

Abstract
The TWIST1 gene has diverse roles in development and pathologic diseases such as cancer. TWIST1 is a dimeric basic helix-loop-helix (bHLH) transcription factor existing as TWIST1-TWIST1 or TWIST1-E12/47. TWIST1 partner choice and DNA binding can be influenced during development by phosphorylation of Thr125 and Ser127 of the Thr-Gln-Ser (TQS) motif within the bHLH of TWIST1. The significance of these TWIST1 phosphorylation sites for metastasis is unknown. We created stable isogenic prostate cancer cell lines overexpressing TWIST1 wild-type, phospho-mutants, and tethered versions. We assessed these isogenic lines using assays that mimic stages of cancer metastasis. In vitro assays suggested the phospho-mimetic Twist1-DQD mutation could confer cellular properties associated with pro-metastatic behavior. The hypo-phosphorylation mimic Twist1-AQA mutation displayed reduced pro-metastatic activity compared to wild-type TWIST1 in vitro, suggesting that phosphorylation of the TWIST1 TQS motif was necessary for pro-metastatic functions. In vivo analysis demonstrates that the Twist1-AQA mutation exhibits reduced capacity to contribute to metastasis, whereas the expression of the Twist1-DQD mutation exhibits proficient metastatic potential. Tethered TWIST1-E12 heterodimers phenocopied the Twist1-DQD mutation for many in vitro assays, suggesting that TWIST1 phosphorylation may result in heterodimerization in prostate cancer cells. Lastly, the dual phosphatidylinositide 3-kinase (PI3K)-mammalian target of rapamycin (mTOR) inhibitor BEZ235 strongly attenuated TWIST1-induced migration that was dependent on the TQS motif. TWIST1 TQS phosphorylation state determines the intensity of TWIST1-induced pro-metastatic ability in prostate cancer cells, which may be partly explained mechanistically by TWIST1 dimeric partner choice.
Abbreviations: EMT, epithelial-mesenchymal transition; bHLH, basic helix-loop-helix; T-T, TWIST1-TWIST1; E12/E47, E2A proteins; T-E, TWIST1-E12; IHC, immunohistochemistry; PI, propidium iodide; PKA, protein kinase A
Introduction
Prostate cancer is diagnosed in one of six men in the United States, making it the most common cancer diagnosed in men and the second leading cause of cancer deaths in men [1]. The natural history of prostate cancer suggests that understanding mechanisms of disease progression from localized to metastatic disease will result in the largest therapeutic gains [2].
One cellular pathway by which cancer cells may become pro-metastatic is the epithelial-mesenchymal transition (EMT). EMT is a transcriptional program, required during development and selectively silenced postnatally, that is reactivated in cancer cells to facilitate aggressive and metastatic behavior [3]. TWIST1 is a basic helix-loop-helix (bHLH) multidomain transcription factor that binds to E-box– and D-box–regulated target genes resulting in the elaboration of an EMT transcriptional program [4], [5], [6], [7], [8], [9]. TWIST1 functions as a dimer, either as a homodimer, TWIST1-TWIST1 (T-T), or as a heterodimer with E2A proteins (E12/E47), TWIST1-E12 (T-E), HAND proteins (HAND1/2), and likely other bHLH superfamily members [7], [9]. During development, TWIST1 dimer partner choice is, in part, regulated by the phosphorylation state of a Thr-Gln-Ser (TQS) motif in helix I of the TWIST1 protein [9], and human mutations in TWIST1 that disrupt TWIST1 phosphoregulation are causative of the human autosomal dominant disease Saethre-Chotzen syndrome [10], [11]. These observations support a model where tight regulation of the phosphorylation state and dimeric partner choice of TWIST1 is essential for normal development.
The role of TWIST pathways in prostate cancer pathogenesis [12], [13] and in prostate cancer disease progression and metastasis is becoming increasingly recognized as potentially important [14], [15], [16], [17], [18]. The critical domains of TWIST1 and dimeric partner required for increased tumorigenicity and aggressive metastatic phenotypes in prostate cancer are understudied [16]. Describing the functional significance of conserved structural domains and identifying critical binding partners of TWIST1 will increase mechanistic insights that can facilitate precise inhibitory strategies for TWIST1-induced cancer progression and metastasis.
Herein, we used a series of phosphorylation mutant and tethered versions of TWIST1 to perform structure-function analysis with assays that are surrogates for aggressive cellular and metastatic phenotypes in prostate cancer cells. By using isogenic androgen-dependent, Myc-CaP [19], and androgen-independent, PC3, cell lines overexpressing TWIST1 or phospho-mutant versions, we demonstrated specific requirements for TWIST1 TQS phosphorylation during TWIST1-induced metastasis of prostate cancer cells in vitro and in vivo. The dual ATP-competitive phosphatidylinositide 3-kinase (PI3K) and mammalian target of rapamycin (mTOR) inhibitor BEZ235 suppressed TWIST1-induced cellular migration. Finally, comparison of the TQS phospho-mutants with tethered TWIST1 overexpressing cells, T-T and T-E, suggested that TWIST1 TQS phosphorylation may be one factor resulting in T-E heterodimerization in prostate cancer cells.
Materials and Methods
Plasmids, Antibodies, and Reagents
pBABE-TWIST1-puro [20] was used to construct the Twist1-AQA and Twist1DQD mutant constructs using the QuikChange Site-Directed Mutagenesis Kit (Stratagene, San Diego, CA) and confirmed by sequencing. The following antibodies were used: Twist (Twist2C1a) (sc-81417; Santa Cruz Biotechnology, Dallas, TX), E-cadherin (ab53033; Abcam, Cambridge, UK), vimentin (ab92547), ZO-1 (5406; Cell Signaling Technology, Beverly, MA), β-actin (A5316; Santa Cruz Biotechnology), c-Myc (N-term) (1472-1; Epitomics, Burlingame, CA), HRP-conjugated secondary antibodies (Invitrogen, Carlsbad, CA), and Alexa Flour 488–conjugated secondary antibodies (Invitrogen). BEZ235 was purchased from Selleckchem, Houston, TX (S1009).
Clone 2088/2089 (tethered Flag–T-E) and 2411 (tethered Flag-TWIST1-Flag-TWIST1) [21] tethered open reading frames were excised from the parental plasmid with HindIII and XbaI digestion, and the ends were made blunt with T4 DNA polymerase and subcloned into pBABE-puro retroviral vector into blunt ended EcoR1 sites. The directionality of the clones was confirmed by restriction digestion. A similar procedure was performed to clone these open reading frames into the multiple cloning sites of the lentiviral vector W118.
Cell Line and Culture Conditions
PC3 and 22RV1 were obtained from American Type Culture Collection (Manassas, VA). Myc-CaP was a kind gift from Dr John Isaacs (Johns Hopkins University [JHU]) [19]. Myc-CaP was grown in Dulbecco's modified Eagle's medium (Invitrogen), PC3 in Hams F12K (Invitrogen), and 22RV1 in RPMI 1640 (Invitrogen). All media were supplemented with 10% FBS, penicillin (100 units/ml), and streptomycin (0.1 mg/ml). Cells were maintained at 37°C in a humidified incubator with 5% CO2. Cell line identity was confirmed by short tandem repeat profiling, and mycoplasma tested.
Retroviral Experiments
We used ecotropic and amphotropic Phoenix packaging lines for retroviral production. Myc-CaP and PC3 cells were transduced with pBABE-Puro vector constructs for two successive times over a 36-hour period followed by selection with 1 mg/ml puromycin and passaged once at 80% confluence. Stable cell lines were made by pooling the selected colonies. All cell lines were maintained under antibiotic selection.
Luciferase Promoter Reporter Assay
Subconfluent cells were transfected using Lipofectamine 2000 (Invitrogen) with 200 ng of firefly luciferase reporter gene construct (100 ng was used for SNAI2 reporter assays), 100 ng of the pRL-SV40 Renilla luciferase construct, and 500 ng of the TWIST1 or TWIST1 phospho-mutant expression constructs. Cell extracts were prepared 36 hours after transfection in passive lysis buffer, and the reporter activity was measured using the Dual Luciferase Reporter Assay System (Promega, Madison, WI).
Wound-Healing Migration Assay
Two-dimensional migration assay was performed using a scratch/wound model. Cells were grown in six-well plates for 24 hours to confluence. Multiple scratch wounds were created using a P-20 micropipette tip, and cells were fed with fresh complete media. Five representative fields of the wound were marked and images were taken at 0 and 24 hours after wounding. Relative wound closure is calculated from the remaining wound area normalized to the initial wound area using ImageJ software (NIH Image, Bethesda, MD).
BEZ235 experiments involved pre-treatment for 24 hours before creating scratch wounds and then proceeded as above.
Biophysical Assays
Fourier transform traction microscopy was used to measure the contractile stress arising at the interface between each adherent cell and its substrate as described [16]. Briefly, cells were plated sparsely on elastic gel blocks coated with type I collagen. Images of fluorescent microbeads (0.2 μm in diameter; Molecular Probes, Eugene, OR) embedded near the gel apical surface were taken before and after cell detachment with trypsin. The fluorescent image of the same region of the gel after trypsin was used as the reference (traction-free) image. The displacement field between a pair of images was then obtained by identifying the coordinates of the peak of the cross-correlation function [22], [23]. From the displacement field and known elastic properties of the gel (Young’s modulus of 1 kPa with a Poisson’s ratio of 0.48), the cell traction field was computed. The computed traction field was used to obtain net contractile moment, which is a scalar measure of the cell’s contractile strength, expressed in pico-Newton meters (pNm).
Matrigel Invasion Assay
The invasion potential was assessed using Chemicon cell invasion assay kit (Millipore, Billerica, MA) as directed by the manufacturer. Transwells with 8 μM pores coated with Matrigel were used for the assay. Serum-starved (0.5 × 106 to 1 × 106) cells (12-16 hours) in 300 μl of 10% FBS complete medium were seeded in the upper chambers, while lower wells were filled with 500 μl of 10% FBS complete medium. Invading cells on the lower surface were fixed and stained. The stain was dissolved in 200 μl of 10% acetic acid and measured at 570 nm. Invasive potential is derived by normalizing with the readings from blank transwell inserts.
Immunohistochemistry and Western Blot Analysis
Immunohistochemistry (IHC), Immunofluorescence (IF), and Western blot analysis were performed as described previously [24].
