

Abstract
The bovine rumen represents a highly specialized bioreactor where plant cell wall polysaccharides (PCWPs) are efficiently deconstructed via numerous enzymes produced by resident microorganisms. Although a large number of fibrolytic genes from rumen microorganisms have been identified, it remains unclear how they are expressed in a coordinated manner to efficiently degrade PCWPs. In this study, we performed a metatranscriptomic analysis of the rumen microbiomes of adult Holstein cows fed a fiber diet and obtained a total of 1,107,083 high-quality non-rRNA reads with an average length of 483 nucleotides. Transcripts encoding glycoside hydrolases (GHs) and carbohydrate binding modules (CBMs) accounted for ∼1% and ∼0.1% of the total non-rRNAs, respectively. The majority (∼98%) of the putative cellulases belonged to four GH families (i.e., GH5, GH9, GH45, and GH48) and were primarily synthesized by Ruminococcus and Fibrobacter. Notably, transcripts for GH48 cellobiohydrolases were relatively abundant compared to the abundance of transcripts for other cellulases. Two-thirds of the putative hemicellulases were of the GH10, GH11, and GH26 types and were produced by members of the genera Ruminococcus, Prevotella, and Fibrobacter. Most (∼82%) predicted oligosaccharide-degrading enzymes were GH1, GH2, GH3, and GH43 proteins and were from a diverse group of microorganisms. Transcripts for CBM10 and dockerin, key components of the cellulosome, were also relatively abundant. Our results provide metatranscriptomic evidence in support of the notion that members of the genera Ruminococcus, Fibrobacter, and Prevotella are predominant PCWP degraders and point to the significant contribution of GH48 cellobiohydrolases and cellulosome-like structures to efficient PCWP degradation in the cow rumen.
INTRODUCTION
In nature, the cow rumen represents a highly specialized bioreactor wherein plant cell wall polysaccharides (PCWPs) are efficiently deconstructed. The extraordinary efficiency results from the concerted action of various enzymes produced by rumen-resident bacteria, archaea, fungi, and protozoa. Three rumen bacteria, i.e., Ruminococcus flavefaciens, Ruminococcus albus, and Fibrobacter succinogenes, which can be isolated and cultivated in the laboratory, have been thought to serve a predominant role in the degradation of cellulosic PCWPs in this niche (1, 2). However, metagenomic quantitations based on 16S rRNA gene analysis indicate that these three species of bacteria account for only less than 5% of the total rumen microorganisms (3). In addition, Koike et al. estimated that ∼77% of the rumen microorganisms attached to solid fibers are uncultured, as their 16S rRNA gene sequences share less than 97% similarity with those of known isolates (4).
To bypass the cultivation step, metagenomic approaches involving the direct analysis of total DNA sequences have been extensively used to investigate the PCWP-degrading gastrointestinal microbes in a variety of herbivores, such as termite hindguts (5); cow (6–8), yak (9), and Svalbard reindeer (10) rumens; and the foreguts of Australian macropods (11). It has been demonstrated that PCWP-degrading enzymes that exist in the rumen are far more diverse than was previously believed. It is somewhat surprising, however, that cellobiohydrolases or exo-type proteins of the glycoside hydrolase 48 (GH48) family were barely detected in the rumen or gut microbiomes and that proteins characteristic of cellulosomes (e.g., dockerins or scaffolding proteins) were also poorly represented in all published metagenomic data sets (7–9). Therefore, it remains far from clear how PCWPs are efficiently degraded in the rumen.
The problem is partially attributable to limitations with the use of metagenomic approaches in understanding fibrolytic activities in the rumen. Although a large number of fibrolytic genes and gene clusters have been identified from rumen microorganisms in metagenomic studies, it has yet to be determined if and how actively these genes are expressed and how the expression of these genes is coordinated in an efficient manner. To help address these questions, we have performed a metatranscriptomic study of plant cell wall hydrolysis by a microbiome in the cow rumen by using the transcriptome sequencing technology. Metatranscriptomics has been widely employed to investigate microbiomes in marine or aquatic environments (12–16), soils (17, 18), the rhizosphere (19), and humans (20, 21). However, to the best of our knowledge, only one report on the metatranscriptomic study of a rumen sample has been published so far (22). In that report, fungal gene expression in the rumen of muskoxen (Ovibos moschatus) was examined through the analysis of a rumen cDNA library.
In the present study, we provide a metatranscriptomic insight into PCWP degradation in the rumen. We have identified actively transcribed genes encoding putative glycoside hydrolases (GHs) involved in cellulose, hemicellulose, and oligosaccharide degradation as well as rumen microorganisms responsible for the expression of these genes. We show that genes encoding GH48 proteins and cellulosome components were transcribed in a relatively high abundance, an observation unexpected from the findings of previous metagenomic analyses of rumen microorganisms.
MATERIALS AND METHODS
Rumen content sampling.
A mixture of rumen fluid and undigested fiber was taken through the rumen fistula 1 h after the morning feeding from two healthy adult Holstein dairy cows (body weight, ∼550 kg each), which had been fed a corn straw (CS)-containing diet (on the basis of the dry matter content, 17.4% crude protein, 2.9% ether extract, 35.6% neutral detergent fiber, 21.7% acid detergent fiber, 0.77% calcium, 0.39% phosphorus) for 3 months. The samples were quickly filtered through four layers of gauze, and the ruminal solids were immediately frozen in liquid nitrogen and transported to the laboratory for total RNA extraction. All animal experiments were carried out following the standard protocols approved by the Institute of Animal Science, Chinese Academy of Agricultural Sciences, Beijing, China (permit number RNL201102).
Total RNA extraction.
The solid rumen samples were crushed and milled in an SPEX 6870 Freezer/Mill (SPEX SamplePrep, Metuchen, NJ) in liquid nitrogen to disrupt the microbial cell wall. Total RNA was extracted from the homogenate and purified using the TRIzol reagent (Invitrogen Inc., USA) according to the manufacturer's protocol. The RNA preparations were treated with RNase-free DNase I (TaKaRa, China) to remove contaminating DNA. The removal of DNA was verified by PCR with primers targeting the 16S rRNA gene. The concentration and the integrity of the RNA preparations were determined with an Agilent 2100 bioanalyzer (Agilent Technologies, USA).
cDNA synthesis and pyrosequencing.
In order to maximize mRNA representation in the metatranscriptomic libraries, a Meta-Bacteria Ribo-Zero rRNA removal kit (Epicentre) was used to remove rRNAs from the total RNA preparations. The double-stranded cDNAs were synthesized by using a SuperScript III double-stranded cDNA synthesis kit (Invitrogen, Carlsbad, CA) with random hexamers as the primers according to the manufacturer's protocol. The resultant cDNAs were converted into single-stranded template DNA (sstDNA) libraries by using a GS DNA library preparation kit (Roche Applied Science, USA). The sstDNA libraries were clonally amplified in a bead-immobilized form by using a GS emPCR kit and sequenced on a 454 genome sequencer FLX instrument (Roche Applied Science).
Removal of rRNA sequences.
The standard flowgram format (SFF) files derived from the 454 sequencing were converted into fasta and quality files using the Roche 454 Sffinfo utility (454 proprietary software) with the default parameters. The adaptor sequences and low-quality reads were removed. Pyrosequencing reads with a length shorter than 100 nucleotides (nt) were filtered out. The filtered sequences were analyzed by BLASTN (23) against Silva small-subunit (SSU) and large-subunit (LSU) rRNA reference databases (SSU Ref and LSU Ref, respectively) (24) using an E-value cutoff of 1 × 10−5 to identify rRNA reads (rRNAs). The sequences assigned to either SSU or LSU rRNA with a bit score of ≥100 were considered rRNAs, and the others were classified as non-rRNA reads (non-rRNAs).
Functional analysis of non-rRNAs.
The sequences of the non-rRNAs were searched against the sequences in the NCBI nonredundant protein database (NR database) (25) using the BLASTX algorithm (23) with an E-value cutoff of 1 × 10−5. The assigned non-rRNAs with a bit score of ≥50 were considered to have putative functions, and the best hits were extracted. In addition, the deduced amino acid sequences of these non-rRNAs were extracted from the best hits with Perl script and were queried against the sequences in the Kyoto Encyclopedia of Genes and Genomes (KEGG) database (26) using BLASTP (23) with an E-value cutoff of 1 × 10−5 and a minimum bit score of 50. Meanwhile, they were queried against the Pfam database (27) employing the HMMER (version 3.0) program (28) with the same E-value cutoff.
Taxonomic binning of assignable non-rRNAs.
The sequences of the non-rRNAs were searched against the sequences in the NR database (25) using the BLASTX algorithm (23) with an E-value cutoff of 1 × 10−5, and the best hits were subjected to analysis with Metagenome Analyzer (MEGAN) (29), a program for taxonomic analysis which has been widely used to assign taxa to sequences obtained from a metagenome (8, 9, 11, 30) or a metatranscriptome (17, 22, 31, 32).
Identification of carbohydrate-degrading enzymes.
Following the Pfam analysis of non-rRNAs, as described above, the deduced amino acid sequences were searched against the sequences of carbohydrate-active enzymes (CAZymes) (33) and carbohydrate binding modules (CBMs) (34) with available Pfam accession numbers in the CAZymes database (CAZy; http://www.cazy.org) (35). Putative PCWP-degrading enzymes were identified and classified on the basis of the CAZy results.
Pyrosequencing data accession number.
The raw standard flowgram format (SFF) data for our samples have been deposited in the NCBI Sequence Read Archive (SRA) under accession number SRP048689.
RESULTS
Pyrosequencing of rumen cDNA libraries.
Two rumen samples, designated CS9007 and CS9036, were taken from two Holstein cows at 1 h after the morning feeding, when active PCWP degradation is known to occur (36, 37). Total RNAs were prepared from the rumen samples. After removing the majority of the rRNAs from the samples, the ribominus RNAs were purified and reverse transcribed into cDNAs. Pyrosequencing of the CS9007 and CS9036 cDNA samples yielded a total of 666,643 and 588,918 high-quality reads, respectively (including 627,883 and 479,200 non-rRNAs, respectively), with average lengths of 525 and 417 nt, respectively (Table 1).
