

Abstract
Secondary metabolites produced by nonribosomal peptide synthetase (NRPS) or polyketide synthase (PKS) pathways are chemical mediators of microbial interactions in diverse environments. However, little is known about their distribution, evolution, and functional roles in bacterial symbionts associated with animals. A prominent example is colibactin, a largely unknown family of secondary metabolites produced by Escherichia coli via a hybrid NRPS-PKS biosynthetic pathway that inflicts DNA damage upon eukaryotic cells and contributes to colorectal cancer and tumor formation in the mammalian gut. Thus far, homologs of this pathway have only been found in closely related Enterobacteriaceae, while a divergent variant of this gene cluster was recently discovered in a marine alphaproteobacterial Pseudovibrio strain. Herein, we sequenced the genome of Frischella perrara PEB0191, a bacterial gut symbiont of honey bees and identified a homologous colibactin biosynthetic pathway related to those found in Enterobacteriaceae. We show that the colibactin genomic island (GI) has conserved gene synteny and biosynthetic module architecture across F. perrara, Enterobacteriaceae, and the Pseudovibrio strain. Comparative metabolomics analyses of F. perrara and E. coli further reveal that these two bacteria produce related colibactin pathway-dependent metabolites. Finally, we demonstrate that F. perrara, like E. coli, causes DNA damage in eukaryotic cells in vitro in a colibactin pathway-dependent manner. Together, these results support that divergent variants of the colibactin biosynthetic pathway are widely distributed among bacterial symbionts, producing related secondary metabolites and likely endowing its producer with functional capabilities important for diverse symbiotic associations.
INTRODUCTION
Characteristic bacterial communities colonize the digestive tracts of almost all animals and influence the health and disease of their hosts (1–4). These communities are typically dominated by specialist bacteria, which are adapted to live in the gut of their host and have evolved specific functions for symbiotic interactions. The honey bee, Apis mellifera, harbors such a characteristic gut microbiota (5). Its simple composition of only eight bacterial species makes the honey bee gut microbiota an ideal model to study the ecology and evolution of gut bacteria and to understand mutualistic, commensal, and parasitic relationships (6). Furthermore, honey bees are important pollinators for agriculture and almost all terrestrial ecosystems. Thus, it is essential to characterize the genomic capabilities of these symbiotic bacteria so as to better understand their impact on the health of their host.
In the anterior part of the honey bee hindgut, two gammaproteobacteria, Gilliamella apicola and Frischella perrara, and one betaproteobacterium, Snodgrassella alvi, are the dominant members of this gut community (7–9). Comparative genomics and functional analyses have recently revealed that S. alvi and G. apicola harbor complementary metabolic pathways, contain diverse sets of genes for symbiotic interactions, and exhibit host-specific colonization patterns (10, 11). In contrast, only little is known about F. perrara. This bacterium is less abundant than the other two species, with fewer bacteria present in the gut of individual bees, and in some cases, the bacterium is not present at all (5, 9, 12). Interestingly, all three bacteria have so far only been found associated with social bees and form deep-branching phylogenetic lineages exclusive of bacteria sampled from other environments (7, 8, 13), supporting longstanding symbiotic associations with their host and among each other.
Bacterial symbionts frequently mediate interactions by using secondary metabolites, such as nonribosomal peptides and polyketides. These natural small molecules harbor a variety of activities, serving as mutualistic factors (14), virulence factors (15), antimicrobials (16), immunomodulators (17), and/or interbacterial exchange factors (e.g., siderophores involved in iron acquisition) (18, 19). The major biosynthetic steps for nonribosomal peptides and polyketides are carried out by nonribosomal peptide synthetases (NRPSs) and polyketide synthases (PKSs), respectively (20). Type I NRPS and PKS biosynthetic systems are large multidomain enzymes organized in modules, catalyzing the covalent attachment of both standard and nonstandard amino acids (in case of NRPSs) or acyl coenzyme A (acyl-CoA) units (in case of PKSs) to a growing peptide or polyketide chain, respectively. Auxiliary domains/proteins and post-assembly line tailoring proteins can introduce further structural complexity (21). The modularity in small molecule synthesis by NRPS, PKS, and hybrid NRPS-PKS systems underlies their remarkable metabolic versatility.
Little is known about natural products involved in symbioses with eukaryotic hosts (14, 22–24). In particular, their roles and distributions among gut communities have mainly remained elusive. Furthermore, natural products of animal-dwelling symbionts often reveal chemical and structural properties distinct from those of free-living microbes and thus hold promise for novel drug discovery (24, 25). In Escherichia coli strains and related coliform Enterobacteriaceae, a hybrid NRPS-PKS biosynthetic gene cluster was found to be involved in symbiotic interactions in the human gut (26–29). This hybrid NRPS-PKS pathway produces a family of largely uncharacterized small molecules termed “colibactin.” The presence of this gene cluster (clb) in E. coli results in DNA damage of eukaryotic cells (28) and contributes to inflammation-induced colorectal cancer in the mammalian gut (30, 31). While a number of small molecules dependent on the clb pathway have been described (32–34), the metabolite or metabolites mediating the genotoxic activity have remained elusive due to its proposed instability. Furthermore, the role of this genotoxic activity for symbioses in the human gut and in other environments has remained unclear. Interestingly, a homologous clb genomic island (GI) was recently identified in an alphaproteobacterial Pseudovibrio strain, FO-BEG1, isolated from a diseased marine coral (35). However, it is not known whether this divergent gene cluster has similar genotoxic capabilities to the clb island of Enterobacteriaceae.
Here, we sequenced the genome of the honey bee gut symbiont F. perrara PEB0191, analyzed its gene content for functions involved in symbiosis, and identified a divergent variant of the clb GI. To determine whether this clb GI homolog has conserved biosynthetic capabilities and in vitro genotoxic activity compared to the pathway described in E. coli, we first analyzed the genomic integration, genetic organization, and domain architecture of the divergent clb GI homologs. We then identified common clb-dependent metabolites in E. coli and F. perrara, and determined the effect of the F. perrara clb pathway on eukaryotic cells. Our results show that the clb pathway has maintained its biosynthetic capabilities and genotoxic activity over the course of evolution, despite its presence in symbionts colonizing distinct environments. This suggests an important role of the clb biosynthetic pathway in diverse microbe-host interactions.
MATERIALS AND METHODS
Genome sequencing, assembly, and annotation.
The complete genome sequence of F. perrara PEB0191 was generated from 64,460 quality-filtered single-molecule real-time (SMRT) DNA sequencing reads (Pacific Biosciences) with an average length of 2.9 kb. A total of 5,411,774 quality-filtered paired-end Illumina reads were used to verify the assembly and to identify sequencing errors by read mapping. A detailed description of the genome sequencing and assembly can be found in the Materials and Methods in the supplemental material. The final assembly of the F. perrara PEB0191 genome was submitted to the IMG pipeline (36) for annotation. tRNA genes were identified with tRNAscan-SE (37).
Comparative genomics and bioinformatics analyses.
