

Abstract
Tumor microenvironments (TMEs) are composed of cancer cells, fibroblasts, extracellular matrix, microvessels, and endothelial cells. Two prolyl endopeptidases, fibroblast activation protein (FAP) and prolyl oligopeptidase (POP), are commonly overexpressed by epithelial-derived malignancies, with the specificity of FAP expression by cancer stromal fibroblasts suggesting FAP as a possible therapeutic target. Despite overexpression in most cancers and having a role in angiogenesis, inhibition of POP activity has received little attention as an approach to quench tumor growth. We developed two specific and highly effective pseudopeptide inhibitors, M83, which inhibits FAP and POP proteinase activities, and J94, which inhibits only POP. Both suppressed human colon cancer xenograft growth > 90% in mice. By immunohistochemical stains, M83- and J94-treated tumors had fewer microvessels, and apoptotic areas were apparent in both. In response to M83, but not J94, disordered collagen accumulations were observed. Neither M83- nor J94-treated mice manifested changes in behavior, weight, or gastrointestinal function. Tumor growth suppression was more extensive than noted with recently reported efforts by others to inhibit FAP proteinase function or reduce FAP expression. Diminished angiogenesis and the accompanying profound reduction in tumor growth suggest that inhibition of either FAP or POP may offer new therapeutic approaches that directly target TMEs.
Abbreviations: FAP, fibroblast activation protein; POP, prolyl oligopeptidase; TME, tumor microenvironment; DPPIV, dipeptidyl peptidase IV; ECM, extracellular matrix; IHC, immunohistochemistry; CAFs, cancer-associated fibroblasts; MMPs, matrix metalloproteinases
Introduction
Tumor microenvironments (TMEs) as small as 1 to 2 mm3 contain parenchymal-derived cancer cells within stroma that is composed of activated fibroblasts, developing microvasculature, and extracellular matrix (ECM); stroma may account for ~ 90% of tumor weight [1], [2]. Fibroblast activation protein (FAP), a type II integral membrane protein and prolyl-specific serine proteinase, is overexpressed on cell membranes of fibroblasts in more than 90% of epithelial cell–derived malignancies, i.e., lung, breast, colon, and so on [3], [4]. FAP is rarely found on adult normal tissues and is essentially absent on benign tumors, features that make it an attractive diagnostic and therapeutic target [5], [6], [7], [8], [9], [10]. It is believed that 1) FAP engages in proteolysis of ECM during tissue invasion [11], [12], [13], [14], [15], 2) FAP-expressing cells appear to foster immune tolerance within TME [16], [17], [18], [19], [20], [21], [22], and 3) FAP/FAP-expressing cells support angiogenesis [9], [10], [23], [24], [25], [26]. Efforts to limit FAP activities that might enhance tumor growth have focused on inhibiting its proteolytic properties [5], [24], [27] or blocking putative FAP+ cell–induced immunotolerance of growing cancer [16], [17], [20], [21], [22].
Commanding less attention has been another prolyl oligopeptidase (POP) normally found in many tissues but commonly overexpressed along with the ubiquitous protein thymosin β4 in a number of malignancies [28], [29], [30], [31], [32], [33], [34], [35], [36]. Following partial cleavage of Tβ4 by an unknown enzyme, its degraded form is digested further by POP to yield the potent angiogenic peptide, Ac-SDKP [37], [38]. POP proteinase activity clearly has a role in angiogenesis [38], [39], [40], [41], but unlike FAP, it resides on cells throughout the tumor and not just on stroma. While inhibition of POP proteolytic activity is reported to arrest the growth of gastric cancer cells in culture [42], in vivo studies of POP inhibition in tumor models are lacking. The individual contribution of either POP or FAP to tumor expansion is difficult to decipher, given their overlapping proteolytic activities for cleaving Z-Gly-Pro-AMC, succinyl-Gly-Pro-AMC, and similar non-specific substrates; in addition, the lack of highly efficient aqueous soluble specific inhibitors of FAP or POP adds to the problem.
Despite lacking specificity, PT-100 (valyl-proline boronic acid; Val-boroPro) and PT-630 (glutamyl-proline boronic acid; Glu-boroPro) have been used to study the effects of FAP proteinase inhibition on cancer growth [24], [43], [44], [45], [46], [47]. Both PT-100 and PT-630, however, also inhibit dipeptidyl peptidase IV (DPPIV) and, to a lesser extent, POP in purified solution. Moreover, PT-100 and PT-630 both rapidly cyclize in physiologic media and lose inhibitory activity [48], [49]. Narra et al. [45] and Santos et al. [24] showed that PT-630 inhibited endogenous lung cancer growth in immunodeficient mice and in syngeneic colon cancer grafts in mice. In both studies, inhibition of FAP or DPPIV by PT-100 or PT-630 appeared to suppress tumor growth [24], [43], [50]. Huang et al. [51], [52] reported that human breast cancer cells transfected with proteolytically inactive recombinant FAP, or breast cancer cells transfected to express wild-type proteolytically active FAP that is inhibitable by PT-630, still formed rapidly growing breast tumors in severe combined immunodeficiency mice. As a consequence, they suggested that FAP proteolytic activity has little or no impact on cancer growth; however, since transfected cancer cells served as FAP+ cells instead of stromal fibroblasts as in human breast cancers, their model differed from established biology of such cancers [51].
In a mouse syngeneic 4T1 mammary carcinoma model, when short hairpin inhibitory RNA (shRNA) targeting FAP was injected intratumorally and peritumorally, FAP expression was knocked down by ~ 50%, tumor growth was reduced, angiogenesis was suppressed, collagen accumulation increased within the tumor, and tumor apoptosis was promoted; apparent side effects were not noted [53]. FAP gene silencing for 17 days did not induce paraneoplastic features such as cachexia, anemia, and lethal bone toxicities that were noted with tumor growth inhibition by immunologic depletion of FAP+ cells within TME [18], [19], [20]. Given the reduction in FAP protein, FAP proteinase activity should also have been significantly reduced. Interestingly, the FAP-knockdown results closely mirrored those yielded by studies in which FAP proteinase activity was inhibited [24], [45]. The sum of studies to date clearly indicates the need for more efficient and predictable FAP inhibition to determine whether simply inhibiting FAP proteolytic activity will circumvent FAP+ cell destruction and thereby avoid perturbing potential FAP+ cell functions that might cause adverse constitutional effects. Moreover, the suggested therapeutic potential for targeted POP inhibition to diminish angiogenesis and reduce tumor growth [40], [54] has not been explored as far as we are aware and deserves direct evaluation. To examine these issues, we designed and synthesized a more stable, specific, and soluble FAP and POP inhibitor that we termed M83 and a highly specific, soluble inhibitor of POP only that we designated as J94 [10], [49].
We used the primary structure surrounding the scissile bond of the only established physiologic substrate for FAP, namely, alpha2-antiplasmin, as a template for designing M83 [49], [55]; similarly, the scissile bond region of POP substrates was used to design J94 [49], [56]. Extensive characterization showed that both inhibitors possessed similar features, i.e., excellent aqueous solubility at neutral pH, low molecular weights [529 (M83) and 554 (J94)], absence of cyclization in aqueous solution, and retention of inhibitory function after prolonged exposure to human plasma. Both are charged and hydrophilic, thereby minimizing intracellular entry; moreover, both M83 and J94 have low nanomolar Ki values for inhibiting FAP or POP or only POP, respectively. J94 does not inhibit FAP and neither M83 nor J94 significantly inhibits DPPIV [49]. When on live cells characteristic of TME, the membrane-associated form of either enzyme can be rapidly and completely inhibited, suggesting easy accessibility to the active site [10]. We now report analyses of substantial growth suppression of human colon cancer xenografts by M83 or J94 in immunodeficient mice without apparent adverse effects.
Methods
Cell Culture
HCT116, a human colon cancer cell line, and H441, a human lung cancer cell line, were obtained from ATCC (Manassas, Virginia) and grown as monolayer cultures in RPMI or Dulbecco's modified Eagle's medium supplemented with 10% fetal calf serum. Cells were maintained in a humid 5% CO2 atmosphere at 37°C.
Drug Design and Synthesis
The FAP/POP dual inhibitor, M83, was synthesized in two steps as previously reported [49]. Briefly, a structure consisting of acetyl-Arg(Pbf)-peg-d-Ala, based on the sequence surrounding the P1-P1′ scissile bond in Met-α2AP, where peg is defined as 8-amino-3,6-dioxaoctanoic acid and Pbf represents NG-2,2,4,6,7-pentamethyldihydrobenzofuran-5-sulfonyl, was synthesized and purified. The second step of the synthesis was linking this protected peptide-like construct to l-boroPro pinanediol ester and subsequently purifying the protected product. Finally, the pinanediol and Pbf groups were chemically removed and M83 was purified by reversed-phase HPLC.
The POP specific inhibitor, J94, was prepared using the equivalent strategy as for M83. The design of the precursive molecule was based on the reported sequence N-terminal to the scissile bond of optimal peptide substrates determined for POP [56]. The protected tripeptide acetyl-Lys(Boc)-Leu-Arg(Pbf)-OH was synthesized and purified, where Boc represents N-tert-butyloxycarbonyl. The second step was identical to that for M83, where the precursor peptide was linked to l-boroPro pinanediol ester and then HPLC purified.
Animals
Five- to six-week-old male athymic nude Foxn1nu mice were purchased from Harlan Laboratories (Houston, Texas). Animals were allowed to acclimate for ~ 5 days before any procedures. All animal protocols used in this study were approved by the Institutional Animal Care and Use Committee of the University of Oklahoma Health Sciences Center.
