzyme (MaeA) but lack a malate dehydrogenase, which should cause aspartate auxotrophy in members of the intestinal cluster. The latter should also be unable to synthesize 2-oxoglutarate and succinyl CoA, resulting in a requirement for glutamate and methionine.
Amino acid and nucleotide biosynthesis.
Although all strains may be unable to form glutamate and/or aspartate de novo, the pathways for the biosynthesis of other amino acids are mostly complete (see Fig. S3 in the supplemental material). The absence of genes encoding threonine aldolase (ItaE) and homoserine O-acetyltransferase (MetX) in all strains except Mmc. luminyensis suggests methionine auxotrophy in the former strains. “Ca. Methanomassiliicoccus intestinalis” is the only strain that lacks genes required for tryptophan synthesis from serine and chorismate. As in all methanogens, the gene coding for histidinol phosphatase (HisJ) remains to be identified. The genes required to operate the pentose phosphate pathway and for biosynthesis of phosphoribosyl pyrophosphate (PRPP) and nucleic acids are present in all strains (see Table S1).
Coenzyme biosynthesis.
All methanogens can synthesize cofactor F430, an Ni porphyrinoid that functions as the prosthetic group of Mcr and is essential for methanogenesis (64, 65). All Methanomassiliicoccales possess the genes for the entire pathway of corrinoid biosynthesis via glutamyl-tRNA reductase via precorrin-2 (HemABCDL and CobA) (66), and the genes required for cobalamin biosynthesis from precorrin-2 are almost complete. Like all other methanogens, they lack the typical pathway for heme biosynthesis via coproporphyrinogen III, but also the genes for the alternative pathway for heme biosynthesis via precorrin-2 (67) are absent in the genomes, underscoring the inability of Methanomassiliicoccales to synthesize cytochromes. Since none of the strains has the capacity to synthesize methionine, the methyl group donor in the biosynthesis of factor F430 (68), neither via the methionine biosynthesis pathway I (which would require succinyl-CoA) nor via one of the other pathways (see Fig. S2 in the supplemental material), they must depend on an external source of this amino acid.
While the genes for the classical pathway of coenzyme M biosynthesis via sulfolactate (69) are absent from the genomes of all Methanomassiliicoccales, the three strains from the human gut encode the genes for the alternative pathway via l-cysteate (70), which explains the requirement of “Ca. Methanoplasma termitum” for 2-mercaptoethanesulfonate. The situation is less clear in the case of methanophenazine, for which the only known enzyme of the biosynthetic pathway is a geranylfarnesyl diphosphate synthase, which is responsible for the addition of the polyprenyl side chain in Methanosarcina mazei (71) but may also be involved in the synthesis of C25 diether lipids, as in other archaea (72). Homologs of the corresponding gene are present in all Methanomassiliicoccales.
Lipid biosynthesis.
Archaea produce unique membrane lipids in which isoprenoid alkyl chains are bound to glycerol moieties via ether linkages (73). Like all Methanomassiliicoccales, “Ca. Methanoplasma termitum” possesses the genes for the mevalonate pathway, the subsequent condensation of isopentenyl diphosphate (IPP) units with dimethylallyl diphosphate (DMAPP) to geranylgeranyl diphosphate (GGPP), and the prenyltransferases that form the ether bonds in geranylgeranylglyceryl phosphate (GGGP) and digeranylgeranylglyceryl phosphate (DGGGP) (see Table S1 in the supplemental material). The enzymes responsible for the activation of the diglyceride, the addition of polar head groups to the glycerol moiety, and the final production of archaeol via the subsequent reduction of the unsaturated isoprenoid chains are also represented.
Ultrastructure.
Negative stains of “Ca. Methanoplasma termitum” strain MpT1 and the closely related strain MpM2 from a millipede showed coccoid cells with diameters between 500 and 800 nm (Fig. 8). No obvious dividing cells were observed. A small number of cells carried appendages, but generally not more than one per cell. Although the diameter (12 nm) of the appendage matches the typical size of an archaellum, none of the Methanomassiliicoccales genomes contain the typical archaellum operon present in other archaea (74). Only “Ca. Methanoplasma termitum,” Mmc. luminyensis, and “Ca. Methanomethylophilus alvus” possess genes that may represent homologs of the archaellum biosynthesis pathway, such as prearchaellin peptidase (FlaK; all three strains), secretion ATPase (FlaI; only Mmc. luminyensis), and a polytopic membrane protein (FlaJ; only Mmc. luminyensis) that interacts with ATPase (74). However, genes encoding archaellin (FlaB), the major filament component of the archaellum, are absent in all strains. Since the same is true also for all genes potentially involved in pilus biosynthesis, the nature of the cell appendages observed in the negative stains remains obscure.
FIG 8.

Ultrastructure of “Ca. Methanoplasma termitum” strain MpT1 (A to C) and the closely related strain MpM2 from a millipede (D to F). Panels A and D show cells negatively stained with uranyl acetate, illustrating the coccoid shape and occasional cell appendages (arrowheads). Ultrathin sections of high-pressure frozen cells at intermediate (B and E) and high magnification (C and F) show the homogenous cytoplasm surrounded by a cytoplasmic membrane (IM) and an additional outermost membrane (OM) that occasionally showed the characteristics of a lipid bilayer (F). Scale bars: 500 nm.