Anoikis Assay and Apoptosis Assessment
Anoikis resistance was measured using a modified protocol [25]. Cells were grown in normal attachment and ultra-low attachment (Corning Inc, Corning, NY) six-well plates. Twenty-four hours later, cells were blocked in 5% FBS and stained with Alexa Fluor 488–conjugated Annexin V followed by propidium iodide (PI) staining (50 μg/ml; Invitrogen). Cells were enumerated on a BD FACSCalibur (BD Biosciences, San Jose, CA), and analysis was done using FloJo analysis software. All conditions were n = 4 and two replicates per experiment.
Clonogenic Survival Assay and Soft Agar Colony Formation Assay
Clonogenic survival was performed as previously described [26]. Soft agar clonogenic assays used six-well plates pre-coated with 1 ml of basal 0.6% agarose in complete media and overlaid with 2 ml of cells (5 × 103 cells/ml) mixed with 0.3% agarose in complete media and allowed to solidify. The wells were constantly fed with complete media to prevent drying of agarose, and then after 10 to 15 days of incubation, colonies were scored under phase contrast microscopy. All conditions were repeated at least twice with three wells per experiment.
Animal Models and Histology
All procedures were carried out in accordance with the Johns Hopkins Animal Care and Use Committee, maintained under pathogen-free conditions, and given food and water ad libitum. For the experimental lung metastasis assay, 100 μl of phosphate-buffered saline containing 5 × 105 cells were injected into athymic nude mice through the tail vein. After 4 weeks, the mice were sacrificed, and necropsies were performed to score surface lung tumors and extra-thoracic metastases.
Microarray Data Acquisition and Analysis
Microarrays were performed using GeneChip WT cDNA Synthesis and Amplification Kit and WT Terminal Labeling Kit (Affymetrix, Santa Clara, CA). The labeled ssDNA was hybridized to the GeneChip Mouse Gene 1.0 ST Array (Affymetrix), washed with the Fluidics Station 450, and performed array scanning as previously described [27]. Arrays were normalized using the Robust Multichip Average in the oligo Bioconductor package at the transcript level [28]. Genes and gene sets with Benjamini-Hochberg [29] P values below .05 were considered statistically significant. Gene set enrichment analysis was performed using the C2 Curated Gene Sets collection from the Molecular Signature Database 3.0 and statistical comparisons by Fisher exact test.
The microarray data have been deposited to the Gene Expression Omnibus (GSE500002).
SYBR Green Quantitative Reverse Transcription–Polymerase Chain Reaction
Total RNA was isolated from cells using the QIAprep RNeasy Kit (Qiagen, Carlsbad, CA) or Trizol (Life Technologies, Carlsbad, CA) according to the manufacturer’s directions. Samples were treated with RQ1 RNase-Free DNase (Promega). The cDNA was generated from 1 μg of total RNA using the Superscript II Kit (Invitrogen Technologies). Control reactions were run without reverse transcription enzyme. Fifty nanograms of cDNA equivalents was amplified for the transcript described below in an ABI Prism 7900 HT for 40 cycles using SYBR Green PCR Master Mix (PerkinElmer Applied Biosystems, Foster City, CA). Polymerase chain reactions (PCRs) were performed in duplicate/triplicate in a final volume of 20 μl. Following amplification, the data were processed with the analysis program Sequence Detection Systems v2.2.2 (PerkinElmer Applied Biosystems). For each sample, the level of RNA for the genes of interest was standardized to a housekeeping gene (ubiquitin or 18S rRNA) within that sample; subsequently, the level of the transcript of interest was normalized to the expression of that transcript from the appropriate comparator sample. Primers for quantitative PCR were reported previously.
Statistical Analyses
Statistical analysis was carried out using GraphPad Prism version 5.04 for Windows (GraphPad Software, La Jolla, CA). Pairwise comparisons were tested using the paired two-tailed t test, Mann-Whitney test, or Fisher exact test. P values < .05 were considered statistically significant, and throughout this study, *P < .05, **P < .01, and ***P < .001.
Results
Study Rationale
The TWIST1 bHLH domain can be phosphorylated at the Thr125-Gln-Ser127 (TQS) motif (Figure 1A). TWIST1 TQS phosphorylation state has been shown to direct TWIST1 dimerization with selective preference for TWIST1 to form T-T homodimers or E12 to form T-E heterodimers [30], [31], but the significance of these phosphorylation states for TWIST1-induced phenotypes in cancer such as metastasis is unknown. Cancer cell metastasis is the culmination of sequential cellular actions that include loss of cell-to-cell adhesion, migration and invasion into the local extracellular matrix, intravasation into the vasculature, resistance to anoikis, extravasation into the parenchyma of distant tissues, and then colonization into a macroscopic metastatic tumor [32]. To assess the significance of the phosphorylation state of the TQS motif for cancer cell metastasis, we generated site-specific TWIST1 mutations at the TQS motif to create hypo-phosphorylation mimetic (Twist1-TQS125AQA) and phospho-mimetic (Twist1-TQS125DQD) versions, referred to as Twist1-AQA and Twist1-DQD, respectively (Figure 1A). We then established stable isogenic cell lines expressing TWIST1, Twist1-AQA, and Twist1-DQD in Myc-CaP and PC3 prostate cancer cells to determine the requirement of TQS motif phosphorylation for these metastatic steps using in vitro and in vivo assays (Figure 1, B and C). We next examined the requirement of phosphorylation on the TWIST1 TQS motif by the PI3K-AKT-mTOR pathway for cell migration using a specific dual PI3K-mTOR small molecule inhibitor. Finally, we created isogenic Myc-CaP lines that overexpressed tethered versions of T-T and T-E dimers and compared them to the TWIST1 phospho-mutant lines to determine the possible functional significance of TQS phosphorylation on TWIST1 dimer preference for cancer cell metastasis.
Figure 1.

Phosphorylation of the TWIST1 TQS motif is required for TWIST1-induced EMT cellular marker profile in prostate cancer cells. (A) A schematic of TWIST1 protein structure and the position 125 threonine (T125) and 127 serine (S127) site-specific mutants examined Twist1-AQA and Twist1-DQD (this schematic is not to scale). The position of the basic domain (in blue) and helix I are represented by bars above the sequence. A red bar below the sequence delineates a PKA recognition site. Western blot analysis was performed for TWIST1 expression in (B) Myc-CaP and (C) PC3 cells stably expressing Vector and overexpressing similar levels of TWIST1 or TWIST1 phospho-mutants with β-actin used as a loading control. The ~ 25 kDa band is the dominant TWIST1 immunoreactive species. The smaller band may be a degradation product or a posttranslationally modified version of Twist1. TWIST1 promoter reporter assays show that the Twist1-DQD mutant is proficient, but the Twist1-AQA mutant is defective for transcriptional regulation of EMT marker genes. Myc-CaP cells were transiently transfected with expression vectors for the firefly luciferase-linked (D) E-cadherin gene (CDH1) promoter construct and a Renilla luciferase reporter vector for normalization of transfection efficiency. After 36 hours, cell extracts were assayed for luciferase and Renilla activity and showed that TWIST1 and Twist1-DQD overexpression repressed transcription from the E-cadherin gene promoter, but the Twist1-AQA mutant was defective for this function. (E) Similar reporter assays were performed using a SLUG gene (SNAI2) promoter construct and showed that the Twist1-AQA mutant had no ability to transactivate transcription compared to wild-type TWIST1 or Twist1-DQD overexpression. Each bar represents values from at least three independent experiments performed in triplicate. Bars represent column mean; error bars ± SEM. Paired two-tailed t test; *P < .05, **P < .01, and ***P < .001. Epithelial and mesenchymal markers were also assessed by Western blot analysis for E-cadherin and vimentin in (F) Myc-CaP and (G) PC3 cells.
Phosphorylation of the TQS Motif Is Required for TWIST1 to Induce EMT Markers
In Myc-CaP prostate cancer cells, transient overexpression of TWIST1 and the Twist1-DQD mutant showed repression of CDH1 (Figure 1D, P < .001 compared to Vector control) and activation of SNAI2 (Figure 1E, P < .05 compared to Vector control) promoter activity (Figure 1, D and E, both P < .001 compared to Vector control). Interestingly, Twist1-AQA was found to be defective for both repression and activation in these assays and was not significantly different from the Vector control (Figure 1, D and E, P > .05 for both). These results suggest that phosphorylation of the TQS motif is required for TWIST1 transcriptional regulation of EMT marker genes.
Stable TWIST1 and Twist1-DQD overexpression led to down-regulation of the epithelial marker E-cadherin and up-regulation of the mesenchymal marker vimentin in Myc-CaP and PC3 cells (Figure 1, F and G). In both Myc-CaP and PC3 cells, the phospho-null mutation, Twist1-AQA, was unable to downregulate E-cadherin or upregulate vimentin (Figure 1, F and G). Taken together, these findings suggest that phosphorylation of the TQS motif is required for TWIST1 to induce EMT phenotype in prostate cancer cells.
Phosphorylation of the TQS Motif Is Required for TWIST-Induced Pro-Metastatic Cellular Behaviors
We then directly assessed the requirement of TQS motif phosphorylation of TWIST1 for conferring pro-metastatic behaviors to prostate cancer cells. The Twist1-AQA mutation appeared to inhibit the migration phenotype of these cells as results were no different than those observed using the Vector control (Figure 2, A and B, P > .1). In contrast, Myc-CaP cells stably overexpressing either TWIST1 or Twist1-DQD migrated faster than both Vector control and Twist1-AQA expressing cells (Figure 2, A and B, all pairwise comparisons P < .01). The Twist1-DQD mutation actually conferred increased migration rate to Myc-CaP cells that exceeded the rate of migration of cells expressing wild-type TWIST1 (Figure 2, A and B, P < .05).
Figure 2.