TABLE 1.
Summary of pyrosequencing results for two cow rumen samples
| Sequencing parameter | Result for sample: |
|
|---|---|---|
| CS9007 | CS9036 | |
| No. of reads with high quality and long length (≥100 nt) | 666,643 | 588,918 |
| Total length (nt) | 349,926,574 | 245,715,015 |
| Avg length (nt) | 525 | 417 |
| No. of rRNAs | 38,760 | 109,718 |
| No. of non-rRNAs | 627,883 | 479,200 |
| Total length of non-rRNAs (nt) | 330,886,052 | 203,358,071 |
| Length range (nt) | 100–1,142 | 100–1,305 |
| Avg length (nt) | 527 | 424 |
| GC content | 48 | 44 |
| % rRNAs | 5.81 | 18.63 |
| % non-rRNAs | 94.19 | 81.37 |
The majority of the reads (∼75% and 66% for samples CS9007 and CS9036, respectively) were assigned to proteins in the NR database (see Table S1 in the supplemental material). The remaining 25% and 34% of the reads from CS9007 and CS9036, respectively, could not be assigned to any proteins in the NR database, suggesting that they may encode unknown proteins or long noncoding RNAs. The amino acid sequences of the best hits of the assigned reads were extracted and searched against the sequences in the KEGG database. Reads were abundantly represented in the category of carbohydrate metabolism, accounting for ∼7% of the total non-rRNAs for the two samples (see Fig. S1 in the supplemental material), indicating that genes responsible for carbohydrate metabolism were robustly transcribed in the rumen. We also determined the abundance of the functional proteins on the basis of the Pfam analysis. As shown in Table S1 and Fig. S2 in the supplemental material, 4,497 protein domains from 359,231 reads and 4,218 domains from 189,727 reads were identified for CS9007 and CS9036, respectively. On average, the level of transcription for a protein domain corresponded to ∼0.01% of the total non-rRNAs in each of the two samples, and only about 20% of the domains showed more than 0.01% abundance (see Fig. S2 in the supplemental material). Therefore, a transcript level of greater than 0.01% of the total non-rRNAs for a given domain was considered abundant in this study.
GHs and CBMs in the cow rumen.
On the basis of the CAZymes search results, ∼2% of the total non-rRNAs were putatively related to members of the CAZymes (E value ≤ 1 × 10−5) (see Tables S1 and S2 in the supplemental material). Among them, sequences encoding glycoside hydrolases (GHs), which hydrolyze the glycosidic bonds in carbohydrates, and carbohydrate binding modules (CBMs), which are a common appendix module of carbohydrate-active enzymes involved in carbohydrate binding, accounted for ∼1% and ∼0.1% of the total non-rRNAs respectively.
Twenty-six of these GH families were predicted to be involved in PCWP degradation (Fig. 1; see also Fig. S3a in the supplemental material). Transcripts coding for GHs possibly involved in cellulose, hemicellulose, and oligosaccharide degradation (designated CHO-GHs) were relatively enriched, representing ∼0.5% of the total non-rRNAs in each of the two metatranscriptomes. Among the CHO-GHs, GH1, GH2, GH3, GH5, GH9, GH10, GH11, GH26, GH43, GH45, GH48, and GH94 were relatively abundant (>0.01% each of the total non-rRNAs).
FIG 1.
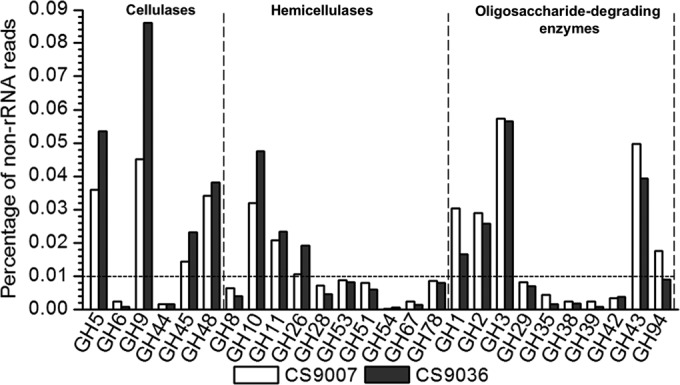
Abundance of sequencing reads encoding members of various GH protein families putatively involved in cellulose, hemicellulose, and oligosaccharide degradation in the cow rumen, as determined by metatranscriptomics. The abundance is defined as the proportion of the number of sequencing reads for a given GH to that for the total non-rRNAs.
Six GH families responsible for cellulose degradation (designated cel-GHs), i.e., GH5, GH6, GH9, GH44, GH45, and GH48, were identified in both metatranscriptomes, and GH5, GH9, GH45, and GH48 each accounted for >0.01% of the total non-rRNAs (Fig. 1). The transcripts for GH9, a family of proteins with soluble or insoluble (amorphous) cellulose as their favored substrate (38), existed in the greatest abundance among the cel-GH transcripts (accounting for 34% and 42% of the total cel-GH reads in CS9007 and CS9036, respectively) (see Fig. S3b in the supplemental material). Transcripts for GH5, GH48, and GH45 were also abundant, being ∼27%, ∼22%, and ∼11% of the cel-GH reads, respectively, in each of the two samples. Notably, the transcripts of GH48, a family of enzymes considered the key component of various cellulolytic systems (39), were among the abundant cel-GH transcripts. Over 95% of the GH48 sequences from both samples were annotated as a putative cellulose, 1,4-β-cellobiosidase (EC 3.2.1.91 or EC 3.2.1.176). More than 50% of the GH9 transcripts, nearly 70% of the GH5 transcripts, and all GH45 transcripts were annotated as endoglucanases (EC 3.2.1.4) in each of the two samples. It is worth noting that the GH48 transcripts were represented in the metatranscriptomes in this study (7.65% of the total GH transcripts targeting PWCP) much more abundantly than expected from previous metagenomic studies (5–9, 11, 30) on herbivore gut microbiomes (0 to 0.16% of the total PWCP-targeting GH genes) (Table 2).
TABLE 2.
Comparison of the PCWP-targeting GHs identified in the present metatranscriptomes with those from other herbivore metatranscriptomes and metagenomesc
| Enzyme and CAZy family | Pfam accession no. | Pfam HMM name | % of each group relative to total PWCP-targeting GHs identified in each data set |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| MT |
MG |
||||||||||
| Holstein cows rumena (this study) | Muskoxen rumen (22) | Angus cow rumenb (6) | Guernsey cow rumen (7) | Jersey cow rumen (8) | Yak rumen (9) | Termite hindgut (5) | Tammar wallaby foregut (11) | Giant panda gut (30) | |||
| Cellulases | |||||||||||
| GH5 | PF00150 | Cellulase | 9.40 | 12.30 | 1.16 | 11.43 | 10.57 | 6.67 | 14.07 | 3.60 | 1.72 |
| GH6 | PF01341 | Glyco_hydro_6 | 0.35 | 9.02 | 0.00 | 0.00 | 0.44 | 0.00 | 0.00 | 0.00 | 0.00 |
| GH7 | PF00840 | Glyco_hydro_7 | 0.00 | 0.00 | 0.00 | 0.01 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| GH9 | PF00759 | Glyco_hydro_9 | 13.71 | 8.52 | 1.06 | 6.26 | 3.52 | 3.93 | 2.26 | 0.00 | 0.00 |
| GH44 | PF12891 | Glyco_hydro_44 | 0.35 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| GH45 | PF02015 | Glyco_hydro_45 | 3.94 | 9.51 | 0.00 | 0.91 | 0.00 | 0.07 | 1.01 | 0.00 | 0.00 |
| GH48 | PF02011 | Glyco_hydro_48 | 7.65 | 8.85 | 0.04 | 0.02 | 0.00 | 0.16 | 0.00 | 0.00 | 0.00 |
| Subtotal | 35.40 | 48.20 | 2.25 | 18.63 | 14.54 | 10.83 | 17.34 | 3.60 | 1.72 | ||
| Endohemicellulases and debranching enzymes | |||||||||||
| GH8 | PF01270 | Glyco_hydro_8 | 1.11 | 0.98 | 0.56 | 2.59 | 0.00 | 0.89 | 1.26 | 0.36 | 1.15 |
| GH10 | PF00331 | Glyco_hydro_10 | 8.48 | 11.64 | 1.10 | 8.07 | 15.42 | 13.64 | 11.56 | 3.96 | 1.15 |
| GH11 | PF00457 | Glyco_hydro_11 | 4.78 | 7.38 | 0.11 | 1.30 | 0.00 | 1.25 | 3.52 | 0.00 | 0.00 |
| GH12 | PF01670 | Glyco_hydro_12 | 0.00 | 0.00 | 0.00 | 0.00 | 0.44 | 0.00 | 0.00 | 0.00 | 0.00 |
| GH26 | PF02156 | Glyco_hydro_26 | 3.12 | 2.13 | 0.84 | 2.91 | 0.44 | 2.75 | 3.77 | 1.80 | 0.00 |
| GH28 | PF00295 | Glyco_hydro_28 | 1.26 | 0.16 | 0.69 | 3.72 | 0.00 | 1.25 | 1.51 | 0.72 | 0.57 |
| GH53 | PF07745 | Glyco_hydro_53 | 1.84 | 0.66 | 3.01 | 0.00 | 7.93 | 5.46 | 3.02 | 3.24 | 0.00 |
| GH51 | PF06964 | Alpha-L-AF_C | 1.50 | 0.16 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 4.68 | 0.00 |
| GH54 | PF09206 | ArabFuran-catal | 0.08 | 0.00 | 0.18 | 0.00 | 0.00 | 0.57 | 0.00 | 0.00 | 0.00 |
| GH62 | PF03664 | Glyco_hydro_62 | 0.00 | 0.00 | 0.00 | 0.01 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| GH67 | PF07488 | Glyco_hydro_67 M | 0.43 | 0.16 | 0.00 | 0.95 | 0.00 | 5.58 | 2.51 | 1.80 | 1.15 |
| GH78 | PF05592 | Bac_rhamnosid | 1.80 | 0.82 | 5.62 | 9.92 | 5.73 | 2.18 | 0.00 | 8.99 | 1.15 |
| Subtotal | 24.41 | 24.10 | 12.11 | 29.46 | 29.96 | 33.57 | 27.14 | 25.54 | 5.17 | ||
| Oligosaccharide-degrading enzymes | |||||||||||
| GH1 | PF00232 | Glyco_hydro_1 | 5.07 | 3.93 | 2.03 | 1.99 | 4.41 | 1.70 | 5.53 | 21.94 | 58.05 |
| GH2 | PF02836 | Glyco_hydro_2_C | 6.01 | 1.97 | 31.60 | 11.31 | 7.05 | 4.82 | 5.78 | 8.63 | 0.57 |
| GH3 | PF00933 | Glyco_hydro_3 | 12.20 | 8.20 | 29.57 | 22.40 | 21.15 | 27.90 | 17.34 | 25.90 | 10.34 |
| GH29 | PF01120 | Alpha_L_fucos | 1.64 | 0.00 | 4.67 | 7.40 | 1.32 | 4.60 | 0.00 | 0.72 | 0.57 |
| GH35 | PF01301 | Glyco_hydro_35 | 0.67 | 0.00 | 2.10 | 1.24 | 0.88 | 2.40 | 0.75 | 1.08 | 2.30 |
| GH38 | PF01074 | Glyco_hydro_38 | 0.47 | 0.16 | 2.90 | 2.14 | 0.44 | 0.46 | 2.76 | 1.08 | 5.75 |
| GH39 | PF01229 | Glyco_hydro_39 | 0.37 | 0.00 | 0.37 | 2.48 | 7.93 | 0.81 | 0.75 | 0.36 | 5.17 |
| GH42 | PF02449 | Glyco_hydro_42 | 0.79 | 0.00 | 2.08 | 2.95 | 0.00 | 1.06 | 6.03 | 2.88 | 10.34 |
| GH43 | PF04616 | Glyco_hydro_43 | 9.53 | 13.28 | 10.32 | 0.00 | 12.33 | 11.85 | 4.02 | 3.60 | 0.00 |
| GH52 | PF03512 | Glyco_hydro_52 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.75 | 0.00 | 0.00 |
| GH94 | PF06165 | Glyco_transf_36 | 3.43 | 0.16 | 0.00 | 0.00 | 0.00 | 0.00 | 11.81 | 4.68 | 0.00 |
| Subtotal | 40.19 | 27.70 | 85.64 | 51.91 | 55.51 | 55.60 | 55.53 | 70.86 | 93.10 | ||
| Total PWCP-targeting GHs | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | ||
Each number is the average for two Holstein cow rumen samples.