In ultrathin sections, both strains showed a homogenous cytoplasm surrounded by a cytoplasmic membrane and an outermost layer that resembled a second membrane (Fig. 8C and F). Although great care was taken to preserve the structure during preparation, the outermost layer was often not present or seemed to be detached from the cells (Fig. 8E). The distance between the two membranes ranged from 10 and 300 nm, often even within the same cell. Since the integrity of its structure was affected by centrifugation, fixation, and freeze substitution, the possibility of artifacts cannot be excluded.
Interestingly, the species description of Mmc. luminyensis (13) also contains evidence for a second membrane system. The transmission electron micrograph of an ultrathin section shows a single cell surrounded by two electron-dense layers, one enclosing the cytoplasm and the other separated from the former by a wide electron-lucent ring. Although this interpretation differs from that of the authors (13), and despite obvious differences to our preparations in structure and contrast, we are confident that the cell envelopes of both “Ca. Methanoplasma termitum” and Mmc. luminyensis do not consist of a single lipid membrane covered by a proteinaceous S-layer, as in most other archaea (75, 76), but that the cells have a two-membrane system. Dual membranes in archaea have so far been restricted to Ignicoccus species (77) and the ultrasmall ARMAN cells (78). However, in view of the sensitivity to manipulation of the outermost membrane of “Ca. Methanoplasma termitum,” it is possible that this structure is more widespread than it appears.
Neither the ultrathin sections of “Ca. Methanoplasma termitum” (this study) nor the image of Mmc. luminyensis (13) shows indications of a proper cell wall. This is in agreement with the absence of most genes involved in the synthesis of UDP-N-acetyl-d-glucosamine, the precursor of pseudomurein (79), from the genomes of “Ca. Methanoplasma termitum” and “Ca. Methanomethylophilus alvus.” Interestingly, both Mmc. luminyensis and Ca. Methanomassiliicoccus intestinalis retain all genes required to synthesize this compound.
Evolution.
Although the Euryarchaeota comprise several nonmethanogenic lineages, the apparent cocladogenesis of phylogenetic (16S rRNA genes) and functional marker (mcrA) genes suggests that methanogens and anaerobic methane oxidizers are a monophyletic group (3, 80). Also, a recent phylogenomic analysis supports the hypothesis that the Methanomassiliicoccales are derived from methanogenic ancestors (14).
Other lineages in the Thermoplasmata obviously lost the capacity for methanogenesis and acquired other modes of energy metabolism. The Thermoplasmatales are facultative anaerobes (81), whereas their closest relatives from deep sea hydrothermal vent group II (which includes “Ca. Aciduliprofundum boonei,” whose complete genome is now available) possess a sulfur-based energy metabolism (82). There is also no evidence for the presence of mcr genes for other deep-branching lineages of Thermoplasmata found in marine sediments or the deep subsurface (12, 14, 83, 84) (Fig. 1).
While the soluble heterodisulfide reductase (HdrABC) is a common feature of all methanogens, its membrane-bound analog is present only in the apical lineages. Interestingly, Methanocellales and Methanomassiliicoccales possess only a homolog of the subunit carrying the catalytic domain (HdrD), whereas the cytochrome-containing membrane anchor (HdrE) must have been acquired at a later stage, since HdrDE is present only in the Methanosarcinales. It is not clear whether the homologs in Archaeoglobales (HmeDC) are derived from their methanogenic ancestor or the result of lateral gene transfer, which would also explain the presence of HdrD in Methanosphaerula palustris (Methanomicrobiales).
Homologs of the 11-subunit complex are present in only a few euryarchaeotal lineages. They are entirely absent from all basal Euryarchaeota but present in the Thermoplasmata (Thermoplasmatales and Methanomassiliicoccales) and the euryarchaeotal crown groups (Archaeoglobales, Methanosarcinales, and Halobacteriales). The phylogeny of the large subunit of the Fpo-like complex of Methanomassiliicoccales is more similar to those of the homologous subunits in the Fpo and Fpo-like complexes of Methanosarcina and Methanosaeta spp. than to those in their closer, nonmethanogenic relatives, the strictly anaerobic “Ca. Aciduliprofundum boonei” (82) and the facultatively anaerobic Thermoplasmatales (85), which suggests that some of them have acquired the complex by lateral gene transfer (Fig. 4). However, the function of related complexes may change by interaction with different electron-accepting modules. This is nicely illustrated by the Fpo-like complexes of Methanomassiliicoccales and Methanosarcinales, which may be of common origin but interact with different electron donors (ferredoxin of cofactor F420) or electron acceptors (HdrD or methanophenazine).
Ecological considerations.
It is striking that obligately methyl-reducing methanogens have so far been isolated only from intestinal tracts (8, 10, 13), although they are apparently not restricted to this habitat (12, 19). The decisive factor limiting their distribution is obviously the simultaneous production of methanol (or methylamines) and hydrogen by the bacterial microbiota, but also, the competition with other microorganisms for one of these substrates should affect their ecological amplitude. For instance, the hydrogen-dependent reduction of methanol to methane (H2 + CH3OH → CH4 + H2O; ΔG°′ = −112.5 kJ per mol of CH4) is thermodynamically more favorable than its disproportionation to methane and CO2 (4 CH3OH → 3 CH4 + CO2 + 2 H2O; ΔG°′ = −103.7 kJ per mol of CH4) under standard conditions (calculated after Thauer et al. [86]). However, the difference becomes smaller with decreasing hydrogen concentrations, and methanol disproportionation would be energetically superior already at moderate hydrogen partial pressures (PH2 < 103 Pa).