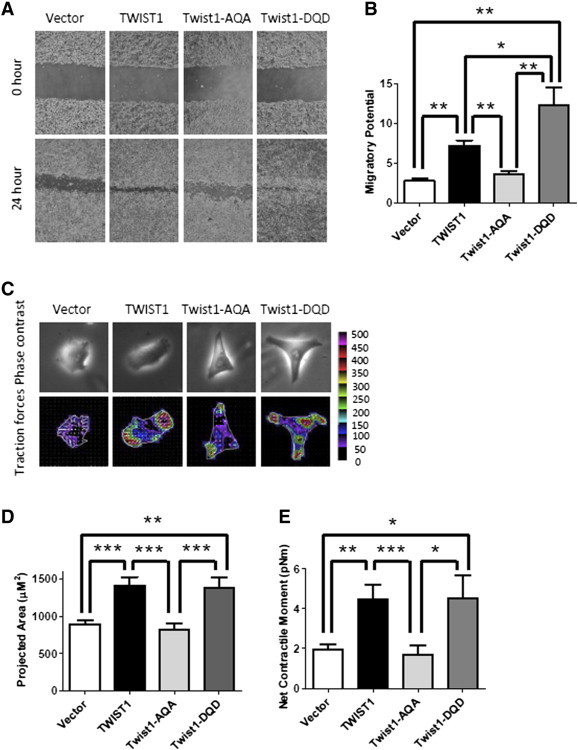
The TWIST1 phospho-null TQS motif mutation is defective for migration in Myc-CaP prostate cancer cells. (A) Representative phase contrast images of Myc-CaP cells overexpressing TWIST1 and TWIST1 phospho-mutants directly following and 24 hours after wound scratch. (B) Relative wound closure is calculated by the remaining wound area normalized to the initial wound area [n ≥ 3 experiments with three fields per experiment by ImageJ software (NIH)] and showed that Myc-CaP cells overexpressing Twist1-AQA were less migratory than wild-type TWIST1 and Twist1-DQD cells. Mann-Whitney test: *P < .05, **P < .01, and ***P < .001. (C) TWIST1 overexpression increases single prostate cancer cell traction forces on the substratum, which is attenuated by the Twist1-AQA mutation. The cell traction forces for individual cells (n = 20) are measured by using Fourier transform traction microscopy. The top panel shows representative phase contrast images of Myc-CaP isogenic cells. The bottom panel shows the traction maps; the colors within the cells represent the absolute magnitude of tractions in Pascal, and the arrows represent the relative magnitude and directions. (D) TWIST1 and Twist1-DQD mutant overexpression increases the mean of the projected area represented in bar graph format, and the Twist1-AQA phospho-mutant isogenic cell line is defective for this phenotype. (E) TWIST1 and Twist1-DQD mutant overexpression increases cell traction force exerted by a single living cell, or net contractile moment, and the Twist1-AQA mutant is completely deficient for this function. Bars represent column mean; error bars are ± SEM. The values are significant by Mann-Whitney test: *P < .05, **P < .01, and ***P < .001.
Cell migration necessitates contractile force generation by cells on their surroundings. Using traction microscopy, we interrogated the force generating capacity of single Myc-CaP overexpressing TWIST1 cells (Figure 2C). Compared with Vector control, the TWIST1 and Twist1-DQD overexpressing Myc-CaP cells showed increased cell spreading area and net contractile moment, a scalar measure of cellular contractile strength (Figure 2, D and E, at least P < .05 for all comparisons). The Twist1-AQA mutation overexpressing Myc-CaP cells displayed less cell spreading and lower net contractile moment compared to TWIST1 and Twist1-DQD (Figure 2, D and E, at least P < .05 for all comparisons) and was indistinguishable from Vector control cells (Figure 2, D and E, P > .1). The single cell biophysical data corroborate our results obtained from the bulk migration assays. Taken together, TWIST1 TQS phosphorylation is required for increasing cytoskeletal force generation and migratory potential in prostate cancer cells.
Myc-CaP and PC3 cells overexpressing TWIST1 and the phospho-mimetic version of TWIST1 showed increased invasion compared to Vector control and Twist1-AQA overexpressing cells using a Matrigel-coated transwell invasion assay (Figure 3, A and B, all pairwise comparisons at least P < .05). The hypo-phosphorylation mimetic TWIST1 mutation was completely defective for conferring TWIST1-induced invasion in Myc-CaP and PC3 cells and was similar to Vector control cells (Figure 3, A and B, P > .3). Similar to migration, the phosphorylation of TWIST1 TQS was required for TWIST1-induced prostate cancer invasion.
Figure 3.
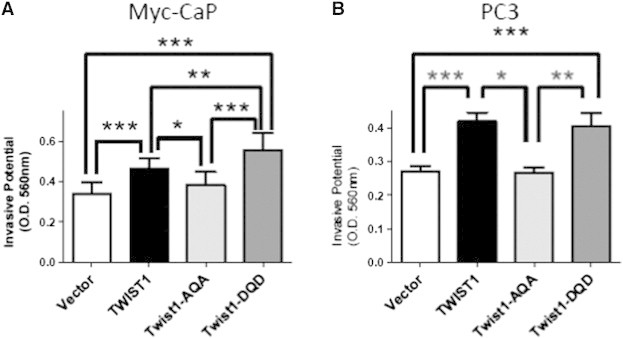
Phosphorylation of the TWIST1 TQS motif is required for TWIST1-induced invasion in prostate cancer cells. Transwell invasion assays with Matrigel were performed with isogenic (A) Myc-CaP (n = 10) and (B) PC3 cells (n = 6). The Myc-CaP cells were allowed to invade for 24 hours and PC3 cells for 60 hours. TWIST1 and Twist1-DQD overexpression increased invasion into Matrigel for both Myc-CaP and PC3 cells, but the Twist1-AQA mutant was no more invasive than Vector (represented by column mean ± SEM; *P < .05, **P < .01, and ***P < .001 by paired two-tailed t test).
The resistance to anoikis facilitates metastasis of cancer cells to distant organs [32]. Both TWIST1 and Twist1-DQD overexpressing Myc-CaP and PC3 cells showed decreased apoptosis when grown in low attachment conditions compared to their isogenic Vector control cells (Figure 4, A and B, all combinations at least P < .001, and Figure S1, A and B). Interestingly, the Twist1-AQA overexpressing Myc-CaP and PC3 cells were able to resist anoikis more than Vector control cells (Figure 4, A and B, both cell lines P < .01 vs Vector, and Figure S1, A and B), suggesting that TQS phosphorylation is not absolutely required for TWIST1-induced anoikis resistance. However, using clonogenic survival assays, we did observe that the phosphorylation of the TQS motif was required to confer radioresistance to prostate cancer cells (Figure S2, A and B). These data suggest that, although phosphorylation of the TWIST1 TQS motif is not generally required for resistance to all cell death signals, phosphorylation of the TQS motif is required for resistance to radiation-induced cell death in prostate cancer cells.
Figure 4.
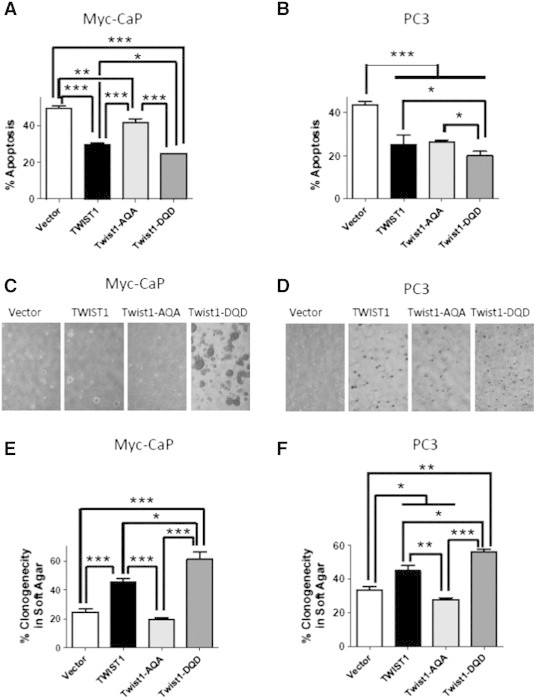
The Twist1-AQA mutant is defective for TWIST1-induced soft agar tumorigenicity. Cells were grown adherent or in suspension using ultra-low attachment dishes. The amount of apoptotic cell death or anoikis for the ultra-low attachment conditions was quantified by Annexin V–Alexa Fluor 488 and PI staining followed by flow cytometric analysis. Percent apoptosis was calculated by normalizing total apoptotic fraction in ultra-low attachment conditions to that of adherent cells and plotted as bar graph ± SEM for (A) Myc-CaP (n = 7) and (B) PC3 (n = 6) TWIST1 isogenic cell lines (*P < .05, **P < .01, and ***P < .001 by paired two-tailed t test); 5 × 105 cells were embedded in soft agar and incubated for 2 weeks. Colonies containing more than 50 cells were scored in at least five random fields. Representative phase contrast images of (C) Myc-CaP and (D) PC3 TWIST1 isogenic cell lines at × 4 magnification are shown. The percent clonogenicity in soft agar is calculated by normalizing the number of colonies to the total number of cells and represented as bar graphs ± SEM for (E) Myc-CaP (n = 6) and (F) PC3 (n = 6) (*P < .05, **P < .01, and ***P < .0001 by Mann-Whitney test).
The anchorage-independent clonogenicity of Myc-CaP and PC3 cells stably overexpressing TWIST1 and the TWIST1 mutations was performed (Figure 4, C–F). Both TWIST1 overexpressing Myc-CaP and PC3 cells showed increased frequency of colonies in soft agar compared to their isogenic Vector control cells (Figure 4, C–F, both cells P < .01). Myc-CaP and PC3 Twist1-AQA overexpressing cells exhibit a decreased ability to form colonies in soft agar (Figure 4, C–F). The Twist1-DQD overexpressing Myc-CaP and PC3 cells formed more colonies in soft agar than even wild-type TWIST1 (Figure 4, C–F, P < .05). In addition, Myc-CaP cells overexpressing Twist1-DQD formed larger sized colonies (Figure 4C). These results with anchorage-independent clonogenicity were confirmed within another prostate cancer cell line, 22Rv1, which also stably overexpressed TWIST1 and Twist1 phospho-mutant versions (Figure S3). Collectively, these data confirm the importance of phosphorylation of the TQS motif for aggressive in vitro prostate cancer cell behavior induced by TWIST1.
TWIST1 Phosphorylation Is Required for TWIST1-Induced Prostate Cancer Metastasis In Vivo
The last and arguably most important step in the metastatic cascade is the colonization of a distant site and growth into a macroscopic metastatic tumor. The potential of wild-type TWIST1 and TWIST1 TQS phospho-mutant overexpressing Myc-CaP cells to colonize and form macrometastatic tumors in vivo was assessed using the experimental lung metastasis assay. TWIST1 overexpression significantly increased the ability of Myc-CaP cells to colonize the lungs and form macroscopic metastases in vivo (Figure 5, A and B, 12 of 22 mice injected with TWIST1 overexpressing Myc-CaP cells vs 2 of 22 mice with isogenic Vector control cells, P < .01). The TWIST1 hypo-phosphorylation mimetic overexpressing Myc-CaP cells exhibited greatly reduced potential to form macroscopic lung metastases in vivo, and metastasis numbers were indistinguishable from Vector control Myc-CaP lungs (Figure 5, A and B, 2 of 12 mice injected with Twist1-AQA overexpressing Myc-CaP cells, P = 1 compared to Vector control; P = .066 compared to TWIST1). Concordant with the in vitro assays above, the TWIST1 phospho-mimetic overexpressing Myc-CaP cells were able to form lung tumors similar to wild-type TWIST1 (Figure 5, A and B, 7 of 12 mice injected with Twist1-DQD overexpressing Myc-CaP cells vs TWIST1, P = 1; vs Vector control, P < .01). The tumor cell morphology from TWIST1 and TWIST1 phospho-mutant overexpressing cells was similar (data not shown). In combination with observations obtained in the in vitro assays, these data strongly support that TWIST1 TQS phosphorylation is required for TWIST1-induced prostate cancer cell colonization of macroscopic metastases in vivo.