Orthologs between analyzed genomes were determined with OrthoMCL (38) as described previously (39). We only considered all-against-all BLASTP hits with protein identities of ≥50% and an alignment length of ≥50% of the length of the query and the hit sequence. Regions with ≥5 F. perrara-specific genes were denoted as GIs. The genome circle of Fig. 1A was visualized with Circos v0.56 (40). Sequence analyses were conducted with Geneious v6.1 using different bioinformatics tools, including MUSCLE (41) to generate sequence alignments and PhyML (42) to infer phylogenetic trees. The species tree was inferred from the concatenated protein alignments of the following eight genes: the alanyl-tRNA synthetase gene (COG0013), uvrC (COG0322), recN (COG0497), the CTP synthase gene (COG0504), the signal recognition particle GTPase gene (COG0544), uvrB (COG0556), radA (COG1066), and a membrane GTPase gene (COG1217). Module analyses and substrate predictions of NRPS and PKS genes were carried out using a combination of BLASTP (43), antiSMASH 2.0 (44), the PKS/NRPS Analysis website (45), and NRPSpredictor2 (46). Predictions of amino acid substrate specificity of adenylation domains and residues in binding pockets were based on the PKS/NRPS Analysis website (45) and NRPSpredictor2 (46). Homology modeling of relict AT domains was conducted with the Phyre2 protein fold recognition server (47).
FIG 1.

(A) Comparison of the genome of F. perrara to other Orbaceae genomes. Starting from outside, the first circle shows the scale of the genome representation of F. perrara in gray and white steps of 100 kb. The second and third circles (green) depict the genes on the plus and minus strands of F. perrara. The fourth circle depicts all tRNA and rRNA genes in blue and black, respectively. The fifth circle highlights F. perrara-specific genomic islands (GIs) compared to other Orbaceae genomes: GI region 1 contains a tellurite resistance operon, GI region 2 contains genes encoding mostly hypothetical proteins and the colibactin biosynthetic gene cluster, GI regions 3 and 4 contain the type I secretion system genes, and GI region 5 contains the type VI secretion system genes. The sixth circle depicts the GC skew over the chromosome of F. perrara with positive values shown in magenta and negative values in peach. The blue circles represent orthologs identified in the genomes of G. apicola wkB1, G. apicola wkB11, G. apicola wkB30, and Orbus hercynius CN3. The blue color range denotes protein identity between these pairwise comparisons, as depicted by the scale in the center of the genome circle. (B) Presence/absence of genes of the TCA cycle (green arrows) and for fermentation (orange arrows) in the genomes of the two honey bee gut symbionts F. perrara and G. apicola wkB1. Semicircles in magenta and blue indicate presence of gene functions in the genomes of F. perrara and G. apicola wkB1, respectively. Other gene functions are either absent or could not be identified (empty semicircles).
Bacterial strains, plasmids, and culture conditions.
All strains, plasmids, and primers used in this study are summarized in Table 1. The clbB transposon mutant (clbB::Tn) of F. perrara PEB0191 was identified from a Himar1 transposon library by screening with different primer pairs for integration into the clb GI. The mutant was verified using PCR and Sanger sequencing. A detailed description of the transposon mutagenesis can be found in the Materials and Methods in the supplemental material. If not otherwise stated, F. perrara PEB0191 and the clbB::Tn mutant were grown on brain heart infusion (BHI) agar at 37°C under anaerobic conditions.
TABLE 1.
Strains, plasmids, and primers used in this study
| Strain, plasmid, or primer | Description or sequence (target)a | Reference or source |
|---|---|---|
| Strains | ||
| F. perrara | ||
| PEB0191 | Type strain of F. perrara isolated from hindgut of a honey bee | 7 |
| clbB::Tn mutant | F. perrara PEB0191 with Himar1 transposon of pBT20 integrated at nucleotide position 6475 of clbB | This study |
| E. coli | ||
| DH10B | F− mcrA (mcrBC-hsdRMS-mrr) [ϕ80dlacZΔM15] lacX74 deoR recA1 endA1 araD139 Δ(ara, leu)7697 galU galK rpsL nupG | Invitrogen |
| Nissle 1917 | Wild type | Ardeypharm GmbH |
| Nissle 1917 Δclb | clb::FRT, complete deletion of clb locus | 34 |
| BL21 | F− dcm ompT hsdS(rB− mB−)gal[malB+] K-12(λS) | Invitrogen |
| β2163 | K-12 strain; F− RP4-2-Tc::Mu ΔdapA::(erm-pir) Emr Kmr | 57 |
| Plasmids | ||
| pBAC-control | pBeloBAC11 without insert | 28 |
| pBAC-PKS | Genomic fragment of E. coli IHE3034 containing complete clb island cloned into pBeloBAC11 | 28 |
| pBT20 | Ori R6Kγ, oriT from pRK2, Mariner C9 transposase, minitransposon with Genr::aaC1 | 58 |
| Primers | ||
| prRND1 | TATAATGTGTGGAATTGTGAGCGG (transposon of pBT20) | |
| prRND1rev | GATGAAGTGGTTCGCATCCTC (transposon of pBT20) | |
| prPE209 | GAAAGAGGTTAATGGTAATGATGC (clbB [20–44]) | |
| prPE210 | CATGACATTTGTGCAATAGATC (clbB [4892–4914]) | |
| prPE211 | GGTATACAATAGTGAAATGACCG (clbC [3–26]) | |
| prPE212 | GCCATCTCAATTACAGCCATC (clbD [354–376]) | |
| prPE213 | GTGTCGCTATCGTAGGTATG (clbI [19–39]) | |
| prPE214 | GTAACCGCTTATGATGCTTTGC (clbJ [1009–1031]) | |
| prPE215 | CGTTATCCAGGAGTTCATAGC (clbK [45–66]) | |
| prPE216 | CTGCATGAAATCCTCGCATTC (clbK [4–17]) | |
| prPE217 | TTCAGTACCGATTGGGCAAGC (clbN [2197–2218]) | |
| prPE245 | CCGGGTTATCCATTTGAACAG (clbB [5791–5811]) | |
| prPE246 | GATAACACTACCCGATTGTATAC (clbB [6530–6552]) |
Positions are shown in brackets.
Organic extractions for metabolomics analysis.
E. coli DH10B/pBAC-PKS, E. coli DH10B/pBAC-control, E. coli Nissle 1917, and E. coli Nissle 1917 Δclb were grown as previously described (34). F. perrara strains were grown for 1 day on gut microbiota medium (GMM) (48), harvested, diluted to an optical density at 600 nm (OD600) of 0.01 in 5 ml GMM, and grown for 16 h at 37°C without shaking in an anaerobic atmosphere. To obtain medium controls, we incubated 5 ml of GMM without bacteria for 16 h under the same conditions. After the designated growth points, whole cultures of E. coli and F. perrara were extracted with 6 ml ethyl acetate (EtOAc) as previously described (34). Five biological replicates were performed for all samples.
Metabolomics data acquisition.
All high-resolution mass spectrometry (HRMS) was performed using an electrospray ionization (ESI) source on an Agilent (Santa Clara, CA) iFunnel 6550 quadrupole time of flight (Q-TOF) mass spectrometer coupled to an Agilent Infinity 1290 high-performance liquid chromatography (HPLC) instrument. Metabolites were analyzed on a Phenomenex Kinetex 1.7-μm C18 100-Å column (100 by 2.10 mm) with a water-acetonitrile (ACN) gradient solvent system containing 0.1% formic acid (FA). Immediately prior to analysis, each extracted sample was dissolved in 500 μl MeOH, and 5 μl of a 1:5 dilution was injected. For F. perrara samples, undiluted injections were also performed to increase identification of the molecular features (MOFs). Collection parameters and MS data acquisitions were conducted as previously reported (34).