It is likely that the hydrogen thresholds of methyl-reducing methanogens differ between members of particular phylogenetic groups. Generally, methanogens with cytochromes have higher hydrogen thresholds than those without cytochromes because they have a more efficient mode of energy conservation and encounter a thermodynamic equilibrium of energy metabolism and ATP synthesis already at relatively high hydrogen partial pressures (2). Unfortunately, only a little is known about the hydrogen thresholds of methanogens during growth on methanol. The hydrogen threshold of the obligately hydrogen-dependent methylotroph Methanomicrococcus blatticola, which is a member of Methanosarcinales and possesses F420 and cytochromes, is only slightly lower than that of Methanosarcina barkeri growing on H2 and CO2 (87). In M. stadtmanae, the only methylotrophic member of Methanobacteriales, the proposed coupling of methanogenesis to energy conservation via an energy-converting hydrogenase (2) would improve with decreasing hydrogen partial pressure, but the hydrogen threshold—determined by the equilibrium point of energy metabolism and ATP synthesis—depends on the number of sodium ions transported by Ehb. The energy metabolism of Methanomassiliicoccales may also serve to increase their affinity for hydrogen. The proposed bifunctional role of heterodisulfide in the production of reduced ferredoxin (via electron bifurcation at the HdrABC complex) and its subsequent oxidation (via the Fpo-like complex and HdrD) would allow only every second event of CH4 production to be coupled with the generation of a membrane potential (Fig. 2), but again, the equilibrium point of energy metabolism and ATP synthesis depends on the stoichiometry of proton translocation (for a discussion of the number of protons transported by the Fpo-like complex, see the review by Welte and Deppenmeier [57]). The presence of lower hydrogen thresholds in Methanomassiliicoccales and M. stadtmanae remains to be experimentally determined, but a similar trade-off between substrate affinity and growth yield is encountered also in aceticlastic methanogens; Methanosaeta spp. achieve an increased affinity for their substrate by investing an additional ATP into acetate activation (57).
The small genome size of “Ca. Methanoplasma termitum” and “Ca. Methanomethylophilus alvus” indicates that the members of the intestinal cluster have experienced a substantial streamlining of their genomes, possibly an adaptation to the rich nutrient supply in the intestinal habitat. So far, none of the strains has been isolated in pure culture, probably due to still-unrecognized dependencies on metabolites provided by bacterial members of the enrichment culture. An interesting aspect is the requirement of “Ca. Methanoplasma termitum” for coenzyme M, which is not a typical bacterial product and probably supplied by other methanogens colonizing the termite gut. This would agree with the observation that Methanomassiliicoccales are never the only methanogens present in the gut microbiota of termites (unpublished results).
The many variations in the metabolic pathways of methylotrophic methanogens may represent adaptations to cope with special environmental conditions. Understanding these strategies will require detailed physiological and biochemical studies of the groups in question.
Description of “Candidatus Methanoplasma termitum.”
Me.tha.no.plas'ma. N.L. n. methanum [from French n. meth(yle) and chemical suffix -ane], methane, N.L. pref. methano-, pertaining to methane, Gr. neut. n. plasma, something formed or molded, a form, N.L. neut. n. Methanoplasma, a methane-producing form.
ter'mi.tum. L. masc. n. termes, termitis (variant of tarmes), a woodworm, a termite, L. masc. n. gen. pl. termitum, of termites, referring to the habitat of the organism.
Short description: roundish cells, 0.5 to 0.8 μm in diameter, without apparent cell wall, surrounded by two membranes, possess archaellum-like cell appendages. Obligate anaerobe. Methanogenic metabolism, obligately methylotrophic, methyl donors: methanol and monomethylamine but not di- or trimethylamine. Obligately hydrogen dependent. Form a monophyletic group within the radiation of the “intestinal cluster” of Methanomassiliicoccales. Habitat: intestinal tracts of termites and cockroaches. Basis of assignment: strain MpT1 from Cubitermes ugandensis (16S rRNA gene sequence JX266068, complete genome sequence CP010070), and 16S rRNA gene sequences of so-far-uncultured representatives (accession numbers JX266062 to JX266070).
Supplementary Material
ACKNOWLEDGMENTS
This study was funded by a grant of the DFG to A.B. in the collaborative research center SFB 987 (Microbial Diversity in Environmental Signal Response). A.K. was supported by the LOEWE program of the state of Hessen (Synmikro).
We are grateful to Rolf Thauer for his support and many helpful discussions and to Bernhard Schink for etymological advice. We thank Uwe Maier for providing the EM facility and Katja Meuser and Marion Debus for technical assistance.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.03389-14.