Figure 5.
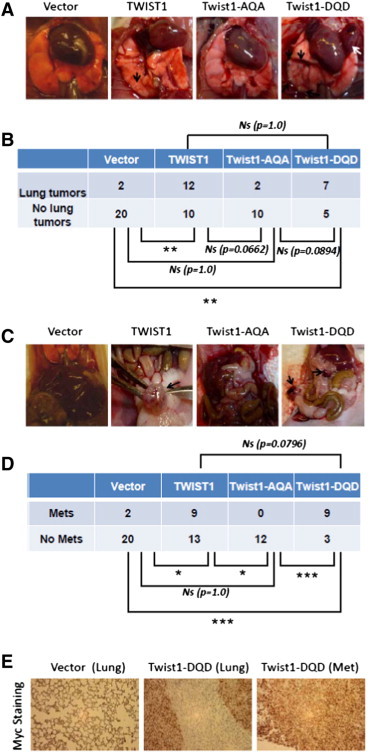
Phosphorylation of the TWIST1 TQS motif is required for TWIST1-induced prostate cancer metastasis in vivo. The experimental lung metastasis assay was performed with Myc-CaP TWIST1 isogenic cell lines; 5 × 105 cells were tail vein injected into 8-week-old athymic nude male mice, sacrificed 4 weeks later and inspected for lung colonization and extra-thoracic metastases. Cohorts of four to six mice were used for each cell line and experiments were performed at least twice. (A) Representative necropsy photographs with lung tumors distinguished by black arrows and a large chest wall lesion in the Twist1-DQD mouse identified with a white arrow. (B) A contingency table comparing the ability of the three isogenic cell lines to colonize lung tumors in vivo from A. TWIST1 and Twist1-DQD overexpressing Myc-CaP cells are able to form macroscopic lung tumors in vivo at a much higher frequency than Vector control cells (P < .01 for both comparisons by Fisher exact test). The Twist1-AQA box mutant Myc-CaP cells had an intermediate phenotype in vivo (P = 1 compared to Vector; P = .0662 compared to wild-type TWIST1; and P = .0894 compared to Twist1-DQD by Fisher exact test). (C) Representative necropsy photographs of extra-thoracic metastases from mice injected with TWIST1 isogenic cells with metastases indicated by black arrows. These extra-thoracic metastases represent the consequence of prostate cancer cells undergoing the full metastatic pathway following tail vein injection. (D) A contingency table comparing the ability of the three isogenic Myc-CaP cell lines to form extra-thoracic metastases from C. TWIST1 and Twist1-DQD overexpression conferred Myc-CaP cells with the ability to form extra-thoracic metastases at a higher frequency than Vector control cells and the Twist1-AQA mutant overexpressing cells (P < .05 for TWIST1 vs Vector; P < .05 for TWIST1 vs Twist1-AQA; P < .001 for Twist1-DQD vs Vector; and P < .001 for Twist1-DQD vs Twist1-AQA by Fisher exact test). (E) Representative anti-Myc IHC images of lungs or extra-thoracic metastases isolated from mouse tail vein injected with Myc-CaP + Vector cells (left) or Myc-CaP + Twist1-DQD (right two panels). The lung tumors (middle) and extra-thoracic metastasis (right panel) stained positive for c-Myc, confirming that the tumor cells were Myc-CaP cells.
Figure 7.

Tethered T-E overexpressing cells phenocopy Twist1-DQD mutant overexpressing cells for pro-metastatic behaviors in vitro. (A) Western blot analysis was performed for TWIST1 and E12 expression in Myc-CaP cells stably expressing Vector and overexpressing similar levels of TWIST1 or tethered versions of TWIST1 with β-actin used as a loading control. (B) Epithelial and mesenchymal markers were also assessed by Western blot analysis for E-cadherin and vimentin. Tethered T-E overexpressing Myc-CaP cells were able to induce an EMT. (C) Using microarray gene expression profiling data (Figure 6), a gene expression signature characteristic of Twist1-DQD expression was chosen, and the expression of these genes was measured by reverse transcriptase–quantitative PCR in isogenic Myc-CaP cells overexpressing tethered and phospho-mutant TWIST1. Relative expression is shown for each gene, with green indicating overexpression and red indicating underexpression. To evaluate the degree of similarity between the samples, hierarchical clustering was performed using a complete linkage algorithm, and the results are represented by the dendrogram. These data suggest that Twist1-DQD and T-E are the most similar based on gene expression profiling. Tethered T-T and T-E overexpressing cells were directly compared against TWIST1 phospho-mutants overexpressing cells for (D) migration, (E) transwell invasion, (F) anoikis resistance, and (G) anchorage-independent growth in soft agar. For all these assays with the exception of anoikis resistance, T-E and Twist1-DQD cells were the most similar.
Cells injected into the venous circulation seed the lungs preferentially and any extra-thoracic metastases produced using this assay must undergo the full metastatic pathway [33]. TWIST1 overexpression significantly increased the frequency of mice with extra-thoracic metastases (Figure 5, C and D, 9 of 22 mice injected with TWIST1 overexpressing Myc-CaP cells compared to 2 of 22 mice with isogenic Vector control cells, P < .05). Twist1-AQA Myc-CaP cells were unable to give rise to extra-thoracic metastases (Figure 5, C and D, 0 of 12 mice injected with Twist1-AQA overexpressing Myc-CaP cells, P < .05 compared to TWIST1 and P = 1 compared to Vector control cells). The Twist1-DQD overexpressing cells demonstrated increased numbers of mice with extra-thoracic metastases, 9 of 12, compared to Vector control (P < .001) and Twist1-AQA (P < .001). Twist1-DQD overexpressing cells produced more mice with extra-thoracic metastases than wild-type TWIST1 cells, which was not statistically significant but trended toward statistical significance (P = .0796). The Myc-CaP identity of lung tumors and extra-thoracic metastases was confirmed by positive Myc IHC (Figure 5E). These results show that the TQS motif phosphorylation of TWIST1 is required for TWIST1-induced metastasis of prostate cancer cells in vivo.
Gene Expression Profiling Reveals that Phosphorylation of the TQS Motif Is Required for the Full TWIST1-Induced Transcriptional Program
Global gene expression analysis was carried out with isogenic Myc-CaP lines, and in each pairwise comparison, we found several genes that were differentially expressed. Compared with Vector control, TWIST1 overexpression altered the expression of 425 genes. Overexpression of the Twist1-AQA and Twist1-DQD phospho-mutants altered the expression of much fewer genes compared to Vector control (Figure 6A). Overexpression of Twist1-AQA altered the expression of 132 genes, and a substantial portion (57 of 132) of these genes was also observed with TWIST1 overexpression in Myc-CaP cells. Overexpression of Twist1-DQD altered the expression of 105 genes, of which 23 were also altered in TWIST1 overexpressing Myc-CaP cells (Figure 6A). This expression pattern is consistent with each of the TWIST1 phospho-mutants inducing a separate altered transcriptional program, each of which is a derivative of the wild-type TWIST1 program, as shown by hierarchical clustering (Figure 6B).
Figure 6.
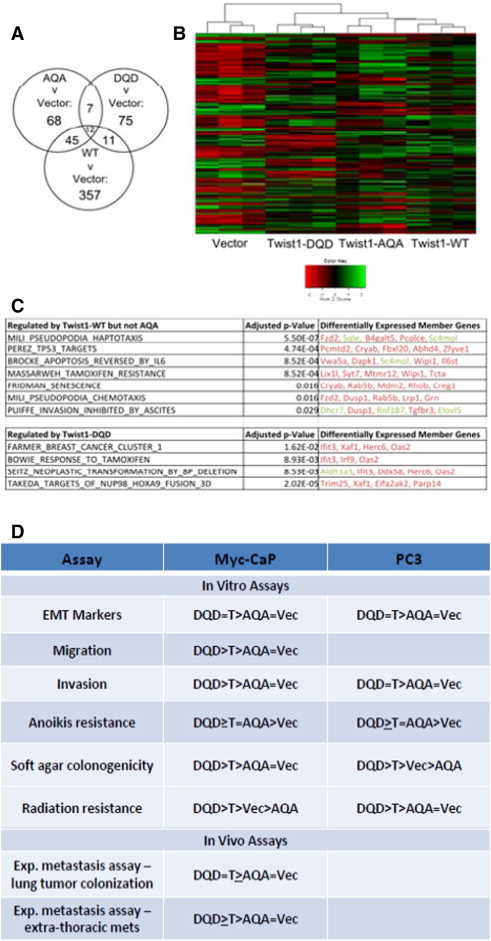
TWIST1 global gene expression profiles are modulated by mutations in the TQS motif in prostate cancer cells. (A) Gene expression analysis of Myc-CaP cells stably expressing Vector, TWIST1 (WT), Twist1-AQA (AQA), and Twist1-DQD (DQD) by microarray revealed a larger set of genes that were differentially expressed between TWIST1 overexpressing cells and Vector control compared to the phospho-mutants and Vector control cells. Gene expression between groups was performed by empirical Bayesian moderated analysis of variance, and genes were considered differentially expressed if B > 0 following Benjamini-Hochberg false discovery rate (FDR). (B) Heat map visualization of supervised clustering analysis of gene expression from Myc-CaP cells expressing Vector, TWIST1 (WT), Twist1-AQA (AQA), and Twist1-DQD (DQD) shows that the TWIST1 phospho-mutants have derivative wild-type TWIST1 gene expression profile. Each column represents a Myc-CaP microarray sample, and each row represents median-centered expression values for a single gene. High expression is indicated in green, intermediate expression in black, and low expression in red. (C) Selected gene sets from the Curated Molecular Signatures database that were overrepresented (P < .05, one-way Fisher exact test, Benjamini-Hochberg FDR) in the set of genes differentially regulated by TWIST1 but not by Twist1-AQA (top table) that is relevant to phenotypic differences between prostate cancer cells overexpressing TWIST1 versus Twist1-AQA as demonstrated in this study (relative overexpression is indicated in green and relative repression in red). Bottom table represents gene sets that were overrepresented in the Twist1-DQD versus TWIST1 transcriptome. (D) Summary of phenotypes for TWIST1 and the TWIST1 phospho-mutants in Myc-CaP and PC3 prostate cancer cells. T, TWIST1; AQA, Twist1-AQA mutant; DQD, Twist1-DQD mutant; Vec, Vector control.
