

Abstract
White spotting variant (Wv) mice are spontaneous mutants attributed to a point mutation in the c-Kit gene, which reduces the tyrosine kinase activity to around 1% and affects the development of melanocytes, mast cells, and germ cells. Homozygous mutant mice are sterile but can live nearly a normal life span. The female Wv mice have a greatly reduced ovarian germ cell and follicle reserve at birth, and the remaining follicles are largely depleted soon after the females reach reproductive stage at around 7 weeks of age. Consequently, ovarian epithelial tumors develop in 100% of Wv females by 3 to 4 months of age. These tumors, called tubular adenomas, are benign but can become invasive in older Wv mice.
We tested if additional genetic mutation(s) could convert the benign ovarian epithelial tumors to malignant tumors by crossing the Wv mutant into the Trp53 knockout background. Surprisingly, we found that global deletion of Trp53 suppressed the development of ovarian tubular adenomas in Wv mice. The ovaries of Wv/Wv; Trp53 (−/−) mice were covered by a single layer of surface epithelium and lacked excessive epithelial proliferation. Rather, the ovaries contained a small number of follicles. The presence of ovarian follicles and granulosa cells, as indicated by Pgc7 and inhibin-alpha expression, correlated with the absence of epithelial lesions. A reduction of Pten gene dosage, as in Wv/Wv; Pten (+/−) mice, produced a similar, though less dramatic, phenotype. We conclude that deletion of Trp53 prolongs the survival of ovarian follicles in Wv mice and consequently prevents the proliferation of ovarian epithelial cells and development of ovarian tubular adenomas. The results suggest that various cell types within the ovary communicate and mutually modulate, and an intact tissue environment is required to ensure homeostasis of ovarian surface epithelial cells. Especially, the current finding emphasizes the importance of ovarian follicles in suppressing the hyperplastic growth of ovarian epithelial cells, dominating over the loss of p53.
Introduction
The white spotting (W) and variant (Wv) mice are spontaneous mutants derived from selective breeding to follow their unique coat color patterning, and several strains were identified to be caused by mutations in the receptor tyrosine kinase c-Kit [1], [2], [3]. Inactivating mutations of c-Kit affect most significantly the development of melanocyte, mast cell, and germ cell lineages and have pleiotropic effects on both embryonic development and hematopoiesis [4]. Homozygous W mutation in mice often leads to perinatal death resulting from a severe anemia [4]. A unique variant, Wv, harbors a c-Kit point mutation that reduces the c-Kit tyrosine kinase activity to around 1%, and the developmental phenotypes are less severe [2], [3]. The heterozygous Wv mice in the C57BL/6 J(B6) background have dorsal and/or ventral white spots, whereas the homozygous mutant mice have a completely white fur coat [4], [5]. The homozygous mutant mice have about 1% to 5% of oocytes/follicles compared with wild type at birth [1], [6], and the follicles are depleted by about 2 months of age when the unaffected (wild type or heterozygous) mice become reproductively mature. Depletion of germ cells/ovarian follicles in females associates with elevation of gonadotropins and development of epithelial lesions [5], [6], [7]. Both male and female Wv/Wv mice are sterile but have a similar life span as wild type. The homozygous Wv female mice present several physiological aspects of menopause found in women [7], [8].
In female mammals, the primordial germ cells are mitotically active during embryogenesis but are stationary following meiosis to produce primary oocytes immediately before birth [9]. Oocytes recruit and become surrounded by granulosa cells to form follicles in late embryogenesis for humans and the immediate neonatal period for mice [9]. Thus, a finite population of oocytes is established at birth, although this paradigm has been challenged and the controversy has not yet been resolved [10], [11], [12]. Nevertheless, after birth, the majority of the oocytes in mammals are progressively lost by atresia, and only a fraction of the follicles in the ovarian reserve develop to maturity and are used in ovulation [13], [14]. The Tp53 family proteins play critical roles in the development and attrition of ovarian follicles [15], [16]. The impact of Tp53 in female reproductive function has been recognized in both mice and women, although the main mechanism is thought to affect embryo implantation [17]. Several reports also noted the influence of Trp53 on oocyte development and attrition [16], [18]. Pten is another tumor suppressor gene known to affect oocyte development and survival [19], [20]. For most mammals in their natural environment, the oocyte reserve and thus the reproductive lifetime generally correlate with life span [21]. Women, especially in modern times, are unique in that they experience an extended life span following reproductive senescence due to ovarian follicle depletion, a phenomenon known as menopause. In this aspect, the female Wv mutant mice also have an extended life span following ovarian follicle depletion and thus bear its biological consequences. The reduced germ cell reserve and a normal life span of the mutant mice make the Wv mice a suitable laboratory model of menopause [8].
The germ cell–deficient phenotype of the W series of mice was characterized in the 1950s before the c-Kit mutation was identified in the strains [1]. It was found that germ cells fail to survive and migrate into the gonadal ridge during embryonic development [1]; for females, the outcome is that few ovarian follicles are present in adults [6]. Ovarian tumors arise following the depletion of germ cell/follicles in the ovaries [5], [6]. The tumors are classified as tubular adenoma, a benign epithelial neoplasm [6]. A fraction of the ovaries develop granulosa cell tumors, which are likely promoted by the elevated gonadotropins in the Wv/Wv mice following follicle depletion [6], [7].
Although activating mutations or gene amplification of c-Kit receptor tyrosine kinase have been found in some cancer types in humans, c-Kit is not commonly expressed or involved in epithelial ovarian cancer [22], [23]. Rather, the formation of ovarian tubular adenomas is attributed to the loss of function of c-Kit in the oocytes and subsequent follicle depletion [5], [6].
More recently, the ovarian tumor phenotype of the Wv mice was studied to understand the relationship between reproductive etiological factors such as menopausal biology and increased ovarian cancer risk [5], [24]. It was observed that the ovarian surface epithelia proliferate and invaginate to form the tubular adenomas following ovarian follicle depletion [5]. A reduction of cyclooxygenase (Cox)-2 gene dosage in the Wv/Wv mice leads to a reduced formation of the tubular adenomas; however, complete Cox-2 knockout does not, because of a compensatory expression of Cox-1 [5]. Suppression of Cox-1 by either genetic knockout or pharmacologic inhibitors prolongs postnatal ovarian follicle life span and also reduces the development of ovarian tubular adenomas in the Wv mice [25].
The benign tubular adenomas in the Wv mice resemble, in morphology, preneoplastic epithelial changes, such as surface invaginations, inclusion cysts, and papillomatosis, found in aged human ovaries [26], [27]. These epithelial changes in ovaries precede cancer development [28], and the increased ovarian epithelial remodeling likely promotes the selection and expansion of cells that have acquired oncogenic mutations such as the inactivation of Trp53 or activation of Ras. The oncogenic mutation(s) confers growth advantage, and ultimately the further expansion and selection of the mutant cells lead to carcinomas. The ovarian tubular adenomas in the ovary of the Wv mice resemble the low-grade ovarian cancer tumors (type I, low–malignant potential tumors), which usually have a wild-type Trp53 and KRas or Raf-1 activating mutation [29]. In high-grade serous ovarian cancer, the tumor suppressor gene TP53 is commonly mutated [29], [30], [31], [32], which presumably allows the cell-autonomous survival and growth of ovarian cancer cells.
In the current study, we tested if the addition of an oncogenic mutation, such as Trp53 deletion or reduction of Pten gene dosage, may convert the benign ovarian epithelial tumors into malignant cancer in this epithelial ovarian tumor–bearing mouse model. The results of the experiments were unexpected, and the findings provide further insight to the etiology and mechanism of ovarian cancer, especially related to cell-cell communication within the ovary and the increased ovarian cancer risk found in peri- and postmenopausal women.
Materials and Methods
Sources, Genotyping, and Characteristics of Genetically Modified Mice
All of the mouse strains used in the study have been kept in the C57BL/6 J-background. Wv mouse (C57BL/6 J-KitW-v) breeding pairs were originally obtained from Jackson Laboratory in 2002. The Wv mouse colony has been maintained by inbreeding in our mouse facility for the last 12 years. The Wv genotypes of the resulting progeny were identified by coat color: white, gray with dorsal and/or ventral spots, or black/agouti represents Wv/Wv homozygous mutant, Wv/+ heterozygous mutant, or wild type, respectively.
The Trp53 (+/−) mice were purchased from Taconic (Hudson, NY) [33], [34]. Inactivation of the Trp53 gene in the mutant allele was achieved by replacing exons 2 to 6, including the start codon, with a neomycin-resistance gene (neo) cassette. The following primer sets were used for polymerase chain reaction (PCR) genotyping that amplifies the wild-type and targeted mutant Trp53 allele: 5′-TGGTG CTTGG ACAAT GTGTT-3′; 5′-CTCCG TCATG TGCTG TGACT-3′; 5′-GGATG ATCTG GACGA AGAGC-3′. All three primers were used simultaneously in the PCR, yielding a 450–base pair wild-type and/or 650–base pair p53 mutant band.