REFERENCES
- 1.Liu Y, Whitman WB. 2008. Metabolic, phylogenetic, and ecological diversity of the methanogenic archaea. Ann N Y Acad Sci 1125:171–189. doi: 10.1196/annals.1419.019. [DOI] [PubMed] [Google Scholar]
- 2.Thauer RK, Kaster A-K, Seedorf H, Buckel W, Hedderich R. 2008. Methanogenic archaea: ecologically relevant differences in energy conservation. Nat Rev Microbiol 6:579–591. doi: 10.1038/nrmicro1931. [DOI] [PubMed] [Google Scholar]
- 3.Bapteste E, Brochier C, Boucher Y. 2005. Higher-level classification of the Archaea: evolution of methanogenesis and methanogens. Archaea 1:353–363. doi: 10.1155/2005/859728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schlegel K, Müller V. 2013. Evolution of Na+ and H+ bioenergetics in methanogenic archaea. Biochem Soc Trans 41:421–426. doi: 10.1042/BST20120294. [DOI] [PubMed] [Google Scholar]
- 5.Costa KC, Wong PM, Wang T, Lie TJ, Dodsworth JA, Swanson I, Burn JA, Hackett M, Leigh JA. 2010. Protein complexing in a methanogen suggests electron bifurcation and electron delivery from formate to heterodisulfide reductase. Proc Natl Acad Sci U S A 107:11050–11055. doi: 10.1073/pnas.1003653107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kaster A-K, Moll J, Parey K, Thauer RK. 2011. Coupling of ferredoxin and heterodisulfide reduction via electron bifurcation in hydrogenotrophic methanogenic archaea. Proc Natl Acad Sci U S A 108:2981–2986. doi: 10.1073/pnas.1016761108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Blaut M, Gottschalk G. 1984. Coupling of ATP synthesis and methane formation from methanol and molecular hydrogen in Methanosarcina barkeri. Eur J Biochem 141:217–222. doi: 10.1111/j.1432-1033.1984.tb08178.x. [DOI] [PubMed] [Google Scholar]
- 8.Miller TL, Wolin MJ. 1985. Methanosphaera stadtmaniae gen. nov., sp. nov.: a species that forms methane by reducing methanol with hydrogen. Arch Microbiol 141:116–122. doi: 10.1007/BF00423270. [DOI] [PubMed] [Google Scholar]
- 9.Fricke WF, Seedorf H, Henne A, Kruer M, Liesegang H, Hedderich R, Gottschalk G, Thauer RK. 2006. The genome sequence of Methanosphaera stadtmanae reveals why this human intestinal archaeon is restricted to methanol and H2 for methane formation and ATP synthesis. J Bacteriol 188:642–658. doi: 10.1128/JB.188.2.642-658.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sprenger WW, van Belzen MC, Rosenberg J, Hackstein JHP, Keltjens JT. 2000. Methanomicrococcus blatticola gen. nov., sp. nov., a methanol- and methylamine-reducing methanogen from the hindgut of the cockroach Periplaneta americana. Int J Syst Evol Microbiol 50:1989–1999. doi: 10.1099/00207713-50-6-1989. [DOI] [PubMed] [Google Scholar]
- 11.Sprenger WW, Hackstein JHP, Keltjens JT. 2005. The energy metabolism of Methanomicrococcus blatticola: physiological and biochemical aspects. Antonie Van Leeuwenhoek 87:289–299. doi: 10.1007/s10482-004-5941-5. [DOI] [PubMed] [Google Scholar]
- 12.Paul K, Nonoh JO, Mikulski L, Brune A. 2012. “Methanoplasmatales,” Thermoplasmatales-related archaea in termite guts and other environments, are the seventh order of methanogens. Appl Environ Microbiol 78:8245–8253. doi: 10.1128/AEM.02193-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dridi B, Fardeau M-L, Ollivier B, Raoult D, Drancourt M. 2012. Methanomassiliicoccus luminyensis gen. nov., sp. nov., a methanogenic archaeon isolated from human faeces. Int J Syst Evol Microbiol 62:1902–1907. doi: 10.1099/ijs.0.033712-0. [DOI] [PubMed] [Google Scholar]
- 14.Borrel G, O'Toole PW, Harris HMB, Peyret P, Brugère J-F, Gribaldo S. 2013. Phylogenomic data support a seventh order of methylotrophic methanogens and provide insights into the evolution of methanogenesis. Genome Biol Evol 5:1769–1780. doi: 10.1093/gbe/evt128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lapage SP, Sneath PHA, Lessel EF, Skerman VBD, Seeliger HPR, Clark WA. 1992. International code of nomenclature of bacteria. American Society for Microbiology, Washington, DC. [PubMed] [Google Scholar]
- 16.Oren A, Garrity GM. 2013. List of new names and new combinations previously effectively, but not validly, published. Int J Syst Evol Microbiol 63:3931–3934. doi: 10.1099/ijs.0.058222-0. [DOI] [PubMed] [Google Scholar]