Tumor Models
HCT116 colon cancer cells (2.5 × 106) or H441 lung cancer cells (2 × 106) were suspended in Matrigel (BD Biosciences, San Jose, California) and injected subcutaneously with a 28G needle on the dorsal side of each thigh of Foxn1nu mice. After 10 days, the mice were randomly assigned to treatment groups. For lung cancer xenografts, animals were treated with 50 μl of saline (vehicle control) through intraperitoneal (i.p.) injection daily in the lower left abdominal quadrant or with 26.5 μg of M83 in saline. For the first experiment with colon cancer xenografts, animals were treated with 50 μl of saline through i.p. injection daily, 50 μg of M83, or 50 μg of J94 in saline. For the second colon cancer xenograft study, the animals were treated with 50 μl of saline through i.p. injection daily, 50 μg of M83, or 100 μg of M83 in saline. Mouse weight and tumor growth were monitored about every 3 days. Tumor volume, indicative of tumor growth, was determined by measuring two perpendicular diameters with calipers and converting to cm3 using the following formula: volume = (width)2 × length/2. Activity, eating, and bowel function were monitored daily. Experiments were performed with six animals per treatment group for lung cancer, six animals per group for the first colon cancer experiment, and three animals per group for the second experiment. Mice were treated for 28 days, after which they were killed. Tumors and selected organs were harvested and placed in 5% buffered formalin or 4% paraformaldehyde or flash frozen in liquid nitrogen for future pathologic or biochemical analyses. FAP activity in control and M83-treated tumors was determined on membranes prepared according to the manufacturer’s instructions (Mem-Per Plus, Pierce) using the fluorescent substrate acetyl-Arg-AEEA-Gly-Pro-AMC [10]. Significance of tumor volume and FAP activity results were analyzed by two-sample t test using the statistical package contained within Origin 9.1 (P < .05 was considered significant).
Histologic and Immunohistochemical Analyses
Tumors and selected organs were harvested and paraffin embedded after fixation with 5% buffered formalin. Tissues were cut in 4-μm slices and stained with hematoxylin and eosin for histologic examination.
For immunohistochemistry (IHC), tissue samples were fixed by immersion in 4% paraformaldehyde in phosphate-buffered saline (PBS) overnight at 4°C, followed by several buffer washes. The samples were then cryoprotected with a solution of 15% sucrose in PBS and embedded in Optimal Cutting Temperature compound. Ten-micrometer cryosections were prepared using a Microm HM500 E microtome cryostat and washed with PBS at 20°C, and free aldehyde groups were quenched with 0.1 M glycine in PBS (15 minutes). The tissue samples were permeabilized with 0.01% saponin in PBS (PBS/SAP; 10 minutes at 20°C) and incubated in 3% BSA in PBS/SAP (1 hour at 20°C). Saponin was kept in all incubation buffers throughout the staining procedure to ensure a proper penetration of the antibodies.
The tissue sections were placed in mixtures of monoclonal antibodies (mAbs) and polyclonal IgGs for 1 hour at 20°C or overnight at 4°C. The antibodies were rabbit anti–FAP-1 IgG (10 μg/ml; Sigma, St. Louis, Missouri; SAB4500839), rat anti-mouse CD31 IgG (1.5 μg/ml; BD Pharmingen, San Jose, California; 550274), mouse anti–single-stranded DNA (anti-ssDNA) IgM (10 μg/ml; Millipore, Billerica, Massachusetts; MAB3299), and goat anti-POP/PREP IgG (5 μg/ml; R&D Systems, Minneapolis, Minnesotta; AF4308). Then, the samples were washed 3 × 10 minutes in PBS/SAP and incubated for 1 hour at 20°C with combinations of appropriate detection antibodies conjugated with fluorescein isothiocyanate or Cy3 diluted 1:100 in 1% BSA in PBS/SAP. Secondary antibodies used were donkey anti-rabbit Cy3 (Jackson ImmunoResearch, West Grove, Pennsylvania; 711-165-152), donkey anti-rat fluorescein isothiocyanate (Jackson ImmunoResearch; 712-095-153), donkey anti-goat Cy3 (Jackson ImmunoResearch; 705-165-147), and F(ab) donkey anti-mouse Cy3 (Jackson ImmunoResearch; 715-165-003). All secondary antibodies were used at 15 μg/ml. After washing as above, the sections were mounted between glass slides and coverslips using VECTASHIELD HardSet Mounting Medium (Vector Laboratories, Burlingame, CA) containing TO-PRO-3 iodine (1 μM; Molecular Probes, Eugene, OR) as a nuclear counterstain. In case of mouse mAbs, the tissue sections were incubated with unconjugated Fab fragment anti-mouse IgG (at 0.1 mg/ml) for 1 hour at room temperature to block the endogenous mouse IgG. As detection antibodies, F(ab) monomeric secondary antibodies were used.
As negative controls for polyclonal antibody staining, the primary antibodies were replaced with equivalent amounts of rabbit or sheep nonimmune serum. Monoclonal antibody against digoxigenin, a hapten antigen that occurs only in plants, was used as control for mAb staining. Specimens were examined by epifluorescence confocal imaging using a Nikon C1 confocal laser-scanning unit equipped with a three-laser launcher (488, 543, and 633 nm emission lines) installed on an Eclipse TE200-U inverted microscope (Nikon, Melville, NY). Images were taken with a × 20 plan achromat objective (NA 0.46). Image collection parameters (neutral density filters, pinhole, and detector gains) were kept constant during image acquisition.
For the collagen staining, tissue samples were fixed by immersion in 4% paraformaldehyde in PBS overnight at 4°C, followed by several buffer washes. The samples were then cryoprotected with a solution of 15% sucrose in PBS and embedded in Optimal Cutting Temperature compound. Ten cryosections were obtained using a Microm HM500 E microtome and then stained with 0.1% sirius red F3B (Sigma) in saturated picric acid (Sigma).
Apoptosis was quantified by integration of the relative area stained by the anti-ssDNA antibody compared to the total tissue area for randomly selected slides from twelve different tumors, four from each M83 and J94-treated mice and four from saline-treated mice. The areas were determined using ImageJ software [57].
Immunoblot Analysis
Tumors were flash frozen and stored in liquid nitrogen before being processed for immunoblot analysis. At the time of assay, tumors were pulverized in a mortar and pestle under liquid nitrogen, weighed, and solubilized in 2 × sodium dodecyl sulfate (SDS) sample buffer. Samples were electrophoresed under reducing conditions on 4% to 12% Bis-Tris SDS–polyacrylamide gel electrophoresis (PAGE) gels (Invitrogen, Grand Island, New York) and transferred to nitrocellulose for Western blot analysis. After blocking with 3% BSA/TBS-Tween, blots were incubated with 0.5 μg/ml rabbit anti-FAP (Sigma) or 0.2 μg/ml goat anti-POP (R&D Systems; #AF4308), all in 3% BSA/TBS-Tween, and then washed and incubated with either 1:105,000 goat anti-rabbit HRP (Thermo Fisher, Carlsbad, California; #31460) or 1:100,000 rabbit anti-goat HRP (R&D Systems; #HAF017). Blots were washed extensively and then developed with ECL-Plus (Thermo Fisher) and were visualized on RPI blue radiographic film (Amersham, Pittsburgh, Pennsylvania).
For protein identification of bands identified as FAP or POP by Western blot analysis, tumors were pulverized as above and then solubilized in immunoprecipitation buffer [1% Triton/150 mM NaCl/10 mM Tris, pH 7.5/1 mM EDTA/1 mM EGTA/0.5% NP-40/10% sucrose with cOmplete ULTRA protease inhibitor cocktail (Roche, Indianapolis, Indiana)]. Lysates were centrifuged to remove detergent-insoluble debris. For FAP, 20 μg/ml polyclonal sheep anti-FAP (R&D Systems), or for POP, 20 μg/ml polyclonal goat anti-POP (R&D Systems), was added and allowed to bind overnight at 4°C. Then, 25 μl of 75% slurry of Protein A/G agarose beads (Thermo Fisher; #20421) in TBS were added and incubated for 3 hours at 4°C. Protein A/G beads were spun down, washed three times with immunoprecipitation buffer, resuspended in loading buffer, and boiled for 5 minutes; the beads were removed by centrifugation and a portion of the supernatant was electrophoresed under reducing conditions on 4% to 12% Bis-Tris SDS-PAGE gels and then Coomassie stained. To confirm the presence of FAP or POP, the regions of each lane corresponding to the molecular weight of FAP or POP were excised and the proteins within each gel slice were reduced with tris[2-carboxyethyl]phosphine, then alkylated with iodoacetamide, and digested with trypsin as described by the In-Gel Tryptic Digestion Kit protocol (Thermo Fisher). Each trypsin digest sample was analyzed by HPLC–tandem mass spectrometry (MS/MS) on a nanoscale Dionex UltiMate 3000 HPLC equipped with an Acclaim PepMap C18 column (75 μm internal diameter × 15 cm length with 3-μm particles) connected to an AB-Sciex QSTAR Elite mass spectrometer. The peptide molecular weights and MS/MS fragment ion spectra observed for each peptide were used to query an Naitonal Center for Biotechnology Information (NCBI) comprehensive non-identical human protein database (February 2012) loaded on an in-house MASCOT database server (version 2.4).