A pair of Trp53 floxed (Trp53 [fl/fl]) mice was provided by Dr Denise Connolly (Fox Chase Cancer Center, Philadelphia, PA). The conditional mutant mice contain a functional Trp53 gene in which exons 2 to 10 are flanked by loxP sites, and the breeding pair was originally obtained from MMRRC (The Mutant Mouse Regional Resource Center, www.mmrrc.org) [35], [36]. The Pten (+/−) mice were a gift from Dr Antony Di Cristofano and were bred and genotyped following the published protocols [37]. The Pten allele was inactivated by replacing exons 4 and 5 with a neo cassette [37]. All animal studies were approved first by the Fox Chase Cancer Center and then the University of Miami Institutional Animal Care and Use Committee, and all experimental procedures were performed following NIH guidelines.
Histology and Immunohistochemistry
Entire ovaries as well as the attached uterine horns from Wv ovarian tumor models were dissected, fixed in 10% formalin, paraffin embedded, and sectioned into 6 μm–thick slices collected on positively charged glass slides. The sections were dewaxed in xylene and hydrated through graded ethanol. Heat-induced antigen retrieval was then carried out in 10-mM sodium citrate (pH 6.0) buffer in a microwave initially set at high-power setting (#10) for 2 minutes, followed by 10 minutes on low-power setting (#2). The endogenous peroxidase activity was blocked by immersing the slides in 3% H2O2 in methanol for 15 minutes. After a 30-minute incubation in blocking serum, slides were incubated with primary antibodies at 4°C overnight at the following dilutions: rat monoclonal anti–Troma-1/cytokeratin-8 (CK8) (Developmental Studies Hybridoma Bank from The University of Iowa, line SP2/0) at 1:600 and mouse monoclonal anti–inhibin-alpha (Serotec, clone 5070) at 1:200. Alternatively, slides were blocked overnight at 4°C and incubated with goat polyclonal anti-Pgc7 (R & D Systems, Cat#: AF2566) at 1:1000 for 1 to 2 hours at room temperature. After extensive washing, the slides were incubated with horseradish peroxidase–labeled secondary antibodies (BD Pharmingen) at 1:100 dilution for CK8 or anti-rabbit labeled polymer horseradish peroxidase (Dako) for 35 minutes at room temperature. Diaminobenzidine was used as the chromogen for the immunoperoxidase reaction. The slides were counterstained with hematoxylin and mounted in 50:50 xylene/Permount.
Preparation and Culturing of Mouse Primary Ovarian Epithelial Cells
Primary cultures of mouse ovarian surface epithelial (MOSE) cells were generated using a slightly modified protocol as described previously [38], [39]. Briefly, ovaries from 2- to 6-month-old female mice were carefully dissected using sterilized surgical instruments. These ovaries were then washed with PBS and incubated in 0.25% Trypsin-EDTA solutions (Invitrogen) at 37°C for 1 hour. The surface cells were washed off from the ovaries by repeated pipetting through a 1-ml pipette with the tip cut to fit the size of the ovaries, and then the ovaries were removed and discarded. Dissociated MOSE cells suspended in the medium were collected by a brief centrifugation and cultured on six-well plates previously coated with 0.1% gelatin. MOSE cells were maintained as monolayers in high-glucose DMEM/F12 medium supplemented with 10% fetal bovine serum, 1 mM glutamine, 10 ng/ml epidermal growth factor, 500 ng/ml hydrocortisone, 5 μg/ml insulin, and 5 μg/ml transferrin. For Wv/Wv ovaries, the epithelial tumor cells were isolated by incubation with DMEM/F12 medium containing 0.5% collagenase (Invitrogen) for 2 hours at 37°C, and the isolated epithelial cells were cultured under the same conditions as wild-type MOSE cells. By analysis of cytokeratin expression using immunofluorescence microscopy, the cell preparations were verified to be more than 90% epithelial cells.
For gene deletion, primary cultures MOSE cells isolated from Trp53 (f/f) or Wv/Wv; Trp53 (f/f) mice were treated with replication-deficient adenovirus carrying Cre coding insert (adenoviral Cre [Adv-Cre]) by addition of the viral vector (200 MOI) to the cells. Adenovirus expressing LacZ was used as a control, and LacZ expression indicated that high efficiency of infection (> 90%) was achieved at 200 MOI. The viral expression vectors were purchased from the Vector Core at the University of Iowa and were used according to the manufacturer’s protocol.
Cell Growth Assay
The growth rate of MOSE cells was analyzed using an MTT-like WST-1 cell proliferation kit from Roche. Briefly, 2 × 104 MOSE cells of various genotypes were seeded on 96-well plates previously coated with 0.1% gelatin. These cells were allowed to grow for 24 to 96 hours. At each time point (24-hour intervals), the cell culture medium was replaced with freshly prepared reaction buffer composed of DMEM/F12 media containing 10% WST-1 solution. These cells were then incubated for 1 hour at 37°C, and the accumulation of the soluble formazan dye produced by viable cells was measured for absorbance at 450 nm using a standard multiwell spectrophotometer. The cell number correlates with the amount of soluble formazan dye.
Western Blot and Antibodies
Cultured MOSE cells were washed with cold PBS and then lysed in SDS gel loading buffer. Lysates were collected and denatured in boiling water bath for 10 minutes. The protein extracts were separated on 8% or 10% SDS polyacrylamide gels, transferred onto nitrocellulose membrane, and subjected to immunoblotting according to standard procedures. The primary antibodies used were: anti–N-cadherin (mouse monoclonal, clone: 32/N-Cadherin, BD Bioscience), anti–E-cadherin (mouse monoclonal, clone: 34/E-Cadherin, BD Bioscience), anti–beta-catenin (mouse monoclonal, clone: 14/Beta-Catenin, BD Bioscience), anti-COX1 (#160105, #160110, Cayman Chemical), anti-COX2 (#160106, Cayman Chemical), anti–claudin-3 (Invitrogen), anti-PCNA (SC56, Santa Cruz), anti-p21 (mouse monoclonal, clone: SXM30, BD Bioscience), anti–phospho-AKT (#9276, Cell Signaling), anti-AKT (#9272, Cell Signaling), anti–phospho-Erk1/2 (mouse monoclonal, clone: E10, Cell Signaling), anti-Erk1/2 (#9102, Cell Signaling), and anti–beta-actin (clone AC-15, Sigma). Chemoluminescence detection was performed using the SuperSignal West Extended Duration Substrate detection kit (Pierce Biotech).
Results
Development of Epithelial Lesions and Tubular Adenomas from Surface and Rete Ovarri in the Ovary of Wv Mice
We have maintained a colony of Wv mice in C57BL/6 J background by inbreeding for the last 12 years. Of the more than 300 mature (3 months or older) Wv/Wv females examined, all exhibited ovarian tubular adenomas, whereas none were observed in the wild-type littermates.
The representative ovarian tumor morphology is shown in Figure 1. At 5 months of age, the wild-type ovary displayed a complex structure composed of various cell types. A single layer of Troma-1/CK8-positive ovarian surface epithelial cells enclosed the ovarian cortex containing follicles at all different stages of development (Figure 1, A and B, arrow). In addition to the ovarian surface, CK8-positive epithelia of rete ovarii were found in the hilus region (Figure 1A, arrowhead) or the mesosalpinx areas (Figure 1B, arrowhead). In the Wv/Wv ovary starting at around 2 months of age, the tissue was replaced with tubular adenomas, shown as CK8-positive epithelial layers mixed with stroma (Figure 1C). At this earlier stage, the epithelial lesions were contiguous with the surface epithelium (Figure 1C, arrow), suggesting that the infolding and invagination of the surface epithelia gave rise to the benign tumor. The tubular adenomas became enlarged for both epithelial and stromal components and appeared more invasive (spreading into stromal area) at later ages, as shown by an example from a 5-month-old Wv/Wv female (Figure 1D).
Figure 1.
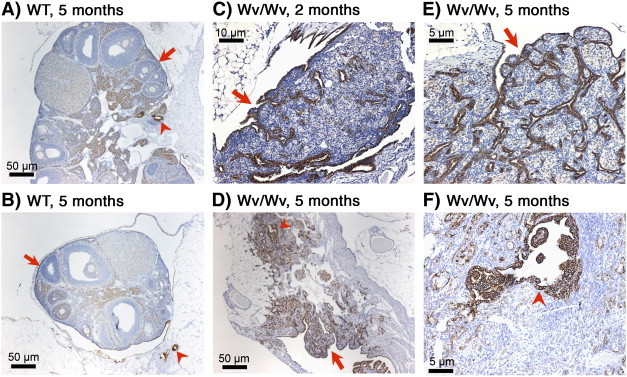
Development of tubular adenomas in Wv/Wv mice. Ovarian morphology was compared between wild-type littermates (A and B) and Wv/Wv (C to F) mice. CK8 (Troma-1) staining was used to highlight epithelial cells. (A, B) Two examples are shown of ovaries from wild-type controls at 5 months of age. Arrows indicate a layer of CK8-positive surface epithelium. Arrowheads indicate the presence of CK8-positive epithelial cells of rete ovarii in the hilus (A) or mesosalpinx (B) areas. (C) Representative examples of ovaries from Wv/Wv mice at the age of 2 months and (D) 5 months. The slides shown are selected from section near the middle of the tissues. An arrow in (C) indicates that the CK8-positive tubular adenoma lesion is contiguous with the ovarian surface epithelium. An arrow in (D) shows peripheral lesions that are likely derived from the surface, and an arrowhead indicates distinct lesions that are likely derived from rete ovarii, which are shown at a higher magnification in (E) and (F), respectively. A detailed time course of the development of the ovarian tubular adenomas has been previously published [5].