Each number is the average for four Angus cow rumen samples.
MT, metatranscriptome; MG, metagenome; PCWP, plant cell wall polysaccharide.
Xylans, mannans, and, in some plants, pectins are the major hemicellulosic polysaccharides in the plant cell wall. Transcripts for proteins of nine GH families (i.e., GH8, GH10, GH11, GH26, GH28, GH51, GH53, GH67, and GH78), which are known to be involved in hemicellulose degradation, were detected in the two metatranscriptomes (Fig. 1; see also Fig. S3c in the supplemental material). These proteins, designated hemi-GHs, include both endo-acting and debranching enzymes. The abundance of GH10, GH11, and GH26 was >0.01% of the total non-rRNAs. GH10 and GH11 represent the two major families of xylan-degrading enzymes. Most of them (i.e., in samples CS9007 and CS9036, 77% and 82% for GH10, respectively, and 94% and 98% for GH11, respectively) encoded putative endo-1,4-β-xylanases (EC 3.2.1.8). The vast majority of GH26 proteins (81% and 89% for the CS9007 and CS9036 samples, respectively) were putative endo-β-1,4-mannanases (EC 3.2.1.78).
Transcripts for a number of GHs responsible for oligosaccharide degradation (designated oligo-GHs) were identified in the metatranscriptomes. They code for enzymes belonging to the GH1, GH2, GH3, GH29, GH35, GH38, GH39, GH42, GH43, and GH94 families. GH1, GH2, GH3, and GH43 transcripts accounted for >0.01% of the non-rRNAs. Among them, GH3 existed in the greatest abundance (∼28% and 35% of the total oligo-GH reads in samples CS9007 and CS9036, respectively) (Fig. 1; see also Fig. S3d in the supplemental material). Transcripts for GH43, GH2, and GH1 were also relatively abundant, accounting for ∼24%, ∼15%, and ∼13% (as the average for two samples) of the oligo-GH reads, respectively. They were primarily annotated as different types of oligosaccharide hydrolases, including β-glucosidase (EC 3.2.1.21), 6-phospho-β-glucosidase (EC 3.2.1.86), β-N-acetylhexosaminidase (EC 3.2.1.52), β-galactosidase (EC 3.2.1.23), glucan 1,4-β-glucosidase (EC 3.2.1.74), xylan 1,4-β-xylosidase/α-N-arabinofuranosidase (EC 3.2.1.37/EC 3.2.1.55), arabinan endo-1,5-α-l-arabinosidase (EC 3.2.1.99), and β-glucuronidase (EC 3.2.1.31). In addition, ∼9% and 6% of the total oligo-GH reads in CS9007 and CS9036, respectively, were annotated as GH94 proteins, among which most (82% and 72%, respectively) were further annotated as putative cellobiose phosphorylases (EC 2.4.1.20), suggesting that cellobiose phosphorylation is a major pathway for cellobiose uptake and metabolism during fibrolytic degradation in the rumen.
CBMs are highly specialized auxiliary domains in PCWP-degrading enzymes. These domains are able to bind to polysaccharides, promoting intimate attachment of the catalytic domains to the substrates and enhancing the activity of the enzymes. Seven CBM families (i.e., CBM2, CBM3, CBM4/9/16/22, CBM6, CBM10, CBM11, and CBM13) with a combined abundance of ∼0.06% in the two samples were predicted to be associated with PCWP-degrading catalytic domains (CHO-CBMs). CBM4/9/16/22 occurred the most frequently (Fig. 2), representing 52% and 64% of the CHO-CBM reads in samples CS9007 and CS9036, respectively (see Fig. S4 in the supplemental material). CBM6 and CBM10 transcripts were also enriched in both samples (Fig. 2; see also Fig. S4 in the supplemental material). CBM6 has been found in hemicellulose- and oligosaccharide-degrading enzymes (40, 41). Transcripts encoding CBM10, a domain which is thought to be linked to the cellulosomes of rumen anaerobic fungi (42–45), were frequently found (0.010% and 0.004% of the non-RNAs in CS9007 and CS9036, respectively). Notably, ∼20% of the CBM10 reads were linked to the GH48 reads. In addition, transcripts for the dockerin and cohesin domains, components of bacterial cellulosomes, were also detected in the rumen metatranscriptomes, representing ∼0.005% and ∼0.03% of the total non-rRNAs in CS9007 and CS9036, respectively (see Table S2 in the supplemental material).
FIG 2.
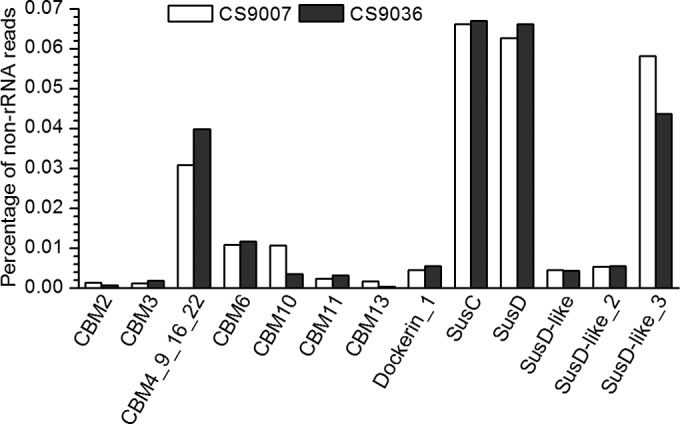
Abundance of sequencing reads encoding the domains of CBMs, dockerin, and SusC/SusD-like proteins, as determined by metatranscriptomics. The abundance is defined as the proportion of the number of sequencing reads for a given domain to that for the total non-rRNAs.
Polysaccharide utilization loci (PULs) are gene clusters encoding multiple proteins that are critical to the utilization of certain complex carbohydrates by some gut bacteria and commonly linked with fibrolytic genes (9). Analysis of our metatranscriptomic data showed a relatively high abundance of the PUL genes. About 0.2% of the total non-rRNAs encoded PUL proteins, i.e., SusC, SusD, SusD-like, SusD-like_2, or SusD-like_3, in both samples (Fig. 2; see also Table S2 in the supplemental material).
Diversity of rumen microorganisms involved in PCWP degradation.
The taxonomic affiliations of metatranscriptomic sequences were determined by using the Metagenome Analyzer (MEGAN) program. Approximately 66% and 75% of the non-rRNAs in samples CS9007 and CS9036, respectively, were assignable taxonomically (see Fig. S5 and Table S1 in the supplemental material). These reads were predominantly affiliated with bacteria (89% and 77% in CS9007 and CS9036, respectively), followed by eukaryotes (6% and 17% in CS9007 and CS9036, respectively), archaea (4% and 5 in CS9007 and CS9036, respectively), and viruses (0.4% and 0.7% in CS9007 and CS9036, respectively).