- 17.Borrel G, Harris HMB, Tottey W, Mihajlosvki A, Parisot N, Peyretaillade E, Peyret P, Gribaldo S, O'Toole PW, Brugère J-F. 2012. Genome sequence of “Candidatus Methanomethylophilus alvus” Mx1201, a methanogenic archaeon from the human gut belonging to a seventh order of methanogens. J Bacteriol 194:6944–6945. doi: 10.1128/JB.01867-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Borrel G, Harris HMB, Parisot N, Gaci N, Tottey W, Mihajlosvki A, Deane J, Gribaldo S, Bardot O, Peyretaillade E, Peyret P, O'Toole PW, Brugère J-F. 2013. Genome sequence of “Candidatus Methanomassiliicoccus intestinalis” Issoire-Mx1, a third Thermoplasmatales-related methanogenic archaeon from human feces. Genome Announc 1(4):e00453-13. doi: 10.1128/genomeA.00453-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Iino T, Tamaki H, Tamazawa S, Ueno Y, Ohkuma M, Suzuki K, Igarashi Y, Haruta S. 2013. Candidatus Methanogranum caenicola: a novel methanogen from the anaerobic digested sludge, and proposal of Methanomassiliicoccaceae fam. nov. and Methanomassiliicoccales ord. nov., for a methanogenic lineage of the class Thermoplasmata. Microbes Environ 28:244–250. doi: 10.1264/jsme2.ME12189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gorlas A, Robert C, Gimenez G, Drancourt M, Raoult D. 2012. Complete genome sequence of Methanomassiliicoccus luminyensis, the largest genome of a human-associated Archaea species. J Bacteriol 194:4745. doi: 10.1128/JB.00956-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brugère J-F, Borrel G, Gaci N, Tottey W, O'Toole PW, Malpuech-Brugère C. 2014. Archaebiotics: proposed therapeutic use of archaea to prevent trimethylaminuria and cardiovascular disease. Gut Microbes 5:5–10. doi: 10.4161/gmic.26749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Borrel G, Parisot N, Harris HM, Peyretaillade E, Gaci N, Tottey W, Bardot O, Raymann K, Gribaldo S, Peyret P, O'Toole PW, Brugère JF. 2014. Comparative genomics highlights the unique biology of Methanomassiliicoccales, a Thermoplasmatales-related seventh order of methanogenic archaea that encodes pyrrolysine. BMC Genomics 15:679. doi: 10.1186/1471-2164-15-679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Boga HI, Brune A. 2003. Hydrogen-dependent oxygen reduction by homoacetogenic bacteria isolated from termite guts. Appl Environ Microbiol 69:779–786. doi: 10.1128/AEM.69.2.779-786.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bubendorfer S, Held S, Windel N, Paulick A, Klingl A, Thormann KM. 2012. Specificity of motor components in the dual flagellar system of Shewanella putrefaciens CN-32. Mol Microbiol 83:335–350. doi: 10.1111/j.1365-2958.2011.07934.x. [DOI] [PubMed] [Google Scholar]
- 25.Peschke M, Moog D, Klingl A, Maier UG, Hempel F. 2013. Evidence for glycoprotein transport into complex plastids. Proc Natl Acad Sci U S A 110:10860–10865. doi: 10.1073/pnas.1301945110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, Peplies J, Glöckner FO. 2013. The SILVA ribosomal RNA gene database project: improved data processing and Web-based tools. Nucleic Acids Res 41:D590–D596. doi: 10.1093/nar/gks1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ludwig W, Strunk O, Westram R, Richter L, Meier H, Yadhukumar, Buchner A, Lai T, Steppi S, Jobb G, Förster W, Brettske I, Gerber S, Ginhart AW, Gross O, Grumann S, Hermann S, Jost R, König A, Liss T, Lüssmann R, May M, Nonhoff B, Reichel B, Strehlow R, Stamatakis A, Stuckmann N, Vilbig A, Lenke M, Ludwig T, Bode A, Schleifer KH. 2004. ARB: a software environment for sequence data. Nucleic Acids Res 32:1363–1371. doi: 10.1093/nar/gkh293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Guindon S, Dufayard JF, Lefort V, Anisimova M, Hordijk W, Gascuel O. 2010. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol 59:307–321. doi: 10.1093/sysbio/syq010. [DOI] [PubMed] [Google Scholar]
- 29.Winnepenninckx B, Backeljau T, De Wachter R. 1993. Extraction of high molecular weight DNA from molluscs. Trends Genet 9:407. doi: 10.1016/0168-9525(93)90102-N. [DOI] [PubMed] [Google Scholar]
- 30.Vollmers J, Voget S, Dietrich S, Gollnow K, Smits M, Meyer K, Brinkhoff T, Simon M, Daniel R. 2013. Poles apart: arctic and antarctic Octadecabacter strains share high genome plasticity and a new type of xanthorhodopsin. PLoS One 8:e63422. doi: 10.1371/journal.pone.0063422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Markowitz VM, Mavromatis K, Ivanova NN, Chen IA, Chu K, Kyrpides NC. 2009. IMG ER: a system for microbial genome annotation expert review and curation. Bioinformatics 25:2271–2278. doi: 10.1093/bioinformatics/btp393. [DOI] [PubMed] [Google Scholar]