Previously, we observed that, in cases of early ovarian lesions in younger (7 to 10 weeks) Wv/Wv mice, the tubular structure is contiguous with the monolayer of the surface epithelium, indicating that the tubular epithelial lesions originate from the surface epithelium [5]. Closer observation showed that the epithelial lesions appeared to be of two different types (Figure 1D). The main tubular adenomas have flat epithelial layers and distribute throughout the ovarian stroma (Figure 1, D and E, arrow). These lesions resemble surface deep invaginations/papillomatosis and likely originate from the surface epithelium. Another distinctive type of CK8-positive lesion was found often around the stem or hilus ligament area (Figure 1, D and F, arrowhead). The cells are columnar-to-stratified epithelial in appearance and often locate deep within the tissues. This second type of lesion likely is derived from rete ovarii because rete ovarii structure and cysts are often observed in the area in the wild-type or younger Wv/Wv ovaries.
Although dysplastic morphology is evident in some epithelial components of the tubular adenomas, the ovarian epithelial tubular structures in the Wv mice are considered nonmalignant tumors, and the lesions are confined to ovarian tissues and do not become metastatic. Histopathological evaluation also indicated their low-grade cytological morphologies without evident mitotic figures.
The oviducts in Wv/Wv mice were not affected because we consistently observed normal morphology of the ducts at all ages despite the persistent presence of ovarian tubular adenomas in the associated ovaries.
Suppression of Ovarian Tubular Adenomas in Wv Mice upon Trp53 Deletion
We attempted to convert the benign ovarian tubular adenomas into more neoplastic tumors by adding additional oncogenic mutations into the Wv model. Because TP53 inactivation is common in high-grade serous ovarian cancer, we first crossed and obtained female Wv/Wv; Trp53 (−/−) mice to analyze for possible ovarian tumor phenotypes.
Our experience in breeding Trp53 mutant mice was similar to that reported [33]. We observed a reduction of the expected ratio of Trp53 (−/−) pups from matings between parents of Trp53 (+/−) genotype to approximately 55% of the expected number. Also, a sex-biased ratio of 5:3 for male-to-female pups was observed. The genotype distribution among progenies was also similar after crossing into the Wv heterozygous background. Because Wv/Wv are sterile and Trp53 (−/−) are poor breeders, we initiated breeding of Wv/+; Trp53 (+/−) parents and kept any Wv/Wv; Trp53 (−/−) female progenies for further analysis. In tabulating the breeding scheme over a 6-year period, of about 1000 progenies genotyped, we only identified 9 females and 14 males of Wv/Wv; Trp53 (−/−) genotype. Thus, the Wv/Wv; Trp53 (−/−) genotypes exhibit an increased embryonic lethality, with a 37% survival rate based on the expected Mendelian ratio. The female Wv/Wv; Trp53 (−/−) mice needed for analysis were even more difficult to generate + because of sex-biased embryonic lethality, and only 29% of the expected number was produced.
The ovaries from mice with the Trp53 (−/−) mutation alone were not noticeably different in appearance and histology compared with those of wild type, as reported previously [40]. We found that crossing Wv mice into mutant p53 did not lead to the development of large ovarian tumors or a malignant tumor phenotype. Instead, surprisingly, the addition of the p53 mutation/deletion suppressed the formation of tubular adenomas in the Wv/Wv mice. The mice were analyzed before reaching 6 months of age because p53 null mice often develop sarcomas and lymphomas after this age [34]. In the 18 ovaries collected from all 9 Wv/Wv; Trp53 (−/−) females at 2 to 6 months of age, we observed an intact ovarian structure without the presence of tumor. Compared with wild-type ovaries that contained follicles of a range of developmental stages (Figure 2A) or to Wv/Wv ovaries that were entirely permeated with tubular adenomas (Figure 2B), the Wv/Wv; Trp53 (−/−) ovaries included no tumor structure, exhibited a more homogeneous cortex, and contained occasional follicles (Figure 2, C and D). Here, two examples with distinctive morphologies are shown. The ovary in (Figure 2D) has a few follicles yet contains corpora lutea–like structures. We speculate that the ovary was harvested immediately following gonadotropin stimulation and ovulation. Staining with CK8 shows that the representative ovary has a smooth layer of epithelium on the surface of the ovaries and no epithelium locates within the ovary (Figure 2, E and F).
Figure 2.
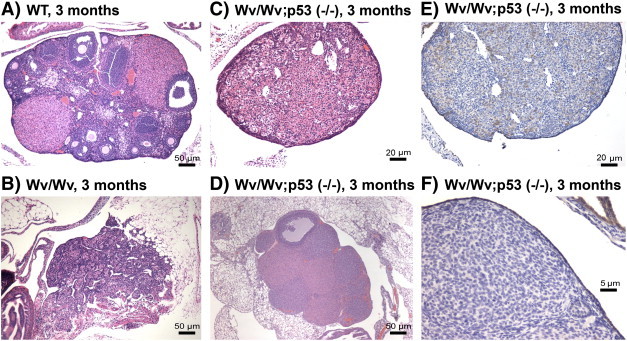
Suppression of tubular adenomas in Wv/Wv; Trp53 (−/−) ovaries. H&E staining of ovaries from 3-month-old wild type (A), Wv/Wv (B), and Wv/Wv; Trp53 (−/−) (C, D) are shown. An immunostaining of the epithelial cell marker CK8 of the ovary in (C) is shown in (E) from a 3-month-old Wv/Wv; Trp53 (−/−) mouse. A higher magnification of the ovary in (E) is shown in (F). The images shown were selected from a section near the middle of the tissue. The staining shows a monolayer of CK8-positive epithelial cells enveloping a representative Wv/Wv; Trp53 (−/−) ovary (C), and no tumor or epithelial lesions were observed in the ovarian cortex.
Subtle Influence on Wv/Wv Ovarian Tumor Phenotypes with Reduction of Pten Gene Dosage
We also undertook another cross to analyze ovaries from Wv/Wv; Pten (+/−) mice with a goal to determine if a reduction of Pten tumor suppressor gene might increase the malignant features of the tubular adenomas in the Wv/Wv mice. Pten is often mutated or its expression is lost in ovarian cancer, though most frequently in the endometrioid subtype [41]. Homozygous Pten deletion causes early embryonic lethality in mice; however, heterozygous Pten mutant mice are predisposed to develop various epithelial tumors, most often the endometrial type [37], [42], [43]. We produced and analyzed 10 Wv/Wv; Pten (+/−) female mice at ages ranging from 3 to 12 months. The addition of Pten mutation into Wv/Wv mice did not significantly alter the tumor phenotype in most of the cases (Figure 3); rather, it reduced the presence of epithelial tumor lesions in about one third of the ovaries (6 out of 20 ovaries analyzed), especially at a younger age (3 to 4 months). Four representative ovaries from the Wv/Wv; Pten (+/−) mice are shown (Figure 3); two represent subtle changes (Figure 3, A and B), and the other two represent a reduction in tumor lesions (Figure 3, C and D). Upon close inspection, we observed the presence of follicles in the Wv/Wv; Pten (+/−) ovaries that contain no tubular epithelial lesions (Figure 3E). Consistently, the presence of follicles correlated with the absence of tubular adenomas in all the Wv/Wv; Pten (+/−) ovaries, and no follicle structures were ever observed in areas containing tubular epithelial structures.
Figure 3.
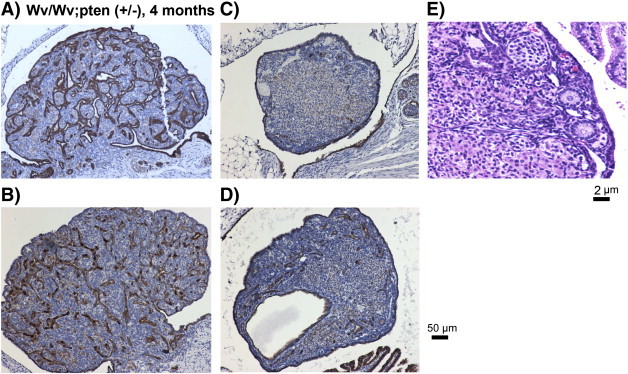
Ovarian morphological features of Wv/Wv; Pten (+/−) mutant mice. Representative CK8 staining (of epithelial cells) of four examples of ovaries from four 4-month-old Wv/Wv; Pten (+/−) mice is shown. Two ovaries (A, B) are similar to those of Wv/Wv mice, and the other two ovaries shown (C, D) exhibit a reduced tumor lesion. An example of H&E staining of the ovary with reduced tumor lesion is shown at higher magnification to visualize the presence of follicle structures (E).