Results
As previously published by us [49], [55], M83 and J94 were each assessed for the ability to inhibit activities of FAP and POP as produced by cultures of cells typically within the TME [10]. Figure 1 shows the molecular design of each inhibitor. For M83, the arginyl group has been shown by us to add binding specificity and aqueous solubility, while the acetyl moiety obviates cleavage by dipeptidases. The insertion of a polyethylene string likewise adds to solubility and increases the length of the inhibitor to position the arginyl for maximal binding; importantly, the additional length eliminates the risk of cyclization as occurs with PT-100 and PT-630 [48], [49]. The d-alanyl residue fulfills the Gly requirement of the -Gly-Pro- motif of the scissile bond–containing peptide in which the P2 group is small, neutral, and essentially unreactive when binding within the FAP active site [55] since the d-alanyl side chain projects into the aqueous environment and away from the restricted space of the S2 pocket. The structure of J94 inhibitor has acetyl attached to the N-terminal lysyl residue, again for protection from dipeptidases. Leucine serves as the requisite hydrophobic residue in P3 position, with positively charged residues in P2 and P4 to enhance binding in the POP active site [56], [58]; lysine in particular was selected for P4 since FAP showed essentially no tolerance for P4 -lysyl-containing substrates [55], thereby eliminating FAP inhibition by J94. The P1 -prolyl- is essential for conjugation of the boronic acid warhead that directly interacts with the active site serine of both FAP and POP. We previously showed that M83 has a Ki of 5.7 nM for FAP and 7.4 nM for POP, while J94 totally inhibits POP at 100 nM and does not inhibit FAP or DPPIV even at 10 μM, thus providing specificity and equivalent efficiency [49].
Figure 1.
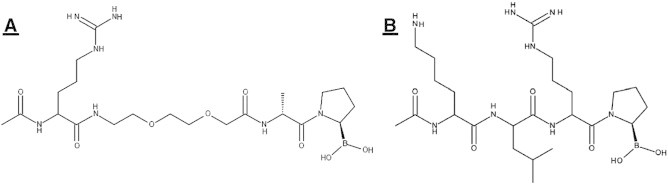
FAP and POP inhibitors. (A) M83, FAP, and POP inhibitor structure: acetyl-arginyl-8-amino-3,6-dioxaoctanoyl-d-alanyl-l-proline boronic acid (MW = 529.4). (B) J94, POP-specific inhibitor structure: acetyl-lysyl-leucyl-arginyl proline boronic acid (MW = 554.5).
Figure 2A shows that M83 inhibitor given as a daily 26.5 μg i.p. dose very effectively suppressed the growth of human lung cancer H441 xenografts in Foxn1nu mice, essentially halting growth of visible, palpable ~ 0.04 cm3 tumors; all mice, untreated or treated, remained alive, and none in the treated group showed signs of toxicity over the entire 28-day treatment period; tumors in the untreated group grew to an average volume of ~ 1.4 cm3 by 28 days at which time the mice were killed. We next assessed treatment with either M83 or J94 in mice with HCT116 human colon cancer xenografts. Each treated mouse was given M83 or J94 inhibitor as a daily i.p. dose of 50 μg when the tumors had grown to ~ 0.06 cm3. Tumors in untreated mice attained a volume of ~ 3 cm3 by the end of the 28-day period, and the mice were killed. As shown in Figure 2B, at treatment doses of 50 μg/day, the M83 and J94 inhibitors each gave about the same extent of tumor suppression over the 28-day treatment course. Although impressive, the tumor growth curve for either inhibitor still indicated continued expansion, and therefore, we repeated this experiment with M83 and compared daily i.p. doses of 50 μg versus 100 μg to see if the increased dose caused more tumor suppression. Indeed, it was evident that a greater limiting effect on tumor growth occurred at 100 μg of M83 per day, with one tumor becoming no longer palpable and another was barely evident; the growth of the remainder were all significantly reduced. Figure 3A shows that the individual tumor growth patterns for the 12 HCT116 human colon cancer xenografts were remarkably similar for all six untreated mice, with each growth curve showing two major inflections of accelerated growth. In control mice, the first came about 7 days after beginning saline i.p. injections, while the second growth burst occurred about day 18; only 1 of the 12 control tumors showed a modest decline in tumor volume, which occurred around day 24. By day 28, all but one tumor had achieved a maximum volume that ranged from 2.2 to 4.9 cm3. Conversely, the 12 treated tumors (Figure 3B) gave curves showing very substantial growth suppression, so that after 28 days, residual tumor volumes ranged between 0.25 and 1.6 cm3 (~ 67-89% tumor volume reduction relative to untreated tumors). At 20 days, most of the treated tumors resumed slow growth, but between days 24 and 28, about half again began showing significant declines in tumor volume that lasted through 28 days of treatment.
Figure 2.
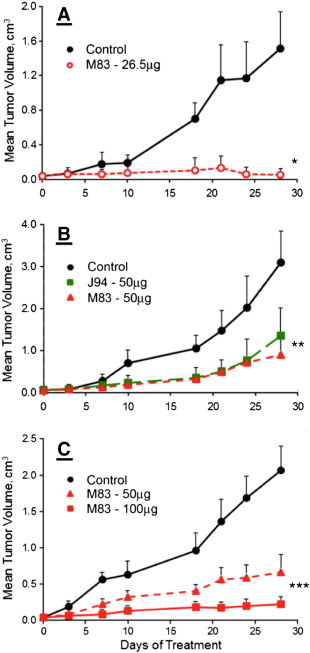
Growth of human xenografts in Foxn1nu mice. Treatment was initiated as i.p. drug injections when tumors reached ~ 0.060 cm3 in size. Tumor volumes were measured over the course of 28 days of treatment. (A) Human H441 lung cancer cell xenograft growth. Treatment consisted of 50-μl saline injections (●) or M83, 26.5 μg in 50 μl of saline (○) per day. *The M83-treated group is statistically different than the saline treatment for days 10 to 28 (P < .001). (B) Human colon cancer HCT116 xenograft growth. Mice were treated with 50 μl of saline (●) or M83 (▲), or J94 (■), 50 μg in 50 μl of saline per day. **Both the M83 and J94 treatment groups are statistically different than the saline-treated group for days 10 to 28 (P < .001). (C) Human colon cancer HCT116 xenograft growth. Mice were treated with 50 μl of saline (●), M83, 50 μg in 50 μl of saline (▲), or M83, 100 μg in 50 μl of saline (■). ***Both M83 treatment groups, 50 μg and 100 μg, are statistically different than the saline group from days 18 to 28 (P < .02 and P < .003).
Figure 3.
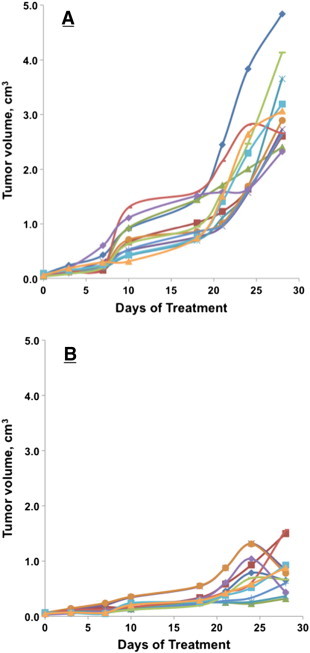
Human HCT116 colon cancer xenograft growth in Foxn1nu mice. The growth of individual tumors was plotted over the course of the 28-day treatment period. Treatment was initiated as i.p. drug injections when tumors reached ~ 0.060 cm3 in size. (A) Tumor growth in control mice treated with daily 50-μl saline injection. (B) Tumor growth curves for mice treated with M83, 50 μg in 50 μl of saline daily.
Figure 4A to C show the xenograft implantation sites in mice given daily i.p. injections of saline control, 50 μg of M83, and 100 μg of M83, respectively, for 28 days. By the time untreated control mice were killed at 28 days, the skin overlying untreated tumors was inflamed (see Figure 4A) and the tumors were clearly beginning to interfere with mobility. Figure 4B shows a typical response of a xenograft tumor on a mouse treated with a daily dose of 50 μg of M83 for 28 days; the marked, progressive reduction in growth of such tumors is clearly evident in Figure 2C. At the end of the treatment period, these tumors usually showed obviously cratered, necrotic centers (6 of 12) with edges that were neither inflamed nor sensitive to moderate pressure. In one mouse receiving 100 μg/day M83 (Figure 4C), one tumor showed complete growth suppression and disappeared entirely, while the residual of another tumor on the same mouse was barely detectable at 28 days. Tumors from untreated mice were excised and flash frozen, after which FAP and POP were each immunoprecipitated from tumor homogenate. Western immunoblot analysis of each immunoprecipitate was performed and protein band identities were confirmed by amino acid sequence determinations. Our sequence results in Figure 5A and B directly confirm that mouse and human FAPs are highly conserved and closely homologous. Since the amino acid sequences of human and mouse FAPs are 93% identical, the inhibitory efficiency we previously established for M83 toward human FAP should be closely similar, if not identical, for mouse FAP; the inhibitory properties of J94 toward POP produced by human colon cancer xenografts should be about the same as what we have shown in purified solutions [49] and tissue culture [10]. Identifications of tryptic peptides of FAP derived from the HCT116 colon cancer indicate that the xenograft stroma is clearly derived from the host, since no peptides unique to human FAP were observed. The homology observed for POP between human and mouse forms, although very significant, was not at the level of that for FAP. Amino acid sequence determination on tryptic peptides of POP from extracts of excised HCT116 xenografts established that tumor-associated POP originated from human cancer cells. Interestingly, by immunoblot analysis (Figure 5C and D), FAP and POP were each present in about the same amount per weight of tumor from both untreated and M83-treated mice, which raises skepticism about whether the mere presence of FAP protein causes immune tolerance in an already immunodeficient mouse and allows further growth of HCT116 tumors, particularly since host-derived stromal fibroblasts growing into the xenograft tumor could easily produce proteolytically active FAP. Our results show that tumor growth is suppressed in response to inhibition of FAP or POP proteolytic activity, thus intimating that these two proteinase functions are critical for tumor growth. With respect to POP, thymosin β4 peptides seem likely substrates, given the apparent role of their derivatives in angiogenesis [29], [30], [31], [32], [33], [34], [35], [36], [37], [38], [39], [40], [41]. Left open, however, is what substrate(s) might be involved, especially for cleavage by FAP, and this prompts conjectures about whether FAP cleavage of minimally degraded or denatured ECM proteins might yield peptides with unique biologic properties that might participate in pro-tumorigenic cell signaling, or whether FAP proteolytic function might be critical in pathways that promote immune tolerance of tumor growth, but if inhibited, immune intolerance is restored and growth becomes diminished. In the present work, tumors from five control mice and five M83-treated mice were analyzed for FAP proteolytic activity. In the M83-treated animals, FAP activity per tissue weight was about 30% decreased when compared with values from tumors in untreated animals (P = .05). Clearly, in the absence of pharmacodynamic/pharmacokinetic studies, and with no defined inhibitor “on/off” values, the implication of reduced, but not absent, FAP activity is difficult to interpret. Others have made similar observations [24], [27], the sum of which may suggest that reduced FAP proteolytic activity equal to or beyond a critical threshold is sufficient to diminish tumor growth by either lessening availability of ECM scaffolding or by reestablishing immune intolerance somehow.