Sample comparisons, data set filtrations, and statistical analysis.
The MS data were processed to extract molecular features using the “common organic molecules” model in MassHunter qualitative analysis. The extracted MS data, set at an intensity cutoff of 1.0 raw count abundance, was statistically analyzed using MassHunter Mass Profiler Professional (MPP version B.12.01; Agilent Technologies). E. coli samples were analyzed as previously described (34). To determine the organic extractable metabolomes of the two F. perrara strains, MOFs present in one out of the five medium controls were removed. MOFs present in F. perrara PEB0191 but either not found or found at reduced levels in the F. perrara clbB::Tn mutant were considered clb pathway-dependent metabolites. The final conservative list was adjusted after manual analysis.
MS2 molecular networking.
Tandem mass spectrometry (MS2) was performed using a targeted auto-MS2 mode as previously described (34). We selected only for the clb pathway-dependent MOFs present in the generated preferred unique ion list acquired for each sample. The MS2 data files were used to build mass spectral networking clusters using the open source software platform Cytoscape version 3.1.0 (http://www.cytoscape.org). Clusters were built based on a cosine cutoff of 0.5, which dictates the connectivity strength between the ion masses (49).
HeLa cell assays.
Cell culturing, bacterial infections, and analysis of the megalocytosis phenotype were performed as previously described (28, 34). E. coli strains used for HeLa cell assays were grown in lysogeny broth (LB) for 16 h. F. perrara strains used for HeLa cell assays were grown on brain heart infusion (BHI) agar for 24 h. γ-H2AX phosphorylation levels in HeLa cells were analyzed 14 h after transient bacterial infection to detect the activation of a double-strand DNA damage response. To this end, cells were immunolabeled with an anti-γ-H2AX primary antibody (clone 20E3; Cell Signaling) followed by a secondary antibody conjugated to fluorescein isothiocyanate (FITC) (goat anti-rabbit AB97199; ABCAM) and analyzed by flow cytometry using a FACSVerse flow cytometer from BD Bioscience. A detailed description of the protocol can be found in the Materials and Methods in the supplemental material.
Nucleotide sequence accession number.
The complete genome of F. perrara PEB0191 has been deposited in GenBank under accession no. CP009056.
RESULTS
Genome sequence of F. perrara and comparative genomics.
The genome of F. perrara PEB0191 consists of a single circular chromosome of ∼2.7 Mb (Fig. 1A), similar to what has been previously observed for the genomes of related Gilliamella apicola isolates from the guts of honey bees and bumble bee species (11). Other genomic features, such as G+C content, percentage of coding content, and number of RNA genes, are also similar (Table 2), reflecting the evolutionary relatedness of F. perrara and G. apicola and suggesting similar patterns of genomic evolution in these bee gut symbionts. Synteny analysis between the two completely sequenced genomes of F. perrara and G. apicola wkB1 revealed little conservation of their genomic backbones. Only a weak X-like synteny pattern could be observed (see Fig. S1 in the supplemental material). This is typical for related genomes and results from frequent inversions around the origin of replication (50).
TABLE 2.
Genome features of F. perrara PEB0191 and comparison to the genomes of the related gut symbiont G. apicola
| Host and organisma | Length (bp) | G+C content (%) | Coding % | No. of CDSsb | No. of tRNA genes | No. of rRNA loci |
|---|---|---|---|---|---|---|
| Honey bee (Apis mellifera) | ||||||
| F. perrara PEB0191 | 2,692,351 | 34.1 | 86.1 | 2,280 | 53 | 4 |
| G. apicola wkB1 | 3,139,412 | 33.6 | 84.1 | 2,809 | 51 | 4 |
| Bumble bee | ||||||
| G. apicola wkB11 (Bombus bimaculatus) | 2,260,992 | 34.4 | 82.4 | 1,997 | 51 | 4 |
| G. apicola wkB30 (Bombus vagans) | 2,320,793 | 34.6 | 84.1 | 2,135 | 48 | 4 |
Bacteria were isolated from the gut of the different bee species shown.
CDS, coding sequences.
F. perrara is a facultative anaerobe (7). Accordingly, its genome lacks many genes of the tricarboxylic acid (TCA) cycle (Fig. 1B) and the respiratory chain (see Fig. S2 in the supplemental material) but encodes the complete pathways for glycolysis and pentose phosphate, as well as several phosphotransferase systems (PTSs) for the uptake of sugars (see Fig. S3 in the supplemental material). Thus, the main energy source of F. perrara may be anaerobic fermentation of carbohydrates. This resembles the primary metabolism of G. apicola (11), suggesting that these bacteria occupy similar nutritional niches in the anterior hindgut of bees.
Ortholog analysis between five genomes of the family Orbaceae (including three genomes of G. apicola and the genome of Orbus hercynius CN3) revealed 586 genes specific to F. perrara (see Table S1). A substantial number of these genes are contained in GIs dispersed over the genome of F. perrara (Fig. 1A). Besides many hypothetical and phage-related protein-encoding genes, the GIs of F. perrara contain a tellurite resistance gene cluster, several type I secretion system genes, and a type VI secretion system locus. The largest GI region of F. perrara measures ∼130 kb and contains only a few genes shared with other sequenced Orbaceae genomes (GI region 2 in Fig. 1A). This island harbors a biosynthetic gene cluster of ∼55 kb, which we identified as a homolog of the clb GI of coliform Enterobacteriaceae and Pseudovibrio strain FO-BEG1. With a few exceptions, the gene order within the clb GI is conserved between F. perrara, the Enterobacteriaceae, and Pseudovibrio FO-BEG1 (Fig. 2). However, the percentages of protein identity of Clb orthologs are relatively low, ranging from 43% to 81% between F. perrara and E. coli and from 27% to 65% between F. perrara and Pseudovibrio FO-BEG1. In comparison, clb orthologs within Enterobacteriaceae reveal >99% protein identities (Fig. 2). Genomic regions flanking the clb GI of F. perrara were distinct from those found in the other bacteria. While transposase and integrase genes are contained adjacent to the clb GIs of the Enterobacteriaceae and Pseudovibrio FO-BEG1 (Fig. 2), no mobile genetic elements could be identified in close proximity to the clb GI of F. perrara, and most flanking genes had no significant hits in the NCBI nonredundant database.
FIG 2.