Thus, again in contrast to our expectation, a reduction of the Pten gene dosage in the Wv mice only had subtle effects on the development of tubular adenomas. Rather, reduced Pten prevented/delayed the development of tubular adenomas in Wv/Wv mice, although the effect was not as dramatic as in the case of Trp53 deletion because only 6 out of 20 ovaries showed a reduced epithelial tubular adenoma lesions compared with 18 out of 18 ovaries from the Wv/Wv; Trp53 (−/−) mice.
NO Increased Cell Proliferation in the Ovaries of Wv/Wv; Trp53 (−/−) Mice
Suppression rather than promotion of tumor development by Trp53 deletion or Pten gene dosage reduction was unexpected. Thus, we examined by immunohistochemistry the effect of Trp53 deletion on cell proliferation in the ovaries (Figure 4A).
Figure 4.
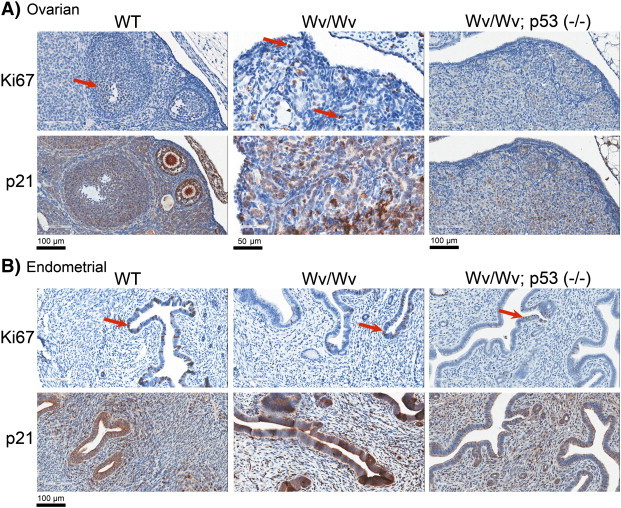
Proliferation and p21 expression in ovarian and endometrial tissues. Ovaries (A) and associated uteri (B) were harvested from 3-month-old wild-type (WT), Wv/Wv, and Wv/Wv; Trp53 (−/−) mice and used for immunostaining with Ki67 as a proliferative index and with p21 as a reporter of Trp53 activity. Adjacent sections were used for p21 and Ki67 stainings, and both the uterine horns and associated ovaries were analyzed together on same slides. Representative images are shown. Arrows indicate examples of Ki67-positive cells.
We used Ki67 staining as an indication of cell proliferation. Because Trp53 protein is difficult to be assayed in normal tissues because of its low abundance, we used the cyclin inhibitor p21, a Trp53-transcriptional target, as a reporter of Trp53 activity. In wild-type ovaries, Ki67-positive cells were limited to a fraction of the granulosa cells (Figure 4A, left panel, arrow), and most of the cell populations, including surface epithelial and granulosa cells and oocytes, were positive for nuclear p21 (Figure 4A, left panel). In the Wv/Wv ovaries, Ki67-positive proliferating cells were abundant in both epithelial and stromal compartments (Figure 4A, middle panel, arrows). The tumor also contained widespread p21-positive cells, which consisted about half of the cell population (Figure 4A, middle panel). Interestingly, few cells in the Wv/Wv; Trp53 (−/−) ovary were positive for Ki67, although the ovary had a much reduced p21 staining, consistent with the loss of p53 activity (Figure 4A, right panel).
The lack of cell proliferation did not apply to the endometrial tissues of the Wv/Wv; Trp53 (−/−) mice (Figure 4B). The uterus of the Wv/Wv; Trp53 (−/−) mice showed a similar extent of Ki67-staining compared with those from wild-type or Wv/Wv mice (Figure 4B, arrows). p21 staining of the endometrial epithelial cells of Wv/Wv; Trp53 (−/−) was slightly reduced compared with those of wild type or Wv/Wv, although the staining appeared largely cytoplasmic. The stromal cells showed similar nuclear staining of p21, suggesting that p21 expression in these cells was not regulated by Trp53. Thus, we conclude that Trp53 deletion suppresses proliferation of all cell types in the Wv/Wv; Trp53 (−/−) ovaries but does not significantly affect uterine cells in the same mice.
Increased Growth of Ovarian Surface Epithelial Cells in Cultures upon Trp53 Deletion
Next, we examined if p53 deletion alters ovarian surface epithelial cell survival and proliferation in culture to determine the possibility that the effect of Trp53 deletion is non–cell autonomous.
We observed that newly isolated epithelial cells from Wv/Wv ovaries had a greater growth rate than ovarian surface epithelial cells isolated from wild-type controls at early passages (Figure 5A). Nevertheless, the growth rate of the Wv/Wv epithelial cells was reduced to the same level as that of the wild type at a later time, after being maintained for four passages, or about 2 weeks, in culture (Figure 5B). Western blot analysis indicated that, in early passages, the cells had similar levels of N-cadherin, E-cadherin, and beta-catenin, but the Wv/Wv epithelial cells had increased COX1, COX2, and claudin-3 expression (Figure 5C). In these cases, the ovarian surface epithelial cells from both the control (wild type) and Wv/Wv mice were kept in a background of Trp53 (f/f). The addition of Trp53 (f/f) made no observable difference in either wild-type or Wv/Wv mice or cells, but it allowed for Trp53 gene deletion upon introducing the expression of Cre. Both Adv-LacZ and reverse transcription PCR of Trp53 alleles indicated high efficiency of infection and gene deletion in the cultured cells. Adv-Cre–mediated Trp53 gene deletion enhanced the growth rates of both Wv/Wv and wild-type ovarian epithelial cells (Figure 5, D and E). The enhanced growth of ovarian surface epithelial cells in culture upon Trp53 deletion has been previously reported [39], [44]. The cells were further analyzed by Western blot for cell cycle markers and activation of signaling pathways (Figure 5F). Proliferating cell nuclear antigen protein levels, an indicator of DNA replication and cell proliferation, were consistent with results obtained from the WST-1 cell growth assay (Figure 5, A, B, D, E, and F). The loss of p21, a Trp53 transcription target, indicated the complete inactivation of Trp53 following the addition of Adv-Cre (Figure 5F). However, Trp53 deletion did not significantly alter Akt or Erk1/2 activation, in either wild-type or Wv/Wv cells, although the level of phospho-Erk1/2 was slightly lower in Wv/Wv than the wild-type cells (Figure 5F).
Figure 5.
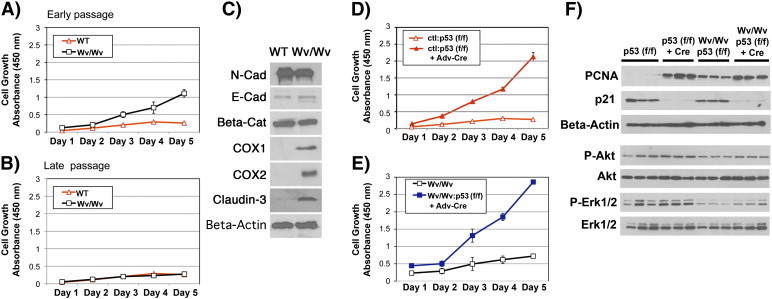
Proliferation and signaling of primary mouse ovarian surface epithelial cells in cultures. Primary ovarian surface epithelial cells were prepared from ovaries of mice of various genotypes. A total of six ovaries from three mice in each group were used to prepare the epithelial cells of wild-type ovarian surface or Wv/Wv ovarian tubular adenomas. The cells were pooled for experiments. The growth rates were measured by WST-1 assay (n = 3) from day 1 to 5. (A) Freshly prepared wild-type control (WT) and Wv/Wv ovarian epithelial cells were compared. (B) Growth of WT and Wv/Wv ovarian epithelial cells was maintained in cultures for four passages or around 2 weeks. The growth rate was then determined by WST-1 assay. (C) Lysates prepared from the cells were analyzed by Western blot for N-cadherin (N-Cad), E-cadherin (E-Cad), beta-catenin (Beta-Cat), COX1, COX2, claudin-3, and beta-actin. (D, E) Ovarian surface epithelial cells were prepared from p53 (f/f) and Wv/Wv; Trp53 (f/f) mice. The cells in primary cultures were treated with either Adv-Cre or Adv-LacZ (ctl), and the growth rate was measured. (F) Lysates were prepared from the cells following treatment with Adv-Cre or Adv-LacZ and were analyzed by Western blot. Three independent cell preparations per group were analyzed.
Thus, Trp53 deletion itself does not impair or slow epithelial proliferation in the Wv/Wv ovarian surface epithelial cells. The lack of epithelial proliferation in the Wv/Wv; Trp53 (−/−) ovaries likely was caused by a cell nonautonomous mechanism, such as growth suppression imposed by stromal cells. We reason that epithelial proliferation was then restored in cultures, which were free of growth suppression imposed by the ovarian stroma.
Prolonged Ovarian Follicle Life Span in Wv Mice with Trp53 Deletion
A clue to the mechanism underlying the unexpected finding that addition of oncogenic mutation in Wv/Wv mice suppressed rather than increased epithelial tumor development was the observation that follicle structures were present in the tumor-free ovaries. Similar to those of Wv/Wv; Pten (+/−), in the Wv/Wv; Trp53 (−/−) ovaries, a number of follicle structures consisting of a circle of granulosa cells surrounding an oocyte could be seen within the cortex (Figure 6). Occasionally, mature follicles were also observed (such as in Figure 2D). Immunostaining of PGC7, a germ cell and oocyte antigen, confirmed the presence of ovarian follicles (Figure 6).