Gene set enrichment analysis [34] was used to identify gene sets that were lost in Twist1-AQA overexpressing Myc-CaP cells compared to TWIST1. Many of the gene sets lost in the Twist1-AQA overexpressing cells were related to phenotypes that we directly assayed, and aggressive cellular behavior and metastasis were observed with overexpression of TWIST1 but not Twist1-AQA (Figure 6C, top table). At the same time, several gene sets related to transformation and cancer aggressiveness were overrepresented in the set of genes regulated by expression of Twist1-DQD compared to TWIST1 (Figure 6C, bottom table). This observation is consistent with the trend toward more aggressive cellular behavior, which we observed in cells overexpressing Twist1-DQD. These findings are highly suggestive of a transcriptional mechanism for the phenotypic differences observed between prostate cancer cells overexpressing TWIST1 and those overexpressing the phospho-mutants. A summary of all in vitro and in vivo phenotypes resulting from stable overexpression of TWIST1 and TWIST1 phospho-mutants is shown in Figure 6D.
T-E Heterodimer Overexpression Phenocopies Phospho-Mimetic Mutant Twist1-DQD Most Closely for Pro-Metastatic Cellular Behaviors
Isogenic Myc-CaP lines that overexpressed tethered versions of T-T and T-E dimers were constructed (Figure 7A). We then analyzed these TWIST1 tethered lines using the in vitro assays used to characterize the TWIST1 phospho-mutants above. Stable T-T tethered overexpressing Myc-CaP cells were able to upregulate vimentin but could not downregulate E-cadherin (Figure 7B). In contrast, T-E tethered Myc-CaP cells expressed a marker profile consistent with induction of an EMT and, at least qualitatively, was able to upregulate vimentin and modestly downregulate E-cadherin compared to wild-type TWIST1 (Figure 7B).
Comparison of the TWIST1 tethered and phospho-mutant cell lines for a gene expression signature characteristic of Twist1-DQD overexpression revealed that Twist1-DQD and T-E gene expression profiles were the most similar based on hierarchal clustering (Figure 7C). Twist1-AQA was most similar to Vector control (Figure 7C), and consistent with the loss-of-function phenotype, this phospho-null mutation displayed in most assays used in this study (Figure 6D). T-T overexpressing cells were the most dissimilar compared to the other TWIST1 isoforms for the gene expression signature (Figure 7C). Thus, T-E dimer overexpression results in a transcriptional program and EMT marker profile most similar to Twist1-DQD overexpression in Myc-CaP cells.
We then assessed the TWIST1 tethered isoforms for pro-metastatic capabilities in vitro. T-E overexpression resulted in increased migration of Myc-CaP cells compared to TWIST1 and Vector control (Figure 7D, P < .01 for pairwise, and Figure S4A). T-T and Vector control cells had the same decreased migratory potential (Figures 7D and S4A). Similarly, the invasive ability of T-E tethered cells was more than that of Vector control (both P < .01), and T-T cells were similar to Vector control (Figure 7E, P = .9). Neither T-T nor T-E tethered overexpressing cells showed any resistance to anoikis compared to Vector control (Figure 7F, P > .2, and Figure S4B). T-E overexpressing cells had the most ability for anchorage-independent growth and increased the soft agar clonogenicity of Myc-CaP cells more than even wild-type TWIST1 (Figure 7G, P < .05, and Figure S4C). T-T overexpressing cells had a slightly increased ability to allow Myc-CaP cell anchorage-independent growth compared to Vector control (Figure 7G, P < .01). Although not a universal correlation, our studies with the tethered TWIST1 isoforms suggested that T-E heterodimers have phenotypes most similar to the Twist1-DQD mutation (compare Figure 2, Figure 3, Figure 4 vs 7, D–G; five of six phenotypes were concordant between T-E and Twist1-DQD).
Pharmacologic Inhibition of the PI3K-AKT-mTOR Pathway Prevents TWIST1-Induced Prostate Cancer Cell Migration
During embryonic development, TWIST1 dimer preference is regulated by protein kinase A (PKA) phosphorylation of the TQS motif. The oncogenic kinase Akt (also known as protein kinase B) is aberrantly activated as part of the PTEN-PI3K-AKT-mTOR pathway in a majority of prostate cancer samples. We treated isogenic Myc-CaP lines stably overexpressing TWIST1 and the isoforms used above with the highly specific PI3K-mTOR dual kinase inhibitor BEZ235 (10 nM) and assayed for effects on TWIST1-induced cellular migration. BEZ235 is reported to have an half maximal inhibitory concentration (IC50) of 4 to 75 nM for different isoforms of PI3K (p110α/γ/δ/β) and 6 nM for mTOR. We observed inhibition of TWIST1-induced cellular migration to background levels or similar to Vector control cells with 24-hour pre-treatment with BEZ235 (Figure 8, A–C, P < .01). Interestingly, the increased cell migration observed in Myc-CaP cells overexpressing the Twist1-DQD phospho-mimetic mutant was not inhibited by BEZ235 treatment (Figure 8, A–C). As expected, migration by Myc-CaP cells overexpressing the Twist1-AQA phospho-null mutant was not affected by BEZ235 treatment (Figure 8, A–C). These effects on cell migration were not due to decreased cell viability or proliferation of TWIST1-overexpressing cells following BEZ235 treatment (Figure S5A). Similar results were observed when treating Myc-CaP cells stably expressing the tethered versions of TWIST1 (Figures S6, A–C, and S5B). Importantly, inhibition of PI3K-mTOR kinase activity did not prevent the T-E isoform from increasing TWIST1-induced cell migration (Figure S6, A–C), similar to Twist1-DQD. Together, these studies suggested that the PI3K-AKT-mTOR pathway phosphorylated TWIST1 on the TQS motif in prostate cancer cells and that this site-specific phosphorylation was required for TWIST1-induced prostate cancer cell migration.
Figure 8.
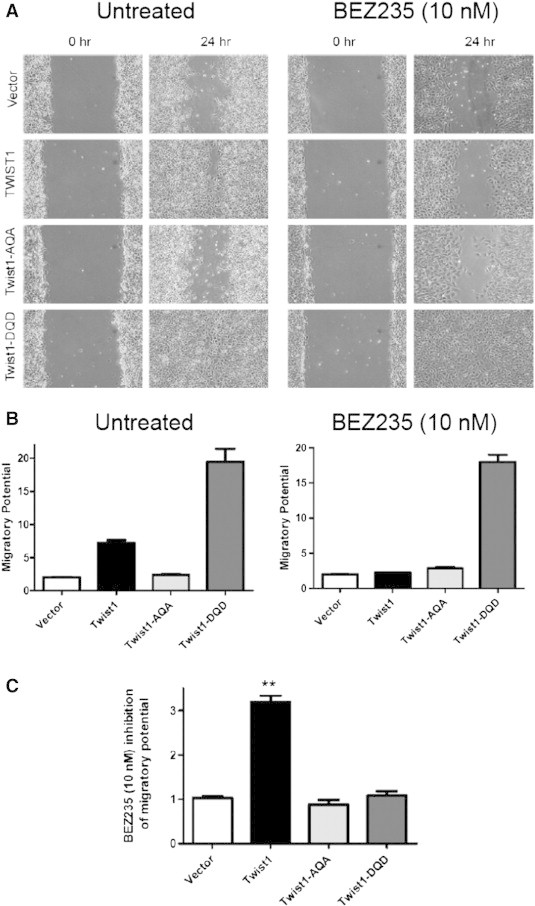
BEZ235 inhibition of PI3K-mTOR kinases prevents TWIST1-induced prostate cancer cell migration. (A) Representative phase contrast images of Myc-CaP cells overexpressing TWIST1 and TWIST1 phospho-mutants that have been treated with 10 nM BEZ235 for 24 hours, then a wound scratch was performed and then 24 hours after wound scratch. (B) Relative wound closure is calculated by the remaining wound area normalized to the initial wound area [n ≥ 3 experiments with three fields per experiment by ImageJ software (NIH)] and showed that BEZ235 treatment only inhibited the migration of Myc-CaP cells overexpressing TWIST1. (C) The relative inhibitory effect of BEZ235 on cell migration was determined by normalizing the migratory potential of untreated cells to that of 10 nM BEZ235-treated cells. Mann-Whitney test: **P < .01.
Discussion
The functional significance of many of the TWIST1 orthologous structural domains for animal development has been well studied [4], [9], [31], [35], [36]. In contrast, the structure-function significance of the TWIST1 TQS motif for cancer metastasis has not been reported. Our study shows that phosphorylation of the TQS motif in helix I of TWIST1 is required for induction of aggressive prostate cancer cell behavior in vitro and, most importantly, for metastasis in vivo. One possible conclusion from our data is that phosphorylation of this TQS motif may result in heterodimerization of TWIST1 with E12 that enables elaboration of specific transcriptional programs that produce a pro-metastatic cellular phenotype.
The TWIST1 TQS motif is highly conserved among the entire Twist subclass of bHLH factors as distantly through evolution as insects, and this domain is shown to be critical for the role of TWIST1 in development and benign pathologic conditions as demonstrated by transgenic mouse studies [21], [30], [31], [37], [38]. Moreover, nearby missense mutations in the TQS region of TWIST1 cause the Saethre-Chotzen syndrome, and these nearby mutations directly affect TWIST1 TQS phosphorylation [10], [11], [31]. Mechanistically, during embryonic development, PKA signal transduction pathways regulate TWIST1 dimerization preference and DNA binding by phosphorylation of this highly conserved TQS motif [21], [31]. At least for limb development, TQS phosphorylation, or the equivalent Twist1-DQD mutation, phenocopies tethered T-T homodimers, while the phospho-null Twist1-AQA mutation has been shown to phenocopy tethered T-E heterodimers [21]. The results of our current study suggest the inverse correlation for prostate cancer cell metastasis. Specifically, Twist1-DQD and tethered T-E appear to be the most similar for pro-metastatic in vitro assays and by gene expression profiling. In addition, whereas TWIST1 TQS phosphorylation was required for prostate cancer metastasis in vivo, TQS phosphorylation was not absolutely required for limb development in mice. In agreement with our findings, T-E tethered heterodimers have been reported to activate transcription from a Tinman reporter, whereas tethered homodimers T-T and E12-E12 homodimers had no activity in this assay [6]. These contrasting findings between embryonic development and cancer metastasis studies suggest that TWIST1 TQS phosphorylation can result in different functional consequences depending on the context. In support of this concept are in vivo data showing that T-T and T-E complexes have antagonistic downstream effects on fibroblast growth factor signaling that determine cranial suture patency [35], [39]. Given these likely context-specific differences of TWIST1 biology, it should not be assumed that PKA phosphorylates the TWIST1 TQS motif in general for all contexts. Protein kinase C [36], as well other serine threonine kinases such as Rho-associated protein kinases, Protein kinase G (PKG) and other members of the protein kinase C family can also phosphorylate related Hand transcription factors [40]. The TWIST1 TQS motif may also be phosphorylated by Akt [38]. Indeed, our pharmacologic data with the specific PI3K-mTOR dual kinase inhibitor, BEZ235, suggest that phosphorylation of the TWIST1 TQS motif by the PI3K-AKT-mTOR pathway is required for TWIST1-induced cell migration. Furthermore, BEZ235 treatment of Myc-CaP cells overexpressing tethered isoforms of TWIST1 suggests that TQS phosphorylation is important to direct T-E heterodimerization. These pharmacologic data need to be validated with complimentary assays in other cancer cell types. Finally, it is worth reiterating that other regulatory mechanisms also influence TWIST1 dimeric partner choice, such as the relative levels of TWIST1, ID, and E-proteins.