Figure 4.
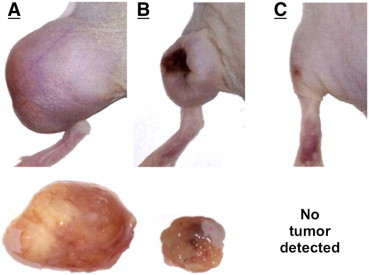
Images of left hindquarters of Foxn1nu mice with HCT116 colon cancer xenografts after 28 days of treatment with daily i.p. injections of (A) saline, (B) 50 μg of M83, or (C) 100 μg of M83. The excised tumors are shown below panels A and B. The saline-treated mouse had a tumor weight of 1.95 g, while the mouse treated with 50 μg of M83 had a tumor weight of 0.26 g. Of the six tumors in the 100 μg M83 treatment group, one tumor was completely suppressed (C), a second tumor was barely detectable, and the remaining four tumors were significantly reduced in size.
Figure 5.

FAP and POP identified from HCT116 mouse xenograft tissue homogenates by excision of protein bands following gel electrophoresis, which were then digested with trypsin, analyzed by HPLC–MS/MS, and identified by MASCOT database search. The complete amino acid sequence of each protein is shown beginning with the amino terminus. (A) Peptides that represented 28% of the amino acid sequence deduced from the DNA sequence were identified. Amino acids in blue are peptides unique to mouse FAP as identified in the tumor tissue, while those in red are peptides that are common to both mouse and human FAPs. (B) Peptides representing 73% of the human POP sequence were observed. Amino acids in green represent peptides unique to human POP that were identified in the tumor tissue, while those in red are peptides that confirm the identification of POP but are common to both mouse and human forms. (C) Pulverized tumors from three control mice and three mice treated with M83 at 100 μg/day were suspended in 2 × SDS sample buffer at equivalent mg/ml concentrations. Equal volumes of each were subjected to SDS-PAGE, transferred to nitrocellulose, and then Western blotted with either rabbit anti-FAP or (D) goat anti-POP. The human FAP reference protein for panel C was 5 ng of the native soluble version termed APCE (94.5 kDa), and the reference for panel D was 1.5 ng of human POP (73 kDa).
Our IHC analyses of untreated and treated tumors are contained in Figure 6. Two representative sections are shown for each staining procedure used for each treatment group: 1) saline control, 2) M83, and 3) J94. FAP staining of tumor sections from untreated control mice occurred largely in highly vascularized stromal areas, frequently appearing parallel to the longitudinal axes of vessels. These findings agree with our past observation that FAP production begins 4 to 6 hours after the onset of tube formation by endothelial cells (ECs) cultured on Matrigel and points to the likelihood that FAP proteinase activity facilitates vessel growth into ECM [10], [23]. Concentrated areas of FAP staining were apparent in the M83-treated group, but vessel formation was reduced when compared to control sections, suggesting that diminished “burrowing” capability of vessels into ECM reduced the density of their usual stromal patterns. In the examples shown for J94-treated tumors, the predilection for FAP depositions to align along microvessels is apparent as is reduced CD31 staining in surrounding areas, indicating less microvasculature development.
Figure 6.
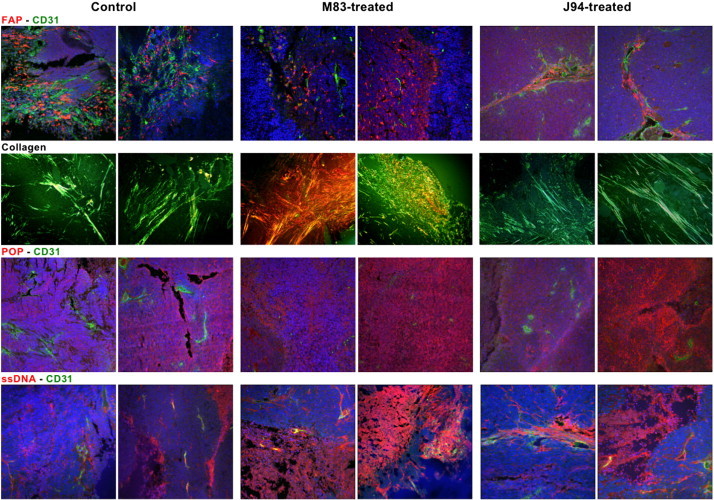
IHC staining of excised HCT116 xenografts. Tissues were double-stained for FAP and CD31 in row one, POP and CD31 in row three, or ssDNA and CD31 in row four. In addition, tissues were stained with picrosirius red for detection of collagen, as shown in row two. The two leftmost columns represent stained images of untreated control tumors, the two center columns show stained tumors from M83-treated mice, and the two rightmost columns show the stained tumor tissue from mice treated with J94. All images were at × 20 magnification.
Picrosirius staining of collagen in the untreated tumor sections showed narrow green-stained filamentous structures in more or less parallel longitudinal arrangements. In the M83-treated group, however, collagen was present in large accumulations of thickened fibers, which frequently manifested a yellow to bright orange staining. These accumulations are consistent with collagen undergoing partial cleavage or modest degradation before extensive digestion by FAP occurs [12]. Hence, it is reasonable to speculate that collagen within M83-treated tumors is initially cleaved by matrix metalloproteinases (MMPs), and since FAP is inhibited by M83, further proteolysis cannot proceed, and consequently, large derivative collagen peptides accumulate. With J94 inhibition of POP, collagen appeared unaffected and gave a filamentous, fibrillar pattern, essentially as in control tumors.
POP is normally distributed throughout virtually all tissues, showing some increased expression in brain, kidney, and testes [59]. The distribution of POP within cells remains unsettled, although it is generally agreed that it is found mostly in cytosol [32]. It has been reported within the nucleus, and while lacking structural features consistent with membrane insertion, it is found on the external surface of cell membranes, conceivably through lipid conjugation [60]. IHC staining showed POP to be distributed in somewhat irregular patterns within control tumors but perhaps less dense and homogeneous than in tumor sections from M83- or J94-treated mice; we cannot explain the latter. Moreover, we found that tumor sections doubly stained for POP and CD31 revealed more microvessels from untreated than from the M83- or J94 inhibitor–treated groups, the latter being consistent with overlapping inhibition of POP and FAP by M83 and only POP inhibition by J94 [10], [49]. The decreased angiogenesis observed with inhibition of POP can be explained by diminished cleavage and, hence, availability of the angiogenic stimulating acetyl-SDKP tetrapeptide from selected thymosin β4 peptides, which would be anticipated to reduce angiogenesis [33], [38], [61].
The most obvious and easiest established metric in our study was the extent of tumor growth suppression by either of our two inhibitors, which suggested that cell death must have occurred by necrosis, apoptosis, or both. In Figure 6, two sections of untreated tumors stained for ssDNA showed small, somewhat indistinct areas of staining that were irregularly distributed throughout both sections. In contrast, M83-treated tumor sections contained large areas of antibody staining consistent with apoptotic zones that covered on average > 40% of each field examined under low-power magnification (differing significantly from the untreated average of 13%, P < .01). Interestingly, sections from the J94-treated tumors showed less extensive apoptosis of 31% per microscopic field (P = .06 vs untreated). Diminished angiogenesis could account for cell death in both the M83- and J94-treated tumors; however, the more extensive apoptosis in the M83-treated group might have also resulted from inhibition of FAP proteolytic activity necessary for degradation of collagen or other substrates within ECM to yield products essential for cell maintenance and survival.