Figure 6.
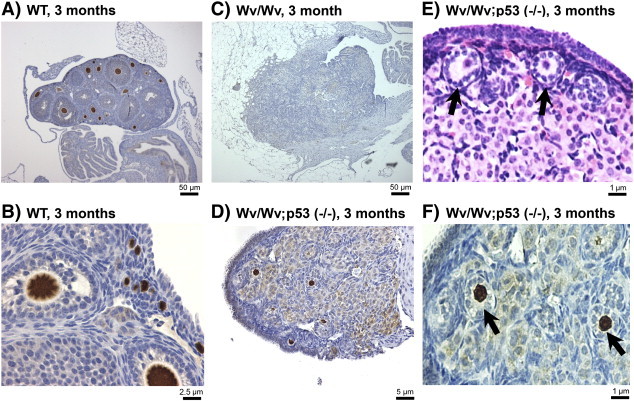
Rescue of germ cells/follicles in Wv/Wv; Trp53 (−/−) ovaries. Immunostaining of the germ cell marker PGC7 was performed on ovaries of various genotypes to identify germ cells and follicles. (A) Three-month-old wild type and (B) at a higher magnification. (C) Three-month-old Wv/Wv. (D) Three-month-old Wv/Wv; Trp53 (−/−). (E) An example of the Wv/Wv; Trp53 (−/−) ovaries is shown at a higher magnification to exemplify the follicle structures (arrow). (F) An example of PGC7 immunostaining of the Wv/Wv; Trp53 (−/−) ovary is shown in higher magnification. Arrows indicate examples of primary follicles.
In the reproductively competent wild types of around 3 months of age, the ovaries contained a large number (hundreds) of follicles at various developmental stages, as revealed by PGC7 staining (Figure 6A). Seen at a higher magnification, the primary follicles were found located immediately underneath the surface epithelium (Figure 6B). However, no ovarian follicles, at germ cell, primary, or mature stage, were observed in the ovaries of Wv/Wv mice, which were occupied by tubular adenomas (Figure 6C). In the Wv/Wv; p53 (−/−) ovaries, PGC7 staining confirmed that the structures indeed contained PGC7-positive oocytes, as shown by a representative image at or near the largest cross section of an ovary (Figure 6D). Similar to those of wild type, primary follicles appeared to align beneath the surface epithelium of the Wv/Wv; Trp53 (−/−) ovaries, as seen by hematoxylin and eosin (H&E) (Figure 6E, arrows) or PGC7 (Figure 6F, arrows) staining shown at a high magnification. From tabulation of the ovaries analyzed, the largest cross section of Wv/Wv; Trp53 (−/−) ovaries had 12 +/− 5 PGC7-positive follicles compared with 300 +/− 56 identified in a section of control ovaries. In contrast, no ovarian follicles were detected in more than 200 Wv/Wv mice analyzed, which is consistent with another recent study from our laboratory reporting the time course of ovarian follicle depletion in the mutant mice [25].
We also confirmed that the tumor-free Wv/Wv; Pten (+/−) ovaries contained follicles (5 +/− 3 in a section), though to a lesser degree than those of Wv/Wv; Trp53 (−/−). No follicles were observed in Wv/Wv; Pten (+/−) ovaries that showed adenoma structures.
Thus, preservation of ovarian follicles may be responsible for a reduced ovarian tumor phenotype in the Wv/Wv mice with Trp53 knockout or Pten heterozygous deletion. The finding supports the hypothesis of follicle depletion as the cause of ovarian epithelial proliferation and formation of tubular adenomas [24].
Inversed Correlation between Inhibin-alpha Expression and the Presence of Epithelial Tubular Adenoma in the Wv Ovaries
We further analyzed the ovarian follicles by staining granulosa cells for inhibin-alpha. In 3-month-old wild types, the ovaries contained follicles at various stages throughout the cortex, and a number of large and mature follicles were very prominent. These follicles contain multiple layers of the inhibin-alpha–positive granulosa cells (Figure 7A). Small primary follicles with only a single layer of inhibin-alpha–positive granulosa cells located mostly at the outer cortex, aligned along and immediately beneath the surface epithelium (Figures 6B and 7A), which showed as a single-layered CK8-positive cells (Figure 7B, arrow). In the follicle-depleted tubular adenomas of Wv/Wv ovaries, inhibin-alpha–positive granulosa cells were greatly reduced (Figure 7C). However, small patches of unorganized inhibin-alpha–positive granulosa cells, which were remnants of degenerated follicles, could be observed (Figure 7C, arrowheads). We recognized a strict inverse correlation between the presence of inhibin-alpha–positive granulosa cells and CK8-positive tubular adenomas at the same location within an ovarian tubular adenoma (Figure 7, C and D, arrowheads). The inverse correlation was confirmed in all 12 cases of Wv/Wv ovaries analyzed for CK-8 and inhibin-alpha: where granulosa cells were present, tubular adenomas were absent. The ovaries from Wv/Wv; Trp53 (−/−) mice in which oocytes and follicles were preserved also had an increased presence of granulosa cells (Figure 7E) and suppression of epithelial tubular adenomas (Figure 7F).
Figure 7.
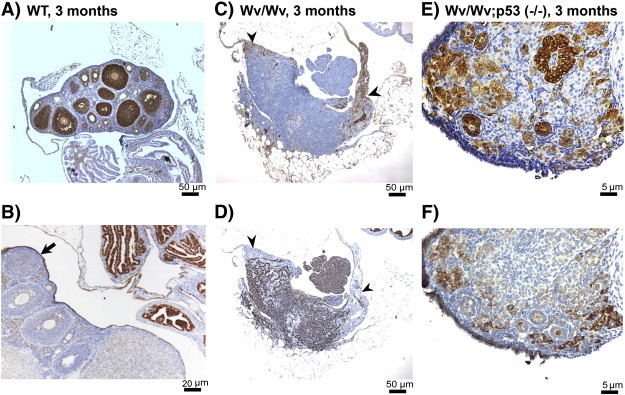
Inverse correlation between the presence of granulosa cells and epithelial lesions in Wv/Wv ovaries. Representative examples of tissue sections stained with the granulosa cell marker inhibin-alpha (A, C, E) and epithelial marker CK8 (B, D, F) in adjacent sections of ovarian tissues from 3-month-old wild-type (A, B), Wv/Wv (C, D), and Wv/Wv; Trp53 (−/−) (E, F) mice. An arrow indicates the single layer of CK8-positive ovarian surface epithelial cells (B). To illustrate the inverse correlation between the presence of granulosa cells and epithelial cells, arrowheads indicate that, in an area which is positive for inhibin-alpha (C), CK8 staining is absent in the adjacent section (D).
Thus, the results suggest that ovarian epithelial cells and granulosa cells are mutually exclusive.
Discussion
Although the p53 gene is the most prominent tumor suppressor, we found in the specific situation of the ovarian tubular adenomas from the Wv mice that deletion of Trp53 actually suppressed the tumor phenotype. The germ cell–deficient Wv/Wv mice develop a benign epithelial ovarian tumor, and the initial goal of our study was to convert the benign tumors into malignant tumors by adding an oncogenic mutation. Instead, we observed that deletion of Trp53 suppressed the ovarian tumor phenotype in the Wv/Wv mice. Although initially perplexed by the results, we observed and realized that the absence of p53 allows the prolonged survival of ovarian germ cells and oocytes. Subsequently, the presence of follicles is critical in maintaining the homeostasis of ovarian surface and rete ovarii epithelia and is essential in preserving the structural arrangement of the multiple cell types within an ovary (Figure 8).
Figure 8.
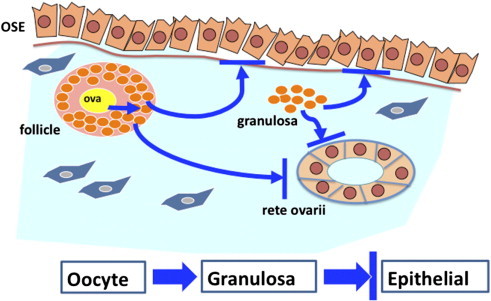
Proposed mechanism for the suppression of epithelial proliferation by follicles in ovaries.
A cartoon illustrates the interactions between cell types within the ovary in maintaining tissue homeostasis. In the premenopausal stage, oocytes (ova) stimulate the proliferation of granulosa cells and the maturation of the follicle structures, which suppress proliferation of both ovarian surface (OSE) and rete ovarii epithelial cells. Based on the inverse correlation, we also speculate that granulosa cells from the remnants of degenerated follicles can also suppress ovarian epithelial proliferation. The model suggests that, in the Wv/Wv ovaries, epithelial proliferation to form tubular adenomas results from the depletion of ovarian follicles and associated granulosa cells and consequently the loss of growth suppression imposed by a paracrine factor(s) produced by granulosa cells. Furthermore, Trp53 deletion rescues oocytes in the Wv/Wv ovaries. Consequently, the oocytes stimulate granulosa cell growth, and the increased presence of granulosa cells suppresses epithelial proliferation and the development of ovarian tubular adenomas.