Equally important are the protein phosphatases that would act to limit TWIST1 metastatic potential. Protein phosphatase 2A (PP2A) containing the B56 delta regulatory subunit has been identified as such a factor [31], [36]. PP2A activity counteracts a number of kinase pathways associated with development of cancer and is often functionally inactivated in many tumors [41]. The large published data set supporting an anti-cancer role for PP2A further supports our conclusions that phosphorylated TWIST1 is mediating advancement of metastatic disease.
The general role of TWIST1 posttranslational modifications in cancer-related functions, predominantly breast cancer, has only recently been appreciated. Hyaluronic acid binding to CD44 promotes c-Src kinase Twist phosphorylation that results in Twist nuclear translocation and transcriptional activation in breast cancer cells leading to increased cellular invasion [42]. Breast tumor cell EMT and cellular invasion can also be increased through p38, c-Jun N-terminal kinase, and extracellular signal–regulated kinase 1/2 phosphorylation of Twist1 at Ser68 that results in Twist1 protein stabilization [43]. Lastly, in response to Interleukin 6 (IL-6) stimulation, Twist stability is also regulated at the posttranslational level in head and neck squamous cell carcinomas through casein kinase 2 phosphorylation of Twist on residues Ser18 and Ser20 [44]. TWIST1 phosphorylation on Ser42 by Akt has been shown to promote EMT and metastasis of breast cancer cells in vivo. Akt phosphorylation of Twist1 Ser42 promotes metastasis by transcriptional up-regulation of transforming growth factor–β2 [45]. Akt phosphorylation of Twist1 Ser42 has also been shown to confer cell death resistance to DNA damage through inhibition of a p53-dependent pathway [46]. In this study, we extend these previous findings, showing that the PI3K-AKT-mTOR pathway is required for TQS motif phosphorylation and can be inhibited by BEZ235 in prostate cancer cells. Thus, our current study is in agreement with the notion that posttranslational modifications of TWIST1 have functional consequences in cancer and extend these general findings to prostate cancer.
Importantly, our study shows for the first time the requirement of the PI3K-AKT-mTOR pathway for TQS motif phosphorylation, which is required for the pro-metastatic functions of TWIST1 in prostate cancer cells in vitro and in vivo.
TWIST1 and Twist1 protein expression levels are correlated with prostate cancer metastasis and clinical factors associated with lethal disease [12], [16], [17]. In addition to being a biomarker of aggressive prostate cancer, Twist1 inhibition may also be a potential novel therapeutic strategy for prostate cancer treatment. Inactivation of Twist1 to only physiological levels may be sufficient for anti-cancer effects [20], [47], [48], [49], and the very limited expression of TWIST1 in normal tissues in the adult mouse suggests that targeting TWIST1 systemically is well tolerated [50]. However, transcription factors have traditionally been difficult to inhibit directly [51]. Our findings suggest that PI3K-AKT-mTOR pathway inhibition with the dual kinase inhibitor BEZ235 may be an attractive indirect approach to target TWIST1-induced pro-metastatic phenotypes. In addition, we observed using both androgen-dependent and androgen-independent cell line models that the major pro-metastatic effects of TWIST1 are independent of the androgen receptor influence on prostate cancer cells. Interestingly, the T-E heterodimer has recently been shown to possess a unique cavity that can be specifically targeted by intercalating molecules for inhibitory purposes [52]. Our study also demonstrates the TQS motif as a critical structural component required for TWIST1-dependent pro-metastatic functions in prostate cancer and points toward targeting the T-E heterodimer as another tractable alternative therapeutic strategy.
Appendix A. Supplementary data
The following are the Supplementary data to this article.
Figure S1. Phosphorylation of the TWIST1 TQS motif is not required for TWIST1-induced resistance to anoikis. Cells were grown adherent or in suspension using ultra-low attachment dishes. The amount of apoptotic cell death or anoikis for the ultra-low attachment conditions was quantified by Annexin V–Alexa Fluor 488 and PI staining followed by flow cytometric analysis. Representative dot plots of (A) Myc-CaP and (B) PC3 Twist1 isogenic cell lines are shown. Percentage of cells in quadrants II (early apoptotic) and III (late apoptotic) that constitute apoptotic fractions are in bold. Percent apoptosis was calculated by normalizing total apoptotic fraction in ultra-low attachment conditions to that of adherent cells and plotted as bar graph ± SEM (Figure 4).
Figure S2. TWIST1 overexpression confers radioresistance to prostate cancer cells, which is attenuated by phospho-null mutations of the TWIST1 TQS motif. Briefly, the clonogenic survival assay was performed (see Materials and Methods section) by plating radiated single cells, 200 to 5000, in 100-mm dishes and stained 2 to 3 weeks later with crystal violet, and distinct colonies (defined as > 50 cells) were scored. The survival fraction at 4 (PC3) or 5 Gy (Myc-CaP) is calculated by total number of colonies normalized to the plating efficiency. Survival fraction is plotted for (A) Myc-CaP (n = 3, five replicates per experiment) and (B) PC3 isogenic cell lines (n = 3, five replicates per experiment). Bars represent column mean; error bars ± SEM; significance by Mann-Whitney test: *P < .05, **P < .01, and ***P < .001.
Figure S3. The Twist1-AQA mutation is deficient for TWIST1-induced soft agar anchorage-independent growth of 22Rv1 prostate cancer cells. (A) Western blot analysis of 22Rv1 cells stably overexpressing similar levels of TWIST1 and TWIST1 phospho-mutants. β-Actin was used as internal control. (B) The representative phase contrast images of soft agar colonies from 22RV1 isogenic cells taken at 4 × objective. (C) Colonies containing more than 50 cells are scored in five random fields from each well (n = 6), and the percentage was determined from the number of soft agar colonies normalized with the total number of cells. Bars represent column mean; error bars ± SEM; significance is by Mann-Whitney test: **P < .01.
Figure S4. Tethered T-E overexpressing cells phenocopy Twist1-DQD mutant overexpressing cells for pro-metastatic behaviors in vitro. (A) Representative phase contrast images of Myc-CaP cells overexpressing TWIST1 and tethered versions of TWIST1 directly following and 24 hours after wound scratch. (B) Representative dot plots of Myc-CaP cells overexpressing TWIST1 and tethered versions of TWIST1 for anoikis levels. See Figure S1 for details. (C) Representative phase contrast images of Myc-CaP cells overexpressing TWIST1 and tethered versions of TWIST1 for anchorage-independent growth in soft agar (see Figure S3 for details).
Figure S5. BEZ235 inhibition of PI3K-mTOR kinases does not change Myc-CaP cell viability. Cell viability assays using CellTiter-Blue as directed by the manufacturer reveal that BEZ235 treatment for 48 hours does not affect cell viability of Myc-CaP cells expressing (A and B) TWIST1, (A) Twist1 phospho-mutant or (B) Twist1 tethered isoforms.
Figure S6. BEZ235 inhibition of PI3K-mTOR kinases prevents TWIST1-induced prostate cancer cell migration. (A) Representative phase contrast images of Myc-CaP cells overexpressing TWIST1 and TWIST1 tethered isoforms that have been treated with 10 nM BEZ235 for 24 hours, then a wound scratch was performed and then 24 hours after wound scratch. (B) Relative wound closure is calculated by the remaining wound area normalized to the initial wound area [n> 3 experiments with three fields per experiment by ImageJ software (NIH)] and showed that BEZ235 treatment only inhibited the migration of Myc-CaP cells overexpressing TWIST1. (C) The relative inhibitory effect of BEZ235 on cell migration was determined by normalizing the migratory potential of untreated cells to that of 10 nM BEZ235-treated cells. Mann-Whitney test: **P < .01.
Footnotes
This article refers to supplementary materials, which are designated by Figures S1 to S6 and are available online at www.neoplasia.com.
Funding: R.D.W. was a Johns Hopkins Laboratory Radiation Oncology Training Fellow (NIH-T32CA121937). A.B.F. was funded by the National Institutes of Health (NIH) (1R0AR061392). S.S.A. was funded by the NIH (P50CA103175, U54CA141868, and HL107361). P.T.T. was funded by the Irene and Bernard L. Schwartz Scholar Award from the Patrick C. Walsh Prostate Cancer Research Fund, the Department of Defense (DoD) (W81XWH-11-1-0272 and W81XWH-13-1-0182), a Kimmel Translational Science Award (SKF-13-021), an ACS Scholar Award (122688-RSG-12-196-01-TBG), and the NIH (R01CA166348). Conflict of interest: The authors declare no conflict of interest.
Supplementary data to this article can be found online at http://dx.doi.org/10.1016/j.neo.2014.10.009.