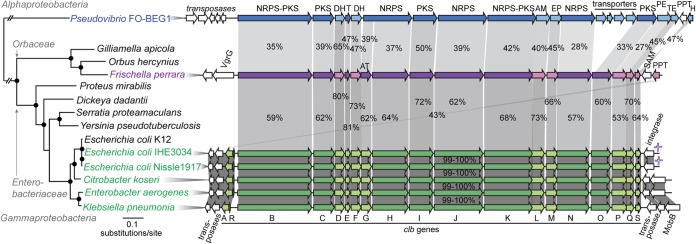
Phylogenetic relationship of bacteria harboring variants of the colibactin (clb) genomic island (GI) and comparison of their genetic organizations. Bacteria containing the clb GI are highlighted in green (Enterobacteriaceae), magenta (Frischella perrara PEB0191), and blue (Pseudovibrio FO-BEG1). For Citrobacter koseri, Enterobacter aerogenes, and Klebsiella pneumoniae, strains 4225-83, EA1509E, and WGLW1, respectively, were analyzed. The maximum likelihood tree is based on the concatenated alignments of eight conserved housekeeping genes. Black circles denote branches with bootstraps of ≥80 (100 replicates). Orthologs are connected via gray blocks. Percentages of protein identities are depicted and reflected by the shading intensity of each block. Genes without a homolog are shown in white. The average G+C contents of the Clb GI are 40.4%, 53.7%, and 51.1% for F. perrara PEB0191, E. coli IHE3034, and Pseudovibrio FO-BEG1, respectively. Genes and gene products are depicted using the following abbreviations: clb, colibactin; IS1351, insertion sequence 1351; MobB, mobilization protein B; VgrG, valine-glycine repeat protein G; NRPS, nonribosomal peptide synthetase; PKS, polyketide synthase; AT, acyl-transferase; T, thiolation sequence of acyl/peptidyl-carrier proteins; DH, dehydrogenase; AM, amidase; EP, efflux protein; PE, peptidase; TE, thioesterase; PPT, phosphopantetheinyl-transferase; SAM, S-adenosylmethionine-binding protein; H, hydrolase.
Conserved biosynthetic assembly line of clb GI homologs.
The presence of a clb homolog in F. perrara prompted us to determine whether the biosynthetic modules for small molecule production are conserved between the clb GIs of F. perrara, E. coli IHE3034 (as a representative of the Enterobacteriaceae), and Pseudovibrio FO-BEG1. Using a bioinformatics approach, we determined that all orthologous NRPS and PKS genes harbor the same domain architecture (Fig. 3). Most residues in the binding pockets of the adenylation (A) domains of Clb proteins are conserved between F. perrara, Enterobacteriaceae, and Pseudovibrio FO-BEG1, and three different bioinformatic tools predicted similar substrates to be incorporated during small molecule synthesis (Fig. 3; see Table S2 in the supplemental material). Furthermore, gene tree analysis shows that ketosynthase (KS) domains of orthologous clb genes form monophyletic clades that are distantly related to each other and belong to a larger group, including KS domains of other hybrid peptide-polyketide biosynthetic pathways (see Fig. S4 in the supplemental material).
FIG 3.

Domain architecture of the Clb NRPS/PKS proteins of F. perrara PEB0191, E. coli IHE3034, and Pseudovibrio FO-BEG1. Predicted amino acid substrate specificities of adenylation (A) domains and residues in binding pockets are depicted. Predictions with NRPSpredictor2 confidence scores (46) of <80% are marked with a question mark. For ClbB and ClbN, the experimentally validated A domain specificities (Ala and Asn, respectively) are depicted (33, 34). For ClbN, this is consistent with the prediction, but for ClbB, the prediction suggested Val. Sequence motifs (GxSxG) of the active sites of AT domains are depicted. Relict cis-AT domains of ClbC, ClbK, and ClbO are shown in white and denoted with an asterisk. Protein identities with a sliding window size of 15 bp are shown (red depicts identity of <30%). Abbreviations of domains are as follows: C, condensation; A, adenylation; T, thiolation sequence of acyl/peptidyl-carrier proteins; KS, ketosynthase; AT, acyl-transferase; KR, ketoreductase; DH, dehydratase; ER, enoyl-reductase; Cy, condensation/cyclase; Ox, oxidase; E, epimerase.
A previous analysis of the Clb proteins of E. coli found that ClbC, CbK, and ClbO each contain a deteriorated cis-acyltransferase (AT) domain (34). We verified their presence in F. perrara and Pseudovibrio FO-BEG1 using structural homology modeling (see Table S3 in the supplemental material). The canonical active site motifs (GxSxG) (51) are mutated and protein identities are relatively low supporting that these AT domains are nonfunctional evolutionary relicts (Fig. 3; see Table S3). In sum, bioinformatics predictions suggest the production of related small molecules by the clb gene clusters of F. perrara, E. coli, and Pseudovibrio, despite high degrees of sequence divergence.
Comparative metabolomics identifies clb pathway-dependent small molecules of F. perrara.
Next, we wanted to confirm the presence of known colibactin molecules. To do this, we compared the organic extractable metabolome of F. perrara to the metabolomes of E. coli Nissle 1917 and E. coli DH10B harboring the E. coli IHE3034 island on a bacterial artificial chromosome (pBAC-PKS), two E. coli strains from which clb pathway-dependent metabolites have previously been identified (34). First, a conservative unique list of 433 F. perrara-specific molecular features (MOFs) was identified in whole-culture ethyl acetate extracts relative to the control medium background. Comparison with clb pathway-dependent metabolites of E. coli Nissle 1917 and E. coli DH10B pBAC-PKS showed that seven out of these 433 organic extractable MOFs were represented in the E. coli colibactin network (see Fig. S5 in the supplemental material). Four MOFs were common to all three bacteria. These included metabolites with the following [M+H]+ m/z: 315.2281 (metabolite 1), 343.2593 (metabolite 2), 341.2440 (metabolite 3), and 369.2749 (metabolite 4) (see Table S4 in the supplemental material). Metabolites 2 to 4 have previously been identified as fatty acyl-d-asparagine cleavage products of the E. coli colibactin gene clusters (34). However, the most abundant ion in F. perrara has an ion mass of m/z 315.2281 (see Table S4), which is only observed as a minor product in the two E. coli strains. ESI-Q-TOF-HRMS analysis, MS2 fragmentation patterns (see Fig. S6 in the supplemental material), structural network analysis, and comparison to previously characterized clb metabolites support the structure of m/z 315.2281 as N-lauryl-d-Asn (metabolite 1 in Fig. 4).
FIG 4.

Colibactin pathway-dependent metabolites in F. perrara (A) and proposed structures for the fatty acyl-Asn metabolites (B) and their production (C). (A) MS2 network analysis between F. perrara and E. coli strains. MOFs in square nodes are specific to F. perrara, those in diamonds are shared among F. perrara and E. coli, and those in oval nodes were detected only in wild-type E. coli strains. (B) Proposed structures for eight metabolites are shown based on network analysis, MS2 fragmentation patterns, and comparison to previously characterized colibactin metabolites. Data for the major metabolite 1 (m/z 315. 2281) support N-lauryl-d-Asn, those for metabolites 2 to 5 have previously been reported (32, 34), and those for metabolites 6 to 8 represent new metabolites produced by F. perrara. (C) Extracted ion chromatogram (EIC) of metabolites 1, 2, 3, and 8, which are produced at a 10:1.2:0.37:0.19 ratio under our experimental conditions. Metabolites 4 to 7 (not shown) were only produced as very minor constituents and were near baseline at the scale shown.