This finding suggests that Trp53 is critical in controlling the survival and life span of ovarian germ cells and follicles in the Wv mice and that the depletion of follicles is a key causal factor for the ovarian tubular tumor phenotype. Trp53 is known to regulate prenatal oogenesis and oocyte attrition [16] and acts in chemical-induced follicle loss [45]. Trp53 deletion in the Wv/Wv mice presumably reduces oocyte apoptosis and prolongs follicle survival, and oocytes then recruit and stimulate granulosa cells. Alternatively, Trp53 is also directly important for controlling the growth and survival of granulosa cells [46]. The impact of Trp53 activity on life span of ovarian follicles observed in the current study adds to the recognized roles of p53 and its family members in female reproduction [15], [16], [17], [47].
The tumor suppressor function of Trp53 is generally executed through its cell autonomous regulation of gene expression of a large collection of effectors in both cell growth arrest and programmed cell death [48], and the tumor suppressor function and mechanisms have been well demonstrated in mouse models [49]. In the current study, as observed in culture, Trp53 deletion enhances proliferation of ovarian surface epithelial cells, which is in agreement with previous reports [36], [44]. However, when globally absent in tissues, Trp53 deletion seems to suppress proliferation of ovarian epithelial cells and reverts the tumor phenotype in the Wv mice. We attribute the observation to a non–cell autonomous effect, that the presence of ovarian follicles due to preservation following the loss of Trp53 in oocytes then suppresses growth of the epithelial cells, despite the simultaneous loss of Trp53 in the epithelial compartment. Indeed, in a preliminary study, we also found that restricted Trp53 deletion in the ovarian surface epithelial cells can greatly enhance tumor growth in the Wv/Wv mice. These findings will be completed and reported in a separate manuscript.
Pten is known to have an active role in controlling ovarian oocyte and follicle activation [19], [20], which may explain our finding that a reduction of Pten gene dosage also lessened ovarian follicle depletion and tumor development in the Wv/Wv; Pten (+/−) mice. Pten deletion in oocytes, or elimination of the downstream transcription factor Foxo3a, also causes stage-specific follicular activation and subsequent follicle depletion; however, no ovarian tubular adenomas have been reported [19], [20], [50], [51]. One possibility is that there are residual follicles in the Foxo3a and conditional pten mutant mice compared with the early and complete deletion in the Wv mice. Additionally, another possibility is that residual granulosa cells may be present following the degeneration of the large number of prematurely activated follicles, and the granulosa cells prevent epithelial proliferation to form tubular adenomas in the Foxo3a or pten mutant mice.
The idea that an intact ovarian tissue structure is required for cell homeostasis has been previously proposed [24], [52]. One recognized mechanism for the presence of ovarian follicles in suppressing ovarian epithelial growth is the regulatory cycle of the ovarian-pituitary axis [53]. Hormones, activin, and inhibin produced from oocyte-associated granulosa cells suppress gonadotropins released from the pituitary [53]. Upon follicle depletion, the increased gonadotropins then stimulate epithelial proliferation, as proposed in the gonadotropin stimulation hypothesis [54], [55], [56], [57], [58]. However, transgenic mice overexpressing follicle-stimulating hormone and luteinizing hormone do not develop ovarian epithelial tumors [59], [60], and systemic gonadotropin stimulation is not sufficient. The inverse relationship between the inhibin-alpha–positive granulosa and the CK8-positive epithelial cells in the same tissues, observed in the current study, suggests that granulosa cells may be part of a negative paracrine signal to suppress ovarian epithelial proliferation. A mutually exclusive expression of inhibin-alpha and cytokeratin has been observed previously in mouse ovarian tumors induced by SV40 T-antigen (FvB/n-Tg(Amhr2-SV40TAg)1Bcv) [61]. Thus, the suppressive activity of granulosa cells, within either intact follicles or clusters from follicle remnants following oocyte degeneration, may be common and may not be limited to tubular adenomas in the Wv/Wv ovaries. Possibly, follicle depletion (and thus granulosa cell degeneration) causes ovarian epithelial outgrowth by both increased systemic gonadotropin stimulation and the loss of a local suppressing signal from granulosa cells.
Another study also reported that additional deletion of Trp53 suppresses ovarian tumors in Pten-deficient and Kras mutant mice [49]. The authors attributed their unusual finding as a new paradigm, that wild-type Trp53 can promote survival and differentiation of the Pten (−/−); Kras mutant tumor cells [49], [62], [63]. From the images shown in the articles, it appears that Trp53 deletion also restored ovarian follicles in the ovaries. However, the studies did not report or discuss the analysis of ovarian follicle status or consider a non–cell autonomous mechanism of Trp53 action.
Based on these observations and analyses, we propose a model for the regulation of homeostasis of the ovarian cell types (Figure 8). It is established that oocytes provide stimulatory signal for proliferation of granulosa cells [9], [64], [65], [66]. We propose that granulosa cells, either associated with intact follicles or as remnants of degenerated follicles, produce a paracrine factor that suppress proliferation of epithelial cells either at the surface or in rete ovarii. The obvious possibility is that the granulosa cell factor may be inhibin-alpha; however, inhibin-alpha knockout does not stimulate epithelial proliferation in ovaries [67], [68]. Transforming growth factor–beta, an epithelial growth suppressor, may be a good candidate, and identification of such factor will be critical to verify the current model. The model suggests that, in the Wv/Wv ovaries, depletion of ovarian follicles leads to the reduced presence of granulosa cells and consequently the absence of the paracrine factor to restrain the growth of both surface and rete ovarii epithelia and the formation of tubular adenomas.
Our finding emphasizes the importance of ovarian follicles in suppressing the hyperplastic growth and transformation of ovarian epithelial cells, dominating over the cell autonomous activity due to loss of Trp53. Perhaps, the follicle depletion hypothesis [24], [69], comprising both the incessant ovulation hypothesis [70], [71] and the gonadotropin stimulation theory [54], [55], [56], [57], [58], is most suitable to explain the epidemiological observation that ovarian cancer risk is associated with menopause (the depletion of ovarian follicle reserve) following incessant ovulation and menopausal-increased gonadotropins when the follicle reserve is depleted.
Acknowledgments
The project was first started at Fox Chase Cancer Center (Philadelphia, PA) and was continued at the Sylvester Comprehensive Cancer Center, University of Miami Miller School of Medicine (Miami, FL). Over the years, several laboratory members contributed work related to this project, including Dr Robert Moore, Wensi Tao, Toni Yeasky, and Malgorzata Rula, and several summer work students. We appreciate the gift of a founder pair of Pten knockout mice from Dr Di Cristofano and p53 flox mice from Dr Connolly. We thank our colleagues and laboratory members, Robert Moore, Wensi Tao, and Santas Rosario, for reading, suggestions, and comments on the manuscript. Especially, the author (X) appreciates the many inspiring discussions with Dr Thomas Hamilton on the relationship between reproductive history and the etiology of ovarian cancer for the development of ideas and hypotheses.
Footnotes
The work was supported by R01 CA095071, CA79716, and CA75389 to X.X. Xu from the National Cancer Institute, National Institutes of Health, and also a grant from DOD (W81XWH-06-1-0095, Ovarian Cancer Research Program Idea Award, “A Mouse Model to Investigate Postmenopausal Biology as an Etiology of Ovarian Cancer Risk”, 11/01/2005-10/31/2008).