References
- 1.Siegel R., Naishadham D., Jemal A. Cancer statistics, 2013. CA Cancer J Clin. 2013;63(1):11–30. doi: 10.3322/caac.21166. [DOI] [PubMed] [Google Scholar]
- 2.Pound C.R., Partin A.W., Eisenberger M.A., Chan D.W., Pearson J.D., Walsh P.C. Natural history of progression after PSA elevation following radical prostatectomy. JAMA. 1999;281(17):1591–1597. doi: 10.1001/jama.281.17.1591. [DOI] [PubMed] [Google Scholar]
- 3.Thiery J.P., Acloque H., Huang R.Y., Nieto M.A. Epithelial-mesenchymal transitions in development and disease. Cell. 2009;139(5):871–890. doi: 10.1016/j.cell.2009.11.007. [DOI] [PubMed] [Google Scholar]
- 4.Spicer D.B., Rhee J., Cheung W.L., Lassar A.B. Inhibition of myogenic bHLH and MEF2 transcription factors by the bHLH protein Twist. Science. 1996;272(5267):1476–1480. doi: 10.1126/science.272.5267.1476. [DOI] [PubMed] [Google Scholar]
- 5.Lee Y.M., Park T., Schulz R.A., Kim Y. Twist-mediated activation of the NK-4 homeobox gene in the visceral mesoderm of Drosophila requires two distinct clusters of E-box regulatory elements. J Biol Chem. 1997;272(28):17531–17541. doi: 10.1074/jbc.272.28.17531. [DOI] [PubMed] [Google Scholar]
- 6.Laursen K.B., Mielke E., Iannaccone P., Fuchtbauer E.M. Mechanism of transcriptional activation by the proto-oncogene Twist1. J Biol Chem. 2007;282(48):34623–34633. doi: 10.1074/jbc.M707085200. [DOI] [PubMed] [Google Scholar]
- 7.Qin Q., Xu Y., He T., Qin C., Xu J. Normal and disease-related biological functions of Twist1 and underlying molecular mechanisms. Cell Res. 2012;22(1):90–106. doi: 10.1038/cr.2011.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ansieau S., Morel A.P., Hinkal G., Bastid J., Puisieux A. TWISTing an embryonic transcription factor into an oncoprotein. Oncogene. 2010;29(22):3173–3184. doi: 10.1038/onc.2010.92. [DOI] [PubMed] [Google Scholar]
- 9.Barnes R.M., Firulli A.B. A twist of insight—the role of Twist-family bHLH factors in development. Int J Dev Biol. 2009;53(7):909–924. doi: 10.1387/ijdb.082747rb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.el Ghouzzi V., Le Merrer M., Perrin-Schmitt F., Lajeunie E., Benit P., Renier D., Bourgeois P., Bolcato-Bellemin A.L., Munnich A., Bonaventure J. Mutations of the TWIST gene in the Saethre-Chotzen syndrome. Nat Genet. 1997;15(1):42–46. doi: 10.1038/ng0197-42. [DOI] [PubMed] [Google Scholar]
- 11.Howard T.D., Paznekas W.A., Green E.D., Chiang L.C., Ma N., Ortiz de Luna R.I., Garcia Delgado C., Gonzalez-Ramos M., Kline A.D., Jabs E.W. Mutations in TWIST, a basic helix-loop-helix transcription factor, in Saethre-Chotzen syndrome. Nat Genet. 1997;15(1):36–41. doi: 10.1038/ng0197-36. [DOI] [PubMed] [Google Scholar]
- 12.Kwok W.K., Ling M.T., Lee T.W., Lau T.C., Zhou C., Zhang X., Chua C.W., Chan K.W., Chan F.L., Glackin C., Wong Y.C., Wang X. Up-regulation of TWIST in prostate cancer and its implication as a therapeutic target. Cancer Res. 2005;65(12):5153–5162. doi: 10.1158/0008-5472.CAN-04-3785. [DOI] [PubMed] [Google Scholar]
- 13.Shiota M., Yokomizo A., Tada Y., Inokuchi J., Kashiwagi E., Masubuchi D., Eto M., Uchiumi T., Naito S. Castration resistance of prostate cancer cells caused by castration-induced oxidative stress through Twist1 and androgen receptor overexpression. Oncogene. 2010;29(2):237–250. doi: 10.1038/onc.2009.322. [DOI] [PubMed] [Google Scholar]
- 14.Xie D., Gore C., Liu J., Pong R.C., Mason R., Hao G., Long M., Kabbani W., Yu L., Zhang H., Chen H., Sun X., Boothman D.A., Min W., Hsieh J.T. Role of DAB2IP in modulating epithelial-to-mesenchymal transition and prostate cancer metastasis. Proc Natl Acad Sci U S A. 2010;107(6):2485–2490. doi: 10.1073/pnas.0908133107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shiota M., Zardan A., Takeuchi A., Kumano M., Beraldi E., Naito S., Zoubeidi A., Gleave M.E. Clusterin mediates TGF-β-induced epithelial-mesenchymal transition and metastasis via Twist1 in prostate cancer cells. Cancer Res. 2012;72(20):5261–5272. doi: 10.1158/0008-5472.CAN-12-0254. [DOI] [PubMed] [Google Scholar]
- 16.Gajula R.P., Chettiar S.T., Williams R.D., Thiyagarajan S., Kato Y., Aziz K., Wang R., Gandhi N., Wild A.T., Vesuna F., Ma J., Salih T., Cades J., Fertig E., Biswal S., Burns T.F., Chung C.H., Rudin C.M., Herman J.M., Hales R.K., Raman V., An S.S., Tran P.T. The Twist box domain is required for Twist1-induced prostate cancer metastasis. Mol Cancer Res. 2013;11(11):1387–1400. doi: 10.1158/1541-7786.MCR-13-0218-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yuen H.F., Chua C.W., Chan Y.P., Wong Y.C., Wang X., Chan K.W. Significance of TWIST and E-cadherin expression in the metastatic progression of prostatic cancer. Histopathology. 2007;50(5):648–658. doi: 10.1111/j.1365-2559.2007.02665.x. [DOI] [PubMed] [Google Scholar]
- 18.Yuen H.F., Kwok W.K., Chan K.K., Chua C.W., Chan Y.P., Chu Y.Y., Wong Y.C., Wang X., Chan K.W. TWIST modulates prostate cancer cell-mediated bone cell activity and is upregulated by osteogenic induction. Carcinogenesis. 2008;29(8):1509–1518. doi: 10.1093/carcin/bgn105. [DOI] [PubMed] [Google Scholar]
- 19.Watson P.A., Ellwood-Yen K., King J.C., Wongvipat J., Lebeau M.M., Sawyers C.L. Context-dependent hormone-refractory progression revealed through characterization of a novel murine prostate cancer cell line. Cancer Res. 2005;65(24):11565–11571. doi: 10.1158/0008-5472.CAN-05-3441. [DOI] [PubMed] [Google Scholar]
- 20.Tran P.T., Shroff E.H., Burns T.F., Thiyagarajan S., Das S.T., Zabuawala T., Chen J., Cho Y.J., Luong R., Tamayo P., Salih T., Aziz K., Adam S.J., Vicent S., Nielsen C.H., Withofs N., Sweet-Cordero A., Gambhir S.S., Rudin C.M., Felsher D.W. Twist1 suppresses senescence programs and thereby accelerates and maintains mutant Kras-induced lung tumorigenesis. PLoS Genet. 2012;8(5):e1002650. doi: 10.1371/journal.pgen.1002650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Firulli B.A., Redick B.A., Conway S.J., Firulli A.B. Mutations within helix I of Twist1 result in distinct limb defects and variation of DNA binding affinities. J Biol Chem. 2007;282(37):27536–27546. doi: 10.1074/jbc.M702613200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Butler J.P., Tolic-Norrelykke I.M., Fabry B., Fredberg J.J. Traction fields, moments, and strain energy that cells exert on their surroundings. Am J Physiol. 2002;282(3):C595–C605. doi: 10.1152/ajpcell.00270.2001. [DOI] [PubMed] [Google Scholar]
- 23.Wang N., Tolic-Norrelykke I.M., Chen J., Mijailovich S.M., Butler J.P., Fredberg J.J., Stamenovic D. Cell prestress. I. Stiffness and prestress are closely associated in adherent contractile cells. Am J Physiol. 2002;282(3):C606–C616. doi: 10.1152/ajpcell.00269.2001. [DOI] [PubMed] [Google Scholar]
- 24.Tran P.T., Bendapudi P.K., Lin H.J., Choi P., Koh S., Chen J., Horng G., Hughes N.P., Schwartz L.H., Miller V.A., Kawashima T., Kitamura T., Paik D., Felsher D.W. Survival and death signals can predict tumor response to therapy after oncogene inactivation. Sci Transl Med. 2011;3(103):03ra99. doi: 10.1126/scitranslmed.3002018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fiucci G., Ravid D., Reich R., Liscovitch M. Caveolin-1 inhibits anchorage-independent growth, anoikis and invasiveness in MCF-7 human breast cancer cells. Oncogene. 2002;21(15):2365–2375. doi: 10.1038/sj.onc.1205300. [DOI] [PubMed] [Google Scholar]
- 26.Zeng J., See A.P., Aziz K., Thiyagarajan S., Salih T., Gajula R.P., Armour M., Phallen J., Terezakis S., Kleinberg L., Redmond K., Hales R.K., Salvatori R., Quinones-Hinojosa A., Tran P.T., Lim M. Nelfinavir induces radiation sensitization in pituitary adenoma cells. Cancer Biol Ther. 2011;12(7):657–663. doi: 10.4161/cbt.12.7.17172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhou G., Wang S., Wang Z., Zhu X., Shu G., Liao W., Yu K., Gao P., Xi Q., Wang X., Zhang Y., Yuan L., Jiang Q. Global comparison of gene expression profiles between intramuscular and subcutaneous adipocytes of neonatal landrace pig using microarray. Meat Sci. 2010;86(2):440–450. doi: 10.1016/j.meatsci.2010.05.031. [DOI] [PubMed] [Google Scholar]
- 28.Carvalho B.S., Irizarry R.A. A framework for oligonucleotide microarray preprocessing. Bioinformatics. 2010;26(19):2363–2367. doi: 10.1093/bioinformatics/btq431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Benjamini Y., Drai D., Elmer G., Kafkafi N., Golani I. Controlling the false discovery rate in behavior genetics research. Behav Brain Res. 2001;125(1–2):279–284. doi: 10.1016/s0166-4328(01)00297-2. [DOI] [PubMed] [Google Scholar]
- 30.Firulli A.B., Conway S.J. Phosphoregulation of Twist1 provides a mechanism of cell fate control. Curr Med Chem. 2008;15(25):2641–2647. doi: 10.2174/092986708785908987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Firulli B.A., Krawchuk D., Centonze V.E., Vargesson N., Virshup D.M., Conway S.J., Cserjesi P., Laufer E., Firulli A.B. Altered Twist1 and Hand2 dimerization is associated with Saethre-Chotzen syndrome and limb abnormalities. Nat Genet. 2005;37(4):373–381. doi: 10.1038/ng1525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chaffer C.L., Weinberg R.A. A perspective on cancer cell metastasis. Science. 2011;331(6024):1559–1564. doi: 10.1126/science.1203543. [DOI] [PubMed] [Google Scholar]
- 33.Price J.E. Xenograft models in immunodeficient animals: I. Nude mice: spontaneous and experimental metastasis models. Methods Mol Med. 2001;58:205–213. doi: 10.1385/1-59259-137-X:205. [DOI] [PubMed] [Google Scholar]