In all, 31 bacterial and 6 archaeal phyla were found in the two rumen metatranscriptomes, among which six bacterial phyla (Firmicutes, Bacteroidetes, Spirochaetes, Proteobacteria, Actinobacteria, and Fibrobacteres) and one archaeal phylum (Euryarchaeota) accounted for more than 60% of the total non-rRNAs (see Table S3 in the supplemental material). At the genus level, over 750 bacterial and 60 archaeal genera were detected (see Table S4 in the supplemental material). The most abundant (>1% of all non-rRNAs in both samples) bacterial genera were Prevotella, Ruminococcus, Clostridium, Butyrivibrio, Eubacterium, Bacteroides, Treponema, Blautia, and Roseburia. Methanobrevibacter (3% in both samples) was the most abundant archaeal genus. The majority of eukaryal reads were affiliated with protozoa (3% and 6% of the total non-rRNAs in samples CS9007 and CS9036, respectively) and fungi (0.6% and 1% in samples CS9007 and CS9036, respectively). However, there was no predominant eukaryal genus in the rumen metatranscriptomic library that accounted for over 0.1% of the total non-rRNAs.
Notably, most of the non-rRNAs related to PCWP degradation were derived from a limited number of genera. GH reads for cellulases, hemicellulases and oligosaccharide-degrading enzymes were primarily from 71 genera (Fig. 3). Over half of the cel-GH reads were from the genera Ruminococcus (40% and 48% for samples CS9007 and CS9036, respectively) and Fibrobacter (13% for both samples) (see Fig. S6 in the supplemental material). Other rumen bacteria that produced >1% of the total cel-GH reads were members of the genera Eubacterium, Prevotella, and Clostridium. Surprisingly, Bacteroides, a genus of the phylum Bacteroidetes well known to be present in the rumen, did not appear to play a predominant role in cellulose degradation, as evidenced by its relatively low representation in the total cel-GH counts. Epidinium and Polyplastron were the major cellulolytic protozoa, contributing 80% and 20% of the protozoan cel-GH transcripts, respectively, whereas the fungi Piromyces and Neocallimastix appeared to be relatively active in cellulose degradation, producing 54% and 41% of the fungal cel-GH reads, respectively. An average of 78% of the cel-GH reads for the two samples encoded homologues from a small group of only 12 species (Table 3). In fact, ∼62% of the cel-GH reads encoded putative cellulases similar to those from R. flavefaciens, Ruminococcus champanellensis, R. albus, F. succinogenes, and the protozoan Epidinium ecaudatum. Since over 85% of these reads encoded proteins with less than 90% similarity at the amino acid sequence level to their homologues from the five known species, it is probably not the well-known culturable representatives of Ruminococcus, Fibrobacter, and Epidinium species but, rather, the unknown relatives of these species that served the most significant roles in cellulose degradation in the rumen. It is worth noting that the GH48 cellobiohydrolases resembled those from Ruminococcus (61% of the total GH48 proteins), the anaerobic fungi Piromyces (8%) and Neocallimastix (6%), as well as uncultured microorganisms (23%), with amino acid sequence similarities ranging from 32% to 100%.
FIG 3.
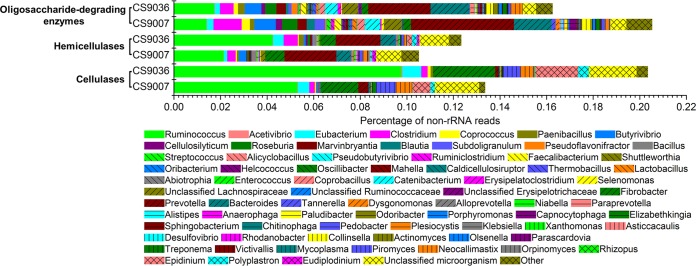
Taxonomic affiliation of putative cellulase, hemicellulase, and oligosaccharide-degrading enzymes. The abundance is defined as the proportion of the number of sequencing reads for a given genus to that for the total non-rRNAs. “Other” represents the taxa detected in only one of the two metatranscriptomes.
TABLE 3.
Profiling of dominant taxa encoding putative cellulasesa
| Domain | Phylum | Genus | Species | Abundanceb (%) | Relative ratioc (%) | % reads with the following % identityd: |
|||
|---|---|---|---|---|---|---|---|---|---|
| <30 | ≥30–<60 | ≥60–<90 | ≥90 | ||||||
| Bacteria | Firmicutes | Ruminococcus | Ruminococcus flavefaciens | 0.0278 | 16.0578 | 0 | 35 | 59 | 6 |
| Ruminococcus champanellensis | 0.0242 | 14.1452 | 0 | 59 | 41 | 0 | |||
| Ruminococcus albus | 0.0234 | 13.6421 | 0 | 19 | 68 | 13 | |||
| Eubacterium | Eubacterium cellulosolvens | 0.0037 | 2.1330 | 0 | 74 | 8 | 18 | ||
| Eubacterium siraeum | 0.0014 | 0.9120 | 0 | 44 | 56 | 0 | |||
| Fibrobacteres | Fibrobacter | Fibrobacter succinogenes | 0.0215 | 12.6428 | 0 | 63 | 34 | 3 | |
| Subtotal for 6 species | 0.1019 | 59.5328 | |||||||
| Eukaryotes | Protozoa | Epidinium | Epidinium ecaudatum | 0.0104 | 5.9223 | 0 | 11 | 81 | 8 |
| Epidinium caudatum | 0.0027 | 1.5107 | 0 | 25 | 71 | 4 | |||
| Polyplastron | Polyplastron multivesiculatum | 0.0033 | 1.8433 | 0 | 23 | 65 | 12 | ||
| Fungi | Neocallimastix | Neocallimastix patriciarum | 0.0061 | 3.8959 | 0 | 13 | 74 | 13 | |
| Piromyces | Piromyces sp. strain E2 | 0.0059 | 3.5314 | 0 | 28 | 66 | 6 | ||
| Piromyces equi | 0.0018 | 1.2945 | 0 | 14 | 82 | 4 | |||
| Subtotal for 6 species | 0.0302 | 17.9981 | |||||||
| Total | 0.1321 | 77.5309 | |||||||
| Unclassified uncultured organisms | 0.0189 | 11.6988 | 0 | 36 | 52 | 12 | |||
Average data for the two cow samples are shown.
Abundance refers to the proportion of cellulase-related reads in the total non-rRNAs. Only organisms with cellulase-related reads with an abundance of ≥0.001% are shown.
The relative ratio refers to the proportion of the number of cellulase-related reads in a given organism in the total number of cellulase-related reads.
The similarities of proteins encoded by transcripts to that of known cellulases from a given organism at the amino acid sequence level are shown.
The majority of the hemi-GH reads in samples CS9007 and CS9036 were derived from the genera Ruminococcus (20% and 34%, respectively), Prevotella (21% and 16%, respectively), Fibrobacter (8% and 5%, respectively), Bacteroides (6% and 5%, respectively), and Clostridium (3% and 5%, respectively) (see Fig. S6 in the supplemental material). About 45% of these reads encoded putative hemicellulases resembling those from the species R. flavefaciens, R. albus, R. champanellensis, Prevotella ruminicola, and F. succinogenes with 60 to 90% similarity at the amino acid sequence level (Table 4).
TABLE 4.
Profiling of the dominant taxa encoding putative hemicellulasesa
| Domain | Phylum | Genus | Species | Abundanceb (%) | Relative ratioc (%) | % reads with the following % identityd: |
|||
|---|---|---|---|---|---|---|---|---|---|
| <30 | ≥30–<60 | ≥60–<90 | ≥90 | ||||||
| Bacteria | Firmicutes | Ruminococcus | Ruminococcus flavefaciens | 0.0172 | 14.6800 | 0 | 35 | 58 | 7 |
| Ruminococcus albus | 0.0089 | 7.7819 | 0 | 22 | 63 | 15 | |||
| Ruminococcus champanellensis | 0.0036 | 3.0999 | 0 | 37 | 60 | 3 | |||
| Ruminococcus sp. | 0.0020 | 1.7640 | 0 | 23 | 73 | 4 | |||
| Clostridium | Clostridium methylpentosum | 0.0017 | 1.5102 | 0 | 100 | 0 | 0 | ||
| Eubacterium | Eubacterium cellulosolvens | 0.0027 | 2.3070 | 0 | 36 | 64 | 0 | ||
| Bacteroidetes | Prevotella | Prevotella ruminicola | 0.0140 | 12.4335 | 0 | 15 | 78 | 7 | |
| Prevotella bergensis | 0.0013 | 1.1137 | 0 | 14 | 79 | 7 | |||
| Bacteroides | Bacteroides eggerthii | 0.0018 | 1.5771 | 0 | 15 | 85 | 0 | ||
| Fibrobacteres | Fibrobacter | Fibrobacter succinogenes | 0.0073 | 6.4952 | 0 | 12 | 70 | 18 | |
| Subtotal for 10 species | 0.0607 | 52.7623 | |||||||
| Eukaryotes | Protozoa | Eudiplodinium | Eudiplodinium maggii | 0.0013 | 1.1806 | 0 | 37 | 73 | 0 |
| Polyplastron | Polyplastron multivesiculatum | 0.0014 | 1.2248 | 0 | 73 | 27 | 0 | ||
| Epidinium | Epidinium ecaudatum | 0.0012 | 0.9975 | 0 | 9 | 58 | 33 | ||
| Subtotal for 3 species | 0.0040 | 3.4029 | |||||||
| Total | 0.0646 | 56.1652 | |||||||
| Unclassified uncultured organisms | 0.0110 | 9.6039 | 0 | 19 | 48 | 33 | |||
Average of data for the two cow samples are shown.
The abundance refers to the proportion of hemicellulase-related reads in the total non-rRNAs. Only organisms with hemicellulase-related reads with an abundance of ≥0.001% are shown.
The relative ratio refers to the proportion of the number of hemicellulase-related reads in a given organism in the total number of hemicellulase-related reads.
Similarities of proteins encoded by transcripts to that of known hemicellulases from a given organism at the amino acid sequence level are shown.