References
- 1.Mintz B. Embryological development of primordial germ-cells in the mouse: influence of a new mutation, Wj. J Embryol Exp Morphol. 1957;5:396–406. [Google Scholar]
- 2.Nocka K, Tan JC, Chiu E, Chu TY, Ray P, Traktman P, Besmer P. Molecular bases of dominant negative and loss of function mutations at the murine c-kit/white spotting locus: W37, Wv, W41 and W. EMBO J. 1990;9:1805–1813. doi: 10.1002/j.1460-2075.1990.tb08305.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Reith AD, Rottapel R, Giddens E, Brady C, Forrester L, Bernstein A. W mutant mice with mild or severe developmental defects contain distinct point mutations in the kinase domain of the c-kit receptor. Genes Dev. 1990;4:390–400. doi: 10.1101/gad.4.3.390. [DOI] [PubMed] [Google Scholar]
- 4.Dubreuil P, Rottapel R, Reith AD, Forrester L, Bernstein A. The mouse W/c-kit locus. A mammalian gene that controls the development of three distinct cell lineages. Ann N Y Acad Sci. 1990;599:58–65. doi: 10.1111/j.1749-6632.1990.tb42364.x. [DOI] [PubMed] [Google Scholar]
- 5.Yang WL, Cai KQ, Smedberg JL, Smith ER, Klein-Szanto A, Hamilton TC, Xu XX. A reduction of Cox-2 gene dosage counters the menopausal ovarian morphological aging and tumor phenotype in Wv mice. Am J Pathol. 2007;170:1325–1336. doi: 10.2353/ajpath.2007.060769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Murphy ED. Hyperplastic and early neoplastic changes in the ovaries of mice after genic deletion of germ cells. J Natl Cancer Inst. 1972;48:1283–1295. [PubMed] [Google Scholar]
- 7.Murphy ED, Beamer WG. Plasma gonadotropin levels during early stages of ovarian tumorigenesis in mice of the Wx -Wv genotype. Cancer Res. 1973;33:721–723. [PubMed] [Google Scholar]
- 8.Smith ER, Yeasky T, Wei JQ, Miki RA, Cai KQ, Smedberg JL, Yang WL, Xu XX. White spotting variant mouse as an experimental model for ovarian aging and menopausal biology. Menopause. 2012;19:588–596. doi: 10.1097/gme.0b013e318239cc53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li R, Albertini DF. The road to maturation: somatic cell interaction and self-organization of the mammalian oocyte. Nat Rev Mol Cell Biol. 2013;14:141–152. doi: 10.1038/nrm3531. [DOI] [PubMed] [Google Scholar]
- 10.Zuckerman S. The number of oocytes in the mature ovary. Recent Prog Horm Res. 1951;6:63–109. [Google Scholar]
- 11.Johnson J, Skaznik-Wikiel M, Lee HJ, Niikura Y, Tilly JC, Tilly JL. Setting the record straight on data supporting postnatal oogenesis in female mammals. Cell Cycle. 2005;4:1471–1477. doi: 10.4161/cc.4.11.2186. [DOI] [PubMed] [Google Scholar]
- 12.Telfer EE, Gosden RG, Byskov AG, Spears N, Albertini D, Andersen CY, Anderson R, Braw-Tal R, Clarke H, Gougeon A. On regenerating the ovary and generating controversy. Cell. 2005;122:821–822. doi: 10.1016/j.cell.2005.09.004. [DOI] [PubMed] [Google Scholar]
- 13.Gosden RG. Biology of Menopause: The Causes and Consequences of Ovarian Aging. In: Gosden RG, editor. Academic Press; 1985. [Google Scholar]
- 14.Lobo RA, Kelsey J, Marcus R, editors. 1st ed. Academic Press; 2000. Menopause: Biology and Pathobiology. [Google Scholar]
- 15.Levine AJ, Tomasini R, McKeon FD, Mak TW, Melino G. The p53 family: guardians of maternal reproduction. Nat Rev Mol Cell Biol. 2011;12:259–265. doi: 10.1038/nrm3086. [DOI] [PubMed] [Google Scholar]
- 16.Ghafari F, Pelengaris S, Walters E, Hartshorne GM. Influence of p53 and genetic background on prenatal oogenesis and oocyte attrition in mice. Hum Reprod. 2009;24:1460–1472. doi: 10.1093/humrep/dep022. [DOI] [PubMed] [Google Scholar]
- 17.Yaron Y, Schwartz D, Evans MI, Aloni R, Kapon A, Rotter V. p53 tumor suppressor gene expression in the mouse ovary during an artificially induced ovulatory cycle. J Reprod Med. 1999;44:107–114. [PubMed] [Google Scholar]
- 18.Hu W, Feng Z, Atwal GS, Levine AJ. p53: a new player in reproduction. Cell Cycle. 2008;7:848–852. doi: 10.4161/cc.7.7.5658. [DOI] [PubMed] [Google Scholar]
- 19.Reddy P, Liu L, Adhikari D, Jagarlamudi K, Rajareddy S, Shen Y, Du C, Tang W, Hämäläinen T, Peng SL. Oocyte-specific deletion of Pten causes premature activation of the primordial follicle pool. Science. 2008;319:611–613. doi: 10.1126/science.1152257. [DOI] [PubMed] [Google Scholar]
- 20.Marx J. Developmental biology. Aging of the ovary linked to PTEN pathway. Science. 2008;319:558–559. doi: 10.1126/science.319.5863.558b. [DOI] [PubMed] [Google Scholar]
- 21.Finn CA. Reproductive ageing and the menopause. Int J Dev Biol. 2001;45:613–617. [PubMed] [Google Scholar]
- 22.Dushkin H, Schilder RJ. Imatinib mesylate and its potential implications for gynecologic cancers. Curr Treat Options Oncol. 2005;6:115–120. doi: 10.1007/s11864-005-0019-9. [DOI] [PubMed] [Google Scholar]
- 23.Miettinen M, Lasota J. KIT (CD117): a review on expression in normal and neoplastic tissues, and mutations and their clinicopathologic correlation. Appl Immunohistochem Mol Morphol. 2005;13:205–220. doi: 10.1097/01.pai.0000173054.83414.22. [DOI] [PubMed] [Google Scholar]
- 24.Smith ER, Xu XX. Ovarian aging, follicle depletion, and cancer: a hypothesis for the etiology of epithelial ovarian cancer concerning follicle depletion. Lancet Oncol. 2008;9:1108–1111. doi: 10.1016/S1470-2045(08)70281-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Smith ER, Yang WL, Yeasky T, Smedberg J, Cai KQ, Xu XX. Cyclooxygenase-1 inhibition prolongs postnatal ovarian follicle lifespan in mice. Biol Reprod. 2013;89:1–8. doi: 10.1095/biolreprod.113.111070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nicosia SV. The aging ovary. Med Clin North Am. 1987;71:1–9. [PubMed] [Google Scholar]
- 27.Cai KQ, Klein-Szanto A, Karthik D, Edelson M, Daly MB, Ozols RF, Lynch HT, Godwin AK, Xu XX. Age-dependent morphological alterations of human ovaries from populations with and without BRCA mutations. Gynecol Oncol. 2006;103:719–728. doi: 10.1016/j.ygyno.2006.03.053. [DOI] [PubMed] [Google Scholar]
- 28.Salazar H, Godwin AK, Daly MB, Laub PB, Hogan WM, Rosenblum N, Boente MP, Lynch HT, Hamilton TC. Microscopic benign and invasive malignant neoplasms and a cancer-prone phenotype in prophylactic oophorectomies. J Natl Cancer Inst. 1996;88:1810–1820. doi: 10.1093/jnci/88.24.1810. [DOI] [PubMed] [Google Scholar]
- 29.Shih IeM, Kurman RJ. Ovarian tumorigenesis: a proposed model based on morphological and molecular genetic analysis. Am J Pathol. 2004;164:1511–1518. doi: 10.1016/s0002-9440(10)63708-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Salani R, Kurman RJ, Giuntoli R, II, Gardner G, Bristow R, Wang TL, Shih IM. Assessment of TP53 mutation using purified tissue samples of ovarian serous carcinomas reveals a higher mutation rate than previously reported and does not correlate with drug resistance. Int J Gynecol Cancer. 2008;18:487–491. doi: 10.1111/j.1525-1438.2007.01039.x. [DOI] [PubMed] [Google Scholar]
- 31.Ahmed AA, Etemadmoghadam D, Temple J, Lynch AG, Riad M, Sharma R, Stewart C, Fereday S, Caldas C, Defazio A. Driver mutations in TP53 are ubiquitous in high grade serous carcinoma of the ovary. J Pathol. 2010;221:49–56. doi: 10.1002/path.2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cancer Genome Atlas Research Network Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474:609–615. doi: 10.1038/nature10166. [Erratum in: Nature 2012; 490, 298] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sah VP, Attardi LD, Mulligan GJ, Williams BO, Bronson RT, Jacks T. A subset of p53-deficient embryos exhibit exencephaly. Nat Genet. 1995;10:175–180. doi: 10.1038/ng0695-175. [DOI] [PubMed] [Google Scholar]
- 34.Jacks T, Remington L, Williams BO, Schmitt EM, Halachmi S, Bronson RT, Weinberg RA. Tumor spectrum analysis in p53-mutant mice. Curr Biol. 1994;4:1–7. doi: 10.1016/s0960-9822(00)00002-6. [DOI] [PubMed] [Google Scholar]
- 35.Jonkers J, Meuwissen R, van der Gulden H, Peterse H, van der Valk M, Berns A. Synergistic tumor suppressor activity of BRCA2 and p53 in a conditional mouse model for breast cancer. Nat Genet. 2001;29:418–425. doi: 10.1038/ng747. [DOI] [PubMed] [Google Scholar]
- 36.Jonkers J, Berns A. Conditional mouse models of sporadic cancer. Nat Rev Cancer. 2002;2:251–265. doi: 10.1038/nrc777. [DOI] [PubMed] [Google Scholar]