- 32.Tatusov RL, Fedorova ND, Jackson JD, Jacobs AR, Kiryutin B, Koonin EV, Krylov DM, Mazumder R, Mekhedov SL, Nikolskaya AN, Rao BS, Smirnov S, Sverdlov AV, Vasudevan S, Wolf YI, Yin JJ, Natale DA. 2003. The COG database: an updated version includes eukaryotes. BMC Bioinformatics 4:41. doi: 10.1186/1471-2105-4-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Punta M, Coggill PC, Eberhardt RY, Mistry J, Tate J, Boursnell C, Pang N, Forslund K, Ceric G, Clements J, Heger A, Holm L, Sonnhammer ELL, Eddy SR, Bateman A, Finn RD. 2012. The Pfam protein families database. Nucleic Acids Res 40:D290–D301. doi: 10.1093/nar/gkr1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Selengut JD, Haft DH, Davidsen T, Ganapathy A, Gwinn-Giglio M, Nelson WC, Richter AR, White O. 2007. TIGRFAMs and genome properties: tools for the assignment of molecular function and biological process in prokaryotic genomes. Nucleic Acids Res 35:D260–D264. doi: 10.1093/nar/gkl1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.The Gene Ontology Consortium. 2010. The Gene Ontology project in 2010: extensions and refinements. Nucleic Acids Res 38:D331–D335. doi: 10.1093/nar/gkp1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gattiker A, Michoud K, Rivoire C, Auchincloss AH, Coudert E, Lima T, Kersey P, Pagni M, Sigrist CJ, Lachaize C, Veuthey AL, Gasteiger E, Bairoch A. 2003. Automated annotation of microbial proteomes in Swiss Prot. Comput Biol Chem 27:49–58. doi: 10.1016/S1476-9271(02)00094-4. [DOI] [PubMed] [Google Scholar]
- 37.Fleischmann A, Darsow M, Degtyarenko K, Fleischmann W, Boyce S, Axelsen KB, Bairoch A, Schomburg D, Tipton KF, Apweiler R. 2004. IntEnz, the integrated relational enzyme database. Nucleic Acids Res 32:D434–D437. doi: 10.1093/nar/gkh119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kanehisa M, Goto S, Sato Y, Kawashima M, Furumichi M, Tanabe M. 2014. Data, information, knowledge and principle: back to metabolism in KEGG. Nucleic Acids Res 42:D199–D205. doi: 10.1093/nar/gkt1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Caspi R, Altman T, Dreher K, Fulcher CA, Subhraveti P, Keseler IM, Kothari A, Krummenacker M, Latendresse M, Mueller LA, Ong Q, Paley S, Pujar A, Shearer AG, Travers M, Weerasinghe D, Zhang P, Karp PD. 2012. The MetaCyc database of metabolic pathways and enzymes and the BioCyc collection of pathway/genome databases. Nucleic Acids Res 40:D742–D753. doi: 10.1093/nar/gkr1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tu J, Zillig W. 1982. Organization of rRNA structural genes in the archaebacterium Thermoplasma acidophilum. Nucleic Acids Res 10:7231–7245. doi: 10.1093/nar/10.22.7231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gaston MA, Zhang L, Green-Church KB, Krzycki JA. 2011. The complete biosynthesis of the genetically encoded amino acid pyrrolysine from lysine. Nature 471:647–650. doi: 10.1038/nature09918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Borrel G, Gaci N, Peyret P, O'Toole PW, Gribaldo S, Brugère J-F. 2014. Unique characteristics of the pyrrolysine system in the 7th order of methanogens: implications for the evolution of a genetic code expansion cassette. Archaea 2014:374146. doi: 10.1155/2014/374146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Thauer RK. 1998. Biochemistry of methanogenesis: a tribute to Marjory Stephenson. Microbiology 144:2377–2406. doi: 10.1099/00221287-144-9-2377. [DOI] [PubMed] [Google Scholar]
- 44.Schwörer B, Thauer R. 1991. Activities of formylmethanofuran dehydrogenase, methylenetetrahydromethanopterin dehydrogenase, methylenetetrahydromethanopterin reductase, and heterodisulfide reductase in methanogenic bacteria. Arch Microbiol 155:459–465. doi: 10.1007/BF00244962. [DOI] [Google Scholar]
- 45.van de Wijngaard WM, Creemers J, Vogels GD, Van der Drift C. 1991. Methanogenic pathways in Methanosphaera stadtmanae. FEMS Microbiol Lett 64:207–211. [DOI] [PubMed] [Google Scholar]
- 46.Paul L, Ferguson DJ, Krzycki J. 2000. The trimethylamine methyltransferase gene and multiple dimethylamine methyltransferase genes of Methanosarcina barkeri contain in-frame and read-through amber codons. J Bacteriol 182:2520–2529. doi: 10.1128/JB.182.9.2520-2529.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hao B, Gong W, Ferguson TK, James CM, Krzycki JA, Chan MK. 2002. A new UAG-encoded residue in the structure of a methanogen methyltransferase. Science 296:1462–1466. doi: 10.1126/science.1069556. [DOI] [PubMed] [Google Scholar]
- 48.Krzycki JA. 2004. Function of genetically encoded pyrrolysine in corrinoid-dependent methylamine methyltransferases. Curr Opin Chem Biol 8:484–491. doi: 10.1016/j.cbpa.2004.08.012. [DOI] [PubMed] [Google Scholar]
- 49.Burke SA, Krzycki JA. 1997. Reconstitution of monomethylamine:coenzyme M methyl transfer with a corrinoid protein and two methyltransferases purified from Methanosarcina barkeri. J Biol Chem 272:16570–16577. [DOI] [PubMed] [Google Scholar]