The oligo-GH reads in samples CS9007 and CS9036 were primarily derived from the genera Prevotella (∼22% and 16%, respectively) and Bacteroides (∼8% and 10%, respectively), followed by Ruminococcus (∼7% and 11%, respectively), Clostridium (∼6% and 4%, respectively), Butyrivibrio (∼4% in both samples), Catenibacterium (∼3% in both samples), and Roseburia (∼3% in both samples) (see Fig. S6 in the supplemental material). Strikingly, Fibrobacter, anaerobic fungi, and protozoa, which were the major producers of cel-GH reads, on the basis of our data, appeared to contribute little to oligo-GH hydrolysis (Fig. 3; see also Fig. S6 in the supplemental material). It is worth noting that oligo-GH reads were produced by more species than either cel-GH or hemi-GH reads, and the major producers of oligo-GHs were less predominant (Table 5; see also Fig. S6 in the supplemental material). The most abundant producer for oligo-GHs was phylogenetically related to P. ruminicola, which produced ∼13% of the total oligo-GH reads.
TABLE 5.
Profiling of the dominant taxa encoding oligosaccharide-degrading enzymesa
| Domain | Phylum | Genus | Species | Abundanceb (%) | Relative ratioc (%) | % reads with the following % identityd: |
|||
|---|---|---|---|---|---|---|---|---|---|
| <30 | ≥30–<60 | ≥60–<90 | ≥90 | ||||||
| Bacteria | Bacteroidetes | Prevotella | Prevotella ruminicola | 0.0245 | 13.1467 | 0 | 3 | 76 | 21 |
| Prevotella bryantii | 0.0023 | 1.2999 | 0 | 4 | 88 | 8 | |||
| Bacteroides | Bacteroides coprocola | 0.0014 | 0.7476 | 0 | 19 | 81 | 0 | ||
| Firmicutes | Catenibacterium | Catenibacterium mitsuokai | 0.0060 | 3.2592 | 0 | 4 | 93 | 3 | |
| Butyrivibrio | Butyrivibrio proteoclasticus | 0.0057 | 3.1174 | 0 | 19 | 79 | 2 | ||
| Butyrivibrio fibrisolvens | 0.0025 | 1.3147 | 0 | 25 | 64 | 11 | |||
| Ruminococcus | Ruminococcus flavefaciens | 0.0060 | 3.3100 | 0 | 6 | 70 | 24 | ||
| Ruminococcus albus | 0.0034 | 1.8549 | 0 | 31 | 61 | 8 | |||
| Ruminococcus champanellensis | 0.0031 | 1.7506 | 0 | 12 | 82 | 6 | |||
| Marvinbryantia | Marvinbryantia formatexigens | 0.0041 | 2.2534 | 0 | 18 | 82 | 0 | ||
| Roseburia | Roseburia intestinalis | 0.0041 | 2.1159 | 0 | 25 | 75 | 0 | ||
| Fibrobacter | Fibrobacter succinogenes | 0.0032 | 1.7520 | 0 | 47 | 53 | 0 | ||
| Faecalibacterium | Faecalibacterium prausnitzii | 0.0026 | 1.4042 | 0 | 31 | 66 | 3 | ||
| Subdoligranulum | Subdoligranulum variabile | 0.0025 | 1.3161 | 0 | 37 | 63 | 0 | ||
| Coprobacillus | Coprobacillus sp. strain 29_1 | 0.0025 | 1.3147 | 0 | 25 | 75 | 0 | ||
| Blautia | Blautia (Ruminococcus) obeum | 0.0016 | 0.8252 | 0 | 17 | 83 | 0 | ||
| Clostridium | Clostridium phytofermentans | 0.0011 | 0.5925 | 0 | 58 | 42 | 0 | ||
| Unclassified Lachnospiraceae | Lachnospiraceae bacterium 3_1_57FAA_CT1 | 0.0047 | 2.5892 | 0 | 36 | 64 | 0 | ||
| Lachnospiraceae bacterium 1_4_56FAA | 0.0012 | 0.6312 | 0 | 54 | 46 | 0 | |||
| Actinobacteria | Collinsella | Collinsella stercoris | 0.0020 | 1.0579 | 0 | 17 | 83 | 0 | |
| Collinsella tanakaei | 0.0017 | 0.9522 | 0 | 0 | 100 | 0 | |||
| Subtotal for 21 species | 0.0861 | 46.6053 | |||||||
| Eukaryotes | Fungi | Neocallimastix | Neocallimastix patriciarum | 0.0034 | 1.9671 | 0 | 66 | 23 | 11 |
| Piromyces | Piromyces sp. strain E2 | 0.0014 | 0.7730 | 0 | 25 | 50 | 25 | ||
| Subtotal for 2 species | 0.0048 | 2.7401 | |||||||
| Total | 0.0909 | 49.3455 | |||||||
| Unclassified uncultured organism | 0.0046 | 2.5116 | 0 | 12 | 78 | 10 | |||
Average data for the two cow samples are shown.
The abundance refers to the proportion of oligosaccharide-degrading enzyme-related reads in the total non-rRNAs. Only organisms with oligosaccharide-degrading enzyme-related reads with an abundance of ≥0.001% are shown.
The relative ratio refers to the proportion of the number of oligosaccharide-degrading enzyme-related reads in a given organism in the total number of oligosaccharide-degrading enzyme-related reads.
Similarities of proteins encoded by transcripts to that of known oligosaccharide-degrading enzymes from a given organism at the amino acid sequence level are shown.
Some of the CBMs showed a restricted distribution (Table 6). For example, CBM10 reads were derived only from anaerobic fungi, whereas CBM11 reads were from both Fibrobacter (93%) and anaerobic fungi (7%). Transcripts for dockerin domains were mostly from Ruminococcus (55%) and Clostridium (31%). Most of the PUL reads were derived from Prevotella (44%) and Bacteroides (36%), as found in the metagenomic analyses of reindeer (10), yak (9), and Bos indicus (46) rumens.
TABLE 6.
Distribution of CHO-GHs, CHO-CBMs, and other related domains among genera active in PCWP degradation
| Domain | Distribution in the following organismsa: |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Bacteria |
Eukaryotes |
||||||||||
|
Firmicutes |
Bacteroidetes |
Fibrobacteres, Fibrobacter | Fungi |
Protozoa |
|||||||
| Ruminococcus | Eubacterium | Clostridium | Prevotella | Bacteroides | Neocallimastix | Piromyces | Epidinium | Polyplastron | Eudiplodinium | ||
| GHs | |||||||||||
| GH5 | ++++ | +++ | +++ | +++ | +++ | +++ | − | − | ++++ | +++ | − |
| GH6 | − | − | − | − | − | − | ++++ | +++ | − | − | − |
| GH9 | ++++ | +++ | +++ | − | ++ | +++ | +++ | +++ | +++ | − | − |
| GH44 | ++++ | − | − | − | − | ++ | − | − | − | − | − |
| GH45 | − | − | − | − | − | ++++ | − | ++ | − | − | − |
| GH48 | ++++ | ++ | − | − | − | − | +++ | +++ | − | − | − |
| GH8 | − | − | − | ++++ | +++ | ++++ | − | − | − | − | − |
| GH10 | +++ | + | +++ | ++++ | +++ | +++ | ++ | − | +++ | + | +++ |
| GH11 | ++++ | − | + | − | − | +++ | +++ | +++ | − | +++ | − |
| GH26 | ++++ | ++++ | +++ | ++++ | +++ | ++++ | − | + | − | − | − |
| GH28 | +++ | − | ++++ | ++++ | +++ | − | − | − | − | − | − |
| GH51 | ++ | − | +++ | ++++ | +++ | − | − | − | − | − | − |
| GH53 | + | − | − | ++++ | +++ | − | − | − | − | − | − |
| GH54 | − | − | − | − | − | +++ | − | − | − | − | − |
| GH67 | ++ | − | ++ | ++++ | ++++ | − | − | − | − | − | − |
| GH78 | − | − | ++++ | ++++ | ++++ | − | − | − | − | − | − |
| GH1 | ++ | − | +++ | − | − | − | +++ | +++ | − | − | − |
| GH2 | +++ | +++ | +++ | ++++ | ++++ | +++ | − | − | − | − | − |
| GH3 | +++ | ++ | +++ | ++++ | +++ | ++ | +++ | +++ | − | − | − |
| GH29 | + | − | +++ | ++++ | ++++ | − | − | − | − | − | − |
| GH35 | − | − | − | ++++ | ++++ | − | − | − | − | − | − |
| GH38 | − | − | ++ | − | ++ | − | ++ | ++ | − | − | − |
| GH39 | ++++ | − | ++ | ++ | ++ | − | − | − | − | − | − |
| GH42 | ++++ | ++ | +++ | ++++ | ++ | − | − | − | − | − | − |
| GH43 | ++++ | ++ | +++ | ++++ | ++++ | +++ | − | − | − | − | − |
| GH94 | ++++ | +++ | +++ | ++++ | − | +++ | − | − | − | − | − |
| CBMs | |||||||||||
| CBM2 | − | ++ | +++ | − | − | − | − | − | − | − | − |
| CBM3 | ++++ | − | − | − | − | − | − | − | ++++ | − | − |
| CBM4/9/16/22 | ++++ | +++ | ++ | +++ | +++ | ++ | − | − | +++ | +++ | − |
| CBM6 | ++++ | − | +++ | +++ | + | ++++ | +++ | − | − | − | − |
| CBM10 | − | − | − | − | − | − | ++++ | ++++ | − | − | − |
| CBM11 | − | − | − | − | − | ++++ | − | − | − | − | − |
| CBM13 | +++ | +++ | − | − | − | − | − | − | − | − | − |
| Other domains | |||||||||||
| Dockerin | ++++ | − | ++++ | − | ++ | − | − | − | − | − | − |
| SusC | − | − | − | ++++ | ++++ | − | − | − | − | − | − |
| SusD | − | − | − | ++++ | ++++ | − | − | − | − | − | − |
| SusD-like | − | − | − | ++++ | ++++ | − | − | − | − | − | − |
| SusD-like_2 | − | − | − | ++++ | ++++ | − | − | − | − | − | − |
| SusD-like_3 | − | − | − | ++++ | ++++ | − | − | − | − | − | − |
The total number of GHs in Ruminococcus, Eubacterium, Clostridium, Prevotella, Bacteroides, Fibrobacter, Neocallimastix, Piromyces, Epidinium, Polyplastron, and Eudiplodinium was 19, 10, 18, 18, 18, 13, 8, 9, 3, 3, and 1, respectively. The total number of CBMs in Ruminococcus, Eubacterium, Clostridium, Prevotella, Bacteroides, Fibrobacter, Neocallimastix, Piromyces, Epidinium, Polyplastron, and Eudiplodinium was 4, 3, 3, 2, 2, 3 2, 1, 1, 1, and 0, respectively. +, 0 < n < 0.1; ++, 0.1 ≤ n < 1; +++, 1 ≤ n < 10; ++++, n ≥ 10; and −, n = 0, where n is the percentage of the domains encoded by the genome of a given genus of the total number of domains.