- 34.Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S., Mesirov J.P. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102(43):15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Connerney J., Andreeva V., Leshem Y., Mercado M.A., Dowell K., Yang X., Lindner V., Friesel R.E., Spicer D.B. Twist1 homodimers enhance FGF responsiveness of the cranial sutures and promote suture closure. Dev Biol. 2008;318(2):323–334. doi: 10.1016/j.ydbio.2008.03.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Castanon I., Baylies M.K. A Twist in fate: evolutionary comparison of Twist structure and function. Gene. 2002;287(1–2):11–22. doi: 10.1016/s0378-1119(01)00893-9. [DOI] [PubMed] [Google Scholar]
- 37.Firulli B.A., Howard M.J., McDaid J.R., McIlreavey L., Dionne K.M., Centonze V.E., Cserjesi P., Virshup D.M., Firulli A.B. PKA, PKC, and the protein phosphatase 2A influence HAND factor function: a mechanism for tissue-specific transcriptional regulation. Mol Cell. 2003;12(5):1225–1237. doi: 10.1016/s1097-2765(03)00425-8. [DOI] [PubMed] [Google Scholar]
- 38.Lu S., Nie J., Luan Q., Feng Q., Xiao Q., Chang Z., Shan C., Hess D., Hemmings B.A., Yang Z. Phosphorylation of the Twist1-family basic helix-loop-helix transcription factors is involved in pathological cardiac remodeling. PLoS One. 2011;6(4):e19251. doi: 10.1371/journal.pone.0019251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Connerney J., Andreeva V., Leshem Y., Muentener C., Mercado M.A., Spicer D.B. Twist1 dimer selection regulates cranial suture patterning and fusion. Dev Dyn. 2006;235(5):1345–1357. doi: 10.1002/dvdy.20717. [DOI] [PubMed] [Google Scholar]
- 40.Kennelly P.J., Krebs E.G. Consensus sequences as substrate specificity determinants for protein kinases and protein phosphatases. J Biol Chem. 1991;266(24):15555–15558. [PubMed] [Google Scholar]
- 41.Perrotti D., Neviani P. Protein phosphatase 2A: a target for anticancer therapy. Lancet Oncol. 2013;14(6):e229–e238. doi: 10.1016/S1470-2045(12)70558-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bourguignon L.Y., Wong G., Earle C., Krueger K., Spevak C.C. Hyaluronan-CD44 interaction promotes c-Src-mediated twist signaling, microRNA-10b expression, and RhoA/RhoC up-regulation, leading to Rho-kinase-associated cytoskeleton activation and breast tumor cell invasion. J Biol Chem. 2010;285(47):36721–36735. doi: 10.1074/jbc.M110.162305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hong J., Zhou J., Fu J., He T., Qin J., Wang L., Liao L., Xu J. Phosphorylation of serine 68 of Twist1 by MAPKs stabilizes Twist1 protein and promotes breast cancer cell invasiveness. Cancer Res. 2011;71(11):3980–3990. doi: 10.1158/0008-5472.CAN-10-2914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Su Y.W., Xie T.X., Sano D., Myers J.N. IL-6 stabilizes Twist and enhances tumor cell motility in head and neck cancer cells through activation of casein kinase 2. PLoS One. 2011;6(4):e19412. doi: 10.1371/journal.pone.0019412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Xue G., Restuccia D.F., Lan Q., Hynx D., Dirnhofer S., Hess D., Ruegg C., Hemmings B.A. Akt/PKB-mediated phosphorylation of Twist1 promotes tumor metastasis via mediating cross-talk between PI3K/Akt and TGF-β signaling axes. Cancer Discov. 2012;2(3):248–259. doi: 10.1158/2159-8290.CD-11-0270. [DOI] [PubMed] [Google Scholar]
- 46.Vichalkovski A., Gresko E., Hess D., Restuccia D.F., Hemmings B.A. PKB/AKT phosphorylation of the transcription factor Twist-1 at Ser42 inhibits p53 activity in response to DNA damage. Oncogene. 2010;29(24):3554–3565. doi: 10.1038/onc.2010.115. [DOI] [PubMed] [Google Scholar]
- 47.Burns T.F., Dobromilskaya I., Murphy S.C., Gajula R.P., Thiyagarajan S., Chatley S.N., Aziz K., Cho Y.J., Tran P.T., Rudin C.M. Inhibition of TWIST1 leads to activation of oncogene-induced senescence in oncogene-driven non-small cell lung cancer. Mol Cancer Res. 2013;11(4):329–338. doi: 10.1158/1541-7786.MCR-12-0456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kwok W.K., Ling M.T., Yuen H.F., Wong Y.C., Wang X. Role of p14ARF in TWIST-mediated senescence in prostate epithelial cells. Carcinogenesis. 2007;28(12):2467–2475. doi: 10.1093/carcin/bgm185. [DOI] [PubMed] [Google Scholar]
- 49.Ansieau S., Bastid J., Doreau A., Morel A.P., Bouchet B.P., Thomas C., Fauvet F., Puisieux I., Doglioni C., Piccinin S., Maestro R., Voeltzel T., Selmi A., Valsesia-Wittmann S., Caron de Fromentel C., Puisieux A. Induction of EMT by twist proteins as a collateral effect of tumor-promoting inactivation of premature senescence. Cancer Cell. 2008;14(1):79–89. doi: 10.1016/j.ccr.2008.06.005. [DOI] [PubMed] [Google Scholar]
- 50.Pan D., Fujimoto M., Lopes A., Wang Y.X. Twist-1 is a PPARdelta-inducible, negative-feedback regulator of PGC-1alpha in brown fat metabolism. Cell. 2009;137(1):73–86. doi: 10.1016/j.cell.2009.01.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Verdine G.L., Walensky L.D. The challenge of drugging undruggable targets in cancer: lessons learned from targeting BCL-2 family members. Clin Cancer Res. 2007;13(24):7264–7270. doi: 10.1158/1078-0432.CCR-07-2184. [DOI] [PubMed] [Google Scholar]
- 52.Bouard C., Terreux R., Hope J., Chemelle J.A., Puisieux A., Ansieau S., Payen L. Interhelical loops within the bHLH domain are determinant in maintaining TWIST1-DNA complexes. J Biomol Struct Dyn. 2014;32(2):226–241. doi: 10.1080/07391102.2012.762722. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Phosphorylation of the TWIST1 TQS motif is not required for TWIST1-induced resistance to anoikis. Cells were grown adherent or in suspension using ultra-low attachment dishes. The amount of apoptotic cell death or anoikis for the ultra-low attachment conditions was quantified by Annexin V–Alexa Fluor 488 and PI staining followed by flow cytometric analysis. Representative dot plots of (A) Myc-CaP and (B) PC3 Twist1 isogenic cell lines are shown. Percentage of cells in quadrants II (early apoptotic) and III (late apoptotic) that constitute apoptotic fractions are in bold. Percent apoptosis was calculated by normalizing total apoptotic fraction in ultra-low attachment conditions to that of adherent cells and plotted as bar graph ± SEM (Figure 4).
Figure S2. TWIST1 overexpression confers radioresistance to prostate cancer cells, which is attenuated by phospho-null mutations of the TWIST1 TQS motif. Briefly, the clonogenic survival assay was performed (see Materials and Methods section) by plating radiated single cells, 200 to 5000, in 100-mm dishes and stained 2 to 3 weeks later with crystal violet, and distinct colonies (defined as > 50 cells) were scored. The survival fraction at 4 (PC3) or 5 Gy (Myc-CaP) is calculated by total number of colonies normalized to the plating efficiency. Survival fraction is plotted for (A) Myc-CaP (n = 3, five replicates per experiment) and (B) PC3 isogenic cell lines (n = 3, five replicates per experiment). Bars represent column mean; error bars ± SEM; significance by Mann-Whitney test: *P < .05, **P < .01, and ***P < .001.
Figure S3. The Twist1-AQA mutation is deficient for TWIST1-induced soft agar anchorage-independent growth of 22Rv1 prostate cancer cells. (A) Western blot analysis of 22Rv1 cells stably overexpressing similar levels of TWIST1 and TWIST1 phospho-mutants. β-Actin was used as internal control. (B) The representative phase contrast images of soft agar colonies from 22RV1 isogenic cells taken at 4 × objective. (C) Colonies containing more than 50 cells are scored in five random fields from each well (n = 6), and the percentage was determined from the number of soft agar colonies normalized with the total number of cells. Bars represent column mean; error bars ± SEM; significance is by Mann-Whitney test: **P < .01.
Figure S4. Tethered T-E overexpressing cells phenocopy Twist1-DQD mutant overexpressing cells for pro-metastatic behaviors in vitro. (A) Representative phase contrast images of Myc-CaP cells overexpressing TWIST1 and tethered versions of TWIST1 directly following and 24 hours after wound scratch. (B) Representative dot plots of Myc-CaP cells overexpressing TWIST1 and tethered versions of TWIST1 for anoikis levels. See Figure S1 for details. (C) Representative phase contrast images of Myc-CaP cells overexpressing TWIST1 and tethered versions of TWIST1 for anchorage-independent growth in soft agar (see Figure S3 for details).
Figure S5. BEZ235 inhibition of PI3K-mTOR kinases does not change Myc-CaP cell viability. Cell viability assays using CellTiter-Blue as directed by the manufacturer reveal that BEZ235 treatment for 48 hours does not affect cell viability of Myc-CaP cells expressing (A and B) TWIST1, (A) Twist1 phospho-mutant or (B) Twist1 tethered isoforms.
Figure S6. BEZ235 inhibition of PI3K-mTOR kinases prevents TWIST1-induced prostate cancer cell migration. (A) Representative phase contrast images of Myc-CaP cells overexpressing TWIST1 and TWIST1 tethered isoforms that have been treated with 10 nM BEZ235 for 24 hours, then a wound scratch was performed and then 24 hours after wound scratch. (B) Relative wound closure is calculated by the remaining wound area normalized to the initial wound area [n> 3 experiments with three fields per experiment by ImageJ software (NIH)] and showed that BEZ235 treatment only inhibited the migration of Myc-CaP cells overexpressing TWIST1. (C) The relative inhibitory effect of BEZ235 on cell migration was determined by normalizing the migratory potential of untreated cells to that of 10 nM BEZ235-treated cells. Mann-Whitney test: **P < .01.