- 50.Janssen PH, Kirs M. 2008. Structure of the archaeal community of the rumen. Appl Environ Microbiol 74:3619–3625. doi: 10.1128/AEM.02812-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Poulsen M, Schwab C, Borg Jensen B, Engberg RM, Spang A, Canibe N, Højberg O, Milinovich G, Fragner L, Schleper C, Weckwerth W, Lund P, Schramm A, Urich T. 2013. Methylotrophic methanogenic Thermoplasmata implicated in reduced methane emissions from bovine rumen. Nat Commun 4:1428. doi: 10.1038/ncomms2432. [DOI] [PubMed] [Google Scholar]
- 52.Tersteegen A, Hedderich R. 1999. Methanobacterium thermoautotrophicum encodes two multisubunit membrane-bound [NiFe] hydrogenases. Eur J Biochem 264:930–943. doi: 10.1046/j.1432-1327.1999.00692.x. [DOI] [PubMed] [Google Scholar]
- 53.Lie TJ, Costa KC, Lupa B, Korpole S, Whitman WB, Leigh JA. 2012. Essential anaplerotic role for the energy-converting hydrogenase Eha in hydrogenotrophic methanogenesis. Proc Natl Acad Sci U S A 109:15473–15478. doi: 10.1073/pnas.1208779109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Meuer J, Bartoschek S, Koch J, Künkel A, Hedderich R. 1999. Purification and catalytic properties of Ech hydrogenase from Methanosarcina barkeri. Eur J Biochem 265:325–335. doi: 10.1046/j.1432-1327.1999.00738.x. [DOI] [PubMed] [Google Scholar]
- 55.Welte C, Krätzer C, Deppenmeier U. 2010. Involvement of Ech hydrogenase in energy conservation of Methanosarcina mazei. FEBS J 277:3396–3403. doi: 10.1111/j.1742-4658.2010.07744.x. [DOI] [PubMed] [Google Scholar]
- 56.Thauer RK, Kaster A-K, Goenrich M, Schick M, Hiromoto T, Shima S. 2010. Hydrogenases from methanogenic archaea, nickel, a novel cofactor, and H2 storage. Annu Rev Biochem 79:507–536. doi: 10.1146/annurev.biochem.030508.152103. [DOI] [PubMed] [Google Scholar]
- 57.Welte C, Deppenmeier U. 2014. Bioenergetics and anaerobic respiratory chains of aceticlastic methanogens. Biochim Biophys Acta 1837:1130–1147. doi: 10.1016/j.bbabio.2013.12.002. [DOI] [PubMed] [Google Scholar]
- 58.Moparthi VK, Hägerhäll C. 2011. The evolution of respiratory chain complex I from a smaller last common ancestor consisting of 11 protein subunits. J Mol Evol 72:484–497. doi: 10.1007/s00239-011-9447-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Welte C, Deppenmeier U. 2011. Membrane-bound electron transport in Methanosaeta thermophila. J Bacteriol 193:2868–2870. doi: 10.1128/JB.00162-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Buan NR, Metcalf WW. 2010. Methanogenesis by Methanosarcina acetivorans involves two structurally and functionally distinct classes of heterodisulfide reductase. Mol Microbiol 75:843–853. doi: 10.1111/j.1365-2958.2009.06990.x. [DOI] [PubMed] [Google Scholar]
- 61.Moparthi VK, Kumar B, Al-Eryani Y, Sperling E, Görecki K, Drakenberg T, Hägerhäll C. 2014. Functional role of the MrpA- and MrpD-homologous protein subunits in enzyme complexes evolutionary related to respiratory chain complex I. Biochim Biophys Acta 1837:178–185. doi: 10.1016/j.bbabio.2013.09.012. [DOI] [PubMed] [Google Scholar]
- 62.van der Oost J, Huynen MA, Verhees CH. 2002. Molecular characterization of phosphoglycerate mutase in archaea. FEMS Microbiol Lett 212:111–120. doi: 10.1111/j.1574-6968.2002.tb11253.x. [DOI] [PubMed] [Google Scholar]
- 63.Say RF, Fuchs G. 2010. Fructose 1,6-bisphosphate aldolase/phosphatase may be an ancestral gluconeogenic enzyme. Nature 464:1077–1081. doi: 10.1038/nature08884. [DOI] [PubMed] [Google Scholar]
- 64.Diekert G, Weber B, Thauer RK. 1980. Nickel dependence of factor F430 content in Methanobacterium thermoautotrophicum. Arch Microbiol 127:273–278. doi: 10.1007/BF00427204. [DOI] [PubMed] [Google Scholar]
- 65.Whitman WB, Wolfe RS. 1980. Presence of nickel in factor F430 from Methanobacterium bryantii. Biochem Biophys Res Commun 92:1196–1201. doi: 10.1016/0006-291X(80)90413-1. [DOI] [PubMed] [Google Scholar]
- 66.Thauer RK, Bonacker LG. 1994. Biosynthesis of coenzyme F430, a nickel porphinoid involved in methanogenesis. Ciba Found Symp 180:210–222. [DOI] [PubMed] [Google Scholar]
- 67.Kühner M, Haufschildt K, Neumann A, Storbeck S, Streif J, Layer G. 2014. The alternative route to heme in the methanogenic archaeon Methanosarcina barkeri. Archaea 2014:327637. doi: 10.1155/2014/327637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Jaenchen R, Diekert G, Thauer RK. 1981. Incorporation of methionine-derived methyl groups into factor F430 by Methanobacterium thermoautotrophicum. FEBS Lett 130:133–136. doi: 10.1016/0014-5793(81)80681-3. [DOI] [Google Scholar]