Taken together, our data indicate that although more than 70 known genera were found to be involved in PCWP degradation, only 11 of them appeared to contribute significantly to the degradation of cellulose and hemicellulose (Tables 3 and 4). These 11 genera were able to produce a large array of CHO-GHs and CHO-CBMs (Table 6). Top on the list were Ruminococcus (19 GHs), Clostridium (18 GHs), Bacteroides (18 GHs), Prevotella (17 GHs), Fibrobacter (13 GHs), and Eubacterium (10 GHs). By comparison, rumen protozoa and fungi synthesized relatively fewer GHs (4 and 10 GHs, respectively) for cellulose, hemicellulose, and oligosaccharide degradation. Members of the CHO-CBM families were synthesized primarily by Ruminococcus (4 CBMs), Clostridium (3 CBMs), and Fibrobacter (3 CBMs) (Table 6).
DISCUSSION
Metatranscriptomic approaches, which entail sequencing of cDNAs derived from gene transcripts and thus permit an in-depth analysis of the contributions and coordinated action of genes of interest, have been carried out on microbiomes in a number of ecosystems (22, 47). In this report, we present a metatranscriptomic analysis of a PCWP-degrading microbiome including both prokaryotes and eukaryotes in the rumen. Our results indicate that genes encoding GH5, GH9, and GH10 cellulases/hemicellulases, as identified in previous studies (6, 7, 9, 10), were actively transcribed in the rumen. Interestingly and intriguingly, a relatively large number of GH48 transcripts were identified in the present study. Since the GH48 genes occurred rarely in rumen metagenomes (6, 7, 9, 10), the potential role of the enzymes of the GH48 family in cellulose degradation might have been overlooked. Most of the GH48 sequences derived from our metatranscriptomes were annotated as putative cellobiohydrolases, which are known to hydrolyze efficiently or help hydrolyze the crystalline regions of cellulose (48). On the basis of the MEGAN analysis, the GH48 enzymes were primarily produced by Ruminococcus, anaerobic fungi, as well as some uncultured microorganisms, suggesting that both prokaryotic and eukaryotic cellulose degraders contribute significantly to crystalline cellulose degradation in the cow rumen. Furthermore, since the prokaryotic GH48 transcripts were relatively abundant and mostly produced by Ruminococcus, which is known to harbor a single copy of the GH48 gene in the genome (49, 50), the GH48 gene appeared to be robustly transcribed in cows fed a fiber diet. Our data support the contention that GH48 cellulases serve a key role in PCWP degradation (51, 52).
We have also identified a relatively large number of transcripts encoding dockerin and cohesin similar to those from Ruminococcus and Clostridium in the present study, raising the possibility that the cellulosome-based fibrolytic activities might play an important part in PCWP degradation in the rumen. Notably, genes encoding CBM10, which is thought to be related to the cellulosomes of rumen anaerobic fungi (45), were among the most actively transcribed CBM genes. Only a few genes for cellulosomal domains, i.e., dockerin and cohesin, were detected, and no CBM10 gene was found in the previous metagenomic studies (5–7, 9, 11). Since some of the CBM10s were linked to a GH48 domain in the metatranscriptomes, it is possible that anaerobic rumen fungi encoding these domains employ GH48-containing cellulosomes in the hydrolysis of PCWPs. The relative abundance of the transcripts encoding dockerin-, cohesin-, and CBM10-like proteins indicates that cellulosomes may play a hitherto underestimated role in PCWP degradation in the rumen.
The PULs were first identified as the starch utilization system (Sus) in the anaerobic human gut bacterium Bacteroides thetaiotaomicron (53) and were later expanded to include cellulose (11), xylan (54, 55), and pectin (56) utilization systems in herbivore gut bacteria. The multiprotein system includes SusC (also called the TonB-dependent receptor), SusD-like proteins, SusE, SusF, SusG, and other components, among which SusC and SusD-like proteins are central to and, thus, typical of this system (10). It has been speculated that, during starch hydrolysis by a bacterium encoding the PUL system, the multiple components of the PUL system simultaneously interact with the same starch molecule, facilitating its binding, degradation, and utilization by the organism (57). In our metatranscriptomes, PULs, represented by the core components, were also relatively abundant, in agreement with the previous findings from the metagenomic studies of ruminants (10, 46). These results suggest that the PUL system is widely used for the assimilation of complex carbohydrates in the cow rumen.
As revealed by metagenomic analyses, the majority of the rumen GH genes for PCWP degradation appeared to be derived from the Bacteroidetes and Firmicutes (7, 9, 11), and very few of these genes were from the Fibrobacteres. The latter observation is somewhat unexpected, since F. succinogenes is believed to be one of the predominant PCWP degraders in the rumen (9, 10). In the present study, the Fibrobacter genes encoding cel-GHs were actively expressed, providing an explanation for the discrepancy described above. F. succinogenes, the best-characterized species in the genus Fibrobacter, is highly efficient in degrading crystalline cellulose in the rumen (58). We found that F. succinogenes synthesized a number of enzymes capable of degrading a wide array of polysaccharides, including both cellulose and hemicellulose. Since the organism utilizes only cellulose and its degradation products, but not pectin, starch, glucomannan, arabinogalactan, or xylans, as the carbon source for growth (59), it is suggested that the ability to degrade hemicellulose allows the organism to gain access to the cellulose portions of the plant cell wall. Our observation that the Fibrobacter transcripts for hemicellulose-degrading GHs were as abundant as those for cellulose-degrading GHs is consistent with the proposal.
Rumen anaerobic fungi and protozoa are capable of efficient hydrolysis of cellulose, as demonstrated by biochemical assays of their cellulase activities (60–62). We show that enzymes of the GH5, GH9, and GH48 families are the predominant cel-GHs from rumen anaerobic fungi and protozoa, as reported previously (22). Interestingly, anaerobic fungi and protozoa appear to contribute little to the pool of oligo-GHs. Therefore, anaerobic eukaryotes may play a greater role in degrading cellulose and hemicellulose than in hydrolyzing oligosaccharides in the rumen.
It has been suggested on the basis of the 16S rRNA gene analysis that most of the rumen microorganisms are uncultured (63). Our data demonstrate that the majority of the PCWP-degrading microorganisms are taxonomically related only to a limited number of known species, such as those of the genera Ruminococcus (R. flavefaciens and R. albus), Fibrobacter (F. succinogenes), and Prevotella (P. ruminicola) as well as a few anaerobic fungi and protozoa. However, most of the putative proteins involved in PCWP degradation, encoded by transcripts identified in the metatranscriptomes, were less than 90% similar at the amino acid sequence level to those of known species, indicating that it is the unknown relatives of the known genera or species that serve predominant roles in cellulose degradation in the rumen.
Supplementary Material
ACKNOWLEDGMENTS
This study was supported by the National Basic Research Program of China (grant no. 2011CB100804) and the National Natural Science Foundation of China (31240050).
We declare no financial competing interests.
X.D., Y.T., J.L., and X.S. carried out the molecular studies, participated in sequence analysis, and drafted the manuscript. X.W., S.Z., and J.W. participated in sampling and RNA extraction. L.L., D.L., and Z.D. conducted the data analysis. Y.L. and H.Z. participated in RNA sequencing. S.H. and L.H. conceived of and coordinated the study and helped draft the manuscript.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.03682-14.