Discussion
The relatively consistent findings of FAP, POP, and thymosin β4 overexpression in cancer TME has prompted efforts to determine whether inhibition of each or selected combinations might serve as a potential therapeutic target for tumor growth suppression [5], [6], [8], [10], [13], [15], [32], [33], [34], [54]. Neither FAP nor POP has a precisely defined biologic function. FAP is believed to cleave minimally degraded or denatured type I collagen into small peptides [12] as ECM becomes remodeled for tumor growth. The proteolytic function of FAP may also support angiogenesis by aiding growth of new microvasculature into ECM [9], [23], [52], [62], [63], [64], [65]. POP is believed to modulate the activities and levels of several biologic peptides < 30 amino acids, most of which lack clearly established functions [66], [67], [68], [69]. Increased amounts of POP have been noted in cell cytosol and on cell membranes of cancers [30], [31], [32]. Tβ4, present in virtually all tissues, undergoes partial cleavage by an unidentified proteinase to generate fragments that POP—but not FAP—cleaves to yield several peptides, including the N-terminal tetrapeptide, acetyl-SDKP, which promotes angiogenesis at subnanomolar levels [33], [38], [61]. When acetyl-SDKP is deficient, diminished angiogenesis appears to be a consequence, as well as the development of fibrosis in selected tissues [37], [70], [71].
We selected a daily dose of M83 or J94 based on 1) the Ki for each inhibitor, 2) inhibition of FAP and POP by M83 or J94 in tissue culture studies [10], and 3) preliminary dose-ranging experiments in a like xenograft cancer model. About the same extent of tumor growth suppression was found for M83 or J94, despite J94 only inhibiting POP (Figure 2). Gel band intensities for FAP or POP per unit weight of tumor tissue (Figure 5C and D) from untreated or M83-treated mice were essentially the same, suggesting that the relative amount of each enzyme to tumor size was not changed by the inhibitor; moreover, specific IHC staining for FAP or POP appeared unchanged in untreated and M83- or J94-responsive tumors. The sum of these observations contravenes the suggestion that FAP protein, even when proteolytically inactive, promotes immune tolerance of cancer [51]. In that study, Huang et al. proposed that tumor growth could still occur whether FAP proteinase activity is present or not, thereby suggesting that immune tolerance is enhanced by the mere presence of FAP protein and that FAP proteolytic activity is non-essential for tumor growth. However, in our tumor model in immunocompromised mice, where immune tolerance is not involved unless through some degree of T-cell leakage as a consequence of the intricacies of large-scale commercial breeding of nude mice, or by diminished natural killer (NK) cell function, inhibition of FAP proteolytic activity resulted in marked tumor suppression. Since Huang et al. [51] used xenografts composed of carcinoma cells transfected with proteolytically inactive FAP to express the mutated FAP, rather than cancer-associated fibroblasts (CAFs), proteolytically active FAP could be produced by CAFs within host stroma as it invades the transfected cancer cell xenografts. Others have suggested that the stroma of cancer xenografts derives from the host [24], [72], but this is now definitively established by FAP and POP amino acid sequence studies (Figure 5A and B) that show xenograft stroma clearly expresses proteolytically active mouse FAP. This and the fact that reduced tumor growth has been shown to result from decreased FAP proteolytic activity, whether by proteinase inhibition [27], [45], [46] or reduced FAP expression [24], despite relatively modest changes in FAP protein levels, supports a role for FAP proteinase function in tumor growth.
During the 28-day treatment period, tumor growth suppression was closely similar in individual M83-treated mice (Figure 3B). Given that 100-μg M83 treatments resulted in complete or nearly complete disappearance of two expanding tumors by day 28, and a second decline in tumor volume between days 24 and 28 in about half of the tumors, the question of whether longer treatment might have resulted in further growth suppression of tumors still apparent at 28 days is raised. The IHC analyses of untreated and treated tumors that showed FAP staining concentrated mainly in highly vascularized areas, and prior observations that FAP production begins and increases with microvessel growth [10], [23], suggest that FAP proteolytic activity might derive from ECs or fibroblast-related pericytes and facilitate the growth of microvessels into ECM.
The accumulation of disorganized collagen in the M83-treated group agrees with reports that collagen is present in excess as FAP proteinase activity is diminished [24], [53]. Previously, we used MMPs in purified systems to show that type I collagen must first undergo partial degradation before FAP can digest it further to small peptides [12]. Tumor expansion is suggested to involve both fibroblast-derived MMPs and FAP in ECM remodeling [23], [51], [62], thereby providing the elements for a similar sequence of collagen digestion in vivo as space is excavated for tumor growth. In M83-treated tumor specimens, picrosirius-stained collagen appeared as large deposits of short, thick brightly orange fluorescent fibrils that were chaotically arranged in contrast to the narrower, more directionally organized, needle-like green fluorescent filaments within untreated tumors. The correlation of collagen thickness with such a staining differential has been described before [73]. The collections of collagen fibrils in M83-treated tumors probably represent MMP-cleaved collagen fragments that cannot be digested further by M83-inhibited FAP [12]. Whether such accumulations of large degraded collagen fragments participate in localized fibrosis that might deter cancer growth, diminish invasion of new microvasculature, or interfere with diffusion of subsequent therapeutic agents remain challenging questions for future study [74]. The putative participation of FAP in angiogenesis remains unclear but probably involves some pathways different from those of POP. Whereas POP proteolytically cleaves thymosin β4 fragments to yield the acetyl-SDKP tetrapeptide that clearly stimulates angiogenesis [33], [38], [39], FAP does not make this cleavage. The overlapping inhibition of both POP and FAP activities by M83, as also reported with some other FAP inhibitors [75], makes assigning a specific angiogenic function for FAP difficult; however, the synchrony with which EC cultures express FAP [10], [23] and the aforementioned POP proteolytic activities suggest that both may participate in regulatory processes important in microvessel organization and growth into underlying matrix. Preliminary data obtained in our laboratory indicate that M83 as well as J94 disrupts in vitro microvessel formation, which agrees with and predicts our present in vivo results that show inhibition of POP suppresses angiogenesis in synchrony with reduced tumor growth. We also suggest, however, that M83-induced tumor growth suppression is not due solely to diminished acetyl-SDKP tetrapeptide production as a consequence of POP inhibition [38], [39], [40], [41] but also to M83 inhibition of FAP proteolytic activity as expressed by those CAFs and pericytes necessary for channeling of microvessels throughout the ECM [26], [65].
M83-treated tumors stained for ssDNA showed large, irregular areas of apoptosis but fewer than expected inflammatory cells. Interestingly, gross inspection of growth-suppressed tumors showed virtually none of the classic findings of inflammation. With J94, apoptosis was also apparent but usually in smaller finger-like zones of cell debris that stained positive for ssDNA and that were often coincident with CD31-positive microvessels. In contrast, tumors from untreated mice displayed significantly less apoptosis. These observations suggest that impaired microvessel development due to M83 or J94 inhibition of POP leads to hypoxia, diminished nutrient supply, enhanced permeability, and ultimately cell death [36], [38], [76]. Santos et al. [24] described minimal apoptosis in tumors treated with Glu-boroPro that may have been less impressive than what we observed in M83- or J94-treated tumors. Whether our finding of large areas of apoptosis is due to M83 functioning as a more specific and sustained inhibitor than Glu-boroPro, or whether M83 has direct apoptotic effects, was not addressed by us. Importantly, however, others [53] have shown that knockdown of FAP expression by shRNA caused three-fold greater apoptosis in syngeneic breast cancer grafts than in control animals, which prompts the conclusion that diminished FAP proteolytic activity, whether by direct inhibition or by decreasing FAP expression, causes extensive apoptosis as tumor growth is suppressed. No gross or microscopic abnormalities, including fibrosis, were apparent in the hearts, livers, lungs, kidneys, and spleens of M83- or J94-treated mice, which suggest effective tumor targeting by either inhibitor without apparent off-targeting effects. It might be speculated that inhibitors of POP that are hydrophobic and enter the cell may interfere with cellular processes that otherwise prevent fibrosis [71], in which case, the hydrophilicity and positively charged structures of M83 and J94 would likely prevent cell entry [77], thereby explaining the lack of fibrosis in our study.
In mouse models of cancer, immunologic [18] or pharmacologic destruction of small numbers of FAP+ cells in normal skeletal muscle and bone marrow [17], [19] was associated with cachexia, anemia, and bone toxicity reminiscent of human paraneoplastic syndromes. It is not clear whether decreased FAP protein, or more specifically, FAP proteinase activity, prompted these changes, or whether they were due to loss of other specific FAP+ cellular functions. In our present study, inhibition of both FAP and POP proteolytic activities by M83, or POP alone by J94, resulted in greater tumor suppression (Figure 7) than other FAP-targeting approaches such as shRNA [53], FAP-gene disruption [24], or dipeptidyl boroPro [27], [45], [46], [51], [78] proteinase inhibitors. Our findings suggest that FAP+ cell destruction may be unnecessary so long as FAP and POP proteinase activities are effectively inhibited. Having demonstrated previously that bone marrow mesenchymal stem cells produced FAP as a constituent membrane protein [10], and being aware that FAP is likewise present on tissue-specific resident stem cell membranes [18], [19], we were also concerned about off-target inhibitory effects and the development of paraneoplastic features during growth suppression of colon cancer xenografts by FAP and POP proteinase inhibitors, but like others [20], [79], [80], we observed none of the adverse effects recently reported by the two groups [18], [19]. In fact, treatment with M83 or J94 was not associated with any apparent toxicities; all treated mice remained lively and socially interactive with cage mates; loss of appetite, weight loss > 10%, listlessness, changes in grooming habits, hunching, or bowel dysfunction was not seen in any mouse during the treatment period. While not substitutive for formal toxicity studies, the continuation of apparent good health in mice during the month of treatment with either inhibitor suggests that neither the pseudopeptide “stem” of our inhibitor constructs nor the boronic acid warhead [44], [81] caused obvious adverse effects. Others have also noted a relative absence of toxicity with in vivo use of boroPro-containing drugs [82]. Clearly, our results and those referred to above underscore some of the dramatic differences sometimes observed with dissimilar therapeutic approaches.