We next generated a clbB transposon mutant (clbB::Tn) of the wild-type (wt) strain of F. perrara and confirmed that the seven shared metabolites are produced in a clb pathway-dependent manner. Comparative metabolomics between the F. perrara wt and clbB::Tn strains allotted 159 clb pathway-dependent MOFs (see Fig. S7 in the supplemental material), including six of the seven shared ions. The most abundant ion of F. perrara, metabolite 1, was found in both the wt and clbB::Tn mutant strains, although it was drastically reduced in the mutant strain (see Table S4 in the supplemental material). Residual production of metabolite 1 can be attributed to assembly line derailment (hydrolysis) of its intermediate thioester from ClbN, which remains intact in this mutant strain. To identify additional F. perrara clb pathway-dependent metabolites, we inspected all wt MOFs either absent or drastically reduced in the mutant strain and manually extracted a conservative unique ion list of 20 putative clb pathway-dependent metabolites (see Table S5 in the supplemental material). For 15 of these 20 metabolites, we could successfully acquire MS2 fragmentation patterns. A network analysis of these data together with the metabolomics data from the E. coli strains identified six metabolites that clustered with metabolites 1 to 4 (Fig. 4), consistent with clb pathway-dependent fatty acyl-d-Asn derivatives. Three of these six represent new metabolites: one shared with E. coli (metabolite 6) and two specific to F. perrara (metabolites 7 and 8). Their MS2 fragmentation data support altered fatty acyl appendages, C10:0 (m/z 287.1970, metabolite 6), C12:1 (m/z 313.2140, metabolite 7), and C13:0 (m/z 329.2443, metabolite 8).
F. perrara causes clb pathway-dependent megalocytosis and DNA damage in eukaryotic cells.
The striking similarities in genetic organization and organic extractable small molecule detection between the clb GIs of E. coli and F. perrara prompted us to test whether F. perrara induces similar clb-dependent phenotypes in eukaryotic cells. Therefore, HeLa cells were exposed transiently to different concentrations of the F. perrara wt or clbB::Tn mutant strain. Similar to E. coli containing clb (28, 29), the F. perrara wt strain induced megalocytosis of HeLa cells in vitro. This occurred in a dosage-dependent manner: i.e., with a higher multiplicity of infection (MOI), the phenotype became more pronounced (Fig. 5). However, in contrast to E. coli, F. perrara did not multiply in the cell culture medium. Therefore, the MOIs necessary to induce the megalocytosis phenotype were higher for F. perrara than for E. coli. Megalocytosis was confirmed to be associated with a functional clb pathway, as the F. perrara clbB::Tn mutant strain did not induce megalocytosis of HeLa cells at any of the tested MOIs (Fig. 5). While HeLa cells were detaching and dying over time when exposed to high MOIs of the F. perrara wt, no cytotoxic effect could be observed when exposed to the same concentration of the F. perrara clbB::Tn mutant. We also tested whether the megalocytosis phenotype inflicted by the F. perrara wt strain upon HeLa cells correlated with DNA damage, as has been shown to be the case for clb-positive E. coli (28). Therefore, we analyzed phosphorylation of the histone H2AX, a sensitive marker for the presence of DNA double-strand breaks in eukaryotic cells. We observed a shift toward higher levels of H2AX phosphorylation in HeLa cells after transient exposure to the F. perrara wt compared to the negative control. No shift was observed after exposure to the F. perrara clbB::Tn mutant (Fig. 5). This implies that F. perrara induces latent DNA double-strand breaks in HeLa cells in a clb pathway-dependent manner.
FIG 5.

F. perrara PEB0191 causes megalocytosis (A and B) and activates a DNA damage response in HeLa cells in vitro (C). (A) Megalocytosis of HeLa cells was analyzed 48 h post-transient infection. HeLa cells were stained with Giemsa as previously described (34). Transient infections with bacteria were carried out for 4 h. Scale bars, 100 μm. (B) Quantification of megalocytosis activity was based on protein content per well using methylene blue staining 48 h postinfection, followed by methylene blue extraction and OD660 measurements as described previously (34). For each condition, three independent wells were quantified. The mean + standard deviation is shown, and P values of two-tailed t tests are indicated: **, P < 0.01; *, P < 0.05. (C) HeLa cells were infected for 4 h at an MOI of 200 for E. coli and 5,000 for F. perrara. γ-H2AX was quantified by flow cytometry after 14 h of incubation. clb+, pBAC-PKS; clb−, pBAC-control; wt, wild type.
DISCUSSION
The importance of the clb GI for host health and disease has been demonstrated for specific E. coli strains. These bacteria typically colonize the gastrointestinal tract of humans, where the putative colibactins are hypothesized to exert genotoxic activity on host cells, resulting in DNA damage linked to tumorigenesis, colorectal cancer, and gut inflammation (28, 30, 31). Recently, a highly divergent variant of the clb gene cluster was identified in the genome of the diseased coral-associated organism Pseudovibrio FO-BEG1, suggesting that this biosynthetic pathway might be more widely distributed among symbionts than previously assumed (35). Indeed, our study discovered a divergent homolog of the clb pathway in F. perrara, a gut symbiont of honey bees. Despite high degrees of sequence divergence, the clb GIs of F. perrara, E. coli, and Pseudovibrio FO-BEG1 have largely maintained a conserved gene synteny (Fig. 2) and biosynthetic module architecture (Fig. 3). Our chemical, functional, and bioinformatic analyses support that F. perrara and E. coli produce a related set of N-acyl-d-Asn metabolites (Fig. 4) and cause similar phenotypes on HeLa cells in vitro, including megalocytosis and DNA damage (Fig. 5). The conservation of the biosynthetic and phenotypic characteristics suggests that the clb pathway mediates similar symbiotic interactions in the distinct gut communities of ecologically distinct hosts.
The occurrence of this biosynthetic pathway in symbionts from diverse environments parallels the evolution of the pederin family of small molecules (24), a group of structurally related polyketides identified in bacteria associated with diverse eukaryotic hosts, including beetles, sponges, and lichens. The biosynthetic gene clusters responsible for the production of these molecules appear to spread via horizontal gene transfer (HGT), facilitating the adoption of functions in distinct symbioses. Several lines of evidence corroborate this hypothesis for the clb GI. In the Enterobacteriaceae and Pseudovibrio FO-BEG1, the island is flanked by mobile genetic elements, and its distribution is limited to specific strains (29, 35). Furthermore, the clb GI has only diverged by a few mutations within the Enterobacteriaceae (Fig. 2), indicating more recent acquisition followed by rapid horizontal dissemination (29). Such characteristic signs of HGT are less evident for the clb gene cluster of F. perrara. While we found an elevated G+C content (40.4%) compared to the average G+C content of the genome (34.1%), no mobile genetic elements are encoded in close proximity (Fig. 2). However, the clb genes of F. perrara are located within a larger genomic region (GI 2 in Fig. 1A) absent from related bacteria. This provides evidence for an ancient HGT event of the clb GI in F. perrara. Mobile genetic elements may have been deleted after integration, while the clb gene cluster was maintained, supporting an important biological role for F. perrara. The low sequence similarity between the clb GIs of F. perrara, the Enterobacteriaceae, and Pseudovibrio further supports ancient divergence points. Thus, it is intriguing to find that they have maintained an almost perfect gene synteny and conserved biosynthetic module architecture, indicating strong purifying selection acting on the Clb assembly line proteins and on the synthesized small molecules. Interestingly, we found relict cis-AT domains to be present in several of the trans-AT PKS genes of all three species. These domains share little sequence similarity with conserved AT domains present in the clb GIs, reveal signs of accelerated evolution (Fig. 3), and harbor mutated active site residues (see Table S3 in the supplemental material), signifying loss of AT enzymatic activity. These relict AT domains are evidence that these Clb trans-AT PKSs have evolved from ancestral cis-AT PKSs; which is in contrast to the previous observation that other trans-AT PKSs have evolved independently from cis-AT PKSs via horizontal transfer of KS domains (25).