REFERENCES
- 1.Krause DO, Denman SE, Mackie RI, Morrison M, Rae AL, Attwood GT, McSweeney CS. 2003. Opportunities to improve fiber degradation in the rumen: microbiology, ecology, and genomics. FEMS Microbiol Rev 27:663–693. doi: 10.1016/S0168-6445(03)00072-X. [DOI] [PubMed] [Google Scholar]
- 2.Russell JB, Rychlik JL. 2001. Factors that alter rumen microbial ecology. Science 292:1119–1122. doi: 10.1126/science.1058830. [DOI] [PubMed] [Google Scholar]
- 3.Sirohi SK, Singh N, Dagar SS, Puniya AK. 2012. Molecular tools for deciphering the microbial community structure and diversity in rumen ecosystem. Appl Microbiol Biotechnol 95:1135–1154. doi: 10.1007/s00253-012-4262-2. [DOI] [PubMed] [Google Scholar]
- 4.Koike S, Yoshitani S, Kobayashi Y, Tanaka K. 2003. Phylogenetic analysis of fiber-associated rumen bacterial community and PCR detection of uncultured bacteria. FEMS Microbiol Lett 229:23–30. doi: 10.1016/S0378-1097(03)00760-2. [DOI] [PubMed] [Google Scholar]
- 5.Warnecke F, Luginbuhl P, Ivanova N, Ghassemian M, Richardson TH, Stege JT, Cayouette M, McHardy AC, Djordjevic G, Aboushadi N, Sorek R, Tringe SG, Podar M, Martin HG, Kunin V, Dalevi D, Madejska J, Kirton E, Platt D, Szeto E, Salamov A, Barry K, Mikhailova N, Kyrpides NC, Matson EG, Ottesen EA, Zhang X, Hernandez M, Murillo C, Acosta LG, Rigoutsos I, Tamayo G, Green BD, Chang C, Rubin EM, Mathur EJ, Robertson DE, Hugenholtz P, Leadbetter JR. 2007. Metagenomic and functional analysis of hindgut microbiota of a wood-feeding higher termite. Nature 450:560–565. doi: 10.1038/nature06269. [DOI] [PubMed] [Google Scholar]
- 6.Brulc JM, Antonopoulos DA, Miller ME, Wilson MK, Yannarell AC, Dinsdale EA, Edwards RE, Frank ED, Emerson JB, Wacklin P, Coutinho PM, Henrissat B, Nelson KE, White BA. 2009. Gene-centric metagenomics of the fiber-adherent bovine rumen microbiome reveals forage specific glycoside hydrolases. Proc Natl Acad Sci U S A 106:1948–1953. doi: 10.1073/pnas.0806191105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hess M, Sczyrba A, Egan R, Kim TW, Chokhawala H, Schroth G, Luo S, Clark DS, Chen F, Zhang T, Mackie RI, Pennacchio LA, Tringe SG, Visel A, Woyke T, Wang Z, Rubin EM. 2011. Metagenomic discovery of biomass-degrading genes and genomes from cow rumen. Science 331:463–467. doi: 10.1126/science.1200387. [DOI] [PubMed] [Google Scholar]
- 8.Wang L, Hatem A, Catalyurek UV, Morrison M, Yu Z. 2013. Metagenomic insights into the carbohydrate-active enzymes carried by the microorganisms adhering to solid digesta in the rumen of cows. PLoS One 8:e78507. doi: 10.1371/journal.pone.0078507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dai X, Zhu Y, Luo Y, Song L, Liu D, Liu L, Chen F, Wang M, Li J, Zeng X, Dong Z, Hu S, Li L, Xu J, Huang L, Dong X. 2012. Metagenomic insights into the fibrolytic microbiome in yak rumen. PLoS One 7:e40430. doi: 10.1371/journal.pone.0040430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pope PB, Mackenzie AK, Gregor I, Smith W, Sundset MA, McHardy AC, Morrison M, Eijsink VG. 2012. Metagenomics of the Svalbard reindeer rumen microbiome reveals abundance of polysaccharide utilization loci. PLoS One 7:e38571. doi: 10.1371/journal.pone.0038571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pope PB, Denman SE, Jones M, Tringe SG, Barry K, Malfatti SA, McHardy AC, Cheng JF, Hugenholtz P, McSweeney CS, Morrison M. 2010. Adaptation to herbivory by the tammar wallaby includes bacterial and glycoside hydrolase profiles different from other herbivores. Proc Natl Acad Sci U S A 107:14793–14798. doi: 10.1073/pnas.1005297107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sheik CS, Jain S, Dick GJ. 2014. Metabolic flexibility of enigmatic SAR324 revealed through metagenomics and metatranscriptomics. Environ Microbiol 16:304–317. doi: 10.1111/1462-2920.12165. [DOI] [PubMed] [Google Scholar]
- 13.Shi Y, Tyson GW, DeLong EF. 2009. Metatranscriptomics reveals unique microbial small RNAs in the ocean's water column. Nature 459:266–269. doi: 10.1038/nature08055. [DOI] [PubMed] [Google Scholar]
- 14.Lehembre F, Doillon D, David E, Perrotto S, Baude J, Foulon J, Harfouche L, Vallon L, Poulain J, Da Silva C, Wincker P, Oger-Desfeux C, Richaud P, Colpaert JV, Chalot M, Fraissinet-Tachet L, Blaudez D, Marmeisse R. 2013. Soil metatranscriptomics for mining eukaryotic heavy metal resistance genes. Environ Microbiol 15:2829–2840. doi: 10.1111/1462-2920.12143. [DOI] [PubMed] [Google Scholar]
- 15.Vila-Costa M, Sharma S, Moran MA, Casamayor EO. 2013. Diel gene expression profiles of a phosphorus limited mountain lake using metatranscriptomics. Environ Microbiol 15:1190–1203. doi: 10.1111/1462-2920.12033. [DOI] [PubMed] [Google Scholar]
- 16.Stewart FJ, Ulloa O, DeLong EF. 2012. Microbial metatranscriptomics in a permanent marine oxygen minimum zone. Environ Microbiol 14:23–40. doi: 10.1111/j.1462-2920.2010.02400.x. [DOI] [PubMed] [Google Scholar]
- 17.de Menezes A, Clipson N, Doyle E. 2012. Comparative metatranscriptomics reveals widespread community responses during phenanthrene degradation in soil. Environ Microbiol 14:2577–2588. doi: 10.1111/j.1462-2920.2012.02781.x. [DOI] [PubMed] [Google Scholar]
- 18.Stewart FJ, Sharma AK, Bryant JA, Eppley JM, DeLong EF. 2011. Community transcriptomics reveals universal patterns of protein sequence conservation in natural microbial communities. Genome Biol 12:R26. doi: 10.1186/gb-2011-12-3-r26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Turner TR, Ramakrishnan K, Walshaw J, Heavens D, Alston M, Swarbreck D, Osbourn A, Grant A, Poole PS. 2013. Comparative metatranscriptomics reveals kingdom level changes in the rhizosphere microbiome of plants. ISME J 7:2248–2258. doi: 10.1038/ismej.2013.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Maurice CF, Haiser HJ, Turnbaugh PJ. 2013. Xenobiotics shape the physiology and gene expression of the active human gut microbiome. Cell 152:39–50. doi: 10.1016/j.cell.2012.10.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McNulty NP, Wu M, Erickson AR, Pan C, Erickson BK, Martens EC, Pudlo NA, Muegge BD, Henrissat B, Hettich RL, Gordon JI. 2013. Effects of diet on resource utilization by a model human gut microbiota containing Bacteroides cellulosilyticus WH2, a symbiont with an extensive glycobiome. PLoS Biol 11:e1001637. doi: 10.1371/journal.pbio.1001637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Qi M, Wang P, O'Toole N, Barboza PS, Ungerfeld E, Leigh MB, Selinger LB, Butler G, Tsang A, McAllister TA, Forster RJ. 2011. Snapshot of the eukaryotic gene expression in muskoxen rumen—a metatranscriptomic approach. PLoS One 6:e20521. doi: 10.1371/journal.pone.0020521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. 1997. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pruesse E, Quast C, Knittel K, Fuchs BM, Ludwig W, Peplies J, Glockner FO. 2007. SILVA: a comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res 35:7188–7196. doi: 10.1093/nar/gkm864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sayers EW, Barrett T, Benson DA, Bolton E, Bryant SH, Canese K, Chetvernin V, Church DM, Dicuccio M, Federhen S, Feolo M, Fingerman IM, Geer LY, Helmberg W, Kapustin Y, Krasnov S, Landsman D, Lipman DJ, Lu Z, Madden TL, Madej T, Maglott DR, Marchler-Bauer A, Miller V, Karsch-Mizrachi I, Ostell J, Panchenko A, Phan L, Pruitt KD, Schuler GD, Sequeira E, Sherry ST, Shumway M, Sirotkin K, Slotta D, Souvorov A, Starchenko G, Tatusova TA, Wagner L, Wang Y, Wilbur WJ, Yaschenko E, Ye J. 2012. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res 40:D13–D25. doi: 10.1093/nar/gkr1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kanehisa M, Goto S. 2000. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res 28:27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Punta M, Coggill PC, Eberhardt RY, Mistry J, Tate J, Boursnell C, Pang N, Forslund K, Ceric G, Clements J, Heger A, Holm L, Sonnhammer EL, Eddy SR, Bateman A, Finn RD. 2012. The Pfam protein families database. Nucleic Acids Res 40:D290–D301. doi: 10.1093/nar/gkr1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Eddy SR. 2009. A new generation of homology search tools based on probabilistic inference. Genome Inform 23:205–211. doi: 10.1142/9781848165632_0019. [DOI] [PubMed] [Google Scholar]
- 29.Huson DH, Mitra S, Ruscheweyh HJ, Weber N, Schuster SC. 2011. Integrative analysis of environmental sequences using MEGAN4. Genome Res 21:1552–1560. doi: 10.1101/gr.120618.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhu L, Wu Q, Dai J, Zhang S, Wei F. 2011. Evidence of cellulose metabolism by the giant panda gut microbiome. Proc Natl Acad Sci U S A 108:17714–17719. doi: 10.1073/pnas.1017956108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dumont MG, Pommerenke B, Casper P. 2013. Using stable isotope probing to obtain a targeted metatranscriptome of aerobic methanotrophs in lake sediment. Environ Microbiol Rep 5:757–764. doi: 10.1111/1758-2229.12078. [DOI] [PubMed] [Google Scholar]