Figure 7.
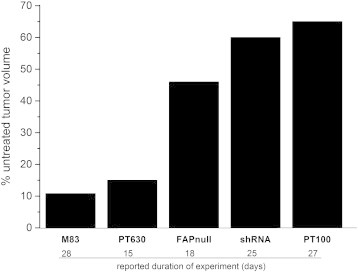
Average remaining tumor volumes after different tumor growth suppression methods. Reported effects of four different treatment approaches are compared with our results for M83 on tumor growth inhibition. Growth inhibition by M83 treatment exceeded that given by the less specific inhibitor, PT630 [24], by the inhibitor, PT100 [51], and by shRNA knockdown of FAP expression [53] and surpassed tumor growth reduction observed in FAP knockout mice [24].
Our results advance knowledge about two inhibitors of potential therapeutic targets for common cancers. Neither cyclize, thereby becoming inactive, nor inhibit DPPIV; both are easily soluble in aqueous media, and since both inhibitors are charged and not hydrophobic, intracellular entry and consequent POP inhibition within the cell is highly unlikely. We conclude that combined inhibition of FAP and POP by M83 is anti-angiogenic, causes buildup of erratically arranged collagen fibers, and results in widespread apoptosis within the tumor—the sum of which suppresses colon cancer xenograft growth. Similar growth inhibition by M83 was noted with human lung cancer xenografts, and while detailed IHC pathologic analyses were not performed in that study, it is reasonable to assume that results would parallel those for human colon cancer xenografts. Inhibition of POP by J94 showed similar anti-angiogenic effects, and despite collagen appearing unaltered, and the presence of apoptotic patterns that differed from those yielded by M83, tumor growth suppression still occurred (Figure 2). Further studies will be necessary to determine if simultaneous inhibition of FAP and POP by M83 is more effective than inhibiting either enzyme alone, and importantly, whether each enzyme is involved in separate, unique mechanisms that promote angiogenesis. Overall, our findings with M83 are mostly in line with those in which FAP enzymatic activity was putatively diminished without apparent FAP+ cell destruction. Finally, we raise the specter of whether tumor growth suppression might continue—even to complete disappearance—with longer administration of M83 or J94 and without evident toxicity. Such an assessment should also be informative as to whether M83 or J94 may be more effective (Figure 2B) over a longer treatment period.
Footnotes
Financial support came from William K. Warren Medical Research Center. The authors have no conflicts of interest.
References
- 1.Connolly J, Schnitt S, Wang H, Dvorak A, Dvorak H. Principles of Cancer Pathology. In: Bast RJ, Kufe D, Pollock R, Welchselbaum R, Holland J, Frei E, editors. Holland-Frei Cancer Medicine. 2000. [Google Scholar]
- 2.Folkman J. What is the evidence that tumors are angiogenesis dependent? J Natl Cancer Inst. 1990;82:4–6. doi: 10.1093/jnci/82.1.4. [DOI] [PubMed] [Google Scholar]
- 3.Park JE, Lenter MC, Zimmermann RN, Garin-Chesa P, Old LJ, Rettig WJ. Fibroblast activation protein, a dual specificity serine protease expressed in reactive human tumor stromal fibroblasts. J Biol Chem. 1999;274:36505–36512. doi: 10.1074/jbc.274.51.36505. [DOI] [PubMed] [Google Scholar]
- 4.Garin-Chesa P, Old LJ, Rettig WJ. Cell surface glycoprotein of reactive stromal fibroblasts as a potential antibody target in human epithelial cancers. Proc Natl Acad Sci U S A. 1990;87:7235–7239. doi: 10.1073/pnas.87.18.7235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cheng JD, Dunbrack RL, Jr., Valianou M, Rogatko A, Alpaugh RK, Weiner LM. Promotion of tumor growth by murine fibroblast activation protein, a serine protease, in an animal model. Cancer Res. 2002;62:4767–4772. [PubMed] [Google Scholar]
- 6.Puré E. The road to integrative cancer therapies: emergence of a tumor-associated fibroblast protease as a potential therapeutic target in cancer. Expert Opin Ther Targets. 2009;13:967–973. doi: 10.1517/14728220903103841. [DOI] [PubMed] [Google Scholar]
- 7.Hayward SW. Preclinical assessment of fibroblast activation protein as a target for antitumor therapy. Future Oncol. 2010;6:347–349. doi: 10.2217/fon.10.8. [DOI] [PubMed] [Google Scholar]
- 8.Brennen WN, Isaacs JT, Denmeade SR. Rationale behind targeting fibroblast activation protein-expressing carcinoma-associated fibroblasts as a novel chemotherapeutic strategy. Mol Cancer Ther. 2012;11:257–266. doi: 10.1158/1535-7163.MCT-11-0340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tchou J, Conejo-Garcia J. Targeting the tumor stroma as a novel treatment strategy for breast cancer: shifting from the neoplastic cell-centric to a stroma-centric paradigm. Adv Pharmacol. 2012;65:45–61. doi: 10.1016/B978-0-12-397927-8.00003-8. [DOI] [PubMed] [Google Scholar]
- 10.Christiansen VJ, Jackson KW, Lee KN, Downs TD, McKee PA. Targeting inhibition of fibroblast activation protein-α and prolyl oligopeptidase activities on cells common to metastatic tumor microenvironments. Neoplasia. 2013;15:348–358. doi: 10.1593/neo.121850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cheng JD, Weiner LM. Tumors and their microenvironments: tilling the soil. Commentary re: A. M. Scott et al., A Phase I dose-escalation study of sibrotuzumab in patients with advanced or metastatic fibroblast activation protein-positive cancer. Clin. Cancer Res., 9: 1639-1647, 2003. Clin Cancer Res. 2003;9:1590–1595. [PubMed] [Google Scholar]
- 12.Christiansen VJ, Jackson KW, Lee KN, McKee PA. Effect of fibroblast activation protein and alpha2-antiplasmin cleaving enzyme on collagen types I, III, and IV. Arch Biochem Biophys. 2007;457:177–186. doi: 10.1016/j.abb.2006.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu R, Li H, Liu L, Yu J, Ren X. Fibroblast activation protein: A potential therapeutic target in cancer. Cancer Biol Ther. 2012;13:123–129. doi: 10.4161/cbt.13.3.18696. [DOI] [PubMed] [Google Scholar]
- 14.Lee HO, Mullins SR, Franco-Barraza J, Valianou M, Cukierman E, Cheng JD. FAP-overexpressing fibroblasts produce an extracellular matrix that enhances invasive velocity and directionality of pancreatic cancer cells. BMC Cancer. 2011;11:245. doi: 10.1186/1471-2407-11-245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jacob M, Chang L, Pure E. Fibroblast activation protein in remodeling tissues. Curr Mol Med. 2012;12:1220–1243. doi: 10.2174/156652412803833607. [DOI] [PubMed] [Google Scholar]
- 16.Liao D, Luo Y, Markowitz D, Xiang R, Reisfeld RA. Cancer associated fibroblasts promote tumor growth and metastasis by modulating the tumor immune microenvironment in a 4T1 murine breast cancer model. PLoS One. 2009;4:e7965. doi: 10.1371/journal.pone.0007965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kraman M, Bambrough PJ, Arnold JN, Roberts EW, Magiera L, Jones JO, Gopinathan A, Tuveson DA, Fearon DT. Suppression of antitumor immunity by stromal cells expressing fibroblast activation protein-alpha. Science. 2010;330:827–830. doi: 10.1126/science.1195300. [DOI] [PubMed] [Google Scholar]
- 18.Tran E, Chinnasamy D, Yu Z, Morgan RA, Lee CC, Restifo NP, Rosenberg SA. Immune targeting of fibroblast activation protein triggers recognition of multipotent bone marrow stromal cells and cachexia. J Exp Med. 2013;210:1125–1135. doi: 10.1084/jem.20130110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Roberts EW, Deonarine A, Jones JO, Denton AE, Feig C, Lyons SK, Espeli M, Kraman M, McKenna B, Wells RJ. Depletion of stromal cells expressing fibroblast activation protein-α from skeletal muscle and bone marrow results in cachexia and anemia. J Exp Med. 2013;210:1137–1151. doi: 10.1084/jem.20122344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kakarla S, Chow KK, Mata M, Shaffer DR, Song XT, Wu MF, Liu H, Wang LL, Rowley DR, Pfizenmaier K. Antitumor effects of chimeric receptor engineered human T cells directed to tumor stroma. Mol Ther. 2013;21:1611–1620. doi: 10.1038/mt.2013.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Reisfeld RA. The tumor microenvironment: a target for combination therapy of breast cancer. Crit Rev Oncog. 2013;18:115–133. doi: 10.1615/critrevoncog.v18.i1-2.70. [DOI] [PubMed] [Google Scholar]