Our HeLa cell experiments showed that F. perrara, like E. coli, produces unknown molecules with genotoxic activity and induces megalocytosis in eukaryotic cells (Fig. 5). The conserved architecture of the biosynthetic assembly line (Fig. 3) indicates that the clb pathways of F. perrara, the Enterobacteriaceae, and Pseudovibrio FO-BEG1 encode related secondary metabolites. This is corroborated by the finding that the specificity-conferring residues in the amino acid binding pockets of the adenylation domains are mostly conserved between the three species (Fig. 3). Furthermore, our comparative metabolomic analysis of F. perrara and E. coli identified a number of common or related clb pathway-dependent fatty acyl-d-Asn metabolites. These small molecules represent peptidase ClbP cleavage products of “precolibactin” precursors (32, 33). One intermediate precolibactin precursor was characterized from E. coli and determined to be an authentic ClbP native substrate (34). However, the structures of advanced precolibactins have not yet been reported. The detection of cleavage products, albeit at various distributions between F. perrara and E. coli, demonstrates that this so-called prodrug activation mechanism (52) is conserved among clb pathways. The difference in metabolite production most plausibly originates from divergent acyl-CoA substrate specificities among ClbN homologs (33). The fragmentation patterns and low molecular weights of the 15 MS2 fragmented clb pathway-dependent molecules from F. perrara did not support the detection of mature precolibactins, although few high-molecular-weight molecules were detected. Most molecules from F. perrara were either identified as accumulated fatty acyl-d-Asn derivatives (Fig. 4) or were below the detection limits for MS2 fragmentation.
An open question concerns the role of the clb GI for bacterial colonization in the gut. Do the similar in vitro phenotypes caused by F. perrara and E. coli (Fig. 5) indicate similar functions of the Clb GI in vivo? For E. coli, it has been shown that the clb GI inflicts DNA damage and chromosome instability in the gut of mice, thereby contributing to inflammation-induced colorectal cancer and senescence-induced tumor growth (30, 31, 53). By contributing to a chronic inflammatory state in the intestine, the clb GI was hypothesized to facilitate long-term persistence of these Enterobacteriaceae. The bacterial growth inhibitory activities of acyl-d-Asn metabolites (34) could also participate in persistence via bacterial competition for niche resources. How would this relate to a potential functional role of the clb GI in the bee gut? F. perrara has so far only been detected in honey bees, where it appears to colonize (together with G. apicola and S. alvi) the anterior part of the hindgut (9). The clb GI of F. perrara might cause phenotypes in the bee gut similar to those caused by E. coli in the human gut—e.g., contributing to niche establishment, persistence, and/or interbacterial competition. Future studies will be necessary to determine whether the in vitro genotoxic activity of F. perrara is directed against host cells in the gut. The fact that F. perrara mediates DNA damage on human cells in vitro suggests that the genotoxic activity is not host specific. Further, a cuticle layer is separating the epithelial cells in the honey bee hindgut from the bacteria in the lumen. This poses the question as to whether the genotoxic activity of the clb pathway could even be mediated to the host cells. In vivo functional studies on the role of the clb GI in the honey bee gut will be necessary to address these questions. Bees are important pollinators, which suffer from a wide range of environmental disturbances, including pathogens and pesticides (54, 55). Thus, it is important to understand to what extent the DNA-damaging activity of the clb GI affects honey bee health. Community analysis showed that relative levels of F. perrara in the gut can vary between individual bees (5, 12). However, no bee pathology has been associated with F. perrara thus far, nor should future efforts to functionally characterize the F. perrara clb GI in vivo be exclusively associated with potential pathogenic attributes. The clb GI of E. coli is present not only in pathogenic strains (28). E. coli Nissle 1917 is a probiotic bacterium used for the treatment of ulcerative colitis (56), and its beneficial effect on the host was shown to be dependent on the presence of the clb GI (26). Understanding the role of the clb biosynthetic pathway in the bee microbiota could also point toward the ecological functions of this GI in more complex communities, such as those present in the human gut or inhabiting corals. Therefore, future studies will focus on the bacterial role of the clb GI in regulating symbiosis, its distribution across different microbiomes, and the molecular mechanisms governing host phenotypic responses.
Supplementary Material
ACKNOWLEDGMENTS
We thank Nancy A. Moran for sponsoring the genome sequencing of F. perrara in her lab and for advice on the work. We also thank Günter Wagner for access to the cell-culturing facility at Yale University and Guilin Wang for conducting the SMRT sequencing and providing the draft assembly of F. perrara. We are grateful to Eric Oswald for providing E. coli pBAC-PKS and E. coli pBAC-control.
This work was funded by Yale University, Swiss NSF fellowships PBBSP3-135986 and PZ00P3_148264, and EMBO fellowship ALTF 1317-2011 (to P.E.), U.S. NSF Dimensions of Biodiversity awards 1046153 and 1415605 (to Nancy A. Moran), and National Institutes of Health grant 1DP2CA186575 (to J.M.C.).
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.03283-14.
REFERENCES
- 1.Blumberg R, Powrie F. 2012. Microbiota, disease, and back to health: a metastable journey. Sci Transl Med 4:137rv7. doi: 10.1126/scitranslmed.3004184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Engel P, Moran NA. 2013. The gut microbiota of insects—diversity in structure and function. FEMS Microbiol Rev 37:699–735. doi: 10.1111/1574-6976.12025. [DOI] [PubMed] [Google Scholar]
- 3.Holmes E, Li JV, Athanasiou T, Ashrafian H, Nicholson JK. 2011. Understanding the role of gut microbiome-host metabolic signal disruption in health and disease. Trends Microbiol 19:349–359. doi: 10.1016/j.tim.2011.05.006. [DOI] [PubMed] [Google Scholar]
- 4.Ley RE, Hamady M, Lozupone C, Turnbaugh PJ, Ramey RR, Bircher JS, Schlegel ML, Tucker TA, Schrenzel MD, Knight R, Gordon JI. 2008. Evolution of mammals and their gut microbes. Science 320:1647–1651. doi: 10.1126/science.1155725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Moran NA, Hansen AK, Powell JE, Sabree ZL. 2012. Distinctive gut microbiota of honey bees assessed using deep sampling from individual worker bees. PLoS One 7:e36393. doi: 10.1371/journal.pone.0036393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Engel P, Moran NA. 2012. Functional and evolutionary insights into the simple yet specific gut microbiota of the honey bee from metagenomic analysis. Gut Microbes 4:60–65. doi: 10.4161/gmic.22517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Engel P, Kwong WK, Moran NA. 2013. Frischella perrara gen. nov., sp. nov., a gammaproteobacterium isolated from the gut of the honey bee, Apis mellifera. Int J Syst Evol Microbiol 63:3646–3651. doi: 10.1099/ijs.0.049569-0. [DOI] [PubMed] [Google Scholar]