- 37.Di Cristofano A, Pesce B, Cordon-Cardo C, Pandolfi PP. Pten is essential for embryonic development and tumour suppression. Nat Genet. 1998;19:348–355. doi: 10.1038/1235. [DOI] [PubMed] [Google Scholar]
- 38.Clark-Knowles KV, Garson K, Jonkers J, Vanderhyden BC. Conditional inactivation of Brca1 in the mouse ovarian surface epithelium results in an increase in preneoplastic changes. Exp Cell Res. 2007;313:133–145. doi: 10.1016/j.yexcr.2006.09.026. [DOI] [PubMed] [Google Scholar]
- 39.Flesken-Nikitin A, Choi KC, Eng JP, Shmidt EN, Nikitin AY. Induction of carcinogenesis by concurrent inactivation of p53 and Rb1 in the mouse ovarian surface epithelium. Cancer Res. 2003;63:3459–3463. [PubMed] [Google Scholar]
- 40.Alexander BM, Van Kirk EA, Naughton LM, Murdoch WJ. Ovarian morphometrics in TP53-deficient mice. Anat Rec (Hoboken) 2007;290:59–64. doi: 10.1002/ar.20409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mills GB, Lu Y, Fang X, Wang H, Eder A, Mao M, Swaby R, Cheng KW, Stokoe D, Siminovitch K. The role of genetic abnormalities of PTEN and the phosphatidylinositol 3-kinase pathway in breast and ovarian tumorigenesis, prognosis, and therapy. Semin Oncol. 2001;28:125–141. doi: 10.1016/s0093-7754(01)90290-8. [DOI] [PubMed] [Google Scholar]
- 42.Suzuki A, de la Pompa JL, Stambolic V, Elia AJ, Sasaki T, del Barco Barrantes I, Ho A, Wakeham A, Itie A, Khoo W. High cancer susceptibility and embryonic lethality associated with mutation of the PTEN tumor suppressor gene in mice. Curr Biol. 1998;8:1169–1178. doi: 10.1016/s0960-9822(07)00488-5. [DOI] [PubMed] [Google Scholar]
- 43.Podsypanina K, Ellenson LH, Nemes A, Gu J, Tamura M, Yamada KM, Cordon-Cardo C, Catoretti G, Fisher PE, Parsons R. Mutation of Pten/Mmac1 in mice causes neoplasia in multiple organ systems. Proc Natl Acad Sci U S A. 1999;96:1563–1568. doi: 10.1073/pnas.96.4.1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mullany LK, Liu Z, King ER, Wong KK, Richards JS. Wild-type tumor repressor protein 53 (Trp53) promotes ovarian cancer cell survival. Endocrinology. 2012;153:1638–1648. doi: 10.1210/en.2011-2131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pru JK, Kaneko-Tarui T, Jurisicova A, Kashiwagi A, Selesniemi K, Tilly JL. Induction of proapoptotic gene expression and recruitment of p53 herald ovarian follicle loss caused by polycyclic aromatic hydrocarbons. Reprod Sci. 2009;16:347–356. doi: 10.1177/1933719108327596. [DOI] [PubMed] [Google Scholar]
- 46.Sirotkin AV, Benco A, Tandlmajerova A, Vasícek D, Kotwica J, Darlak K, Valenzuela F. Transcription factor p53 can regulate proliferation, apoptosis and secretory activity of luteinizing porcine ovarian granulosa cell cultured with and without ghrelin and FSH. Reproduction. 2008;136:611–618. doi: 10.1530/REP-08-0229. [DOI] [PubMed] [Google Scholar]
- 47.Feng Z, Zhang C, Kang HJ, Sun Y, Wang H, Naqvi A, Frank AK, Rosenwaks Z, Murphy ME, Levine AJ. Regulation of female reproduction by p53 and its family members. FASEB J. 2011;25:2245–2255. doi: 10.1096/fj.10-180166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pietsch EC, Sykes SM, McMahon SB, Murphy ME. The p53 family and programmed cell death. Oncogene. 2008;27:6507–6521. doi: 10.1038/onc.2008.315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lozano G, Zambetti GP. What have animal models taught us about the p53 pathway? J Pathol. 2005;205:206–220. doi: 10.1002/path.1704. [DOI] [PubMed] [Google Scholar]
- 50.Castrillon DH, Miao L, Kollipara R, Horner JW, DePinho RA. Suppression of ovarian follicle activation in mice by the transcription factor Foxo3a. Science. 2003;301:215–218. doi: 10.1126/science.1086336. [DOI] [PubMed] [Google Scholar]
- 51.Jagarlamudi K, Liu L, Adhikari D, Reddy P, Idahl A, Ottander U, Lundin E, Liu K. Oocyte-specific deletion of Pten in mice reveals a stage-specific function of PTEN/PI3K signaling in oocytes in controlling follicular activation. PLoS One. 2009;4:e6186. doi: 10.1371/journal.pone.0006186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Vanderhyden BC. Loss of ovarian function and the risk of ovarian cancer. Cell Tissue Res. 2005;322:117–124. doi: 10.1007/s00441-005-1100-1. [DOI] [PubMed] [Google Scholar]
- 53.Gregory SJ, Kaiser UB. Regulation of gonadotropins by inhibin and activin. Semin Reprod Med. 2004;22:253–267. doi: 10.1055/s-2004-831901. [DOI] [PubMed] [Google Scholar]
- 54.Cramer DW, Welch WR. Determinants of ovarian cancer risk. II. Inferences regarding pathogenesis. J Natl Cancer Inst. 1983;71:717–721. [PubMed] [Google Scholar]
- 55.Mohle J, Whittemore A, Pike M, Darby S. Gonadotrophins and ovarian cancer risk. J Natl Cancer Inst. 1985;75:178–180. [PubMed] [Google Scholar]
- 56.Lukanova A, Kaaks R. Endogenous hormones and ovarian cancer: epidemiology and current hypotheses. Cancer Epidemiol Biomarkers Prev. 2005;14:98–107. [PubMed] [Google Scholar]
- 57.Smith ER, Xu XX. Etiology of epithelial ovarian cancer: a cellular mechanism for the role of gonadotropins. Gynecol Oncol. 2003;91:1–2. doi: 10.1016/s0090-8258(03)00463-3. [DOI] [PubMed] [Google Scholar]
- 58.Choi JH, Wong AS, Huang HF, Leung PC. Gonadotropins and ovarian cancer. Endocr Rev. 2007;28:440–461. doi: 10.1210/er.2006-0036. [DOI] [PubMed] [Google Scholar]
- 59.Rulli SB, Huhtaniemi I. What have gonadotrophin overexpressing transgenic mice taught us about gonadal function? Reproduction. 2005;130:283–291. doi: 10.1530/rep.1.00661. [DOI] [PubMed] [Google Scholar]
- 60.Huhtaniemi I. Are gonadotrophins tumorigenic—a critical review of clinical and experimental data. Mol Cell Endocrinol. 2010;329:56–61. doi: 10.1016/j.mce.2010.04.028. [DOI] [PubMed] [Google Scholar]
- 61.Garson K, Gamwell LF, Pitre EM, Vanderhyden BC. Technical challenges and limitations of current mouse models of ovarian cancer. J Ovarian Res. 2012;5:39. doi: 10.1186/1757-2215-5-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mullany LK, Liu Z, Wong KK, Deneke V, Ren YA, Herron A, Richards JS. Tumor repressor protein 53 and steroid hormones provide a new paradigm for ovarian cancer metastases. Mol Endocrinol. 2014;28:127–137. doi: 10.1210/me.2013-1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mullany LK, Fan HY, Liu Z, White LD, Marshall A, Gunaratne P, Anderson ML, Creighton CJ, Xin L, Deavers M. Molecular and functional characteristics of ovarian surface epithelial cells transformed by KrasG12D and loss of Pten in a mouse model in vivo. Oncogene. 2011;30:3522–3536. doi: 10.1038/onc.2011.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Emori C, Sugiura K. Role of oocyte-derived paracrine factors in follicular development. Anim Sci J. 2014;85:627–633. doi: 10.1111/asj.12200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gilchrist RB, Ritter LJ, Armstrong DT. Oocyte-somatic cell interactions during follicle development in mammals. Anim Reprod Sci. 2004;82–83:431–446. doi: 10.1016/j.anireprosci.2004.05.017. [DOI] [PubMed] [Google Scholar]
- 66.Eppig JJ. Oocyte control of ovarian follicular development and function in mammals. Reproduction. 2001;122:829–838. doi: 10.1530/rep.0.1220829. [DOI] [PubMed] [Google Scholar]
- 67.Matzuk MM, Finegold MJ, Su JG, Hsueh AJ, Bradley A. Alpha-inhibin is a tumour-suppressor gene with gonadal specificity in mice. Nature. 1992;360:313–319. doi: 10.1038/360313a0. [DOI] [PubMed] [Google Scholar]
- 68.Myers M, Middlebrook BS, Matzuk MM, Pangas SA. Loss of inhibin alpha uncouples oocyte-granulosa cell dynamics and disrupts postnatal folliculogenesis. Dev Biol. 2009;334:458–467. doi: 10.1016/j.ydbio.2009.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Smith ER, Wang Y, Xu XX. Development of a mouse model of menopausal ovarian cancer. Front Oncol. 2014;4:36. doi: 10.3389/fonc.2014.00036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Fathalla MF. Incessant ovulation—a factor in ovarian neoplasia? Lancet. 1971;2:163. doi: 10.1016/s0140-6736(71)92335-x. [DOI] [PubMed] [Google Scholar]
- 71.Godwin AK, Testa JR, Handel LM, Liu Z, Vanderveer LA, Tracey PA, Hamilton TC. Spontaneous transformation of rat ovarian surface epithelial cells: association with cytogenetic changes and implications of repeated ovulation in the etiology of ovarian cancer. J Natl Cancer Inst. 1992;84:592–601. doi: 10.1093/jnci/84.8.592. [DOI] [PubMed] [Google Scholar]