- 22.Feig C, Jones JO, Kraman M, Wells RJ, Deonarine A, Chan DS, Connell CM, Roberts EW, Zhao Q, Caballero OL. Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti–PD-L1 immunotherapy in pancreatic cancer. Proc Natl Acad Sci U S A. 2013;110:20212–20217. doi: 10.1073/pnas.1320318110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Huang Y, Wang S, Kelly T. Seprase promotes rapid tumor growth and increased microvessel density in a mouse model of human breast cancer. Cancer Res. 2004;64:2712–2716. doi: 10.1158/0008-5472.can-03-3184. [DOI] [PubMed] [Google Scholar]
- 24.Santos AM, Jung J, Aziz N, Kissil JL, Puré E. Targeting fibroblast activation protein inhibits tumor stromagenesis and growth in mice. J Clin Invest. 2009;119:3613–3625. doi: 10.1172/JCI38988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zimmerlin L, Donnenberg VS, Donnenberg AD. Pericytes: a universal adult tissue stem cell? Cytometry A. 2012;81:12–14. doi: 10.1002/cyto.a.21168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jung YD, Ahmad SA, Liu W, Reinmuth N, Parikh A, Stoeltzing O, Fan F, Ellis LM. The role of the microenvironment and intercellular cross-talk in tumor angiogenesis. Semin Cancer Biol. 2002;12:105–112. doi: 10.1006/scbi.2001.0418. [DOI] [PubMed] [Google Scholar]
- 27.Cheng JD, Valianou M, Canutescu AA, Jaffe EK, Lee HO, Wang H, Lai JH, Bachovchin WW, Weiner LM. Abrogation of fibroblast activation protein enzymatic activity attenuates tumor growth. Mol Cancer Ther. 2005;4:351–360. doi: 10.1158/1535-7163.MCT-04-0269. [DOI] [PubMed] [Google Scholar]
- 28.Sedo A, Krepela E, Kasafirek E. Dipeptidyl peptidase IV, prolyl endopeptidase and cathepsin B activities in primary human lung tumors and lung parenchyma. J Cancer Res Clin Oncol. 1991;117:249–253. doi: 10.1007/BF01625433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Goossens F, De Meester I, Vanhoof G, Scharpé S. Distribution of prolyl oligopeptidase in human peripheral tissues and body fluids. Eur J Clin Chem Clin Biochem. 1996;34:17–22. doi: 10.1515/cclm.1996.34.1.17. [DOI] [PubMed] [Google Scholar]
- 30.Larrinaga G, Perez I, Blanco L, Lopez JI, Andrés L, Etxezarraga C, Santaolalla F, Zabala A, Varona A, Irazusta J. Increased prolyl endopeptidase activity in human neoplasia. Regul Pept. 2010;163:102–106. doi: 10.1016/j.regpep.2010.03.012. [DOI] [PubMed] [Google Scholar]
- 31.Myöhänen TT, Pyykkö E, Männistö PT, Carpen O. Distribution of prolyl oligopeptidase in human peripheral tissues and in ovarian and colorectal tumors. J Histochem Cytochem. 2012;60:706–715. doi: 10.1369/0022155412453051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Larrinaga G, Perez I, Blanco L, Sanz B, Errarte P, Beitia M, Etxezarraga MC, Loizate A, Gil J, Irazusta J. Prolyl endopeptidase activity is correlated with colorectal cancer prognosis. Int J Med Sci. 2014;11:199–208. doi: 10.7150/ijms.7178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu JM, Garcia-Alvarez MC, Bignon J, Kusinski M, Kuzdak K, Riches A, Wdzieczak-Bakala J. Overexpression of the natural tetrapeptide acetyl-N-ser-asp-lys-pro derived from thymosin β4 in neoplastic diseases. Ann N Y Acad Sci. 2010;1194:53–59. doi: 10.1111/j.1749-6632.2010.05488.x. [DOI] [PubMed] [Google Scholar]
- 34.Cha HJ, Jeong MJ, Kleinman HK. Role of thymosin β4 in tumor metastasis and angiogenesis. J Natl Cancer Inst. 2003;95:1674–1680. doi: 10.1093/jnci/djg100. [DOI] [PubMed] [Google Scholar]
- 35.Nemolato S, Restivo A, Cabras T, Coni P, Zorcolo L, Orru G, Fanari M, Cau F, Gerosa C, Fanni D. Thymosin β-4 in colorectal cancer is localized predominantly at the invasion front in tumor cells undergoing epithelial mesenchymal transition. Cancer Biol Ther. 2012;13:191–197. doi: 10.4161/cbt.13.4.18691. [DOI] [PubMed] [Google Scholar]
- 36.Kim NS, Kang YJ, Jo JO, Kim HY, Oh YR, Kim YO, Jung MH, Ock MS, Cha HJ. Elevated expression of thymosin β4, vascular endothelial growth factor (VEGF), and hypoxia inducible factor (HIF)-1α in early-stage cervical cancers. Pathol Oncol Res. 2011;17:493–502. doi: 10.1007/s12253-010-9327-x. [DOI] [PubMed] [Google Scholar]
- 37.Cavasin MA, Rhaleb NE, Yang XP, Carretero OA. Prolyl oligopeptidase is involved in release of the antifibrotic peptide Ac-SDKP. Hypertension. 2004;43:1140–1145. doi: 10.1161/01.HYP.0000126172.01673.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Myohanen TT, Tenorio-Laranga J, Jokinen B, Vazquez-Sanchez R, Moreno-Baylach MJ, Garcia-Horsman JA, Mannisto PT. Prolyl oligopeptidase induces angiogenesis both in vitro and in vivo in a novel regulatory manner. Br J Pharmacol. 2011;163:1666–1678. doi: 10.1111/j.1476-5381.2010.01146.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang D, Carretero OA, Yang XY, Rhaleb NE, Liu YH, Liao TD, Yang XP. N-acetyl-seryl-aspartyl-lysyl-proline stimulates angiogenesis in vitro and in vivo. Am J Physiol Heart Circ Physiol. 2004;287:H2099–H2105. doi: 10.1152/ajpheart.00592.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Smart N, Rossdeutsch A, Riley PR. Thymosin β4 and angiogenesis: modes of action and therapeutic potential. Angiogenesis. 2007;10:229–241. doi: 10.1007/s10456-007-9077-x. [DOI] [PubMed] [Google Scholar]
- 41.Grant DS, Rose W, Yaen C, Goldstein A, Martinez J, Kleinman H. Thymosin beta4 enhances endothelial cell differentiation and angiogenesis. Angiogenesis. 1999;3:125–135. doi: 10.1023/a:1009041911493. [DOI] [PubMed] [Google Scholar]
- 42.Suzuki K, Sakaguchi M, Tanaka S, Yoshimoto T, Takaoka M. Prolyl oligopeptidase inhibition-induced growth arrest of human gastric cancer cells. Biochem Biophys Res Commun. 2014;443:91–96. doi: 10.1016/j.bbrc.2013.11.051. [DOI] [PubMed] [Google Scholar]
- 43.Adams S, Miller GT, Jesson MI, Watanabe T, Jones B, Wallner BP. PT-100, a small molecule dipeptidyl peptidase inhibitor, has potent antitumor effects and augments antibody-mediated cytotoxicity via a novel immune mechanism. Cancer Res. 2004;64:5471–5480. doi: 10.1158/0008-5472.CAN-04-0447. [DOI] [PubMed] [Google Scholar]
- 44.Nemunaitis J, Vukelja SJ, Richards D, Cunningham C, Senzer N, Nugent J, Duncan H, Jones B, Haltom E, Uprichard MJ. Phase I trial of PT-100 (PT-100), a cytokine-inducing small molecule, following chemotherapy for solid tumor malignancy. Cancer Invest. 2006;24:553–561. doi: 10.1080/07357900600894732. [DOI] [PubMed] [Google Scholar]
- 45.Narra K, Lee HO, Lerro A, Valvardi J, Azeez O, Jesson MI, Aziz N, Jones B, Cheng JD. 2006. Inhibitors of the stromal protease fibroblast activation protein attenuate tumor growth in vivo; p. 1029. (AACR Meeting Abstracts). [Google Scholar]
- 46.Narra K, Mullins SR, Lee HO, Strzemkowski-Brun B, Magalong K, Christiansen VJ, McKee PA, Egleston B, Cohen SJ, Weiner LM. Phase II trial of single agent Val-boroPro (Talabostat) inhibiting Fibroblast Activation Protein in patients with metastatic colorectal cancer. Cancer Biol Ther. 2007;6:1691–1699. doi: 10.4161/cbt.6.11.4874. [DOI] [PubMed] [Google Scholar]
- 47.Walsh MP, Duncan B, Larabee S, Krauss A, Davis JP, Cui Y, Kim SY, Guimond M, Bachovchin W, Fry TJ. Val-boroPro accelerates T cell priming via modulation of dendritic cell trafficking resulting in complete regression of established murine tumors. PLoS One. 2013;8:e58860. doi: 10.1371/journal.pone.0058860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kelly T, Adams J, Bachovchin W, Barton R, Campbell S, Courts S, Kennedy C, Snow R. Immunosuppressive boronic acid dipeptides: correlation between conformation and activity. J Am Chem Soc. 1993;115:12637–12638. [Google Scholar]
- 49.Lee KN, Jackson KW, Christiansen VJ, Dolence EK, McKee PA. Enhancement of fibrinolysis by inhibiting enzymatic cleavage of precursor α2-antiplasmin. J Thromb Haemost. 2011;9:987–996. doi: 10.1111/j.1538-7836.2011.04195.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Maes M, Goossens F, Scharpe S, Calabrese J, Desnyder R, Meltzer HY. Alterations in plasma prolyl endopeptidase activity in depression, mania, and schizophrenia: effects of antidepressants, mood stabilizers, and antipsychotic drugs. Psychiatry Res. 1995;58:217–225. doi: 10.1016/0165-1781(95)02698-v. [DOI] [PubMed] [Google Scholar]