- 8.Kwong WK, Moran NA. 2012. Cultivation and characterization of the gut symbionts of honey bees and bumble bees: Snodgrassella alvi gen. nov., sp. nov., a member of the Neisseriaceae family of the Betaproteobacteria; and Gilliamella apicola gen. nov., sp. nov., a member of Orbaceae fam. nov., Orbales ord. nov., a sister taxon to the Enterobacteriales order of the Gammaproteobacteria. Int J Syst Evol Microbiol 63:2008–2018. doi: 10.1099/ijs.0.044875-0. [DOI] [PubMed] [Google Scholar]
- 9.Martinson VG, Moy J, Moran NA. 2012. Establishment of characteristic gut bacteria during development of the honeybee worker. Appl Environ Microbiol 78:2830–2840. doi: 10.1128/AEM.07810-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Engel P, Martinson VG, Moran NA. 2012. Functional diversity within the simple gut microbiota of the honey bee. Proc Natl Acad Sci U S A 109:11002–11007. doi: 10.1073/pnas.1202970109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kwong WK, Engel P, Koch H, Moran NA. 2014. Genomics and host specialization of honey bee and bumble bee gut symbionts. Proc Natl Acad Sci U S A 111:11509–11514. doi: 10.1073/pnas.1405838111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sabree ZL, Hansen AK, Moran NA. 2012. Independent studies using deep sequencing resolve the same set of core bacterial species dominating gut communities of honey bees. PLoS One 7:e41250. doi: 10.1371/journal.pone.0041250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Martinson VG, Danforth BN, Minckley RL, Rueppell O, Tingek S, Moran NA. 2011. A simple and distinctive microbiota associated with honey bees and bumble bees. Mol Ecol 20:619–628. doi: 10.1111/j.1365-294X.2010.04959.x. [DOI] [PubMed] [Google Scholar]
- 14.Nakabachi A, Ueoka R, Oshima K, Teta R, Mangoni A, Gurgui M, Oldham NJ, van Echten-Deckert G, Okamura M, Hongoh Y, Miyagishima S-Y, Hattori M, Piel J, Fukatsu T. 2013. Defensive bacteriome symbiont with a drastically reduced genome. Curr Biol 23:1478–1484. doi: 10.1016/j.cub.2013.06.027. [DOI] [PubMed] [Google Scholar]
- 15.Walton JD. 2006. HC-toxins. Phytochemistry 67:1406–1413. doi: 10.1016/j.phytochem.2006.05.033. [DOI] [PubMed] [Google Scholar]
- 16.He H, Silo-Suh LA, Handelsman J, Clardy J. 1994. Zwittermicin A, an antifungal and plant protection agent from Bacillus cereus. Tetrahedron Lett 38:2499–2502. [Google Scholar]
- 17.Faulds D, Goa KL, Benfield P. 1993. Cyclosporin. A review of its pharmacodynamic and pharmacokinetic properties, and therapeutic use in immunoregulatory disorders. Drugs 145:953–1040. [DOI] [PubMed] [Google Scholar]
- 18.D'Onofrio Crawford JM, Stewart EJ, Witt K, Gavrish E, Epstein S, Clardy J, Lewis K. 2010. Siderophores from neighboring organisms promote the growth of uncultured bacteria. Chem Biol 17:254–264. doi: 10.1016/j.chembiol.2010.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hider RC, Kong X. 2010. Chemistry and biology of siderophores. Nat Prod Rep 27:637. doi: 10.1039/b906679a. [DOI] [PubMed] [Google Scholar]
- 20.Fischbach MA, Walsh CT. 2006. Assembly-line enzymology for polyketide and nonribosomal peptide antibiotics: logic, machinery, and mechanisms. Chem Rev 106:3468–3496. doi: 10.1021/cr0503097. [DOI] [PubMed] [Google Scholar]
- 21.Walsh CT, Chen H, Keating TA, Hubbard BK, Losey HC, Luo L, Marshall G, Miller DA, Patel HM. 2001. Tailoring enzymes that modify nonribosomal peptides during and after chain elongation on NRPS assembly lines. Curr Opin Chem Biol 5:525–534. doi: 10.1016/S1367-5931(00)00235-0. [DOI] [PubMed] [Google Scholar]
- 22.Crawford JM, Clardy J. 2011. Bacterial symbionts and natural products. Chem Commun (Camb) 47:7559–7566. doi: 10.1039/c1cc11574j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Crawford JM, Portmann C, Zhang X, Roeffaers MBJ, Clardy J. 2012. Small molecule perimeter defense in entomopathogenic bacteria. Proc Natl Acad Sci U S A 109:10821–10826. doi: 10.1073/pnas.1201160109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kampa A, Gagunashvili AN, Gulder TAM, Morinaka BI, Daolio C, Godejohann M, Miao VPW, Piel J, Andrésson O. 2013. Metagenomic natural product discovery in lichen provides evidence for a family of biosynthetic pathways in diverse symbioses. Proc Natl Acad Sci U S A 110:E3129–E3137. doi: 10.1073/pnas.1305867110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nguyen T, Ishida K, Jenke-Kodama H, Dittmann E, Gurgui C, Hochmuth T, Taudien S, Platzer M, Hertweck C, Piel J. 2008. Exploiting the mosaic structure of trans-acyltransferase polyketide synthases for natural product discovery and pathway dissection. Nat Biotechnol 26:225–233. doi: 10.1038/nbt1379. [DOI] [PubMed] [Google Scholar]
- 26.Olier M, Salvador-Cartier C, Secher T, Dobrindt U, Boury M, Bacquié V, Pénary M, Gaultier E, Nougayrede JP, Fioramonti J, Oswald E. 2012. Genotoxicity of Escherichia coli Nissle 1917 strain cannot be dissociated from its probiotic activity. Gut Microbes 3:501–509. doi: 10.4161/gmic.21737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Buc E, Dubois D, Sauvanet P, Raisch J, Delmas J, Darfeuille-Michaud A, Pezet D, Bonnet R. 2013. High prevalence of mucosa-associated E. coli producing cyclomodulin and genotoxin in colon cancer. PLoS One 8:e56964. doi: 10.1371/journal.pone.0056964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nougayrede J-P, Homburg S, Taieb F, Boury M, Brzuszkiewicz E, Gottschalk G, Buchrieser C, Hacker J, Dobrindt U, Oswald E. 2006. Escherichia coli induces DNA double-strand breaks in eukaryotic cells. Science 313:848–851. doi: 10.1126/science.1127059. [DOI] [PubMed] [Google Scholar]
- 29.Putze J, Hennequin C, Nougayrède J-P, Zhang W, Homburg S, Karch H, Bringer M-A, Fayolle C, Carniel E, Rabsch W, Oelschlaeger TA, Oswald E, Forestier C, Hacker J, Dobrindt J. 2009. Genetic structure and distribution of the colibactin genomic island among members of the family Enterobacteriaceae. Infect Immun 77:4696–4703. doi: 10.1128/IAI.00522-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Arthur JC, Perez-Chanona E, Mühlbauer M, Tomkovich S, Uronis JM, Fan T-J, Campbell BJ, Abujamel T, Dogan B, Rogers AB, Rhodes JM, Stintzi A, Simpson KW, Hansen JJ, Keku TO, Fodor AA, Jobin C. 2012. Intestinal inflammation targets cancer-inducing activity of the microbiota. Science 338:120–123. doi: 10.1126/science.1224820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cuevas-Ramos G, Petit CR, Marcq I, Boury M, Oswald E, Nougayrède J-P. 2010. Escherichia coli induces DNA damage in vivo and triggers genomic instability in mammalian cells. Proc Natl Acad Sci U S A 107:11537–11542. doi: 10.1073/pnas.1001261107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bian X, Fu J, Plaza A, Herrmann J, Pistorius D, Stewart AF, Zhang Y, Müller R. 2013. In vivo evidence for a prodrug activation mechanism during colibactin maturation. Chembiochem 14:1194–1197. doi: 10.1002/cbic.201300208. [DOI] [PubMed] [Google Scholar]