- 32.Sanders JG, Beinart RA, Stewart FJ, DeLong EF, Girguis PR. 2013. Metatranscriptomics reveal differences in in situ energy and nitrogen metabolism among hydrothermal vent snail symbionts. ISME J 7:1556–1567. doi: 10.1038/ismej.2013.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lombard V, Golaconda Ramulu H, Drula E, Coutinho PM, Henrissat B. 2014. The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res 42:D490–D495. doi: 10.1093/nar/gkt1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Boraston AB, Bolam DN, Gilbert HJ, Davies GJ. 2004. Carbohydrate-binding modules: fine-tuning polysaccharide recognition. Biochem J 382:769–781. doi: 10.1042/BJ20040892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cantarel BL, Coutinho PM, Rancurel C, Bernard T, Lombard V, Henrissat B. 2009. The Carbohydrate-Active EnZymes database (CAZy): an expert resource for glycogenomics. Nucleic Acids Res 37:D233–D238. doi: 10.1093/nar/gkn663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Koike S, Pan J, Kobayashi Y, Tanaka K. 2003. Kinetics of in sacco fiber-attachment of representative ruminal cellulolytic bacteria monitored by competitive PCR. J Dairy Sci 86:1429–1435. doi: 10.3168/jds.S0022-0302(03)73726-6. [DOI] [PubMed] [Google Scholar]
- 37.Weng X, Bu D, Li F, Zhang Y, Wang D. 2012. Responses in milk yield, milk composition and rumen fermentation in lactating cows receiving a corn straw or mixed forage diet. J Anim Vet Adv 11:4678–4683. doi: 10.3923/javaa.2012.4678.4683. [DOI] [Google Scholar]
- 38.Ravachol J, Borne R, Tardif C, de Philip P, Fierobe HP. 2014. Characterization of all family-9 glycoside hydrolases synthesized by the cellulosome-producing bacterium Clostridium cellulolyticum. J Biol Chem 289:7335–7348. doi: 10.1074/jbc.M113.545046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sukharnikov LO, Alahuhta M, Brunecky R, Upadhyay A, Himmel ME, Lunin VV, Zhulin IB. 2012. Sequence, structure, and evolution of cellulases in glycoside hydrolase family 48. J Biol Chem 287:41068–41077. doi: 10.1074/jbc.M112.405720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Michel G, Barbeyron T, Kloareg B, Czjzek M. 2009. The family 6 carbohydrate-binding modules have coevolved with their appended catalytic modules toward similar substrate specificity. Glycobiology 19:615–623. doi: 10.1093/glycob/cwp028. [DOI] [PubMed] [Google Scholar]
- 41.Pires VM, Henshaw JL, Prates JA, Bolam DN, Ferreira LM, Fontes CM, Henrissat B, Planas A, Gilbert HJ, Czjzek M. 2004. The crystal structure of the family 6 carbohydrate binding module from Cellvibrio mixtus endoglucanase 5a in complex with oligosaccharides reveals two distinct binding sites with different ligand specificities. J Biol Chem 279:21560–21568. doi: 10.1074/jbc.M401599200. [DOI] [PubMed] [Google Scholar]
- 42.Raghothama S, Eberhardt RY, Simpson P, Wigelsworth D, White P, Hazlewood GP, Nagy T, Gilbert HJ, Williamson MP. 2001. Characterization of a cellulosome dockerin domain from the anaerobic fungus Piromyces equi. Nat Struct Biol 8:775–778. doi: 10.1038/nsb0901-775. [DOI] [PubMed] [Google Scholar]
- 43.Steenbakkers PJ, Li XL, Ximenes EA, Arts JG, Chen H, Ljungdahl LG, Op Den Camp HJ. 2001. Noncatalytic docking domains of cellulosomes of anaerobic fungi. J Bacteriol 183:5325–5333. doi: 10.1128/JB.183.18.5325-5333.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Youssef NH, Couger MB, Struchtemeyer CG, Liggenstoffer AS, Prade RA, Najar FZ, Atiyeh HK, Wilkins MR, Elshahed MS. 2013. The genome of the anaerobic fungus Orpinomyces sp. strain C1A reveals the unique evolutionary history of a remarkable plant biomass degrader. Appl Environ Microbiol 79:4620–4634. doi: 10.1128/AEM.00821-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nagy T, Tunnicliffe RB, Higgins LD, Walters C, Gilbert HJ, Williamson MP. 2007. Characterization of a double dockerin from the cellulosome of the anaerobic fungus Piromyces equi. J Mol Biol 373:612–622. doi: 10.1016/j.jmb.2007.08.007. [DOI] [PubMed] [Google Scholar]
- 46.Rosewarne CP, Pope PB, Cheung JL, Morrison M. 2014. Analysis of the bovine rumen microbiome reveals a diversity of Sus-like polysaccharide utilization loci from the bacterial phylum Bacteroidetes. J Ind Microbiol Biotechnol 41:601–606. doi: 10.1007/s10295-013-1395-y. [DOI] [PubMed] [Google Scholar]
- 47.Wang P, Qi M, Barboza P, Leigh MB, Ungerfeld E, Selinger LB, McAllister TA, Forster RJ. 2011. Isolation of high-quality total RNA from rumen anaerobic bacteria and fungi, and subsequent detection of glycoside hydrolases. Can J Microbiol 57:590–598. doi: 10.1139/w11-048. [DOI] [PubMed] [Google Scholar]
- 48.Teixeira RS, da Silva AS, Kim HW, Ishikawa K, Endo T, Lee SH, Bon EP. 2013. Use of cellobiohydrolase-free cellulase blends for the hydrolysis of microcrystalline cellulose and sugarcane bagasse pretreated by either ball milling or ionic liquid [Emim][Ac]. Bioresour Technol 149:551–555. doi: 10.1016/j.biortech.2013.09.019. [DOI] [PubMed] [Google Scholar]
- 49.Berg Miller ME, Antonopoulos DA, Rincon MT, Band M, Bari A, Akraiko T, Hernandez A, Thimmapuram J, Henrissat B, Coutinho PM, Borovok I, Jindou S, Lamed R, Flint HJ, Bayer EA, White BA. 2009. Diversity and strain specificity of plant cell wall degrading enzymes revealed by the draft genome of Ruminococcus flavefaciens FD-1. PLoS One 4:e6650. doi: 10.1371/journal.pone.0006650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Suen G, Stevenson DM, Bruce DC, Chertkov O, Copeland A, Cheng JF, Detter C, Detter JC, Goodwin LA, Han CS, Hauser LJ, Ivanova NN, Kyrpides NC, Land ML, Lapidus A, Lucas S, Ovchinnikova G, Pitluck S, Tapia R, Woyke T, Boyum J, Mead D, Weimer PJ. 2011. Complete genome of the cellulolytic ruminal bacterium Ruminococcus albus 7. J Bacteriol 193:5574–5575. doi: 10.1128/JB.05621-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Devillard E, Goodheart DB, Karnati SK, Bayer EA, Lamed R, Miron J, Nelson KE, Morrison M. 2004. Ruminococcus albus 8 mutants defective in cellulose degradation are deficient in two processive endocellulases, Cel48A and Cel9B, both of which possess a novel modular architecture. J Bacteriol 186:136–145. doi: 10.1128/JB.186.1.136-145.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Olson DG, Tripathi SA, Giannone RJ, Lo J, Caiazza NC, Hogsett DA, Hettich RL, Guss AM, Dubrovsky G, Lynd LR. 2010. Deletion of the Cel48S cellulase from Clostridium thermocellum. Proc Natl Acad Sci U S A 107:17727–17732. doi: 10.1073/pnas.1003584107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Martens EC, Koropatkin NM, Smith TJ, Gordon JI. 2009. Complex glycan catabolism by the human gut microbiota: the Bacteroidetes Sus-like paradigm. J Biol Chem 284:24673–24677. doi: 10.1074/jbc.R109.022848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dodd D, Moon YH, Swaminathan K, Mackie RI, Cann IK. 2010. Transcriptomic analyses of xylan degradation by Prevotella bryantii and insights into energy acquisition by xylanolytic bacteroidetes. J Biol Chem 285:30261–30273. doi: 10.1074/jbc.M110.141788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dodd D, Mackie RI, Cann IK. 2011. Xylan degradation, a metabolic property shared by rumen and human colonic Bacteroidetes. Mol Microbiol 79:292–304. doi: 10.1111/j.1365-2958.2010.07473.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Martens EC, Lowe EC, Chiang H, Pudlo NA, Wu M, McNulty NP, Abbott DW, Henrissat B, Gilbert HJ, Bolam DN, Gordon JI. 2011. Recognition and degradation of plant cell wall polysaccharides by two human gut symbionts. PLoS Biol 9:e1001221. doi: 10.1371/journal.pbio.1001221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cameron EA, Maynard MA, Smith CJ, Smith TJ, Koropatkin NM, Martens EC. 2012. Multidomain carbohydrate-binding proteins involved in Bacteroides thetaiotaomicron starch metabolism. J Biol Chem 287:34614–34625. doi: 10.1074/jbc.M112.397380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Weimer PJ. 1993. Effects of dilution rate and pH on the ruminal cellulolytic bacterium Fibrobacter succinogenes S85 in cellulose-fed continuous culture. Arch Microbiol 160:288–294. doi: 10.1007/BF00292079. [DOI] [PubMed] [Google Scholar]
- 59.Suen G, Weimer PJ, Stevenson DM, Aylward FO, Boyum J, Deneke J, Drinkwater C, Ivanova NN, Mikhailova N, Chertkov O, Goodwin LA, Currie CR, Mead D, Brumm PJ. 2011. The complete genome sequence of Fibrobacter succinogenes S85 reveals a cellulolytic and metabolic specialist. PLoS One 6:e18814. doi: 10.1371/journal.pone.0018814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Findley SD, Mormile MR, Sommer-Hurley A, Zhang XC, Tipton P, Arnett K, Porter JH, Kerley M, Stacey G. 2011. Activity-based metagenomic screening and biochemical characterization of bovine ruminal protozoan glycoside hydrolases. Appl Environ Microbiol 77:8106–8113. doi: 10.1128/AEM.05925-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Harhangi HR, Akhmanova A, Steenbakkers PJ, Jetten MS, van der Drift C, Op den Camp HJ. 2003. Genomic DNA analysis of genes encoding (hemi-)cellulolytic enzymes of the anaerobic fungus Piromyces sp. E2. Gene 314:73–80. doi: 10.1016/S0378-1119(03)00705-4. [DOI] [PubMed] [Google Scholar]
- 62.Ljungdahl LG. 2008. The cellulase/hemicellulase system of the anaerobic fungus Orpinomyces PC-2 and aspects of its applied use. Ann N Y Acad Sci 1125:308–321. doi: 10.1196/annals.1419.030. [DOI] [PubMed] [Google Scholar]
- 63.Stevenson DM, Weimer PJ. 2007. Dominance of Prevotella and low abundance of classical ruminal bacterial species in the bovine rumen revealed by relative quantification real-time PCR. Appl Microbiol Biotechnol 75:165–174. doi: 10.1007/s00253-006-0802-y. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.