- 51.Huang Y, Simms AE, Mazur A, Wang S, León NR, Jones B, Aziz N, Kelly T. Fibroblast activation protein-α promotes tumor growth and invasion of breast cancer cells through non-enzymatic functions. Clin Exp Metastasis. 2011;28:567–579. doi: 10.1007/s10585-011-9392-x. [DOI] [PubMed] [Google Scholar]
- 52.Kelly T, Huang Y, Simms AE, Mazur A. Fibroblast activation protein-α: a key modulator of the microenvironment in multiple pathologies. Int Rev Cell Mol Biol. 2012;297:83–116. doi: 10.1016/B978-0-12-394308-8.00003-0. [DOI] [PubMed] [Google Scholar]
- 53.Cai F, Li Z, Wang C, Xian S, Xu G, Peng F, Wei Y, Lu Y. Short hairpin RNA targeting of fibroblast activation protein inhibits tumor growth and improves the tumor microenvironment in a mouse model. BMB Rep. 2013;46:252–257. doi: 10.5483/BMBRep.2013.46.5.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Xiao Y, Chen Y, Wen J, Yan W, Zhou K, Cai W. Thymosin β4: a potential molecular target for tumor therapy. Crit Rev Eukaryot Gene Expr. 2012;22:109–116. doi: 10.1615/critreveukargeneexpr.v22.i2.30. [DOI] [PubMed] [Google Scholar]
- 55.Lee KN, Jackson KW, Terzyan S, Christiansen VJ, McKee PA. Using substrate specificity of antiplasmin-cleaving enzyme for fibroblast activation protein inhibitor design. Biochemistry. 2009;48:5149–5158. doi: 10.1021/bi900257m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gorrao SS, Hemerly JP, Lima AR, Melo RL, Szeltner Z, Polgar L, Juliano MA, Juliano L. Fluorescence resonance energy transfer (FRET) peptides and cycloretro-inverso peptides derived from bradykinin as substrates and inhibitors of prolyl oligopeptidase. Peptides. 2007;28:2146–2154. doi: 10.1016/j.peptides.2007.08.018. [DOI] [PubMed] [Google Scholar]
- 57.Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Van ER, Lambeir AM. Structure and function relationship in prolyl oligopeptidase. CNS Neurol Disord Drug Targets. 2011;10:297–305. doi: 10.2174/187152711794653814. [DOI] [PubMed] [Google Scholar]
- 59.Goossens F, De MI, Vanhoof G, Scharpé S. A sensitive method for the assay of serum prolyl endopeptidase. Eur J Clin Chem Clin Biochem. 1992;30:235–238. [PubMed] [Google Scholar]
- 60.Tenorio-Laranga J, Venäläinen JI, Männistö PT, García-Horsman JA. Characterization of membrane-bound prolyl endopeptidase from brain. FEBS J. 2008;275:4415–4427. doi: 10.1111/j.1742-4658.2008.06587.x. [DOI] [PubMed] [Google Scholar]
- 61.Liu JM, Kusinski M, Ilic V, Bignon J, Hajem N, Komorowski J, Kuzdak K, Stepien H, Wdzieczak-Bakala J. Overexpression of the angiogenic tetrapeptide AcSDKP in human malignant tumors. Anticancer Res. 2008;28:2813–2817. [PubMed] [Google Scholar]
- 62.Cavallo-Medved D, Rudy D, Blum G, Bogyo M, Caglic D, Sloane BF. Live-cell imaging demonstrates extracellular matrix degradation in association with active cathepsin B in caveolae of endothelial cells during tube formation. Exp Cell Res. 2009;315:1234–1246. doi: 10.1016/j.yexcr.2009.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Newman AC, Nakatsu MN, Chou W, Gershon PD, Hughes CC. The requirement for fibroblasts in angiogenesis: fibroblast-derived matrix proteins are essential for endothelial cell lumen formation. Mol Biol Cell. 2011;22:3791–3800. doi: 10.1091/mbc.E11-05-0393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Fukumura D, Jain RK. Tumor microvasculature and microenvironment: targets for anti-angiogenesis and normalization. Microvasc Res. 2007;74:72–84. doi: 10.1016/j.mvr.2007.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Noma K, Smalley KS, Lioni M, Naomoto Y, Tanaka N, El-Deiry W, King AJ, Nakagawa H, Herlyn M. The essential role of fibroblasts in esophageal squamous cell carcinoma-induced angiogenesis. Gastroenterology. 2008;134:1981–1993. doi: 10.1053/j.gastro.2008.02.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Polgár L. The prolyl oligopeptidase family. Cell Mol Life Sci. 2002;59:349–362. doi: 10.1007/s00018-002-8427-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.García-Horsman JA, Männistö PT, Venäläinen JI. On the role of prolyl oligopeptidase in health and disease. Neuropeptides. 2007;41:1–24. doi: 10.1016/j.npep.2006.10.004. [DOI] [PubMed] [Google Scholar]
- 68.Tenorio-Laranga J, Männistö PT, García-Horsman JA. Hunting for peptide substrates of prolyl oligopeptidase: classical versus non-classical bioactive peptides. CNS Neurol Disord Drug Targets. 2011;10:319–326. doi: 10.2174/187152711794653841. [DOI] [PubMed] [Google Scholar]
- 69.Myöhänen TT, García-Horsman JA, Tenorio-Laranga J, Männistö PT. Issues about the physiological functions of prolyl oligopeptidase based on its discordant spatial association with substrates and inconsistencies among mRNA, protein levels, and enzymatic activity. J Histochem Cytochem. 2009;57:831–848. doi: 10.1369/jhc.2009.953711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Cavasin MA, Liao TD, Yang XP, Yang JJ, Carretero OA. Decreased endogenous levels of Ac-SDKP promote organ fibrosis. Hypertension. 2007;50:130–136. doi: 10.1161/HYPERTENSIONAHA.106.084103. [DOI] [PubMed] [Google Scholar]
- 71.Zuo Y, Chun B, Potthoff SA, Kazi N, Brolin TJ, Orhan D, Yang HC, Ma LJ, Kon V, Myöhänen T. Thymosin β4 and its degradation product, Ac-SDKP, are novel reparative factors in renal fibrosis. Kidney Int. 2013;84:1166–1175. doi: 10.1038/ki.2013.209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Spector I, Zilberstein Y, Lavy A, Nagler A, Genin O, Pines M. Involvement of host stroma cells and tissue fibrosis in pancreatic tumor development in transgenic mice. PLoS One. 2012;7:e41833. doi: 10.1371/journal.pone.0041833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Rich L, Whittaker P. Collagen and picrosirius red staining: a polarized light assessment of fibrillar hue and spatial distribution. Braz J Morphol Sci. 2005;22:97–104. [Google Scholar]
- 74.Netti PA, Berk DA, Swartz MA, Grodzinsky AJ, Jain RK. Role of extracellular matrix assembly in interstitial transport in solid tumors. Cancer Res. 2000;60:2497–2503. [PubMed] [Google Scholar]
- 75.Jansen K, Heirbaut L, Verkerk R, Cheng JD, Joossens J, Cos P, Maes L, Lambeir AM, De Meester I, Augustyns K. Extended structure-activity relationship and pharmacokinetic investigation of (4-quinolinoyl)glycyl-2-cyanopyrrolidine inhibitors of fibroblast activation protein (FAP) J Med Chem. 2014;57:3053–3074. doi: 10.1021/jm500031w. [DOI] [PubMed] [Google Scholar]
- 76.Jo JO, Kim SR, Bae MK, Kang YJ, Ock MS, Kleinman HK, Cha HJ. Thymosin β4 induces the expression of vascular endothelial growth factor (VEGF) in a hypoxia-inducible factor (HIF)-1α-dependent manner. Biochim Biophys Acta. 2010;1803:1244–1251. doi: 10.1016/j.bbamcr.2010.07.005. [DOI] [PubMed] [Google Scholar]
- 77.Cooper G. The Cell: A Molecular Approach. Sinauer Associates; Sunderland, MA: 2000. Transport of small molecules. [www.ncbi.nlm.nih.gov/books/NBK9847/] [Google Scholar]
- 78.Niedermeyer J, Garin-Chesa P, Kriz M, Hilberg F, Mueller E, Bamberger U, Rettig WJ, Schnapp A. Expression of the fibroblast activation protein during mouse embryo development. Int J Dev Biol. 2001;45:445–447. [PubMed] [Google Scholar]
- 79.Wang LC, Lo A, Scholler J, Sun J, Majumdar RS, Kapoor V, Antzis M, Cotner CE, Johnson LA, Durham AC. Targeting fibroblast activation protein in tumor stroma with chimeric antigen receptor T cells can inhibit tumor growth and augment host immunity without severe toxicity. Cancer Immunol Res. 2014;2:154–166. doi: 10.1158/2326-6066.CIR-13-0027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Brennen WN, Rosen DM, Chaux A, Netto GJ, Isaacs JT, Denmeade SR. Pharmacokinetics and toxicology of a fibroblast activation protein (FAP)-activated prodrug in murine xenograft models of human cancer. Prostate. 2014;74:1308–1319. doi: 10.1002/pros.22847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Hall DG. Wiley-VCH; 2011. Boronic Acids. [Google Scholar]
- 82.Connolly BA, Sanford DG, Chiluwal AK, Healey SE, Peters DE, Dimare MT, Wu W, Liu Y, Maw H, Zhou Y. Dipeptide boronic acid inhibitors of dipeptidyl peptidase IV: determinants of potency and in vivo efficacy and safety. J Med Chem. 2008;51:6005–6013. doi: 10.1021/jm800390n. [DOI] [PubMed] [Google Scholar]