- 33.Brotherton CA, Balskus EP. 2013. A prodrug resistance mechanism is involved in colibactin biosynthesis and cytotoxicity. J Am Chem Soc 135:3359–3362. doi: 10.1021/ja312154m. [DOI] [PubMed] [Google Scholar]
- 34.Vizcaino MI, Engel P, Trautman E, Crawford JM. 2014. Comparative metabolomics and structural characterizations illuminate colibactin pathway-dependent small molecules. J Am Chem Soc 136:9244–9247. doi: 10.1021/ja503450q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bondarev V, Richter M, Romano S, Piel J, Schwedt A, Schulz-Vogt HN. 2013. The genus Pseudovibrio contains metabolically versatile bacteria adapted for symbiosis. Environ Microbiol 15:2095–2113. doi: 10.1111/1462-2920.12123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Markowitz VM, Szeto E, Palaniappan K, Grechkin Y, Chu K, Chen I-MA, Dubchak I, Anderson I, Lykidis A, Mavromatis K, Ivanova NN, Kyprides NC. 2007. The integrated microbial genomes (IMG) system in 2007: data content and analysis tool extensions. Nucleic Acids Res 36:D528–D533. doi: 10.1093/nar/gkm846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schattner P, Brooks AN, Lowe TM. 2005. The tRNAscan-SE, snoscan and snoGPS web servers for the detection of tRNAs and snoRNAs. Nucleic Acids Res 33:W686–W689. doi: 10.1093/nar/gki366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li L, Stoeckert CJ, Roos DS. 2003. OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res 13:2178–2189. doi: 10.1101/gr.1224503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Engel P, Stepanauskas R, Moran NA. 2014. Hidden diversity in honey bee gut symbionts detected by single-cell genomics. PLoS Genet 10:e1004596. doi: 10.1371/journal.pgen.1004596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Krzywinski M, Schein J, Birol I, Connors J, Gascoyne R, Horsman D, Jones SJ, Marra MA. 2009. Circos: an information aesthetic for comparative genomics. Genome Res 19:1639–1645. doi: 10.1101/gr.092759.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Edgar RC. 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Guindon S, Dufayard J-F, Lefort V, Anisimova M, Hordijk W, Gascuel O. 2010. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol 59:307–321. doi: 10.1093/sysbio/syq010. [DOI] [PubMed] [Google Scholar]
- 43.Altschul SF, Gish W, Miller W, Myers EW. 1990. Basic local alignment search tool. J Mol Biol 215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 44.Blin K, Medema MH, Kazempour D, Fischbach MA, Breitling R, Takano E, Weber T. 2013. antiSMASH 2.0—a versatile platform for genome mining of secondary metabolite producers. Nucleic Acids Res 41:W204–W212. doi: 10.1093/nar/gkt449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bachmann BO, Ravel J. 2009. Chapter 8. Methods for in silico prediction of microbial polyketide and nonribosomal peptide biosynthetic pathways from DNA sequence data. Methods Enzymol 458:181–217. doi: 10.1016/S0076-6879(09)04808-3. [DOI] [PubMed] [Google Scholar]
- 46.Röttig M, Medema MH, Blin K, Weber T, Rausch C, Kohlbacher O. 2011. NRPSpredictor2—a web server for predicting NRPS adenylation domain specificity. Nucleic Acids Res 39:W362–W367. doi: 10.1093/nar/gkr323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kelley LA, Sternberg MJE. 2009. Protein structure prediction on the Web: a case study using the Phyre server. Nat Protoc 4:363–371. doi: 10.1038/nprot.2009.2. [DOI] [PubMed] [Google Scholar]
- 48.Goodman AL, Kallstrom G, Faith JJ, Reyes A, Moore A, Dantas G, Gordon JI. 2011. Extensive personal human gut microbiota culture collections characterized and manipulated in gnotobiotic mice. Proc Natl Acad Sci USA 108:6252–6257. doi: 10.1073/pnas.1102938108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Watrous J, Roach P, Alexandrov T, Heath BS, Yang JY, Kersten RD, van der Voort M, Pogliano K, Gross H, Raaijmakers JM, Moore BS, Laskin J, Bandeira N, Dorrestein PC. 2012. Mass spectral molecular networking of living microbial colonies. Proc Natl Acad Sci U S A 109:E1743–E1752. doi: 10.1073/pnas.1203689109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Eisen JA, Heidelberg JF, White O, Salzberg SL. 2000. Evidence for symmetric chromosomal inversions around the replication origin in bacteria. Genome Biol 1:research0011.1. doi: 10.1186/gb-2000-1-6-research0011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Haydock SF, Aparicio JF, Molnár I, Schwecke T, Khaw LE, König A, Marsden AFA, Galloway IA, Staunton J, Leadlay PF. 1995. Divergent sequence motifs correlated with the substrate specificity of (methyl)malonyl-CoA:acyl carrier protein transacylase domains in modular polyketide synthases. FEBS Lett 374:246–248. doi: 10.1016/0014-5793(95)01119-Y. [DOI] [PubMed] [Google Scholar]
- 52.Reimer D, Bode HB. 2014. A natural prodrug activation mechanism in the biosynthesis of nonribosomal peptides. Nat Prod Rep 31:154–159. doi: 10.1039/c3np70081j. [DOI] [PubMed] [Google Scholar]
- 53.Secher T, Samba-Louaka A, Oswald E, Nougayrède J-P. 2013. Escherichia coli producing colibactin triggers premature and transmissible senescence in mammalian cells. PLoS One 8:e77157. doi: 10.1371/journal.pone.0077157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Evans JD, Lopez DL. 2004. Bacterial probiotics induce an immune response in the honey bee (Hymenoptera: Apidae). J Econ Entomol 97:752–756. doi: 10.1603/0022-0493(2004)097[0752:BPIAIR]2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- 55.Mullin CA, Frazier M, Frazier JL, Ashcraft S, Simonds R, vanEngelsdorp D, Pettis JS. 2010. High levels of miticides and agrochemicals in North American apiaries: implications for honey bee health. PLoS One 5:e9754. doi: 10.1371/journal.pone.0009754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Schultz M. 2008. Clinical use of E. coli Nissle 1917 in inflammatory bowel disease. Inflamm Bowel Dis 14:1012–1018. doi: 10.1002/ibd.20377. [DOI] [PubMed] [Google Scholar]
- 57.Demarre G, Guérout A-M, Matsumoto-Mashimo C, Rowe-Magnus D, Marlière P, Mazel D. 2005. A new family of mobilizable suicide plasmids based on broad host range R388 plasmid (IncW) and RP4 plasmid (IncPα) conjugative machineries and their cognate Escherichia coli host strains. Res Microbiol 156:245–255. doi: 10.1016/j.resmic.2004.09.007. [DOI] [PubMed] [Google Scholar]
- 58.Kulasekara HD, Ventre I, Kulasekara BR, Lazdunski A, Filloux A, Lory S. 2005. A novel two-component system controls the expression of Pseudomonas aeruginosa fimbrial cup genes. Mol Microbiol 55:368–380. doi: 10.1111/j.1365-2958.2004.04402.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.