- 69.Graham DE, Xu H, White RH. 2002. Identification of coenzyme M biosynthetic phosphosulfolactate synthase: a new family of sulfonate-biosynthesizing enzymes. J Biol Chem 277:13421–13429. doi: 10.1074/jbc.M201011200. [DOI] [PubMed] [Google Scholar]
- 70.Graham DE, Taylor SM, Wolf RZ, Namboori SC. 2009. Convergent evolution of coenzyme M biosynthesis in the Methanosarcinales: cysteate synthase evolved from an ancestral threonine synthase. Biochem J 424:467–478. doi: 10.1042/BJ20090999. [DOI] [PubMed] [Google Scholar]
- 71.Ogawa T, Yoshimura T, Hemmi H. 2010. Geranylfarnesyl diphosphate synthase from Methanosarcina mazei: different role, different evolution. Biochem Biophys Res Commun 393:16–20. doi: 10.1016/j.bbrc.2010.01.063. [DOI] [PubMed] [Google Scholar]
- 72.Tachibana A. 1994. A novel prenyltransferase, farnesylgeranyl diphosphate synthase, from the haloalkaliphilic archaeon, Natronobacterium pharaonis. FEBS Lett 341:291–294. doi: 10.1016/0014-5793(94)80475-3. [DOI] [PubMed] [Google Scholar]
- 73.Villanueva L, Sinninghe Damsté JS, Schouten S. 2014. A re-evaluation of the archaeal membrane lipid biosynthetic pathway. Nat Rev Microbiol 12:438–448. doi: 10.1038/nrmicro3260. [DOI] [PubMed] [Google Scholar]
- 74.Jarrell KF, Albers S-V. 2012. The archaellum: an old motility structure with a new name. Trends Microbiol 7:307–312. doi: 10.1016/j.tim.2012.04.007. [DOI] [PubMed] [Google Scholar]
- 75.König H, Rachel R, Claus H. 2007. Proteinaceous surface layers of Archaea: ultrastructure and biochemistry, p 315–340. In Cavicchioli R. (ed), Archaea: molecular and cellular biology. ASM Press, Washington, DC. [Google Scholar]
- 76.Klingl A, Flechsler J, Heimerl T, Rachel R. 2013. Archaeal cells. In Encylopedia of Life Sciences. John Wiley & Sons Ltd, Chichester, United Kingdom. doi: 10.1002/9780470015902.a0000383.pub2. [DOI] [Google Scholar]
- 77.Rachel R, Wyschkony I, Riehl S, Huber H. 2002. The ultrastructure of Ignicoccus: evidence for a novel outer membrane and for intracellular vesicle budding in an archaeon. Archaea 1:9–18. doi: 10.1155/2002/307480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Comolli LR, Baker BJ, Downing KH, Siegerist CE, Banfield JF. 2009. Three-dimensional analysis of the structure and ecology of a novel, ultra-small archaeon. ISME J 3:159–167. doi: 10.1038/ismej.2008.99. [DOI] [PubMed] [Google Scholar]
- 79.Kandler O, König H. 1978. Chemical composition of the peptidoglycan-free cell wall of methanogenic bacteria. Arch Microbiol 118:141–152. doi: 10.1007/BF00415722. [DOI] [PubMed] [Google Scholar]
- 80.Knittel K, Boetius A. 2009. Anaerobic oxidation of methane: progress with an unknown process. Annu Rev Microbiol 63:311–334. doi: 10.1146/annurev.micro.61.080706.093130. [DOI] [PubMed] [Google Scholar]
- 81.Huber H, Stetter KO. 2006. Thermoplasmatales, p 101–112. In Dworkin M, Falkow S, Rosenberg E, Schleifer K-H, Stackebrandt E (ed), The prokaryotes, vol 3 Springer, New York, NY. [Google Scholar]
- 82.Reysenbach A-L, Liu Y, Banta AB, Breveridge TJ, Kirshtein JD, Schouten S, Tivey MK, Von Damm KL, Voytek MA. 2006. A ubiquitous thermoacidophilic archaeon from deep-sea hydrothermal vents. Nature 442:444–447. doi: 10.1038/nature04921. [DOI] [PubMed] [Google Scholar]
- 83.Ragon M, Van Driessche AES, García-Ruíz JM, Moreira D, López-García P. 2013. Microbial diversity in the deep-subsurface hydrothermal aquifer feeding the giant gypsum crystal-bearing Naica Mine, Mexico. Front Microbiol 4:37. doi: 10.3389/fmicb.2013.00037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Lloyd KG, Schreiber L, Petersen DG, Kjeldsen KU, Lever MA, Steen AD, Stepanauskas R, Richter M, Kleindienst S, Lenk S, Schram A, Jørgensen BB. 2013. Predominant archaea in marine sediments degrade detrital proteins. Nature 496:215–218. doi: 10.1038/nature12033. [DOI] [PubMed] [Google Scholar]
- 85.Segerer A, Langworthy TA, Stetter KO. 1988. Thermoplasma acidophilum and Thermoplasma volcanium sp. nov. from solfatara fields. Syst Appl Microbiol 10:161–171. doi: 10.1016/S0723-2020(88)80031-6. [DOI] [Google Scholar]
- 86.Thauer RK, Jungermann K, Decker K. 1977. Energy conservation in chemotrophic anaerobic bacteria. Bacteriol Rev 41:100–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Sprenger WW, Hackstein JH, Keltjens JT. 2007. The competitive success of Methanomicrococcus blatticola, a dominant methylotrophic methanogen in the cockroach hindgut, is supported by high substrate affinities and favorable thermodynamics. FEMS Microbiol Ecol 60:266–275. doi: 10.1111/j.1574-6941.2007.00287.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.